

Summary
Induced pluripotent stem cells (iPSCs) represent an innovative source for the standardized in vitro generation of macrophages (Mφ). We here describe a robust and efficient protocol to obtain mature and functional Mφ from healthy as well as disease-specific murine iPSCs. With regard to morphology, surface phenotype, and function, our iPSC-derived Mφ (iPSC-Mφ) closely resemble their counterparts generated in vitro from bone marrow cells. Moreover, when we investigated the feasibility of our differentiation system to serve as a model for rare congenital diseases associated with Mφ malfunction, we were able to faithfully recapitulate the pathognomonic defects in GM-CSF signaling and Mφ function present in hereditary pulmonary alveolar proteinosis (herPAP). Thus, our studies may help to overcome the limitations placed on research into certain rare disease entities by the lack of an adequate supply of disease-specific primary cells, and may aid the development of novel therapeutic approaches for herPAP patients.
Keywords: macrophages, PAP, iPSC, disease modeling, hematopoiesis
Graphical Abstract
Highlights
-
•
Myeloid differentiation of murine iPSCs into mature Mφ (iPSC-Mφ)
-
•
Functional iPSC-Mφ similar to bone marrow lineage-negative cell-derived Mφ
-
•
Disease modeling of defects in GM-CSF signaling and Mφ function present in herPAP
In this article, Lachmann and colleagues describe a highly efficient and robust protocol to obtain mature Mφ (iPSC-Mφ) from healthy as well as disease-specific murine iPSCs, which display characteristic morphology, surface phenotype, and functionality. Moreover, the protocol was applied to successfully model the pathognomonic defects in GM-CSF signaling and Mφ function present in hereditary pulmonary alveolar proteinosis (herPAP).
Introduction
Following the groundbreaking work of Takahashi and Yamanaka in 2006 (Takahashi et al., 2007, Takahashi and Yamanaka, 2006), induced pluripotent stem cells (iPSCs) and their in vitro differentiation into appropriate cell types or tissues have become a powerful tool to model defined disease entities as well as to study pathophysiology and new therapeutic options. With regard to rare diseases, this approach also allows us to overcome the limited availability of primary patient cells, a problem still hampering research in this area to a considerable extent. In this respect, iPSCs and derived progeny thereof constitute a valuable and potentially highly standardized source for the derivation of patient- and disease-specific cells for medical research. This also applies to diseases caused by defects in progenitor or mature effector cells of the hematopoietic system, such as lymphocytes (ADA-SCID, X-SCID) (Menon et al., 2015, Park et al., 2008), granulocytes and myeloid progenitors (chronic granulomatous disease, juvenile myelomonocytic leukemia) (Gandre-Babbe et al., 2013, Jiang et al., 2012, Ye et al., 2009), erythrocytes (thalassemias, sickle cell disease) (Ma et al., 2013, Tubsuwan et al., 2013, Zou et al., 2011), or megakaryocytes (congenital amegakaryocytic thrombocytopenia) (Hirata et al., 2013), which all have been successfully recapitulated in vitro utilizing iPSC-based models.
Macrophages (Mφ) constitute another highly interesting cell population in this context. In the past years, our understanding of Mφ biology has been considerably extended, especially with regard to tissue-resident populations, such as osteoclasts, Langerhans' and Kupffer cells, microglia, or peritoneal and alveolar Mφ (Davies et al., 2013). While traditionally tissue-resident Mφ (TRM) have been regarded as being continuously replenished from the hematopoietic stem cells via the intermediate step of peripheral blood monocytes, this concept now has been challenged for many tissues. Thus, it has recently been demonstrated that substantial populations of TRMs are already seeded prenatally, exhibit considerable longevity (months to years), and have, at least in part, self-renewal potential (Gomez Perdiguero et al., 2015, Hashimoto et al., 2013). Furthermore, a high degree of plasticity has been demonstrated for Mφ and TRMs, allowing them to adjust to the specific functional requirements of the surrounding tissues, e.g., following organ-specific transplantation (Lavin et al., 2014).
For several of these Mφ and TRM populations, a direct and causative role has been identified in the pathophysiology of distinct congenital diseases, including heme oxygenase 1 deficiency (Kovtunovych et al., 2014), adrenoleukodystrophy (Biffi et al., 2011, Cartier et al., 2014), autosomal recessive osteopetrosis (Neri et al., 2015), Gaucher disease (Sgambato et al., 2015), and mucopolysaccharidosis type I (Viana et al., 2016). Another example in this context is hereditary pulmonary alveolar proteinosis (herPAP), a life-threatening, rare pulmonary disease caused by mutations in CSF2RA or CSF2RB encoding the α or β chain of the granulocyte macrophage colony-stimulating factor (GM-CSF) receptor, respectively (Suzuki et al., 2008, Suzuki et al., 2011). These mutations lead to receptor dysfunction and altered GM-CSF response. As GM-CSF signaling is especially important for alveolar Mφ differentiation, maturation, and function, the corresponding signaling defect leads to an impaired capacity of alveolar Mφ to clear the alveolar spaces from proteins and phospholipids (Hansen et al., 2008, Trapnell and Whitsett, 2002). Clinically this deficiency leads to progressive, life-threatening respiratory insufficiency and an increased susceptibility to pulmonary infections. Current treatment options of herPAP are purely symptomatic and comprise vigorous treatment of infections and repetitive bilateral whole lung lavage, an invasive procedure requiring general anesthesia and associated with significant cardiovascular risks (Trapnell et al., 2003, Trapnell et al., 2009).
