

Abstract
Platelets bind to exposed vascular matrix at a wound site through a highly specialized surface receptor, glycoprotein (GP) Ib-IX-V complex, which recognizes von Willebrand factor (VWF) in the matrix. GPIb-IX-V is a catch bond for it becomes more stable as force is applied to it. After attaching to the wound site, platelets generate cytoskeletal forces to compact and reinforce the hemostatic plug. Here, we evaluated the role of the GPIb-IX-V complex in the transmission of cytoskeletal forces. We used arrays of flexible, silicone nanoposts to measure the contractility of individual platelets on VWF. We found that a significant proportion of cytoskeletal forces were transmitted to VWF through GPIb-IX-V, an unexpected finding given the widely held notion that platelet forces are transmitted exclusively through its integrins. In particular, we found that the interaction between GPIbα and the A1 domain of VWF mediates this force transmission. We also demonstrate that the binding interaction between GPIbα and filamin A is involved in force transmission. Furthermore, our studies suggest that cytoskeletal forces acting through GPIbα are involved in maintaining platelet adhesion when external forces are absent. Thus, the GPIb-IX-V/VWF bond is able to transmit force, and uses this force to strengthen the bond through a catch-bond mechanism. This finding expands our understanding of how platelets attach to sites of vascular injury, describing a new, to the best of our knowledge, mechanism in which the catch bonds of GPIb-IX-V/VWF can be supported by internal forces produced by cytoskeletal tension.
Introduction
Receptors involved in the adhesion of platelets are essential for the cessation of bleeding (hemostasis). The interaction between glycoprotein (GP) Ib-IX-V complex and von Willebrand factor (VWF) facilitates the arrest of circulating platelets at sites of vascular injury (1, 2). Additional platelets attach on top of surface-bound platelets to form a platelet-rich hemostatic plug (3, 4). During adhesion and aggregation, platelets use integrins such as αIIbβ3 and α2β1 to mediate interactions with other platelets and with the injured vessel wall (5, 6). Integrins are able to transmit contractile forces that arise from cytoskeletal tension generated by actin and myosin filaments. These cytoskeletal forces help to improve the adhesion of platelets to the injured vascular site (7) and shrink the gaps between platelets in the hemostatic plug (8). Individuals with reduced expression of integrins are predisposed to bleeding because of their inability to form stable hemostatic plugs (9, 10). Nevertheless, aggregates of platelets can still form, albeit more slowly and less efficiently than in normal individuals (11). Thus, there appears to be an alternative mechanism for stabilizing the adhesion of platelets at a wound site.
GPIb-IX-V complex is an obvious candidate to stabilize the adhesion and aggregation of platelets because it is abundant on the surface of a platelet and it associates with actin filaments in the cytoskeleton (2, 12, 13). GPIb-IX-V is composed of four polypeptides—GPIbα, GPIbβ, GPIX, and GPV—in a 2:4:2:1 stoichiometry (14). GPIbα contains a binding site for the A1 domain of VWF in its extracellular domain and another binding site for filamin A in its cytoplasmic domain. Filamin A mediates the association of GPIb-IX-V with actin filaments (15), which we propose could provide a means for transmitting cytoskeletal forces to VWF.
The bond between GPIbα and the A1 domain is regarded as a catch bond, i.e., the lifetime of the bond increases as force is applied to it (16). These specialized bonds allow platelets to remain attached to surfaces and withstand the effect of strong detachment forces acting on them due to high shear flow. Therefore, we reasoned that in a manner similar to external hydrodynamic forces, a platelet’s catch bonds could be strengthened through internal cytoskeletal forces transmitted through the GPIb-IX-V/A1 domain bond.
Here, we used arrays of flexible nanoposts to measure the cytoskeletal forces of platelets transmitted through GPIb-IX-V. We determined the contribution of GPIb-IX-V versus αIIbβ3 by blocking the receptor on the platelet surface or its binding site on VWF. We confirmed our results using Chinese hamster ovary cells expressing a recombinant GPIb-IX complex. In addition, we found that the binding site on GPIbα for filamin A regulates the transmission of cytoskeletal forces and these forces are required for the adhesion of platelets to A1 domain.
Materials and Methods
Nanopost array fabrication and VWF coating
Nanopost arrays were replicated in polydimethylsiloxane (PDMS) from a silicon master, which was manufactured by electron-beam lithography and inductively coupled plasma etching (Fig. S1). Details of the fabrication are provided in Supporting Methods in the Supporting Material. To coat the tips of the nanoposts, we placed a 300 μL droplet of VWF solution (10 μg/mL in phosphate buffered saline (PBS)) on the tips of nanopost arrays and let the protein adsorb for 2 h. VWF was purified from cryoprecipitate as previously described (17). We observed that VWF from the droplets formed fiber-like strands between the tips of the nanoposts (Fig. S2).
Nanoposts were then treated with Alexa Fluor 594-conjugated bovine serum albumin (BSA) (20 μg/mL in PBS; Sigma, Bloomington, MN) for 1 h to facilitate the fluorescent visualization of the nanoposts and block the adsorption of other proteins. For additional blocking, we submerged the nanoposts in a solution of 0.2% Pluronic F-127 (BASF Ludwigshafen, Germany) in PBS for 1 h. The quality of the blocking layer was evaluated by incubating platelets with nanoposts treated with Alexa Fluor 594-conjugated BSA and Pluronic F-127, but with no VWF coating. Platelets did not attach to these coatings.
