

Summary
Bone marrow niches for hematopoietic progenitor cells are not well defined despite their critical role in blood homeostasis. We previously found that cells expressing osteocalcin, a marker of mature osteolineage cells, regulate the production of thymic-seeding T lymphoid progenitors. Here, using a selective cell deletion strategy, we demonstrate that a subset of mesenchymal cells expressing osterix, a marker of bone precursors in the adult, serve to regulate the maturation of early B lymphoid precursors by promoting pro-B to pre-B cell transition through insulin-like growth factor 1 (IGF-1) production. Loss of Osx+ cells or Osx-specific deletion of IGF-1 led to a failure of B cell maturation and the impaired adaptive immune response. These data highlight the notion that bone marrow is a composite of specialized niches formed by pairings of specific mesenchymal cells with parenchymal stem or lineage committed progenitor cells, thereby providing distinctive functional units to regulate hematopoiesis.
Graphical Abstract
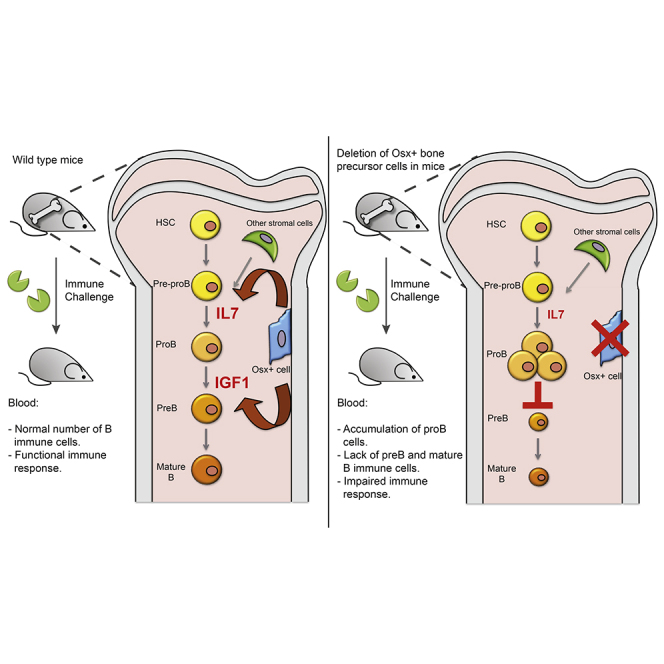
Highlights
-
•
Loss of Osx+ osteolineage cells halted B cell maturation and caused immune failure
-
•
Mice with Osx+ cell-specific deletion of IGF-1 phenocopied Osx+ cell ablated mice
-
•
Osx+ cell promotes pro-B to pre-B cell transition through IGF-1 production
-
•
Specialized niche cell-hematopoietic progenitor pairings regulate hematopoiesis
In this article, Scadden, Ferraro, and colleagues show that cells expressing osterix, a marker of bone precursors, serves to regulate the maturation of early B lymphoid precursors through IGF-1 production. Together with the previous finding that mature osteolineage cells regulate the production of thymic-seeding T lymphoid precursors, these data highlight the notion that niche cells and parenchymal cells form distinct cell pairings to create specialized functional units to regulate different processes of hematopoiesis.
Introduction
Characterization of the niche has provided insight into molecular pathways modulating stem cell function for tissue regeneration and cancer. Although the multitude of cells comprising bone marrow stroma remains incompletely defined (Yu and Scadden, 2016a), recent studies indicate that while primitive mesenchymal progenitors are more critical for hematopoietic stem cell (HSC) function, lineage-restricted mesenchymal cells control more committed hematopoietic progenitors (Mendez-Ferrer et al., 2010). Specifically, it was described that the deletion of a range of osteolineage cells or CXCL12 from these cells alters B cell progenitors (Greenbaum et al., 2013, Visnjic et al., 2004) or B and T cell counts (Ding and Morrison, 2013). Furthermore, deletion of osteocytes created systemic disruption of metabolism and affected thymic function (Sato et al., 2013).
These data suggest that niches are not limited to the governance of stem cells (Wu et al., 2009, Yu et al., 2015a), and the definition of “progenitor niches” may offer new ways to understand the regulation of tissue composition and function. We recently demonstrated that selective deletion of cells expressing the mature osteolineage cell marker Osteocalcin (Ocn) results in decreased T competent common lymphoid progenitors (CLPs) with a minimal effect on B cell-biased CLPs (Ly6D+) (Yu et al., 2015a) due to a defective generation of thymic-seeding progenitor cells. The mutant phenotype was recapitulated by selective deletion of DLL4 in Ocn+ cells, or of its receptor and downstream signaling molecules in primitive hematopoietic cells; therefore Ocn+ cells constitute a regulatory niche guiding the production and differentiation of hematopoietic cells contributing to T cell neogenesis.
Given that others using genetic constructs to disrupt a broad range of osteolineage cells showed a decrease in mature B cells (Ding and Morrison, 2013, Visnjic et al., 2004, Wu et al., 2008), that B cells are adjacent to CXCL12-abundant reticular (CAR) cells (Omatsu et al., 2010), and that continuous deletion of CXCL12 in development using Osx-Cre leads to reduced B cell production (Greenbaum et al., 2013), we hypothesized that Osx+ cells may serve as a B cell differentiation modulator in adult animals. We sought to determine this using a cell-type-specific in vivo cell-ablation strategy.
The perceived hierarchy of bone cell differentiation begins from the multipotent mesenchymal stem cell, which matures to become the osteoprogenitor, the pre-osteoblast, the mature osteoblast and/or the lining cell, and eventually the terminally differentiated osteocyte. While less is known regarding the heterogeneity of the mesenchymal stem cell population (Yu and Scadden, 2016b) and multiple markers such as Leptin receptor (Zhou et al., 2014), Nestin (Ono et al., 2014), and Gremlin1 (Worthley et al., 2015), and Mx1(Park et al., 2012) among others (Chan et al., 2015) had been recently proposed to label mesenchymal stem cell populations of various regenerative properties, markers for osteoprogenitor and mature osteoblast are more clearly defined. Osterix (Osx/Sp7) is a zinc finger-containing transcription factor expressed in osteoprogenitors and serves as a key regulator of osteolineage differentiation (Mizoguchi et al., 2014, Nakashima et al., 2002). Osteocalcin (Ocn) is the most well-characterized extracellular matrix protein expressed by mature osteoblasts. However, these markers were mostly used in independent studies, rendering it difficult to study the dynamic relationship of osteolineage subsets in vivo. Here, we generated a triple transgenic mouse that differentially labels Osx+ and Ocn+ cells. We used this model to interrogate whether distinct osteolineage subsets serve to regulate different hematopoietic processes.