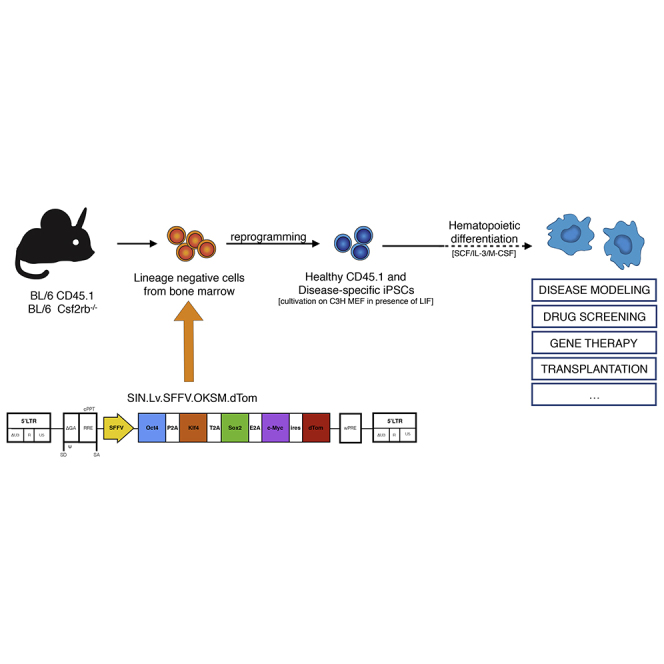
Against this background, we have investigated the feasibility of utilizing murine iPSCs and their hematopoietic differentiation as a model system for congenital diseases caused by deficiencies in the Mφ compartment. In this regard, and extending the previous work of our group (Pfaff et al., 2012), we have established a robust protocol allowing for the differentiation of functional murine Mφ from iPSCs. Using this protocol, we then employed iPSC lines derived from a mouse model of herPAP to model the GM-CSF receptor dysfunction pathognomonic of this disease.
Results
Generation and Characterization of CD45.1iPSCs
Initial studies were performed with iPSCs generated from BMlin− cells of healthy CD45.1 BL/6 mice (B6.SJL-PtprcaPepcb/BoyJ). CD45.1 cells were used for reprogramming to allow for the easy tracking of respective progeny in classical C57BL/6-based murine transplant models including Csf2rb−/− PAP mice (Nishinakamura et al., 1995), which are based on CD45.2 (C57BL/6J) recipients. Reprogramming was achieved by transduction with a polycistronic third-generation SIN lentiviral vector expressing the classical “Yamanaka” factors OCT3/4, KLF4, SOX2, and c-MYC (SIN.Lv.SFFV.OKSM.dTom) (Kuehle et al., 2014). Several clones were generated and, based on its hematopoietic differentiation potential, one clone was selected for further characterization. This clone, termed CD45.1(10.3) iPSCs, showed classical embryonic stem cell (ESC)-like morphology (Figure 1A) and displayed all major characteristics of pluripotency such as alkaline phosphatase activity (Figure 1B), endogenous expression of the transcription factors Nanog, Oct4, and Sox2 (Figures 1C and 1D), expression of the stage-specific embryonic antigen 1 (SSEA-1; Figure 1E), and, most importantly, formation of teratomas that comprised tissues of all three embryonic germ layers upon subcutaneous transplantation into NOD/SCID/γcnull mice (Figure 1F). Two further clones named CD45.1(1.1) and CD45.1(10.4) iPSC were characterized for ESC-like morphology, alkaline phosphatase activity, and expression of SSEA-1 as well as NANOG, SOX2, and OCT4 expression (Figures S1A–S1F).
Figure 1.

Characterization of iPSCs Derived from CD45.1 C57BL/6 Mice
(A and B) ESC-like morphology in bright-field images (A) and positive alkaline phosphatase staining of CD45.1(10.3) iPSCs (B). Scale bar, 200 μm.
(C and D) NANOG, OCT4, and SOX2 expression by immunofluorescence staining (C) (scale bar, 50 μm) as well as by (D) qRT-PCR using murine specific primers (independent experiments, n = 3, mean ± SD). ns, not significant compared with ESCs, two-way ANOVA.
(E) Representative flow cytometry plot revealing expression of the SSEA-1 surface marker (unstained, orange filled; SSEA-1 stained CD45.1(10.3) iPSCs, green).
(F) Representative pictures of CD45.1(10.3) iPSC-derived teratomas containing tissues of all three embryonic germ layers. Scale bar, 50 μm for ectoderm and endoderm; 100 μm for mesoderm.