Blood collection and platelet isolation
Blood from healthy donors was collected into vacuum tubes containing acid citrate dextrose solution (BD Medical, North Bend, WA) and used within 1 h of collection. Platelets were isolated from the blood by centrifugation, washed in a CGS buffer (120 mM NaCl, 13 mM sodium citrate, and 30 mM glucose at pH 7) and resuspended to the original volume of platelet-rich plasma in Tyrode’s buffer (10 mM HEPES, 138 mM NaCl, 5.5 mM glucose, 12 mM NaHCO3, 0.36 mM Na2HPO4, 2.9 mM KCl, 0.4 mM MgCl2, and 0.8 mM CaCl2, pH 7.5). All reagents for CGS and Tyrode’s buffer were from Sigma (Bloomington, MN).
Antibody treatment
We incubated platelets in Tyrode’s buffer with antibody AK2 (5 μg/mL; Abcam, Cambridge, MA), 7E3 (20 μg/mL; courtesy of Barry Coller, Rockefeller University), or both AK2 and 7E3 for 20 min with gentle rocking. We assessed whether the antibodies activated the platelets by flow cytometry (Fig. S5). The forward and side scatter profiles of untreated and antibody-treated platelets were similar, indicating that the antibodies did not induce platelet activation.
Platelets were added to dishes containing nanoposts submerged in Tyrode’s buffer. Platelets adhered to the VWF coating on the tips of the nanoposts. After 10 min of incubation, we gently washed the nanoposts in PBS to remove unattached platelets and then incubated the nanoposts in Tyrode’s buffer for an additional 30 min to allow the platelets to spread and contract. We fixed the platelets on the nanoposts with 4% paraformaldehyde, permeabilized them with 0.1% Triton X-100 (Sigma), and then stained them with Alexa Fluor 488-conjugated phalloidin (Life Technologies, Carlsbad, CA) for 1 h. After staining, the samples were mounted on glass slides with Fluoromount-G (VWR International, Radnor, PA) for confocal microscopy.
Recombinant GPIbα-300 G233V (Ibα300gof)
A partial cDNA encoding amino acids 1–300 of GPIbα that contains a gain-of-function G233V mutation (18) was amplified by polymerase chain reaction and cloned into the NheI and XhoI sites of pBIG-4f (described in the Supplemental Methods section). This vector was transfected into Chinese hamster ovary (CHO) Tet-On cells (Clontech, Mountain View, CA) along with pUC19-puromycin to enable selection of stably expressing cell lines. Cells secreting the recombinant GPIbα-300 G233V peptide (Ibα300gof) were incubated for 48 h in serum-free medium (EX-CELL 302; Sigma) containing 10 μM biotin (Sigma) and 2 μg/mL doxycycline (Sigma). To harvest Ibα300gof, the medium was concentrated and desalted to remove free biotin using a PD-10 column (Sigma). For experiments, the nanoposts were coated with VWF as described previously and then incubated with Ibα300gof (170 μg/mL) for 20 min to block the A1 domain of VWF and inhibit platelet adhesion.
Recombinant A1 domain of VWF
A cDNA encoding amino acids 1260–1471 of VWF that comprise its A1 domain was polymerase chain reaction amplified and cloned into the PstI and XhoI sites of pBIG-4e (see Supporting Methods in the Supporting Material). CHO Tet-On cells (Clontech, Mountain View, CA) were stably transfected with pBIG-4e-A1 and pUC19-puromycin to enable stable selection. Recombinant A1 domain was produced by incubating the cells in serum-free medium (EX-CELL 302; Sigma) containing 10 μM biotin and 2 μg/mL doxycycline for 48 h, and then concentrating and desalting the medium using a PD-10 column to remove free biotin. To determine protein concentration, serial dilutions of the recombinant protein and biotinylated BSA standards were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis, detected using streptavidin-horseradish peroxidase, and the protein concentration determined by densitometry assuming one biotin per molecule (Fig. S6).
A1 domain coating
We applied the biotinylated recombinant A1 domain to the tips of the nanoposts by coating the tips with streptavidin (10 μg/mL in PBS; Calbiochem, Darmstadt, Germany) using microcontact printing, as described previously (19). After treating the nanoposts with Alexa Fluor 594-conjugated BSA and 0.2% Pluronic F-127, the nanoposts were submerged in a solution containing recombinant A1 domain (52 μg/mL) for 20 min. Experiments with platelets seeded onto nanoposts coated with recombinant A1 domain were conducted in an identical manner to the experiments with full-length VWF coatings described previously. We used nanoposts coated with only streptavidin or coated with A1 domain, but blocked with Ibα300gof as negative controls. The results of negative control experiments indicate that platelets bind specificity to the A1 coating on the nanoposts.
Micropost arrays fabrication and coating
Micropost arrays were replicated in PDMS from a master, which was manufactured by photolithography of SU-8 on a silicon wafer. Details of the fabrication are provided in Supporting Methods in the Supporting Material. A microcontact printing technique was used to transfer VWF (10 μg/mL in PBS) onto the tips of the microposts as described previously (19). We then treated the micropost arrays with Alexa Fluor 594-conjugated BSA and 0.2% Pluronic F-127 as described previously for nanoposts. To capture the biotinylated A1 domain at the tips of the microposts, we first stamped streptavidin (10 μg/mL in PBS) onto the tips. After treating the microposts with Alexa Fluor 594-conjugated BSA and 0.2% Pluronic F-127, we submerged them in a solution containing recombinant A1 domain (52 μg/mL) for 20 min.