Results
We examined whether the Osx+ cells could be distinguished from Ocn+ cells in vivo for an interval sufficient to test their distinctive biological function. Mice carrying a fusion of Cre and modified estrogen receptor under the control of the Osterix promoter (Osx-CreERt2 [Maes et al., 2010]), hereafter called OsxCre, were crossed with mice bearing a Rosa26-loxP-stop-loxP-mCherry (Rosa-mCh) transgene (OsxCre;Rosa-mCh) (Strecker et al., 2013). Administration of 4-hydroxy-tamoxifen (4-OHT) to OsxCre+;Rosa-mCh+ mice resulted in Cre activation in Osx+ cells followed by excision of the stop cassette and production of the mCherry fluorophore. Upon 4-OHT injection, the red fluorescence marks cells expressing OSX as well as their progeny. These mice were crossed with mice expressing the GFP, Topaz, driven by the Osteocalcin promoter (Ocn:Topaz) (Bilic-Curcic et al., 2005). In this triple transgenic model (OsxCre+;Rosa-mCh+;Ocn:Topaz+), the OCN-expressing cells are green, the OSX-expressing cells (and their descendants) are red, and cells expressing both markers are yellow (Figure 1A). According to osteolineage ontology, we anticipated that the Osx+ osteoprogenitors initially labeled red would become yellow as they express OCN. Six-week-old OsxCre+;Rosa-mCh+ mice were pulsed with an injection of tamoxifen (day 0) and fluorescent cells were quantified over time. In a 6-week chase, a modest number of dual-labeled (++) cells emerged (0.02% of total bone cells) (Figure 1B), but the majority of cells were either mCherry (OSX+) or Topaz (OCN+) single positive (Figure 1C). Cells labeled as ++ were found at the metaphyseal region, located near the endosteal surface. These data show that Osx+ cells do not necessarily transition to Ocn+ cells over a 6-week period, although a modest number of cells do. It may be that limited efficiencies of fluorophore expression underestimate the cells transitioning from Osx to Ocn expression. Also possible is that some OSX labeling occurs in cells that do not proceed to osteoblasts expressing OCN or that dually labeled cells are lost due to disadvantageous characteristics from dual fluorophore production. Nonetheless, this triple transgenic system enables us to isolate distinct subpopulations of the osteolineage within the same animal by flow cytometry and allows subsequent characterization of their molecular and functional profiles.
Figure 1.

A Triple Transgenic Mouse to Study the Dynamics of Osteolineage Subpopulations
(A) Osterix-Cre (OsxCre) mice were crossed with the Rosa26-loxP-stop-loxP-mCherry reporter mice (Rosa-mCh), which express mCherry fluorescent protein upon Cre-mediated excision of a stop sequence. OsxCre+;Rosa-mCh+ mice were then crossed with the osteocalcin-Topaz (Ocn:Topaz) mice. Upon tamoxifen injection into the OsxCre+;Rosa-mCh+;Ocn:Topaz+ mice, the Osx+ cells and their progeny are labeled red, whereas the Ocn+ cells are labeled green. Cells that express osterix following tamoxifen injection and produce osteocalcin are yellow.
(B) Osx+, Ocn+, and ++ cells were enumerated in femur sections of OsxCre+;Rosa-mCh+;Ocn:Topaz mice at the indicated time points following tamoxifen injection.
(C) Representative sections of an OsxCre+;Rosa-mCh+;Ocn:Topaz+ mouse 4–6 weeks after tamoxifen injection showing the location of the different osteolineage subsets. Blue indicates DAPI, red mCherry (Osx+ cells), and green Topaz (Ocn+ cells). Yellow arrow indicates ++ cells and red arrow points to Osx+ cells. BT, bone, trabecular; BM, bone marrow cavity. Note the punctated appearance of the mCherry fluorescence. Scale bar, 75 μm.
Experiment repeated once, n = 4/group/experiment (B and C).
We then assessed the three labeled populations. At day 4 after 4-OHT treatment, Osx+, ++, and Ocn+ cells were purified by fluorescence-activated cell sorting from bone cell suspensions isolated from the triple transgenic model, and subjected to gene-expression profiling by microarray. Principal component analysis of the 2,509 top genes clustered together Osx+ and ++ cells as more similar compared with Ocn+ cells (data not shown). Restrictive filtering revealed the top 25 differentially expressed genes among the three populations (Table S1). Gene ontology analysis revealed that genes upregulated in Ocn+ cells were related to cell adhesion and cytokines, whereas Osx+ cells highly transcribe genes involved in extracellular matrix interaction and Hedgehog-dependent pathways (Table S2). Interestingly, the ++ population represented a well-defined population with distinctive expression of pro-inflammatory cytokines and macrophage-related surface and secreted molecules. Microarray results were validated by RT-PCR on sorted Osx+, ++, and Ocn+ cells (Figure S1A).
We investigated whether Osx+, Ocn+, or ++ cells have unique effects on hematopoietic stem and progenitor cell (HSPC) function ex vivo. Six days following 4-OHT injection into 4- to 6-week-old triple transgenic mice, FACS sorted Osx+, ++, or Ocn+ cells were separately co-cultured for 5 days with purified HSPCs (LINEAGElo, C-KIT+, SCA-1+, CD150+, CD48−) isolated from GFP+ mice. Following co-culture, cells were transplanted into lethally irradiated recipients (Figure S1B). HSPCs co-cultured with each osteolineage subset maintained their long-term reconstitution and multilineage potential (Figure S1C). However, HSPCs co-cultured with Osx+ cells displayed increased total chimerism compared with those co-cultured with Ocn+ and ++ cells at 4 weeks, and improved chimerism compared with ++ cells at 16 weeks (Figure S1C). B lymphoid cells (B220+ cells) were the major contributors to the increased engraftment from HSPCs co-cultured with Osx+ cells (Figure S1D), while there was short-term enhanced reconstitution of T cells (CD4+ and CD8+ cells) at 8 weeks, and this effect disappeared at 12 and 16 weeks (Figure S1E). No difference was observed for the Mac/Gr1+ subset (Figure S1F).
We next postulated whether Osx+ cell regulates B cell populations in vivo using a selective cell-depletion model. We crossed the Osx1-GFP::Cre mice with the iDTR mice (hereafter OsxCre;iDTR), in which ubiquitous expression of the diphtheria toxin receptor (iDTR) is blocked by a LoxP-flanked STOP sequence. Cre-mediated excision of the STOP sequence allows expression of the iDTR in select cell populations that then become susceptible to killing upon peritoneal administration of diphtheria toxin (DT). We began daily DT injections into both control and mutant mice starting at 4 weeks of age. At 6 weeks, there was a striking difference of skeletal size when we compared the OsxCre+;iDTR+/+ controls with the OsxCre+;iDTRFl/+ mutants (Figures 2A and 2B). OsxCre+;iDTRFl/+ mutants showed reduced bone mass (Figures 2C and 2D) and osteoblastic functions including reduced bone formation, serum levels of osteocalcin, and type I procollagen production (Figure 2D). Flow cytometry revealed a ∼50% reduction of Osx+ cells in mutants (Figure 2E). To assure targeted cell deletion, we examined DTR in mutant animals without toxin injection by immunohistochemistry. Expression of the DTR strongly overlapped with staining of OSX in the OsxCre+;iDTRFl/+ animals (Figures S2A–S2J). Of note, OsxCre+;iDTRFl/+ animals did not display extensive overlapping with OCN staining, confirming our observations from the lineage-tracing experiments. Staining of bone sections with OSX-specific antibodies in combination with TUNEL staining confirmed that the targeted osteolineage cells were correctly deleted (Figure 2F). Tartrate-resistant acid phosphatase (TRAP) staining and measurement of degraded collagen confirmed that osteoclastogenesis was not affected (Figure 2G). These data indicate that a distinct subset of osteolineage cells were depleted in the OsxCre+;iDTRFl/+ mouse and that we were able to selectively assess the impact of these Osx+ cells for a time lapse following cell deletion. We asked whether selective deletion of Osx+ cells might lead to a compensatory expansion of more primitive bone progenitor cells, thereby confounding our interpretation. Colony-forming unit osteoblast (CFU-Ob) assays failed to detect any increase in primitive mesenchymal progenitors between controls and mutants (Figure S2K). However, other non-hematopoietic cell types might be affected by Osx+ cell depletion and influence the hematopoietic phenotype observed.
Figure 2.

Osx+ Cell-Specific Deletion In Vivo without Altering Osteoclastogenesis
(A and B) Skeletal development in the OsxCre;iDTR mutants compared with control littermates at 6 weeks of age. Three independent experiments; n = 9–15/group. ∗∗∗p < 0.001.