Differentiation of CD45.1iPSCs into Mφ
Differentiation studies of murine CD45.1iPSCs into Mφ utilized an embryoid body (EB)-based protocol previously established by our group and employing the cytokines murine stem cell factor (mSCF) and murine interleukin-3 (mIL-3) to drive hematopoietic differentiation and generate immature CD41+ hematopoietic stem/progenitor cells (Pfaff et al., 2012). For the current studies, the protocol was modified to perform suspension cultures of EBs on an orbital shaker, and to achieve terminal Mφ differentiation in secondary cultures employing macrophage colony-stimulating factor (M-CSF) treatment of CD41+ cells from day 8 onward (Figures 2A and 2B). Secondary cultures were maintained for at least another 10 days, at which time point cells displayed plastic adherence and Mφ-typical morphology. Following this protocol, expression of the early hematopoietic marker CD41, traditionally used to define a primitive hematopoietic population in the aorta-gonad-mesonephros region of mice (Rybtsov et al., 2011), was detected in 25%–70% of cells on day 8 for the CD45.1(10.3) iPSC clone. Of note, most of CD41+ cells displayed classical hematopoietic progenitor (blast-like) morphology on May-Grünwald/Giemsa-stained cytospins (Figure 2C). When secondary cultures set up from these CD41+ cells were harvested after another 10–20 days of differentiation in the presence of M-CSF, an approximately 10-fold increase in cell numbers was observed (Figure 2D). Mφ characteristics of almost all cells (>95% purity) were verified by typical, spread-out morphology upon bright-field microscopy and strong adherence to non-tissue culture-treated plates as well as a classical Mφ phenotype on May-Grünwald/Giemsa-stained cytospins resembling BMlin−-Mφ used as controls (Figures 2E and 2F). CD45.1(10.3) iPSC-Mφ displayed the classical Mφ marker F4/80 in association with the myeloid marker CD11b on their surface (Figures 2G and 2H). Moreover, cells expressed high levels of the pan-hematopoietic marker CD45, here specifically CD45.1 (Ly5.1), whereas markers of alternative hematopoietic lineages such as Gr-1, CD11c, CD3, and B220 were low or absent.
Figure 2.

Differentiation of iPSCs into Mφ
(A) Schematic representation of the three step EB-based protocol employed for hematopoietic differentiation. (1) embryoid bodies (EBs) are formed for 8 days in the presence of 10 ng/ml IL-3 with 30 ng/ml SCF added from day 5 onward; (2) EBs are dissociated to single cells and CD41+ cells are isolated by fluorescence-activated cell sorting; and (3) terminal differentiation is achieved by 10–15 days of adherent culture in the presence of 30% M-CSF containing supernatant (L929 cells).
(B) Representative picture of EBs at day 8. Scale bar, 500 μm.
(C) Flow cytometric analysis of CD41 expression on day 8 and representative picture of May-Grünwald/Giemsa-stained cytospins of CD41+ cells. Scale bar, 50 μm.
(D) Approximately 10-fold increase in cell number during differentiation (independent experiments, n = 3, mean ± SD).
(E and F) Representative pictures of (E) BMlin−-Mφ and (F) CD45.1 iPSC-Mφ in bright-field and in May-Grünwald/Giemsa-stained cytospins. Scale bars, 100 μm and 20 μm.
(G and H) Exemplary flow cytometry plots for expression of surface markers F4/80, CD11b, CD14, CD11c, Gr-1, CD45.1, CD45.2, CD41, CD3, and B220 on BMlin−-Mφ (G) and CD45.1iPSC-Mφ (H).
To assess the robustness of our differentiation protocol, we compared hematopoietic differentiation of the CD45.1(10.3) iPSC wild-type control clone with two further CD45.1 iPSC clones (clones 1.1 and 10.4), two iPSC clones derived from BMlin− cells (lin-7 and -9) as well as a murine ESC clone (OG2 ESCs). Of note, for all these clones our protocol yielded reliable macrophage differentiation, which was confirmed by classical macrophage morphology in cultures and on May-Grünwald/Giemsa-stained cytospins (Figures S2A–S2E) as well as a CD45+, CD11b+, and F4/80+ surface phenotype associated with low or absent expression of other hematopoietic lineage markers such as CD14, CD11c, Gr-1, CD3, B220, and CD41 (Figures S2F–S2J). However, considerable clone-specific differences were detected with regard to the numbers of CD41+ cells and macrophages obtained for a given iPSC input (Figure S3).
When compared with Mφ derived from different in vivo sources (blood, peritoneal cavity, bronchoalveolar lavage fluid) or generated in vitro from BMlin− cells, iPSCs-Mφ [here represented by cells derived from clone CD45.1(10.3) iPSC] most closely resembled their in vitro-generated counterparts (Table 1). While all cells analyzed expressed high levels of CD45 and were positive for the common Mφ markers F4/80, in accordance with the current literature, CD11b was expressed highest on monocytes and peritoneal Mφ, and CD11c and SIGLEC-F was detected primarily on alveolar Mφ (Feng and Mao, 2012, Hussell and Bell, 2014).