CHO cell lines
CHOαβIX, CHOαΔ534βIX, and CHOαFW-AAβIX cells were produced as described (20, 21, 22). Briefly, wild-type and mutant GPIbα constructs and pREP4 (Invitrogen, Carlsbad, CA), which carries a hygromycin-resistant marker, were cotransfected into CHO cells stably expressing GPIbβ and GPIX (CHOβIX). We evaluated the expression of GPIX and GPIbα subunits by flow cytometry after staining the cells with anti-CD42a and anti-CD42b monoclonal antibodies (BD Biosciences, Franklin Lakes, NJ), respectively (Fig. S7). As a control, the two cell lines were stained with mouse IgG (BD Biosciences). The cell lines were maintained in Dulbecco’s modified Eagle medium (Sigma), 10% fetal bovine serum (Sigma), L-glutamine (2 mM, Sigma), 1% penicillin/streptomycin (Sigma), G418 (400 μg/mL, Sigma), methotrexate (80 μM, Sigma), and hygromycin (100 μg/mL, Sigma).
CHO cell experiments
CHOαβIX, CHOβIX, CHOαΔ534βIX, and CHOαFW-AAβIX cells were seeded onto the microposts at a concentration of 5 × 104/mL in serum-free medium and incubated for 16 h at 37°C in 5% CO2 in the presence of ristocetin (1.5 mg/mL, American Biochemical and Pharmaceutical, Marlton, NJ). The cells were washed with PBS, fixed, and permeabilized for 30 min in PBS containing 4% paraformaldehyde and 0.1% Triton X-100, and stained for 30 min with FITC-phalloidin (1:200 in PBS). In the blebbistatin experiments, CHOαβIX cells on the microposts were incubated with blebbistatin (10 μM; Sigma) overnight at 37°C. CHOαβIX did not adhere to micropost arrays that were coated with Alexa Fluor 594-conjugated BSA and 0.2% Pluronic F-127. In addition, CHOβIX cells, lacking GPIbα, failed to adhere to microposts coated with A1 domain or VWF. Together, these results indicated a specific binding of GPIbα in CHOαβIX to the A1 domain.
Platelet wash-off experiments
Substrates with thin films of PDMS were incubated with A1 domain for 2 h and then washed with PBS and blocked with 2% BSA in PBS for 1 h at room temperature. These substrates were then incubated with 0.2% Pluronic F-127 in PBS for 1 h. Washed platelets were treated with blebbistatin (10 μM) for 20 min or left untreated and then seeded at low density onto the substrates, ensuring that they did not aggregate. Substrates of PDMS coated with only 2% BSA and Pluronic F-127 and not A1 served as a control for nonspecific adhesion. Platelets did not attach to this coating. 40 min after seeding the platelets, unbound or loosely bound platelets were removed by washing the substrates with PBS. Samples were then fixed, permeabilized, and stained as previously described. Platelet adhesion was visualized using a 60× oil immersion lens on an Olympus (Tokyo, Japan) microscope. Images were collected at 10 arbitrary locations on the substrate. The number of platelets per each 60× field of view was counted for each substrate.
Statistical analysis
R software was used for the statistical analysis. A linear regression model was developed to assess differences in platelet experiments. This model accounts for donor-to-donor variability as a random factor. The same model was used to assess the statistical differences in all CHO cell experiments. Statistical significance was defined as a p-value < 0.05. Error bars shown in the figures represent the standard error of the mean.
Results
To assess whether cytoskeletal forces in platelets can be transmitted to the external environment via the GPIb-IX-V complex, we measured the contractility of individual platelets using arrays of flexible nanoposts coated with GPIb-IX-V ligands. Each nanopost in an array acts as a cantilever beam that bends in proportion to the force applied at its tip (Fig. S3). Platelets attached to the tips of the nanoposts, spread over multiple nanoposts, and deflected them using cytoskeletal forces (Fig. 1 A). We measured the deflections of the nanoposts from confocal microscopy images (Fig. 1, B and C). The force on each nanopost was calculated according to Hooke’s law , where k is the bending stiffness of the nanoposts in the array (15.7 nN/μm). We noted that platelets on nanoposts (Fig. 2 A) had morphologies that were similar to those of platelets on flat surfaces coated with VWF (Fig. S4 A), which indicated that the topology of the array did not impede the ability of the platelets to change shape or spread.
Figure 1.

Nanopost arrays measure single platelet forces. (A) Scanning electron microscope image of a fixed and dehydrated platelet on an array of silicone nanoposts. The tips of the nanoposts were coated with VWF by letting the protein adsorb from a droplet of solution placed on the array. VWF strands formed between the nanoposts due to the droplet adsorption method (Fig. S4). Platelets in Tyrode’s buffer were seeded onto the arrays, allowed to adhere onto the tips, and then fixed. Platelets that attached to the nanoposts were observed to generate forces that cause the nanoposts to deflect. It should be noted that the deflections of the nanoposts were more dramatic when observed using electron microscopy than with confocal microscope due to the dehydration steps needed to prepare the samples for electron microscopy (37). (B) Side view and (C) top view of an individual platelet on an array of nanoposts taken with confocal microscopy (green: actin, red: post). The force vectors on each nanopost were calculated according to Hooke’s law based upon measurements of their deflections. Scale bars represent 2 μm, force scale bars represent 3 nN. To see this figure in color, go online.