(C) Histology of femurs from OsxCre;iDTR control and mutant mice. Three independent experiments; n = 9–15/group.
(D and E) Bone histomorphometric measurement on trabecular number, trabecular separation, number of osteoblasts, bone formation rate, serum production of osteocalcin, and type I procollagen production. Three independent experiments; n = 9–15/group. ∗p < 0.05, ∗∗p < 0.01. (E) Flow cytometric quantification of Osx+ cells in control and mutant groups. Three independent experiments; n = 9–15/group.
(F) Bone sections were stained with osterix-specific antibody overlapped with apoptotic TUNEL staining to confirm targeted cell deletion. Three independent experiments; n = 9–15/group.
(G) Measurement of osteoclast number as indicated by TRAP staining, and osteoclast activity, as indicated by the rate of collagen breakdown in sera. Two independent experiments; n = 22–24/group.
Error bars represent ± SEM.
Despite the dramatic changes in bone morphology in OsxCre;iDTR mutants, bone marrow cellularity was unaffected compared with their control littermates (Figure 3A). Red blood cell and platelet numbers were unchanged in the blood of the OsxCre;iDTR mutant, but there was a 50% decrease in the white blood cell count due to severe lymphopenia (Figure 3B). The mutant bone marrow had fewer mature B220+IgM+ B cells, increased monocytes and granulocytes (Figure 3A), and a corresponding increase in granulocyte and macrophage progenitors (GMPs) (Figure 3C). Given the importance of the spleen for B lymphopoiesis and extramedullary hematopoiesis, we analyzed the spleens and noted no difference in weight, but similarly increased Mac1+Gr1+ cells in the OsxCre;iDTR mutants (Figure 3D). In summary, Osx+ cell deletion caused a loss of mature B cells and an increase in myeloid cells in both the bone marrow and spleen of mutant animals.
Figure 3.

Osx+ Cell Ablation Causes B lymphopenia and an Increase in Monocytes and Macrophages
(A) Flow cytometric measurement of bone marrow cellularity and multiple mature lineages in OsxCre;iDTR mutants compared with control littermates.
(B) White blood cell, red blood cell, and platelet counts in the peripheral blood of OsxCre;iDTR mutants versus controls.
(C) Enumeration of different progenitor subtypes in the bone marrow.
(D) Examination of the spleen as a major site for B lymphopoiesis and extramedullary hematopoiesis showed a similar increase in Mac1+Gr1+ myeloid cells and attenuated mature B cell number.
Experiment repeated 2–3 times; n = 4–12/group. ∗p < 0.05, ∗∗∗p < 0.001. Error bars represent ± SEM.
We assessed whether the GMP increase in the OsxCre;iDTR mutants was secondary to increased proliferation or decreased death of GMPs. There was change in neither cell-cycle status (Figure S3A) nor apoptosis (Figure S3B). The accumulation of GMP and Mac1+Gr1+ cells could either be an osteoprogenitor-mediated hematopoietic effect or a consequence of inflammatory phagocytic-type immune response triggered by the death of osteolineage cells. To test this hypothesis, we co-injected indomethacin, a non-steroidal anti-inflammatory drug, together with DT in OsxCre;DTR mutant mice for 1 week. Bone marrow analysis revealed normalization of the GMP, Mac1+, and Mac1+Gr1+ cell populations in indomethacin-treated animals (Figure S3C). Therefore, the increase in Mac1+Gr1+ myeloid cells could be due to inflammatory response to osteolineage cell death, although we cannot exclude an osteolineage cell-mediated myeloid effect.
We asked whether HSC function was affected by the short-term deletion of Osx+ cells. We observed no differences between the OsxCre;iDTR mutants and controls in long-term HSC (LINloC-KIT+SCA-1+CD48−CD150+) number (Figure S4A), proliferation (Figure S4B), or apoptosis (Figure S4C). There was an increase in LINloC-KIT+SCA-1− progenitor cell number and cell cycle (Figures S4A and S4B). However, no defect in HSC function was detected when bone marrow cells from were transplanted into primary and secondary recipients in a 1:1 ratio with congenic SJL competitor cells (Figure S4D). These data suggest that short-term deletion of Osx+ cells did not affect HSC function.
Given the diminished production of mature B cells, we assessed CLPs and found no difference (Figure 3C). However, intermediate stages of B cell development were affected. Specifically, a decrease in both pre-B and mature B populations were noted while pro-B cells, especially during the later C′ pro-B and C″ pro-B stages, were significantly increased (Figure 4A). These data strongly suggest that B cell differentiation was impaired at the pro-B to pre-B juncture.
Figure 4.

Osx+ Cell Deletion Leads to Blockade of pro-B to pre-B Transition and Impairs Adaptive Immunity
(A) Analysis of intermediate stages of B cell maturation in the OsxCre;iDTR mutants compared with control littermates.
(B and C) Animals were challenged with NP-Ficoll to produce immunoglobulins in a T cell-independent manner. Production of IgG (B) and IgM (C) were assessed over 28 days.
Three independent experiments; n = 12–16/group. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. Error bars represent ± SEM.
We next tested whether these changes affected the animal's immune response. Mice were challenged with NP-Ficoll to trigger a T cell-independent (Maizels et al., 1988) immune response and followed for 28 days. Data revealed that OsxCre;iDTR mutants could not sustain immunoglobulin G (IgG) and M (IgM) production over time compared with their control littermates (Figures 4B and 4C). These data indicate that the perturbation in B cell development was not simply immunophenotypic, but there was also a functional reduction in Ig production and immune response.
To determine whether the B cell differentiation defect was microenvironment-dependent, we transplanted 1 × 106 bone marrow cells from congenic SJL mice into lethally irradiated control or mutant OsxCre;iDTR recipients. A similar rate of reconstitution was seen in both control and mutants (Figure 5A). As wild-type SJL cells repopulated both control and mutant OsxCre;iDTR recipients over the next 12 weeks, we observed a gradual recapitulation of the OsxCre+/−;DTRFl/+ hematopoietic phenotype in the mutant recipients but not in the controls (Figures 5B–5F). These data strongly suggest that the incomplete B cell differentiation observed in OsxCre;iDTR was caused by an altered bone microenvironment.
Figure 5.

Osx+ Cell Controls B Lymphopoiesis through a Microenvironment-Dependent Effect
(A–F) Transplantation of wild-type cells into mutant hosts. 1 × 106 total bone marrow cells from wild-type congenic SJL mice (CD45.1) were transplanted into lethally irradiated control or mutant OsxCre;iDTR recipients (CD45.2). (A) A similar rate of reconstitution was seen in both control and mutant groups. (B–F) As wild-type hematopoietic cells repopulated the new hosts over the next 12 weeks, we observed a gradual re-creation of the OsxCre;iDTR mutant hematopoietic phenotype in the mutant recipients but not in the control group.
(G–L) Transplantation of mutant cells into wild-type hosts. Bone marrow cells (5 × 105) from either control or mutant OsxCre;iDTR mice were competed with wild-type 5 × 105 SJL bone marrow competitors in a 1:1 ratio, and transplanted into lethally irradiated SJL hosts. (G) No functional disadvantage was seen in reconstitution when OsxCre;iDTR mutant cells competed with wild-type cells. (H–L) When hematopoietic cells from OsxCre;iDTR mutant donors were placed into a wild-type microenvironment, the hematopoietic defect was normalized over time.
Two independent experiments; n = 10/group/experiment. ∗p < 0.05, ∗∗p < 0.01. Error bars represent ± SEM.