Table 1.
Marker Expression of Different Monocyte/Macrophage Populations
| In Vivo Derived Cell Types |
In Vitro Derived Cell Types |
||||
|---|---|---|---|---|---|
| Blood Monocytes | Peritoneal Mφ | Alveolar Mφ | BMlin-Mφ | CD45.1iPSC Mφ | |
| CD45 | +++ | +++ | ++ | ++ | +++ |
| CD11b | ++ | +++ | – | + | + |
| CD11c | + | – | ++ | + | + |
| CD200R | – | + | + | ++ | + |
| F4/80 | + | ++ | + | +++ | + |
| SIGLEC-F | – | – | + | + | + |
Mean fluorescence intensities (MFIs) lower than 3 × 102 are – (no expression), MFIs between 3 × 102 and 104 are + (low expression), MFIs between 104 and 5 × 104 are ++ (intermediate expression), and MFIs greater than 5 × 104 are +++ (high expression).
In addition, we analyzed the functionality of our CD45.1(10.3) iPSC-Mφ with regard to the release of chemokines in response to extracellular stimuli as well as their phagocytic capacity. In these studies, CD45.1(10.3) iPSC-Mφ produced chemokines typically secreted by Mφ upon bacterial endotoxin stimulation and mediating crucial functions in the inflammatory response such as monocyte chemoattractant protein 1 (MCP-1), MCP-3, macrophage inflammatory protein 1β (MIP-1β), and RANTES. Chemokine levels induced in CD45.1(10.3) iPSC-Mφ in response to lipopolysaccharide (LPS) stimulation were similar to the levels induced in their BMlin− cell-derived counterparts (Figure 3A). To assess the phagocytic capacity of CD45.1(10.3) iPSC-Mφ, we tested their ability to internalize yeasts using the number of ingested yeast particles per cell as a readout. These studies also demonstrated similar functionality of CD45.1iPSC- and BMlin−-Mφ (Figure 3B). Of note, immature cells such as iPSCs were not able to take up yeast cells.
Figure 3.

Functional Characterization of iPSC-Mφ
(A) Secretion of monocyte chemoattractant protein 1 (MCP-1), -3 (MCP-3), macrophage inflammatory protein 1β (MIP-1β), and RANTES upon stimulation of BMlin−- and CD45.1(10.3) iPSC-Mφ with LPS (technical replicates n = 2, mean ± SD).
(B) Phagocytosis of yeast particles by undifferentiated iPSCs, BMlin−-, and CD45.1iPSC-Mφ (technical replicates n = 2, mean ± SD).
To rule out the presence of residual undifferentiated iPSCs that could give rise to teratomas and thus may be problematic in future in vivo experiments employing our iPSC-Mφ, we performed transplant experiments in immunodeficient NSG mice. In these studies, no teratomas were detected 5 weeks after subcutaneous injection of 3 × 106 CD45.1(10.3) iPSC-Mφ into the flanks or 4 weeks after intratracheal application of 1.8–2.3 × 106 CD45.1(10.3) iPSC-Mφ into the airways of NSG mice (both n = 2). However, we did detect a substantial population of donor-derived cells in the lungs of recipient animals following the intratracheal transplant procedure.
Moreover, to further assess the feasibility to perform in vivo studies with our iPSC-Mφ we investigated the pulmonary engraftment and survival of CD45.1 iPSC(10.3)-Mφ in our murine herPAP disease model 8 weeks following intratracheal application of 4 × 106 cells (n = 1). In this experiment the presence of a substantial population of CD45.1+ donor type (i.e., CD45.1(10.3) iPSC-Mφ) cells was detected, contributing 3.1% and 1.2% to the respective populations of the bronchoalveolar fluid and the lungs (Figures S4A–S4C).
Generation and Hematopoietic Differentiation of Murine PAP-Specific iPSCs from Csf2rb−/− Mice
As a next step, we wanted to investigate the feasibility of our in vitro differentiation system to serve as a model for rare congenital diseases associated with Mφ malfunction. In this regard, disease-specific miPAP were generated from BMlin− cells of Csf2rb−/− mice (Nishinakamura et al., 1995), a mouse strain that reliably recapitulates the disease phenotype of herPAP patients (Happle et al., 2014). Two clones (miPAP1 and miPAP2) were established that showed characteristic morphology (Figures 4A and S5A) and pluripotency features such as alkaline phosphatase activity, endogenous expression of the pluripotency-associated transcription factors Oct4 and Nanog, SSEA1 expression on the cell surface, and the ability to form teratomas in immunodeficient mice (Figures 4B–4F and S5B–S5F). The knockout of the GM-CSF receptor β-chain gene (Csf2rb) in these clones was confirmed by PCR on genomic DNA (Figure 4G). When we subjected these cells to our differentiation protocol, we again obtained typical EBs giving rise to CD41+ cells on day 8 of differentiation (Figures 5A, 5B, S5G, and S5H). In the presence of M-CSF these CD41+ cells could be further differentiated to Mφ with classical morphology and an F4/80+ CD11b+ CD45.2+ surface phenotype (Figures 5C, 5D, and S5I–S5K). In this regard, the data mimicked those obtained with the different CD45.1 iPSCs clones generated from healthy cells. In quantitative terms, however, efficacy of macrophage differentiation for the two miPAP clones was rather low, resulting in a markedly (at least 1–2 log) decreased macrophage output when compared with input iPSCs.