Figure 2.

Integrin αIIbβ3 and GPIb-IX-V can transmit cytoskeletal forces in platelets. Representative image of individual platelets that received (A) no treatment (control), (B) AK2 antibody to block the GPIbα, (C) 7E3 antibody to block integrin αIIbβ3, or (D) both blocking antibodies together. (E) Platelet spread areas are shown relative to the control condition in each experiment. (F) Platelet contractile forces were normalized by the average force of the control condition in each experiment (N = 5 replicate experiments). Scale bars represent 2 μm, force scale bars represent 3 nN, and the error bars represent the standard error of the mean. Asterisk denotes p-value <0.05. To see this figure in color, go online.
VWF is a multidomain protein that contains binding sites for both the GPIb-IX-V complex within its A1 domain and integrin αIIbβ3 within its C1 domain (23, 24). To isolate the effect of each receptor, we treated platelets with monoclonal antibodies: AK2 to block GPIbα, 7E3 to block αIIbβ3, and AK2 and 7E3 to block both receptors. For each experimental cohort, we analyzed platelet morphology, spread area, and contractile forces (Table S1). Contractile forces on nanoposts coated with VWF were higher than forces reported previously for single platelets on fibrinogen coatings (25, 26). For each donor, we normalized the data with respect to the results for untreated platelets to account for donor-to-donor variability in platelet function. Inhibition of GPIbα with AK2 did not affect platelet morphology (Fig. 2, A and B), but it did significantly decrease the spread area of the platelets (Fig. 2 E). Treatment of the platelets with AK2 reduced the transmission of cytoskeletal forces by ∼20% (Fig. 2 F). In contrast, inhibition of αIIbβ3 with 7E3 caused platelets to develop a dense actin core, numerous filopodia, and defective lamellipodia (Fig. 2 C). This morphology was expected given that lamellipodial formation in platelets requires the engagement of αIIbβ3 with its extracellular ligands (27). Platelets treated with 7E3 displayed a significantly reduced spread area (Fig. 2 E) and cytoskeletal force transmission (Fig. 2 F) than untreated platelets or AK2-treated platelets. However, the contractile forces were not abolished completely with αIIbβ3 inhibition since platelets still generated ∼50% of the force produced by untreated platelets. When platelets were treated with both AK2 and 7E3, many of the platelets failed to attach to the nanoposts. The few platelets that did attach were rounded (Fig. 2 D), poorly spread (Fig. 2 E), and transmitted less force than untreated platelets, AK2-treated platelets, or 7E3-treated platelets (Fig. 2 F). These results suggest that in addition to αIIbβ3, GPIb-IX-V supports a portion of the cytoskeletal forces transmitted to VWF.
To occupy the A1 domain of VWF so that GPIb-IX-V binding is inhibited, we used an N-terminal GPIbα polypeptide (Ibα300gof) that contains a gain-of-function mutation (G233V), which has been found previously to allow GPIbα to bind VWF spontaneously (18). Blocking the A1 domain of VWF with Ibα300gof did not affect the morphology of the platelets (Fig. 3, A and B), but it did reduce their spread area (Fig. 3 D; Table S2) and ability to transmit cytoskeletal forces (Fig. 3 E; Table S2).
Figure 3.

GPIbα-A1 bond transmits platelet cytoskeletal forces. Representative image of platelets on nanopost arrays coated with (A) VWF (control), (B) VWF and recombinant peptide for GPIbα (Ibα300gof), or (C) recombinant A1 domain (green: actin, red: post). (D) Platelet spread areas are shown relative to the control condition in each experiment. (E) Platelet contractile forces were normalized by the average force of the control condition in each experiment (N = 5 replicate experiments). Scale bars represent 2 μm, force scale bars represent 3 nN, and the error bars represent the standard error of the mean. Asterisk denotes p-value <0.05. To see this figure in color, go online.
As another approach to measuring cytoskeletal forces associated with GPIb-IX-V, we immobilized recombinant A1 domain of VWF onto the tips of the nanoposts using biotin and streptavidin. Platelets that adhered onto recombinant A1 domain had morphologies (Fig. 3 C) that were similar to platelets on PDMS surfaces coated with recombinant A1 domain in a similar manner (Fig. 4 B). The morphology of platelets on recombinant A1 domain resembled that of platelets treated with 7E3 on VWF (Fig. 2 C), which indicates that interactions between αIIbβ3 and VWF were sufficiently inhibited. Platelets spread less on recombinant A1 domain than on multimeric VWF (Fig. 3 D; Table S2). The contractile forces of platelets on recombinant A1 domain were 15% of those obtained on full-length VWF (Fig. 3 E; Table S2), which further shows that forces are transmitted through the GPIb-IX-V complex independent of integrin αIIbβ3.
Figure 4.

CHO cells confirm the role of GPIbα in force transmission. Representative images of CHOαβIX cells on microposts coated with (A) VWF, (B) recombinant A1 domain, or (C) CHOαΔ534βIX cell, or (D) CHOαFW-AAβIX cell on VWF-coated microposts (green: actin, red: post, blue: nucleus). (E) Spread area and (F) contractile forces for CHOαβIX and CHOβIX cells (N = 5 replicate experiments). (G) Spread area and (H) contractile forces of CHOαβIX cells treated with or without blebbistatin (N = 3 replicate experiments). (I) Spread area and (J) contractile forces for CHOαβIX, CHOαΔ534βIX, or CHOαFW-AAβIX cells (N = 4 replicate experiments). Scale bars represent 6 μm, force scale bars represent 30 nN, and the error bars represent the standard error of the mean. Asterisk denotes p-value <0.05. To see this figure in color, go online.