To confirm this, we transplanted mutant cells into wild-type hosts in a reverse transplant setting. Either 5 × 105 OsxCre;iDTR mutant or control bone marrow cells, competed with 5 × 105 SJL bone marrow cells in a 1:1 ratio, were transplanted into lethally irradiated SJL hosts. When cells from OsxCre;iDTR mutant mice were competed with cells from wild-type mice, no functional disadvantage was seen during reconstitution (Figure 5G). In addition, when hematopoietic cells from OsxCre;iDTR mutant donors were transplanted into a wild-type host, the hematopoietic defect was normalized over time (Figures 5H–5L). Thus the hematopoietic defect observed in the OsxCre;iDTR mutants was not cell autonomous, but caused by alteration in the microenvironment.
To assess which secreted factors released by osteoprogenitors mediate B cell differentiation, we compared local cytokines within the bone marrow of OsxCre;iDTR mutant and control mice by cytokine array. Since we observed a pro-B to pre-B differentiation blockade in the OsxCre;iDTR mutants, and interleukin-7 (IL-7) is critical for commitment to B cell fate (Nagasawa, 2006), we initially focused on changes in IL-7 production. Decreased Il7 transcription was noted in OsxCre;iDTR mutant bones by qPCR (Figure S5A); however, IL-7 protein in the bone marrow sera of mutants was not statistically decreased (Figure S5B). This suggests that although Osx+ cell expresses IL-7, other stromal cell types within the bone marrow likely produce IL-7 when Osx+ cells are absent.
Among the measured cytokines, insulin-like growth factor 1 (IGF-1) was notably decreased in our screen (Figure S5B). We confirmed that OsxCre;iDTR mutant bones had decreased Igf1 transcripts compared with controls (Figure S5A). To determine what kind of hematopoietic cells were the targets of the IGF-1 released by Osx+ cells, we assessed flow sorted hematopoietic populations including LINloSCA-1+C-KIT+ (LKS), CLP, common myeloid progenitor (CMP), GMP, megakaryocyte-erythroid progenitor (MEP), pro-B, pre-B, mature B, and LIN+ mature cells by qPCR for IGF-1 receptor (IGF-1R) expression. IGF-1R showed higher expression in LKS, CLP, pro-B, pre-B, mature B, and LIN+ cells but not in CMP, GMP, and MEP (Figure S5C), although the difference was not statistically significant.
To further assess whether IGF-1 promotes B cell differentiation, we evaluated hematopoietic cells from femurs of the OsxCre;iDTR mutant and control mice by methylcellulose assays. Cells were grown with (1) no supplement, (2) IL-7, (3) IGF-1, or (4) IL-7 + IGF-1 for a period of 14 days and scored for CFU-G, CFU-M, CFU-GM, CFU-GEMM, burst-forming unit (BFU)-E, and CFU-Pre-B (Figures S5D–S5I). CFU-Pre-B assays showed that IL-7 promoted pre-B colony formation, and the effect was further enhanced when IGF-1 was added (Figure S5I). No positive effect on the differentiation of other hematopoietic progenitors was observed even when the combination of IL-7 and IGF-1 was added (Figures S5D and S5F–S5H). However, the addition of IL-7 and IGF-1 had a negative effect on CFU-M (Figure S5E).
Our data suggest that Osx+ cells express IGF-1 to mediate B cell differentiation. To validate this hypothesis in vivo, we crossed the Osx1-GFP::Cre mouse with the B6.129(FVB)-Igf1tm1Dlr/J (IGF-1F/F) strain, which has LoxP sites flanking exon 4 of the Igf1 gene. Expression of Cre under the Osx promoter in Osx1-GFP::Cre+;IGF-1F/F mice specifically abolishes IGF-1 in Osx+ cells (Figure S6). Interestingly, Osx1-GFP::Cre+;IGF-1F/F mutants showed increased myeloid and decreased B lymphoid cells in the bone marrow (Figure 6A), and decreased white blood cell counts in the blood (Figure 6B), a phenotype reminiscent of the Osx+ cell deletion mouse model. Strikingly, examination of B cell development showed that cell maturation was arrested at the pro-B to pre-B transition, a phenocopy of the OsxCre;iDTR mutants, and the effect was even more pronounced (Figure 6C). These data confirm that Osx+ cell regulates B cell development by providing IGF-1 to enable B cell maturation.
Figure 6.

Deletion of IGF-1 in Osx+ Cells Phenocopied OsxCre;iDTR Lymphoid Defects
To delete IGF-1 in Osx+ cells, we crossed the Osx1-GFP::Cre mouse with the IGF-1F/F strain.
(A) Osx1-GFP::Cre+;IGF-1F/F mutants showed increased Mac1+ and Gr1+ myeloid cells, and decreased B lymphoid cells in the bone marrow. Three independent experiments; n = 8–14/group.
(B) Lymphopenia was also reflected in the peripheral blood. Experiment repeated once; n = 4–6/group.
(C) Osx1-GFP::Cre+;IGF-1F/F mutants showed a loss of mature B cells due to differentiation arrest at the pro-B to pre-B transition. Analysis of B cell differentiation showed accumulation of cells at the B′, C′, and C″ pro-B cell stages, but decrease in number at the pre-B, immature B, and mature B stages. Three independent experiments; n = 8–14/group.
∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. Error bars represent ± SEM.
Finally, we tested whether the B cell differentiation defect in the OsxCre;iDTR mutants could be rescued in vivo by intravenous administration of recombinant IL-7 and IGF-1. Both controls and mutants were subjected to daily injections of DT, DT + IL-7, or DT + IGF-1 for 12 days. At the end of the rescue regimen, bone marrow cells were evaluated by flow cytometry (Figures 7A–7H). IL-7 administration increased the C′ and C″ pro-B populations (Figures 7C and 7D) but did not rescue later B stages (Figures 7F–7H). In contrast, IGF-1 had no effect at the early pro-B stages (Figures 7A–7E) but rescued the pre-B and mature B levels (Figures 7F–7H). These results suggest that while IL-7 supports the differentiation of early B precursors, IGF-1 is required for downstream B cell maturation and that Osx+ cells regulate this process via production of IGF-1.
Figure 7.

Osx+ Cell Produces IL-7 and IGF-1 to Regulate B Cell Differentiation
(A–H) In vivo rescue experiment of OsxCre;iDTR lymphoid phenotype. OsxCre;iDTR control and mutant animals were subjected to either: DT, DT + IL-7, or DT + IGF-1 daily injections for 12 days. Bone marrow cells were harvested the next day following the last injection for flow cytometric analysis of B cell differentiation. Injection of IL-7 augmented the base level of C′ and C″ pro-B (C and D), but failed to rescue pre-B and downstream B cell maturation (F–H). In contrast, IGF-1 had no effect on earlier B cell differentiation (A–E), but rescued the pre-B and mature B cell loss in the mutant group (F and H). Three independent experiments; n = 6–9/group. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗∗p < 0.0001.
(I) Model of regulation of B and T lymphopoiesis by Osx+ and Ocn+ osteolineage cells. Osx+ cell produces IL-7 and IGF-1, and both molecules are required to support full B lineage differentiation. While IL-7, also likely produced by other stomal cell types, mediates hematopoietic differentiation from early B cell precursors to pre-B cells, Osx+ cell secretion of IGF-1 is necessary for further B cell maturation from pre-B to mature B. In comparison, Ocn+ cell expresses the Notch ligand DLL4, which binds to cell-surface Notch receptor on the T cell-competent Ly6D− CLP population (Yu et al., 2015a). This engagement ensures T cell progenitor production, and expression of the chemotactic molecules CCR7 and PSGL1 for subsequent thymic seeding. The precise balance of Osx+ and Ocn+ osteolineage cells within the bone marrow niche is critical for both B and T lymphopoiesis, as perturbation of this balance leads to specific loss of B or T immune cells.