Figure 4.

Characterization of Disease-Specific iPSCs from Csf2rb−/− Mice
(A and B) ESC-like morphology in bright-field images (A) and positive alkaline phosphatase staining of miPAP1 iPSCs (B). Scale bar, 200 μm.
(C) NANOG, OCT4, and SOX2 expression by immunofluorescence staining (C; scale bar, 50 μm) as well as by (D) qRT-PCR using murine-specific primers (independent experiments, n = 3, mean ± SD). ns, not significant compared with ESCs, two-way ANOVA.
(E) Representative flow cytometry plot revealing expression of the SSEA-1 surface marker.
(F) Representative pictures of miPAP1-derived teratomas containing tissues of all three embryonic germ layers. Scale bar, 50 μm for ectoderm and endoderm; 100 μm for mesoderm.
(G) Scheme and gel electrophoresis of PCR on genomic DNA for genotyping miPAP clones.
Figure 5.

Differentiation of miPAP1 into Mφ
(A and B) Representative picture of (A) bright-field EBs at day 8 of differentiation (scale bar, 500 μm) and (B) flow cytometric analysis of CD41 expression.
(C) Representative pictures of (upper picture; scale bar, 100 μm) bright-field microscopy and (lower picture; scale bar, 20 μm) May-Grünwald/Giemsa-stained cytospins of miPAP1-Mφ.
(D) Flow cytometric analysis of surface marker expression (data for F4/80, CD11b, CD14, CD11c, Gr-1, CD45.1, CD45.2, CD41, CD3, and B220).
miPAP-Derived Macrophages Display Perturbed GM-CSF-Mediated Functionalities
To investigate whether our differentiation procedure was able to recapitulate the defect in the GM-CSF receptor functionality typical for herPAP, we first tested the capacity of CD41+ progenitors generated from miPAPs to form colonies in the presence of GM-CSF. Here, CD41+ cells derived from miPAP clones did not form colonies in the presence of GM-CSF only (Figure 6A). This was in contrast to CD45.1(10.3) iPSC-derived CD41+ cells, which gave rise to GM-CSF-dependent white colonies. Of note, control cultures with a combination of IL-3, SCF, IL-6, erythropoietin, granulocyte colony-stimulating factor (G-CSF), and M-CSF (complete medium) served as positive controls for both CD45.1iPSC- and miPAP-derived CD41+ cells. Similar results were obtained when miPAP-derived terminally differentiated Mφ were evaluated (miPAP-Mφ). When these cells were tested for the ability to clear GM-CSF from the medium, the cytokine concentration remained constant in the supernatant of miPAP-Mφ, whereas healthy CD45.1(10.3) iPSC-Mφ reduced GM-CSF concentrations over a time period of 30 hr (Figure 6B). Moreover, to study signaling from the GM-CSF receptor in our model in more detail, we evaluated the activation of downstream targets. Here we focused on the phosphorylation state of the transcription factor STAT5, one of the most important downstream targets of GM-CSF receptor signaling. As depicted for an exemplary experiment (Figure 6C) and summarized in Figures 6D and 6A, significant upregulation of the phosphoSTAT5 signal was detected in Mφ derived from CD45.1(10.3) iPSCs employed as a positive control. In contrast, perturbed upregulation was detectable in Mφ derived from the two miPAP clones, again underscoring the suitability of our in vitro differentiation system to model the defective Mφ functionality associated with herPAP.
Figure 6.

Disease Modeling of herPAP Using miPAP iPSCs
(A) Number of CD41+ progenitor-derived colonies in methylcellulose-based clonogenic assays employing complete (IL-6, erythropoietin, SCF, IL-3, and supplemented with 20 ng/ml human G-CSF + 20 ng/ml murine GM-CSF) or basic medium (50 ng/ml murine GM-CSF only; independent experiments, n = 3 miPAP1 and 2, n = 2 CD45.1 iPSC, mean ± SD) and representative pictures of colonies. Scale bars, 500 μm.
(B) GM-CSF clearance assay comparing miPAP-Mφ with CD45.1(10.3) iPSC-Mφ, BMlin−-Mφ, and no cells incubated with 2 ng/ml GM-CSF: at indicated time points (0, 4, 10, 24, and 30 hr) GM-CSF concentrations in supernatants were analyzed by ELISA, normalized to 0 hr (independent experiments, n = 3 BMlin− and CD45.1 iPSC, n = 2 miPAP1 and 2, mean ± SD).