We used CHO cells expressing GPIbα, GPIbβ, and GPIX (CHOαβIX cells) to determine if GPIbα expression in another context is able to transmit force to VWF. There is no native adhesion to VWF in CHO cells, but expression of GPIbα, GPIbβ, and GPIX in CHO cells has been found to enable them to bind to VWF like platelets (20). Because CHO cells are much larger than platelets, we used arrays of PDMS microposts, which are larger and more widely spaced than nanoposts (28, 29). CHOαβIX cells were able to adhere and spread on microposts that were coated with either VWF (Fig. 4 A) or recombinant A1 domain (Fig. 4 B). CHOαβIX cells did not spread to the same degree on the two coatings (Fig. 4 E), but generated contractile forces of similar magnitude (Fig. 4 F), indicating that GPIbα transmits forces to VWF. CHOβIX cells, which express only GPIbβ and GPIX and not GPIbα, did not adhere to the microposts, confirming that GPIbα is the essential GPIb-IX-V subunit for adhesion and force transmission on VWF. We tested whether the contractile forces produced by CHOαβIX cells were generated by myosin activity by treating the cells with blebbistatin, an inhibitor of myosin’s ATPase activity (30). Blebbistatin increased the spreading of CHOαβIX cells (Fig. 4 G) and significantly lowered the forces they produced on VWF-coated microposts (Fig. 4 H). Together, these results indicate that myosin-dependent forces imparted onto the A1 domain of VWF act through the GPIbα subunit of GPIb-IX-V complex.
We tested whether the actin-binding protein, filamin A, serves as an intermediary in transmitting cytoskeletal forces through GPIb-IX-V to VWF using two different cell lines expressing GPIbα mutants. In one cell line (CHOαΔ534βIX), we expressed a mutant GPIbα truncated after residue 534, which lacks the binding site for filamin A (21). In the other cell line (CHOαFW-AAβIX), we expressed a double mutant of GPIb with F568A and W570A, which had been found previously to be essential for filamin A binding (22). Both mutant cell lines adhered to the microposts coated with VWF (Fig. 4, C and D), but in comparison to CHOαβIX cells, CHOαΔ534βIX cells spread poorly and CHOαFW-AAβIX spread to a greater extent (Fig. 4 I). Both mutant cell lines exerted contractile forces that were significantly lower than the forces produced by CHOαβIX cells and were statistically equivalent to each other (Fig. 4 J). These studies confirmed that the binding site on GPIbα for filamin A is needed for the transmission of cytoskeletal forces to the GPIb-IX-V complex.
The catch bond interaction between GPIbα and the VWF A1 domain is known to play an important role in the attachment of platelets to sites of vascular injury (31). We evaluated the possibility that GPIbα-A1 domain catch bonds serve another purpose: to allow cytoskeletal forces to strengthen platelet adhesion to VWF in the absence of external forces. Consistent with this possibility, we found that in the absence of flow, inhibition of myosin with blebbistatin significantly reduced platelet adhesion to surfaces coated with recombinant A1 domain (Fig. 5; Table S3). This evidence might be suggesting that internal forces from actin-myosin interactions enhance the bond between GPIbα and A1 domain, which therefore leads to an overall stronger adhesion. Thus, cytoskeletal forces generated inside platelets and transmitted through GPIb-IX-V might support the adhesion of platelets to VWF, particularly under conditions where external forces are absent.
Figure 5.
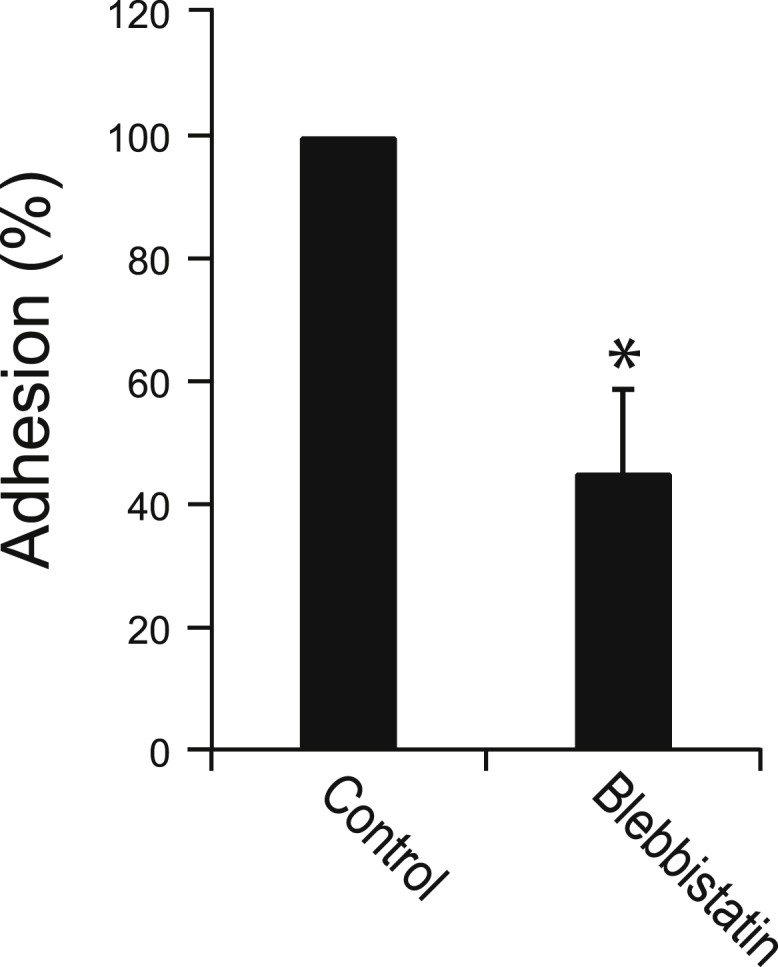
Cytoskeletal forces are needed for platelet adhesion on A1 domain. Platelets adhered onto PDMS substrates coated with recombinant A1 domain had weaker adhesion strength when treated with blebbistatin as compared to untreated platelets (N = 4 replicate experiments). Asterisk denotes p-value <0.05.