Error bars represent ± SEM.
Discussion
These and our recent publication (Yu et al., 2015a) demonstrate that mesenchymal cell populations in the niche have stage-specific functional interactions with the hematopoietic system. Specifically, while primitive mesenchymal cells regulate HSC, we have recently shown that Ocn+ cells modulate production of T cells through the regulation of thymus-seeding progenitors. Those data indicate that T lymphoid progenitor specification and production were diminished with Ocn+ deletion; DLL4 was the identified Ocn+ cell product responsible for the production of these T cell precursors. Here we show that specific steps of B cell differentiation are affected by the partial loss of Osx+ cells. The importance of osteolineage cells in regulating B cells within the bone marrow has been known from reports documenting the roles of IL-7 and CXCL12, among other factors (Ding and Morrison, 2013, Greenbaum et al., 2013, Visnjic et al., 2004, Wu et al., 2009, Zhu et al., 2007). Previous studies reported that IGF-1 promotes B cell expansion in vitro (Taguchi et al., 2006) and that activation of the IGF-1 receptor is needed for immunoglobulin production (Baudler et al., 2005). Local IGF-1 production may be most important for marrow B lymphopoiesis, as prior studies of either liver or pituitary-specific IGF-1 deficiency only observed altered B cell numbers in the spleen and not within the bone marrow (Montecino-Rodriguez et al., 1997, Welniak et al., 2004). Our data indicate that osteoprogenitors secrete IL-7 and IGF-1; both are necessary to support full B lineage differentiation in the bone marrow (Figure 7I). Deletion of Osx+ cells lead to minimal changes in IL-7 serum levels despite significant knockdown of Il7 transcripts. It is likely that other cell types compensate for the production of IL-7 in the niche. Rescue experiments showed that IGF-1 is required together with IL-7 to restore the full differentiation program of B cells. These data suggest that IL-7 controls early stages of B cell differentiation, in agreement with previous data (Tokoyoda et al., 2004), while IGF-1 is necessary downstream of IL-7 for the differentiation into pro-B and mature B cells. Taken together, our data suggest that marrow B lymphopoiesis requires localized IGF-1 production while extramedullary maturation is more affected by systemic IGF-1.
Having defined through the triple transgenic model that Osx+ cells are distinctive molecularly from Ocn+ cells, we tested their selective function. The model we used was intentionally of short duration to avoid the progression of Osx+ cells to Ocn-expressing, ++ cells. In vivo, these cells are dynamic and are lost after ∼90 days (Park et al., 2012). Our data demonstrate that the Osx+ cells have no effect on CLP or the T competent progenitors, Ly6D−CLP (Figure 4C) in contrast to the Ocn+ cells. Nor do they produce DLL4 at levels comparable with those in Ocn+ cells. However, they do express abundant IL-7 and IGF-1 and through these molecules regulate the maturation of B cell through the pro-B cell stage. This maturation step is critical, as blunting B cell progression by deleting Osx+ support cells resulted in B lymphopenia and functional compromise of immunoglobulin production.
Others have reported both B and T cell effects after osteolineage cell deletion (Ding and Morrison, 2013) but the promoter used in some of those studies, Col1(2.3), extends through many cell stages of osteolineage progression. Here we define that these osteolineage cell states provide distinctive functions in support of hematopoiesis through secretion of specific regulatory molecules. Some of these effects are in specific lineage production as noted in Ocn+ cell deletion (Yu et al., 2015a) and some are in lineage differentiation as in the Osx+ deletion model.
It was notable that hematopoietic cell abnormalities in deletion of either of these osteolineage subsets appeared to be predominantly in the lymphoid compartment with little or no perturbation of HSC function. Nevertheless, we encourage caution in interpreting the HSC data. Prior reports by us (Calvi et al., 2003, Ferraro et al., 2011, Fleming et al., 2008, Raaijmakers et al., 2010) and others (Visnjic et al., 2004, Zhang et al., 2003) indicate a functional role for osteolineage cells in HSPC regulation. A possible explanation for the observed differences is that our cell depletion was not 100% efficient. It is also possible that the lack of functional defects in HSC, as demonstrated by the serial transplantation experiment, could depend upon the limited time donor HSCs were exposed to the mutated microenvironment. Finally, we cannot rule out the possibility that other cell types might have compensated for the osteolineage cell loss, thus mitigating any HSC phenotype. In summary, whether HSC depend on osteolineage cells deserves further clarification.
Several markers, including LEPTIN receptor (LEPR), NESTIN, PDGFRβ, CXCL12, PRX-1, CD105, and OSX, have been described to label overlapping mesenchymal stem cell populations with broad differentiation potential in vivo (Yu and Scadden, 2016a, Yu and Scadden, 2016b). In published studies using OsxCreERt2, Osx+ cells did not overlap with Nestin+ or LepR+ cells after short induction, but overlapped highly after several weeks (Liu et al., 2013, Mizoguchi et al., 2014, Ono et al., 2014). In addition, Osx+ cells have been shown to contribute to osteogenic, adipogenic, and chondrogenic lineages upon injury in vivo and in the neonate, but are restricted to the osteolineage in the adult (Mizoguchi et al., 2014). These inconsistent reports suggest that these markers, rather than being cell specific, might reflect functional states of mesenchymal cells that are dynamic during development and tissue regeneration. We examined the expression profile of these markers on Osx+ cells in our model by microarray and flow cytometry. Osx+ cells isolated from 6- to 8-week-old adults expressed low levels of LEPR, NESTIN, and CD105, and low to intermediate levels of PRX-1, CXCL12, and PDGFRβ (Figures S7A and S7B). We did not find extensive overlap in the Osx+ cells with LepR+ cells that might be considered CAR cells (Figures S7A and S7B), despite others (Greenbaum et al., 2013) reporting that some Osx+ cells overlaps with CAR cells as defined by the expression of CXCL12 (the age of the mice was not reported). Nonetheless, our data cannot exclude the possibility of Osx+ cells being adipogenic in models with a longer time chase, after injury or during perinatal life, although we observed no evidence of adipocyte generation under the conditions we studied.
There is a growing appreciation of a woven functional architecture to the bone marrow space with mesenchymal stem cells providing support for HSC (Mendez-Ferrer et al., 2010), mature osteoblasts affecting HSC mobilization by granulocyte colony-stimulating factor (Ferraro et al., 2011), osteoprogenitors requiring microRNA processing to maintain integrity of early and late hematopoiesis (Raaijmakers et al., 2010), and Osx+ and Ocn+ cells having key roles in B lymphoid differentiation and thymic homing progenitors (Yu et al., 2015a). The emerging model is one of a highly interrelated system with “intermediate” populations of both skeleton and blood having very specific interactions. Whether these interactions are perturbed is of particular relevance in settings where specific subsets of cells are deficient, such as in particular blood disorders, or in the adaptive immune deregulation seen after allogeneic bone marrow transplantation. Furthermore, these heterologous cell interactions can now be explored in malignant processes such as lymphoma, leukemia, and bone metastatic processes. If the complexity of the relationship between mesenchymal subsets of bone and parenchymal subsets of blood is true in other organ types, defining mesenchymal cells that comprise organ “stroma” may provide insight into parenchymal regulation and dysfunction.