(C and D) Flow cytometry plots of STAT5 phosphorylation levels upon stimulation with mGM-CSF (C) and (D) summary of mean fluorescence intensity (MFI) data (independent experiments, n = 3 BMlin− and CD45.1 iPSC, n = 2 miPAP1 and 2, mean ± SD).
ns, not significant; ∗∗p ≤ 0.01, ∗∗∗p ≤ 0.001, ∗∗∗∗p ≤ 0.0001, two-way ANOVA.
Discussion
Extending on previously established protocols initially designed to generate CD41+ hematopoietic progenitor cells (Ma et al., 2008, Pfaff et al., 2012), we here describe a robust and highly reproducible hematopoietic differentiation protocol allowing for the efficient generation of mature and functional Mφ from murine iPSC and ESC sources. The Mφ obtained recapitulate the morphology, surface phenotype, and functionality of their counterparts differentiated in vitro from BMlin− hematopoietic progenitor cells. Differentiation was associated with profound cell expansion, as in our hands up to 1.5 × 105 Mφ were generated from a starting population of 1,000 iPSCs from our reference CD45.1(10.3) iPSC clone. This efficacy appears to compare favorably with data from other groups, including a recently published study investigating the generation of AM-type macrophages from iPSCs sources reporting an approximately one-to-one ratio of starting iPSCs to AM φ generated (Litvack et al., 2016), although a direct comparison turns out to be difficult because of different starting populations as well as the diversity of methods employed (Neri et al., 2015, Senju et al., 2009, Zhuang et al., 2012). However, we observed marked differences in the efficacy of Mφ generation when we investigated a variety of iPSC and ESC clones. While some of these clones performed even better than the CD45.1(10.3) iPSC reference clone, in particular our disease-specific miPAP clones demonstrated a markedly reduced output of CD41+ cells as well as macrophages. Most likely this can be explained by the lack of the common GM-CSF receptor β chain (βc) in the miPAP clones, as the β chain also is involved in conventional IL-3 signaling and our differentiation protocol primarily employs the cytokines SCF and IL-3 to drive hematopoietic specification. Even though murine cells have an additional “private” IL-3 signal transduction chain independent from the common GM-SCF/IL-3/IL-5 receptor complex (Nishinakamura et al., 1995), the lack of the β chain may contribute to the reduced expansion efficacy observed for the miPAP clones. On the other hand, these differences also may reflect the considerable genetic and epigenetic differences present between individual iPSC and ESC clones and sometimes even between different passage cells of the same clone (Liang and Zhang, 2013).
We believe that our iPSC-Mφ hold considerable potential for disease modeling, in particular for disease entities associated with Mφ malfunction. Therefore, as a proof of concept, we generated herPAP-specific iPSCs from mice deficient for the GM-CSF receptor βc chain, differentiated these cells into Mφ, and analyzed GM-CSF-dependent functions in these cells. As previously described for primary Mφ of these mice including alveolar Mφ (Happle et al., 2014, Kleff et al., 2008), these in vitro studies recapitulated the characteristic defects in the GM-CSF response of herPAP patient-derived Mφ. Specifically, defects in GM-CSF-driven colony formation and terminal differentiation capacity, GM-CSF uptake, as well as the phosphorylation of STAT-5 downstream of the GM-CSF receptor upon GM-CSF stimulation were demonstrated. Given the extreme rarity of CSF2RB-deficient PAP patients (Suzuki et al., 2011) and the ensuing lack of primary patient material for research in this disease entity, the murine iPSC-based in vitro herPAP model presented here may have immediate relevance. Provided that the number of macrophages obtained from our miPAP clones can be increased, the model presented here may be employed for pathophysiologic studies as well as for drug testing. An example would be a screen for pharmaceutical compounds potentially ameliorating the PAP phenotype such as inducers of the transcription factor PU.1 or the nuclear receptor PPARγ (Malur et al., 2011, Shibata et al., 2001).
Recently, profound therapeutic efficacy of intratracheally transplanted bone marrow-derived Mφ has been demonstrated in two murine disease models of herPAP, indicating a therapeutic potential also of iPSC-derived Mφ (Happle et al., 2014, Suzuki et al., 2014a). In this context genetically repaired human herPAP patient-specific iPSCs lines already have been generated, and functional Mφ derived from these lines potentially represent an autologous cell source for herPAP therapy (Lachmann et al., 2014, Suzuki et al., 2014b). Moreover, in a murine disease model of adenosine deaminase deficiency, therapeutic benefit has been demonstrated following the intrapulmonary administration of iPSC-derived AM-like cells with a characteristic F4/80+ CD11c+, SIGLEC-F+ surface phenotype and an AM-typical gene-expression pattern (Litvack et al., 2016). On this background pulmonary macrophage transfer of autologous gene-corrected iPSC-Mφ may represent an innovative therapeutic option for other disease entities associated with malfunction of AMs such as chronic obstructive pulmonary disease (Hodge et al., 2003), bronchopulmonary dysplasia (Blackwell et al., 2011), or cystic fibrosis (Bruscia et al., 2009, Di et al., 2006). Moreover, site-specific transplantation of iPSC-Mφ into organs such as the liver or the brain may turn out to be relevant also for other diseases associated with Mφ malfunction such as heme oxygenase-1 deficiency (Kovtunovych et al., 2014) or adrenoleukodystrophy (Okamura et al., 2009).