Discussion
Platelets rely on catch bonds formed between GPIb-IX-V and VWF to maintain their adhesion at a wound site (16). External hemodynamic forces are able to engage these catch bonds and thereby arrest platelets from circulating in the blood flow (16, 31). Here, we report that GPIb-IX-V transmits a significant portion of cytoskeletal forces on VWF. Furthermore, the internal forces acting through GPIb-IX-V might be reinforcing catch bonds from the inside of the platelet, which may lead to stronger temporal adhesions. Force transmission through GPIb-IX-V may become increasingly important after a platelet has attached to a surface, where external forces acting on it become reduced due to flattening of a platelet as it spreads on a surface. This finding may be relevant for patients with type 2B von Willebrand disease, where mutations in VWF prevent the formation of a catch bond with GPIb-IX-V (31). In these patients, platelets can bind rapidly to VWF, but are not able to maintain their adhesion at a wound site.
It is widely held that platelets transmit force to VWF via their αIIbβ3 receptors, a process that underlies clot retraction, among other phenomena (32). The GPIb-IX-V complex is involved in the initial capture of platelets on VWF, which allows for αIIbβ3 activation and leads to aggregation (33). Our findings indicate that the GPIb-IX-V complex also contributes in the transmission of cytoskeletal forces, and does so in a manner that is independent of αIIbβ3. Previous force assays for platelets have overlooked the role of GPIb-IX-V complex in force transmission (25, 26, 32, 34, 35). This omission may be because αIIbβ3 and fibrin dominate the process of clot retraction. In addition, these systems may not be able to resolve the contribution of each receptor individually. To address this knowledge gap, we used nanoposts with controlled surface coatings and found that the force transmission for αIIbβ3 versus GPIb-IX-V is 50% versus 20%, a 2.5/1 ratio. This ratio is close to the 3:1 ratio that has been reported for the number of copies for each receptor on the surface of a platelet (4). This finding indicates that a GPIb-IX-V/VWF bond could be transmitting as much force as an αIIbβ3/VWF bond.
Our work with CHO cells demonstrates that GPIb-IX-V complex is capable of force transmission in a context other than platelets. We show that CHOαβIX cells were able to produce contractile forces on either the VWF or A1 domain that were statistically equivalent. Together with the observation that CHOβIX cells are unable to bind to either coating, these studies agree with previous observations that the binding of GPIb-IX-V complex to VWF is through the GPIbα subunit specifically. These results also indicate that GPIbα is essential for force transmission through GPIb-IX-V.
The mutant forms of GPIbα in CHOαΔ534βIX and CHOαFW-AAβIX cells have a lower capacity for filamin A binding (21, 22). We observed that these cells exerted contractile forces on VWF that were significantly lower than those produced by CHOαβIX, implicating a role for filamin A in force transmission through GPIb-IX-V. However, we had expected that neither CHOαΔ534βIX nor CHOαFW-AAβIX cells would be able to transmit cytoskeletal forces, because filamin A is the only protein known to link the GPIb-IX-V complex to the actin cytoskeleton (15). Consequently, the transmission of forces by CHOαΔ534βIX and CHOαFW-AAβIX cells indicates that filamin A may bind to other regions of GPIbα, which has been suggested previously (22), or that another mechanism exists to link the GPIb-IX-V complex to the cytoskeleton.
In summary, we have described a new, to the best of our knowledge, role for the GPIb-IX-V complex in transmission of cytoskeletal forces from the interior of the platelet to its external environment. GPIb-IX-V has many well-described functions, such as the initial capture of the platelets on VWF at the vessel wall through catch bond interactions (1, 16), signaling pathways that initiate with GPIb-IX-V engagement to VWF that lead to platelet spreading and αIIbβ3 activation, and aggregation of multiple platelets in a thrombus through multivalent ligands like VWF (4, 36). It is as yet not clear how force transmission through GPIb-IX-V affects these functions of the receptor, but it is possible that it is a mechanism for supporting the GPIb-IX-V/VWF catch bonds through internal force generation during the formation of a thrombus.
Author Contributions
S.F., A.D.M., and W.W.T. performed the experiments; A.D.M. made the CHOαFW-AAβIX cell lines; J.D.K. constructed the pBIG-4e and pBIG-4f vectors; S.F., W.W.T., A.M.F., and S.R. analyzed the data; S.F., A.D.M., J.A.L., and N.J.S. designed the research; and S.F., J.A.L., and N.J.S. wrote the article.