Experimental Procedures
Osteolineage Cell Deletion Mouse Models
The Osx1-GFP::Cre mouse strain (Rodda and McMahon, 2006) was crossed to the iDTR strain (Buch et al., 2005) to achieve Osx cell deletion. These primers were used for genotyping: OsxCreF-CTCTTCATGAGGAGGACCCT, OsxCreR-CAGGCAGGTGCCTGGACAT, oIMR8052-GCGAAGAGTTTGTCCTCAACC, oIMR8545-AAAGTCGCTCTGAGTTGTTAT, oIMR8546-GGAGCGGGAGAAATGGATATG. OsxCre+;iDTR+/+ injected with DT or OsxCre+;iDTRFl/+ injected with PBS were used as controls while OsxCre+;iDTRFl/+ injected with DT were mutants. For most experiments, 100 ng of DT/g body weight (BW) was injected daily into both controls and mutants from 4 to 6 weeks old to achieve an acute deletion of specific osteolineage subsets. Mice were harvested the next day after the last dose of DT injection. Injection regimen had been prolonged to 4 weeks for phenotype comparison. For all experiments, littermates were used as controls. C57BL/6J and B6.SJL-PtprcaPepcb/BoyJ (SJL) strains were obtained from Jackson Laboratories. All animal use and procedures performed were approved by the Institutional Animal Care and Use Committee of Massachusetts General Hospital.
Bone Histomorphometry
Paraffin sections of long bones were stained with H&E, von Kossam, and Goldner trichrome, and analyzed using BioQuant software. Statistical analysis was performed using the non-parametric Mann-Whitney test. TRAP staining was used to reveal osteoclasts. Osteolineage activity was assessed by measuring serum levels of OCN and N-terminal propeptide of type I procollagen (P1NP) using RatLaps ELISA kits. Bone resorption was assessed by measuring the C-terminal telopeptides of collagen type I fragments in mouse serum using the RatLaps EIA kit.
Immunohistochemistry
Expression of the DT receptor was detected by immunohistochemistry using an anti-human heparin-binding EGF-like growth factor (anti-hHB-EGF) antibody. In brief, paraffin sections were treated with 0.1% trypsin and 3% H2O2, and blocked with reagent containing 5% animal serum in PBS + Tween 20 for 1 hr. Anti-hHB-EGF antibody (R&D Systems MAB391, 1:25 dilution) was used as primary antibody coupled with biotinylated IgG secondary antibody (Vector Laboratories BA-9500, 1:400 dilution). After staining, sections were treated with a Vectastain ABC Kit for biotin-streptavidin signal amplification and visualized by a DAB Peroxidase Substrate Kit. Osx+ cells were detected by staining sections with anti-SP7 antibody (Abcam Ab22552, 1:2,000 dilution) coupled with secondary antibody conjugated to Alexa Fluor 488 (Thermo Fisher Scientific A27034, 1:1,000 dilution). Ocn+ cells were recognized by anti-OCN antibody (Santa Cruz Biotechnology SC18322, 1:50 dilution) followed by secondary antibody conjugated to Alexa Fluor 488 (Thermo Fisher A11078, 1:1,000 dilution). Green fluorescent OSX- or OCN-antibody staining was then individually superimposed with red fluorescent TUNEL staining (In Situ Cell Death Detection Kit TMR Red, Roche Applied Science).
Microarray Data
Microarray analysis was performed as previously described (Yu et al., 2015b). The Accession number is GEO: GSE66042.
CFU-Ob Assay
The CFU-Ob assay was performed as previously described (Yu et al., 2015a).
Flow Cytometry
Peripheral blood from each mouse was subjected to complete blood count. Tibias, femurs, iliac crests, spines, ulnae, radii, and humeri were harvested for bone marrow cells. Spleen and thymus were collected for lymphocyte staining. Flow cytometry staining for all hematopoietic subpopulations including T cell developmental stages was performed as previously described (Yu et al., 2015a). For B cell development, the following scheme was used: B220-Pacific Blue (BD Biosciences 558108), IgM-PE-Cy5 (eBiosciences 15-5790-82), CD43-fluorescein isothiocyanate (FITC) (eBiosciences 11-0431-82), CD24-APC (Biolegend 101814), and BP1-PE (eBiosciences 12-5891-83). Definitions of stages of B cell maturation were as follows: A′ pre-pro-B (IgM−B220+CD43+BP1−CD24−), B′ pro-B (IgM−B220+CD43+BP1−CD24+), C′ pro-B (IgM−B220+CD43+BP1+CD24lo), C″ pro-B (IgM−B220+CD43+BP1+CD24hi), pro-B (IgM−B220+CD43+), pre-B (IgM−B220+CD43−), B progenitors (IgM−B220+), immature B (IgM+B220lo), and mature B (IgM+B220hi). For cell-cycle analysis, bromodeoxyuridine-FITC and 7AAD, or Ki67-FITC and DAPI staining were coupled with staining of specific populations to reveal their cell-cycle status.
Anti-inflammatory Assay
Indomethacin was injected at 2.5 mg/kg BW daily along with DT throughout the deletion regimen. Mice were euthanized the next day following the last injection. Bone marrow was harvested from femurs, tibias, iliac crests, and spines. After lysis of red blood cells, bone marrow cells were stained with antibodies against monocytes and granulocytes (Mac1, Gr1), macrophages (Mac1), and granulocyte macrophage progenitors (CD127, LIN, SCA-1, C-KIT, CD34, CD16/32) for flow cytometric measurement as previously described (Yu et al., 2015a).
Immune Response Assay
After 1 week of DT injection (day 0), mice were injected with 0.1 μg/μl NP28-AECM-Ficoll/PBS intraperitoneally to trigger T cell-independent Ig production. In this assay, DT injection continued throughout the next 28 days. Mice were re-immunized with a second boost of NP28-AECM-Ficoll at day 14. Mice were bled at days 0, 7, 14, 21, and 28. Sera collected from control and mutant animals were subjected to measurement of IgG and IgM levels by ELISA following standard protocol.
Transplantation
For transplantation of wild-type cells into mutant environment, 1 × 106 bone marrow cells from 6-week-old SJL donors were transplanted into each of ten age-matched 9.5-Gy lethally irradiated OsxCre+;iDTR+/+ control or OsxCre+;iDTRFl/+ mutant recipients. DT injection into both control and mutant recipients began at 7 days before transplantation and were maintained every alternate day until 12 weeks post transplant. Recipients were bled at 4, 8, and 12 weeks post transplant. Peripheral blood was subjected to complete blood cell count and reconstitution analysis as previously published (Yu et al., 2015a). All recipients were euthanized at 12 weeks post transplant. Bone marrow cells were harvested from femurs, tibias, and iliac crests, and subjected to lineage reconstitution analysis as described previously (Yu et al., 2015a). For transplantation of mutant cells into wild-type recipients, 5 × 105 bone marrow cells from 6-week-old mutant OsxCre+;iDTRFl/+ or control OsxCre+;iDTRFl/+ donors were competed with 5 × 105 bone marrow cells from age-matched SJL mice. For both controls and mutants, a total of 1 × 106 mixed donor cells were transplanted into each of ten 9.5-Gy irradiated SJL recipients. Recipients were bled at 4, 8, 12, and 16 weeks post transplant. Peripheral blood was subjected to complete blood cell count and reconstitution analysis by flow cytometry as reported by Yu et al. (2015a). At 16 weeks post transplant, all recipients were euthanized. Bone marrow cells were subjected to lineage reconstitution analysis as described by Yu et al. (2015a).