In summary, we here describe a highly robust and efficient protocol for the generation of murine iPSC-Mφ, which phenotypically and functionally show high similarity to their primary Mφ counterparts. Moreover, we employed this protocol to obtain disease-specific iPSC-Mφ from herPAP mice and recapitulated in vitro the hallmark Mφ phenotype of this disorder. Thus, this study introduces a unique tool for the study of Mφ and iPSC-Mφ biology in healthy and disease contexts, provides an in vitro disease model to study CSF2RB-deficient herPAP, and may eventually contribute a platform upon which to develop advanced therapeutic approaches in herPAP or other severe Mφ-related disease entities.
Experimental Procedures
Bone Marrow Isolation and Lineage Depletion
Femurs and tibias of wild-type B6.SJL-PtprcaPepcb/BoyJ (CD45.1 BL/6) and C57BL/6 Csf2rb−/− (Nishinakamura et al., 1995) mice were flushed with PBS supplemented with 2% heat-inactivated fetal calf serum (FCS; Millipore) and 2 mM EDTA (Sigma). Subsequently, bone marrow lineage-negative cells (BMlin−) were purified using immunomagnetic separation (mouse lineage depletion kit, Miltenyi Biotech) as recommended by the manufacturer. BMlin− cells were maintained in serum-free STIF medium as described previously (Brennig et al., 2015).
Lentiviral Particles Production
Production and titration of third-generation self-inactivating (SIN) lentiviral vectors was performed as previously described (Lachmann et al., 2015) and titers of 105–106 TU/ml were achieved.
Reprogramming of BMlin− Cells
5 × 105 BMlin− cells were transduced with an established third-generation monocistronic SIN lentiviral vector containing the four pluripotency factors OCT4, SOX2, KLF4, and c-MYC expressed form the viral promoter SFFV (Kuehle et al., 2014). Transduction was performed on retronectin-coated plates (Takara Bio) according to the manufacturer's instructions. Subsequently, cells were cultured in STIF medium supplemented with 2 mM valproic acid (Sanofi). After 7 days, cells were moved onto C3H murine embryonic fibroblasts (MEF), and an equal amount of mESC medium (see below) containing 103 U/ml leukemia inhibitory factor (LIF) (kindly provided by the Institute of Technical Chemistry, Hannover Medical School) was added to the culture at this time point and completely substituted after 14 days. Approximately 21 days later, the first ESC-like colonies were picked from the original plate and single colonies were trypsinized and expanded on C3H MEF.
Murine iPSC Cell Culture
Murine ESCs and iPSCs (OG2 ESCs [Szabó et al., 2002], CD45.1 iPSCs clones 10.3, 1.1, and 10.4, miPAP 1 and 2, and iPSC lin-7 and lin-9 [Pfaff et al., 2012]) were maintained in Knockout DMEM medium (Thermo Fisher Scientific) supplemented with 15% FCS, 1 mM penicillin-streptomycin, 1 mM L-glutamine, 0.05 mM β-mercaptoethanol, 1 mM non-essential amino acids, and 103 U/ml LIF on C3H MEF as previously described (Ackermann et al., 2014).
Immunofluorescence and Alkaline Phosphatase Staining
Cells were plated in a 24-well plate at low confluence, fixed in 4% paraformaldehyde (Sigma-Aldrich), permeabilized in 0.7% Triton X-100 (Carl Roth), and blocked with the appropriate serum. Nuclei were stained with DAPI (dilution 1:4,000) for 45 min and primary as well as subsequent secondary antibodies were incubated for 1 hr at room temperature. Primary antibodies: anti-hOCT3/4 (mouse monoclonal antibody [mAb] C-10, sc-5279, Santa Cruz Biotechnology, dilution 1:250), anti-hSOX2 (goat primary antibody [pAb] Y-17, sc17320, Santa Cruz, dilution 1:100), and anti-NANOG (Rb pAb, ab80892, Abcam, dilution 1:500). Secondary antibodies: donkey anti-mouse immunoglobulin G (IgG)-Alexa 488, rabbit anti-mouse IgG-Alexa546, goat anti-rabbit IgG-Alexa 488 (715-545-150/711-545-152, all Jackson Immune Research Europe, dilution 1:400). Cells were stained with the Alkaline Phosphatase Detection Kit (Millipore) according to the manufacturer's instructions.
qRT-PCR
Total RNA was isolated using TRIzol-chloroform and subsequently treated with DNaseI (DNase I, RNase-free [1 U/μl]; Thermo Fisher Scientific). RNA was reverse transcribed using RevertAid reverse transcriptase and random hexamer primers (both Thermo Fisher Scientific). 100 ng of cDNA was used as input for quantification using the following TaqMan primers: mOct4 (Mm00658129_gH), mSox2 (Mm00488369), and mNanog (Mm02384862_g1) (Thermo Fischer Scientific). Real-time PCR was performed on a StepOnePlus thermocycler (Applied Biosystems).