Acknowledgments
The authors declare that N. J. Sniadecki is a cofounder of, and N. J. Sniadecki and S. Feghhi have equity in, Stasys Medical Corporation.
We thank Kristopher Lawler and Richard Bojko at the University of Washington Nanofabrication Facility for fabricating the silicon master, Junmei Chen, Assaf Oron, and Wendy Thomas for comments, Nikita Taparia for her assistance, and Barry Coller for providing the 7E3 antibody.
This work was supported in part by grants to N.J.S. from the National Institutes of Health (NIH) (HL097284), National Science Foundation (NSF) CAREER award program (CMMI-0846780), Defense Advanced Research Projects Agency (DARPA) Young Faculty Award program (N66001-11-1-4129), and the Coulter Foundation’s Translational Research Award program; a fellowship to S.F. by an NIH T32 program award (EB001650); and funding from NIH (HL091153) to J.A.L. and from the Bloodworks Research Institute to A.D.M. and J.A.L.
Editor: Margaret Gardel.
Footnotes
Supporting Methods, seven figures, and three tables are available at http://www.biophysj.org/biophysj/supplemental/S0006-3495(16)30469-6.
Supporting Material
References
- 1.Chen J., López J.A. Interactions of platelets with subendothelium and endothelium. Microcirculation. 2005;12:235–246. doi: 10.1080/10739680590925484. [DOI] [PubMed] [Google Scholar]
- 2.Andrews R.K., Gardiner E.E., Berndt M.C. Glycoprotein Ib-IX-V. Int. J. Biochem. Cell Biol. 2003;35:1170–1174. doi: 10.1016/s1357-2725(02)00280-7. [DOI] [PubMed] [Google Scholar]
- 3.Stalker T.J., Traxler E.A., Brass L.F. Hierarchical organization in the hemostatic response and its relationship to the platelet-signaling network. Blood. 2013;121:1875–1885. doi: 10.1182/blood-2012-09-457739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Michelson A.D. Academic Press/Elsevier, Amsterdam; Boston: 2007. Platelets. [Google Scholar]
- 5.Moroi M., Jung S.M. Integrin-mediated platelet adhesion. Front. Biosci. 1998;3:d719–d728. doi: 10.2741/a315. [DOI] [PubMed] [Google Scholar]
- 6.Savage B., Almus-Jacobs F., Ruggeri Z.M. Specific synergy of multiple substrate-receptor interactions in platelet thrombus formation under flow. Cell. 1998;94:657–666. doi: 10.1016/s0092-8674(00)81607-4. [DOI] [PubMed] [Google Scholar]
- 7.Boettiger D. Mechanical control of integrin-mediated adhesion and signaling. Curr. Opin. Cell Biol. 2012;24:592–599. doi: 10.1016/j.ceb.2012.07.002. [DOI] [PubMed] [Google Scholar]
- 8.Brass L.F., Zhu L., Stalker T.J. Minding the gaps to promote thrombus growth and stability. J. Clin. Invest. 2005;115:3385–3392. doi: 10.1172/JCI26869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nurden A.T., Fiore M., Pillois X. Glanzmann thrombasthenia: a review of ITGA2B and ITGB3 defects with emphasis on variants, phenotypic variability, and mouse models. Blood. 2011;118:5996–6005. doi: 10.1182/blood-2011-07-365635. [DOI] [PubMed] [Google Scholar]
- 10.Nieuwenhuis H.K., Akkerman J.W., Sixma J.J. Human blood platelets showing no response to collagen fail to express surface glycoprotein Ia. Nature. 1985;318:470–472. doi: 10.1038/318470a0. [DOI] [PubMed] [Google Scholar]
- 11.Simon D., Kunicki T., Nugent D. Platelet function defects. Haemophilia. 2008;14:1240–1249. doi: 10.1111/j.1365-2516.2008.01898.x. [DOI] [PubMed] [Google Scholar]
- 12.Auton M., Zhu C., Cruz M.A. The mechanism of VWF-mediated platelet GPIbalpha binding. Biophys. J. 2010;99:1192–1201. doi: 10.1016/j.bpj.2010.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang W., Deng W., Li R. Identification of a juxtamembrane mechanosensitive domain in the platelet mechanosensor glycoprotein Ib-IX complex. Blood. 2015;125:562–569. doi: 10.1182/blood-2014-07-589507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Clemetson K.J., Clemetson J.M. Platelet GPIb-V-IX complex. Structure, function, physiology, and pathology. Semin. Thromb. Hemost. 1995;21:130–136. doi: 10.1055/s-2007-1000387. [DOI] [PubMed] [Google Scholar]
- 15.Nakamura F., Pudas R., Ylänne J. The structure of the GPIb-filamin A complex. Blood. 2006;107:1925–1932. doi: 10.1182/blood-2005-10-3964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Andrews R.K., Berndt M.C. Platelet adhesion: a game of catch and release. J. Clin. Invest. 2008;118:3009–3011. doi: 10.1172/JCI36883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen J., Fu X., López J.A. Oxidative modification of von Willebrand factor by neutrophil oxidants inhibits its cleavage by ADAMTS13. Blood. 2010;115:706–712. doi: 10.1182/blood-2009-03-213967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dong J., Schade A.J., López J.A. Novel gain-of-function mutations of platelet glycoprotein IBalpha by valine mutagenesis in the Cys209-Cys248 disulfide loop. Functional analysis under statis and dynamic conditions. J. Biol. Chem. 2000;275:27663–27670. doi: 10.1074/jbc.M909952199. [DOI] [PubMed] [Google Scholar]