Cytokine Array
Bones with both ends excised were centrifuged at 12,000 rpm for 1 min for bone marrow extraction. Each sample was further centrifuged at 6,000 rpm for 10 min for serum isolation, and stored at −20°C until use. For measurement of cytokines in circulation, 200 μl of peripheral blood was collected, centrifuged at 6,000 rpm for 10 min for serum isolation, and stored at −20°C until use. A 1/5 dilution of each serum sample was subjected to RayBio Mouse Cytokine Antibody Array G Series 4 (RayBiotech, AAM-CYT-G4-4) (n = 24 per group).
qPCR
Positive hits of cytokine array were validated by qPCR using the GAPDH gene for normalization. Various sources were used for RNA extraction: total bone marrow cells of mutant or control mice, flushed femoral bone of mutant or control mice, and flow sorted cells of stained hematopoietic populations including LKS, CLP, CMP, GMP, MEP, Pro-B, Pre-B, Mature B, and Lineage+ cells.
Rescue Experiments
For in vitro rescue experiments, bone marrow cells were harvested from femurs and tibias of OsxCre;iDTR controls or mutants and plated at 2 × 104 in each well of a six-well plate with methylcellulose-containing medium, supplemented with 100 ng/ml IL-7, 100 ng/ml IGF-1, or both IL-7 and IGF-1. Cells were incubated at 5% CO2 at 37°C undisturbed and enumerated for progenitor colonies by day 8 or day 14. For in vivo rescue experiments, control and mutant OsxCre;iDTR mice were intraperitoneally injected with (1) 100 ng/g BW DT alone, (2) DT and 250 ng/g BW recombinant murine IL-7, and (3) DT and 1 μg/g BW recombinant mouse IGF-1 daily for 12 days. IL-7 and IGF-1 were injected intravenously. Mice were euthanized for bone marrow cell harvest. Cells were stained for GMP- and B cell development-specific antibodies for flow cytometric analysis.
Statistical Analysis
Statistical analysis of all paired experiments was analyzed by two-tailed Student's t test, unless otherwise stated. Data in all graphs represent the mean and SEM where ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001. Statistical comparison of multiple parameters were analyzed by one-way ANOVA followed by Bonferroni post test, where ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001.
Author Contributions
V.W.C.Y. designed, executed, and interpreted data for the majority of experiments in this project and wrote the manuscript. S.L. designed, and together with F.F., A.J., P.S., and R.V. carried out and analyzed the experiments involving the triple fluorescent mouse model. D.T.S. supervised the whole project and was involved in experimental design, data interpretation and manuscript editing. V.W.C.Y., S.L., and F.F. provided editorial input for the manuscript.
Acknowledgments
We thank Dr. Andrew P. McMahon at Harvard University for his generosity in providing the Osx1-GFP::Cre mouse strain. We are thankful to Dr. Rene Maher who provided the Rosa 26-loxP-STOP-loxP-membrane Cherry (Rosa-mCh) mouse. We thank the Harvard Stem Cell Institute Flow Cytometry Core for assistance in flow cytometry. This work was supported by NIH HL044851, HL096372, EB014703 to D.T.S. who was also supported by the Gerald and Darlene Jordan Chair of Medicine.
Published: July 21, 2016
Footnotes
Supplemental Information includes seven figures and two tables and can be found with this article online at http://dx.doi.org/10.1016/j.stemcr.2016.06.009.
Contributor Information
Francesca Ferraro, Email: f.ferraro.mgh@gmail.com.
David T. Scadden, Email: david_scadden@harvard.edu.
Supplemental Information
References
- Baudler S., Baumgartl J., Hampel B., Buch T., Waisman A., Snapper C.M., Krone W., Bruning J.C. Insulin-like growth factor-1 controls type 2 T cell-independent B cell response. J. Immunol. 2005;174:5516–5525. doi: 10.4049/jimmunol.174.9.5516. [DOI] [PubMed] [Google Scholar]
- Bilic-Curcic I., Kronenberg M., Jiang X., Bellizzi J., Mina M., Marijanovic I., Gardiner E.M., Rowe D.W. Visualizing levels of osteoblast differentiation by a two-color promoter-GFP strategy: type I collagen-GFPcyan and osteocalcin-GFPtpz. Genesis. 2005;43:87–98. doi: 10.1002/gene.20156. [DOI] [PubMed] [Google Scholar]
- Buch T., Heppner F.L., Tertilt C., Heinen T.J., Kremer M., Wunderlich F.T., Jung S., Waisman A. A Cre-inducible diphtheria toxin receptor mediates cell lineage ablation after toxin administration. Nat. Methods. 2005;2:419–426. doi: 10.1038/nmeth762. [DOI] [PubMed] [Google Scholar]
- Calvi L.M., Adams G.B., Weibrecht K.W., Weber J.M., Olson D.P., Knight M.C., Martin R.P., Schipani E., Divieti P., Bringhurst F.R. Osteoblastic cells regulate the haematopoietic stem cell niche. Nature. 2003;425:841–846. doi: 10.1038/nature02040. [DOI] [PubMed] [Google Scholar]
- Chan C.K., Seo E.Y., Chen J.Y., Lo D., McArdle A., Sinha R., Tevlin R., Seita J., Vincent-Tompkins J., Wearda T. Identification and specification of the mouse skeletal stem cell. Cell. 2015;160:285–298. doi: 10.1016/j.cell.2014.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding L., Morrison S.J. Haematopoietic stem cells and early lymphoid progenitors occupy distinct bone marrow niches. Nature. 2013;495:231–235. doi: 10.1038/nature11885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferraro F., Lymperi S., Mendez-Ferrer S., Saez B., Spencer J.A., Yeap B.Y., Masselli E., Graiani G., Prezioso L., Rizzini E.L. Diabetes impairs hematopoietic stem cell mobilization by altering niche function. Sci. Transl. Med. 2011;3:104ra101. doi: 10.1126/scitranslmed.3002191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming H.E., Janzen V., Lo Celso C., Guo J., Leahy K.M., Kronenberg H.M., Scadden D.T. Wnt signaling in the niche enforces hematopoietic stem cell quiescence and is necessary to preserve self-renewal in vivo. Cell Stem Cell. 2008;2:274–283. doi: 10.1016/j.stem.2008.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenbaum A., Hsu Y.M., Day R.B., Schuettpelz L.G., Christopher M.J., Borgerding J.N., Nagasawa T., Link D.C. CXCL12 in early mesenchymal progenitors is required for haematopoietic stem-cell maintenance. Nature. 2013;495:227–230. doi: 10.1038/nature11926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y., Strecker S., Wang L., Kronenberg M.S., Wang W., Rowe D.W., Maye P. Osterix-cre labeled progenitor cells contribute to the formation and maintenance of the bone marrow stroma. PLoS One. 2013;8:e71318. doi: 10.1371/journal.pone.0071318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maes C., Kobayashi T., Selig M.K., Torrekens S., Roth S.I., Mackem S., Carmeliet G., Kronenberg H.M. Osteoblast precursors, but not mature osteoblasts, move into developing and fractured bones along with invading blood vessels. Dev. Cell. 2010;19:329–344. doi: 10.1016/j.devcel.2010.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maizels N., Lau J.C., Blier P.R., Bothwell A. The T-cell independent antigen, NP-ficoll, primes for a high affinity IgM anti-NP response. Mol. Immunol. 1988;25:1277–1282. doi: 10.1016/0161-5890(88)90042-9. [DOI] [PubMed] [Google Scholar]
- Mendez-Ferrer S., Michurina T.V., Ferraro F., Mazloom A.R., Macarthur B.D., Lira S.A., Scadden D.T., Ma'ayan A., Enikolopov G.N., Frenette P.S. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature. 2010;466:829–834. doi: 10.1038/nature09262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizoguchi T., Pinho S., Ahmed J., Kunisaki Y., Hanoun M., Mendelson A., Ono N., Kronenberg H.M., Frenette P.S. Osterix marks distinct waves of primitive and definitive stromal progenitors during bone marrow development. Dev. Cell. 2014;29:340–349. doi: 10.1016/j.devcel.2014.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montecino-Rodriguez E., Clark R.G., Powell-Braxton L., Dorshkind K. Primary B cell development is impaired in mice with defects of the pituitary/thyroid axis. J. Immunol. 1997;159:2712–2719. [PubMed] [Google Scholar]
- Nagasawa T. Microenvironmental niches in the bone marrow required for B-cell development. Nat. Rev. Immunol. 2006;6:107–116. doi: 10.1038/nri1780. [DOI] [PubMed] [Google Scholar]
- Nakashima K., Zhou X., Kunkel G., Zhang Z., Deng J.M., Behringer R.R., de Crombrugghe B. The novel zinc finger-containing transcription factor osterix is required for osteoblast differentiation and bone formation. Cell. 2002;108:17–29. doi: 10.1016/s0092-8674(01)00622-5. [DOI] [PubMed] [Google Scholar]
- Omatsu Y., Sugiyama T., Kohara H., Kondoh G., Fujii N., Kohno K., Nagasawa T. The essential functions of adipo-osteogenic progenitors as the hematopoietic stem and progenitor cell niche. Immunity. 2010;33:387–399. doi: 10.1016/j.immuni.2010.08.017. [DOI] [PubMed] [Google Scholar]
- Ono N., Ono W., Mizoguchi T., Nagasawa T., Frenette P.S., Kronenberg H.M. Vasculature-associated cells expressing nestin in developing bones encompass early cells in the osteoblast and endothelial lineage. Dev. Cell. 2014;29:330–339. doi: 10.1016/j.devcel.2014.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park D., Spencer J.A., Koh B.I., Kobayashi T., Fujisaki J., Clemens T.L., Lin C.P., Kronenberg H.M., Scadden D.T. Endogenous bone marrow MSCs are dynamic, fate-restricted participants in bone maintenance and regeneration. Cell Stem Cell. 2012;10:259–272. doi: 10.1016/j.stem.2012.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raaijmakers M.H., Mukherjee S., Guo S., Zhang S., Kobayashi T., Schoonmaker J.A., Ebert B.L., Al-Shahrour F., Hasserjian R.P., Scadden E.O. Bone progenitor dysfunction induces myelodysplasia and secondary leukaemia. Nature. 2010;464:852–857. doi: 10.1038/nature08851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodda S.J., McMahon A.P. Distinct roles for Hedgehog and canonical Wnt signaling in specification, differentiation and maintenance of osteoblast progenitors. Development. 2006;133:3231–3244. doi: 10.1242/dev.02480. [DOI] [PubMed] [Google Scholar]
- Sato M., Asada N., Kawano Y., Wakahashi K., Minagawa K., Kawano H., Sada A., Ikeda K., Matsui T., Katayama Y. Osteocytes regulate primary lymphoid organs and fat metabolism. Cell Metab. 2013;18:749–758. doi: 10.1016/j.cmet.2013.09.014. [DOI] [PubMed] [Google Scholar]
- Strecker S., Fu Y., Liu Y., Maye P. Generation and characterization of Osterix-Cherry reporter mice. Genesis. 2013;51:246–258. doi: 10.1002/dvg.22360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taguchi T., Takenouchi H., Matsui J., Tang W.R., Itagaki M., Shiozawa Y., Suzuki K., Sakaguchi S., Ktagiri Y.U., Takahashi T. Involvement of insulin-like growth factor-I and insulin-like growth factor binding proteins in pro-B-cell development. Exp. Hematol. 2006;34:508–518. doi: 10.1016/j.exphem.2006.01.009. [DOI] [PubMed] [Google Scholar]
- Tokoyoda K., Egawa T., Sugiyama T., Choi B.I., Nagasawa T. Cellular niches controlling B lymphocyte behavior within bone marrow during development. Immunity. 2004;20:707–718. doi: 10.1016/j.immuni.2004.05.001. [DOI] [PubMed] [Google Scholar]
- Visnjic D., Kalajzic Z., Rowe D.W., Katavic V., Lorenzo J., Aguila H.L. Hematopoiesis is severely altered in mice with an induced osteoblast deficiency. Blood. 2004;103:3258–3264. doi: 10.1182/blood-2003-11-4011. [DOI] [PubMed] [Google Scholar]
- Welniak L.A., Karas M., Yakar S., Anver M.R., Murphy W.J., LeRoith D. Effects of organ-specific loss of insulin-like growth factor-I production on murine hematopoiesis. Biol. Blood. Marrow. Transplant. 2004;10:32–39. doi: 10.1016/j.bbmt.2003.09.008. [DOI] [PubMed] [Google Scholar]
- Worthley D.L., Churchill M., Compton J.T., Tailor Y., Rao M., Si Y., Levin D., Schwartz M.G., Uygur A., Hayakawa Y. Gremlin 1 identifies a skeletal stem cell with bone, cartilage, and reticular stromal potential. Cell. 2015;160:269–284. doi: 10.1016/j.cell.2014.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J.Y., Purton L.E., Rodda S.J., Chen M., Weinstein L.S., McMahon A.P., Scadden D.T., Kronenberg H.M. Osteoblastic regulation of B lymphopoiesis is mediated by Gs{alpha}-dependent signaling pathways. Proc. Natl. Acad. Sci. USA. 2008;105:16976–16981. doi: 10.1073/pnas.0802898105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J.Y., Scadden D.T., Kronenberg H.M. Role of the osteoblast lineage in the bone marrow hematopoietic niches. J. Bone Miner. Res. 2009;24:759–764. doi: 10.1359/jbmr.090225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu V.W., Scadden D.T. Hematopoietic stem cell and its bone marrow niche. Curr. Top. Dev. Biol. 2016;118:21–44. doi: 10.1016/bs.ctdb.2016.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu V.W., Scadden D.T. Heterogeneity of the bone marrow niche. Curr. Opin. Hematol. 2016;23:331–338. doi: 10.1097/MOH.0000000000000265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu V.W., Saez B., Cook C., Lotinun S., Pardo-Saganta A., Wang Y.H., Lymperi S., Ferraro F., Raaijmakers M.H., Wu J.Y. Specific bone cells produce DLL4 to generate thymus-seeding progenitors from bone marrow. J. Exp. Med. 2015;212:759–774. doi: 10.1084/jem.20141843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu V.W.C., Lymperi S., Ferraro F., Scadden D.T. Transcriptome comparison of distinct osteolineage subsets in the hematopoietic stem cell niche using a triple fluorescent transgenic mouse model. Genomics Data. 2015;5:318–319. doi: 10.1016/j.gdata.2015.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J., Niu C., Ye L., Huang H., He X., Tong W.G., Ross J., Haug J., Johnson T., Feng J.Q. Identification of the haematopoietic stem cell niche and control of the niche size. Nature. 2003;425:836–841. doi: 10.1038/nature02041. [DOI] [PubMed] [Google Scholar]
- Zhou B.O., Yue R., Murphy M.M., Peyer J.G., Morrison S.J. Leptin-receptor-expressing mesenchymal stromal cells represent the main source of bone formed by adult bone marrow. Cell Stem Cell. 2014;15:154–168. doi: 10.1016/j.stem.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J., Garrett R., Jung Y., Zhang Y., Kim N., Wang J., Joe G.J., Hexner E., Choi Y., Taichman R.S. Osteoblasts support B-lymphocyte commitment and differentiation from hematopoietic stem cells. Blood. 2007;109:3706–3712. doi: 10.1182/blood-2006-08-041384. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.