- 19.Sniadecki N.J., Chen C.S. Microfabricated silicone elastomeric post arrays for measuring traction forces of adherent cells. Methods Cell Biol. 2007;83:313–328. doi: 10.1016/S0091-679X(07)83013-5. [DOI] [PubMed] [Google Scholar]
- 20.López J.A., Leung B., Fox J.E. Efficient plasma membrane expression of a functional platelet glycoprotein Ib-IX complex requires the presence of its three subunits. J. Biol. Chem. 1992;267:12851–12859. [PubMed] [Google Scholar]
- 21.Dong J.F., Li C.Q., López J.A. The cytoplasmic domain of glycoprotein (GP) Ibalpha constrains the lateral diffusion of the GP Ib-IX complex and modulates von Willebrand factor binding. Biochemistry. 1997;36:12421–12427. doi: 10.1021/bi970636b. [DOI] [PubMed] [Google Scholar]
- 22.Cranmer S.L., Pikovski I., Jackson S.P. Identification of a unique filamin A binding region within the cytoplasmic domain of glycoprotein Ibalpha. Biochem. J. 2005;387:849–858. doi: 10.1042/BJ20041836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Girma J.P., Kalafatis M., Meyer D. Mapping of distinct von Willebrand factor domains interacting with platelet GPIb and GPIIb/IIIa and with collagen using monoclonal antibodies. Blood. 1986;67:1356–1366. [PubMed] [Google Scholar]
- 24.Naimushin Y.A., Mazurov A.V. Von Willebrand factor can support platelet aggregation via interaction with activated GPIIb-IIIa and GPIb. Platelets. 2004;15:419–425. doi: 10.1080/09537100410001721333. [DOI] [PubMed] [Google Scholar]
- 25.Lam W.A., Chaudhuri O., Fletcher D.A. Mechanics and contraction dynamics of single platelets and implications for clot stiffening. Nat. Mater. 2011;10:61–66. doi: 10.1038/nmat2903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schwarz Henriques S., Sandmann R., Köster S. Force field evolution during human blood platelet activation. J. Cell Sci. 2012;125:3914–3920. doi: 10.1242/jcs.108126. [DOI] [PubMed] [Google Scholar]
- 27.McCarty O.J.T., Calaminus S.D.J., Watson S.P. von Willebrand factor mediates platelet spreading through glycoprotein Ib and alpha(IIb)beta3 in the presence of botrocetin and ristocetin, respectively. J. Thromb. Haemost. 2006;4:1367–1378. doi: 10.1111/j.1538-7836.2006.01966.x. [DOI] [PubMed] [Google Scholar]
- 28.Tan J.L., Tien J., Chen C.S. Cells lying on a bed of microneedles: an approach to isolate mechanical force. Proc. Natl. Acad. Sci. USA. 2003;100:1484–1489. doi: 10.1073/pnas.0235407100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.du Roure O., Saez A., Ladoux B. Force mapping in epithelial cell migration. Proc. Natl. Acad. Sci. USA. 2005;102:2390–2395. doi: 10.1073/pnas.0408482102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kovács M., Tóth J., Sellers J.R. Mechanism of blebbistatin inhibition of myosin II. J. Biol. Chem. 2004;279:35557–35563. doi: 10.1074/jbc.M405319200. [DOI] [PubMed] [Google Scholar]
- 31.Yago T., Lou J., Zhu C. Platelet glycoprotein Ibalpha forms catch bonds with human WT vWF but not with type 2B von Willebrand disease vWF. J. Clin. Invest. 2008;118:3195–3207. doi: 10.1172/JCI35754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Carr M.E., Jr., Carr S.L., Braaten J. Glycoprotein IIb/IIIa blockade inhibits platelet-mediated force development and reduces gel elastic modulus. Thromb. Haemost. 1995;73:499–505. [PubMed] [Google Scholar]
- 33.Varga-Szabo D., Pleines I., Nieswandt B. Cell adhesion mechanisms in platelets. Arterioscler. Thromb. Vasc. Biol. 2008;28:403–412. doi: 10.1161/ATVBAHA.107.150474. [DOI] [PubMed] [Google Scholar]
- 34.Liang X.M., Han S.J., Sniadecki N.J. Platelet retraction force measurements using flexible post force sensors. Lab Chip. 2010;10:991–998. doi: 10.1039/b918719g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Carr M.E., Jr., Zekert S.L. Measurement of platelet-mediated force development during plasma clot formation. Am. J. Med. Sci. 1991;302:13–18. doi: 10.1097/00000441-199107000-00004. [DOI] [PubMed] [Google Scholar]
- 36.Kroll M.H., Harris T.S., Schafer A.I. von Willebrand factor binding to platelet GpIb initiates signals for platelet activation. J. Clin. Invest. 1991;88:1568–1573. doi: 10.1172/JCI115468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gusnard D., Kirschner R.H. Cell and organelle shrinkage during preparation for scanning electron microscopy: effects of fixation, dehydration and critical point drying. J. Microsc. 1997;110(1):51–57. doi: 10.1111/j.1365-2818.1977.tb00012.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.