Teratoma Formation
Undifferentiated iPSCs (2 × 106) or CD45.1(10.3) iPSC-Mφ (3 × 106) were injected subcutaneously into the right or left flank of NOD/SCID/γcnull (NSG) mice. Two to five weeks after injection, tumors derived from undifferentiated iPSCs were palpable and the mice were euthanized. Isolated teratomas were fixed in 4% formaldehyde and paraffin sections were stained with H&E for histologic analysis. In addition, 1.8–2.3 × 106 CD45.1(10.3) iPSC-derived macrophages were transplanted intratracheally into NSG mice.
Mφ Differentiation of iPSCs
iPSC-derived Mφ were generated using a modified version of a previously established protocol (Pfaff et al., 2012). In brief, iPSCs were passaged on a gelatinized flask before undergoing single-cell-based EB formation. 5,000–30,000 cells/ml were seeded in differentiation medium (IMDM medium; Thermo Fisher Scientific) with 15% pre-tested FCS (ES-Cult, Stem Cell Technologies), 1 mM penicillin-streptomycin, 1 mM L-glutamine, 50 ng/ml ascorbic acid (Sigma-Aldrich), and 150 mM monothioglycerol (Sigma-Aldrich) in a 10-cm suspension dish and grown for 8 days on a rotator shaker (100 rpm). At day 5, medium was supplemented with 10 ng/ml murine IL-3 and 30 ng/ml mSCF (Peprotech). After 8 days EBs were dissociated in collagenase IV (Life Technologies) and CD41-expressing cells were isolated via fluorescence-activated cell sorting. The purified population was further differentiated into Mφ for 10–20 days in RPMI medium (Thermo Fisher Scientific) supplemented with 10% FCS, 1 mM L-glutamine, 1 mM penicillin-streptomycin, and 30% supernatant of L929 producer cells as a source of M-CSF (Zanoni et al., 2009).
Intrapulmonary Transplantation of CD45.1(10.3) iPSC-Mφ into Csf2rb−/− and NSG Mice
Csf2rb-/-, NSG and B6.SJL-Ptprca-Pep3b/BoyJ (CD45.1) donor mice were obtained from the central animal facility of Hannover Medical School. Mice were kept under specific pathogen free conditions and had free access to food and water. All animal experiments were approved by the Lower Saxony State animal welfare committee and performed according to their guidelines. CD45.1(10.3) iPSC-Mφ (4 × 106) were resuspended in a 60-μl volume and instilled through the trachea. Eight weeks after transplantation into Csf2rb−/− mice bronchoalveolar lavage fluid, lungs, bone marrow, lymph nodes, spleen, and liver were isolated, and flow cytometry analysis as well as genomic DNA isolation was performed.
Similarly, 4 weeks after intratracheal administration of 1.8–2.3 × 106 CD45.1(10.3) iPSC-Mφ into NSG mice, genomic DNA was isolated, and the presence of CD45.1(10.3) iPSC-derived progenies in the lung, liver, spleen, bone marrow, and lymph nodes was investigated by PCR.
Cytospin
Mφ or CD41+ cells (20,000–40,000) were spun on glass slides at 600 × g for 7 min and then stained according to the May-Grünwald/Giemsa staining protocol.
Flow Cytometry
For detection of surface markers and intracellular proteins, cells were harvested after trypsin treatment and resuspended in PBS supplemented with 2% FCS. Cells were pretreated with CD16/CD32 Fc-Block (eBioscience) for 20 min at 4°C. The following antibodies were used in concentrations according to instructions: SSEA-1-APC (51-8813-71), CD41-APC (17-0411-82), F4/80-APC (17-4801-80), CD11b-PE/eFluor 450 (12-0112-82/48-0112), CD14-FITC (11-0141-81), CD11c-PE-Cy7 (11-0141-81), Gr-1-eFluor450 (48-5931-82), CD45.1-PE-Cy7/APC (25-0453-81/17-0453), CD45.2-PE (12-0454-81), CD3-FITC (11-0031-81), B220-eFluor450 (48-0452-82), CD45-PE-Cy7/eFluor450 (25-0451-82/48-0451), CD200R-APC (17-5201), CD68-PE-Cy7 (25-0681) (all from eBioscience), SIGLEC-F-PE (552126, Becton Dickinson), Phospho-STAT5 (Tyr694) (C71E5) rabbit mAb (Alexa Fluor 647 conjugate) (9365, Cell Signaling), or Anti-Human/Mouse phospho-STAT5 (Y694) APC (17-9010, eBioscience). Fluorescence was measured using a BD FACSCalibur or BD LSR II and Fc-Block treated unstained sample was used as negative control.
Yeast Phagocytosis Assay
For testing of phagocytic activity, BMlin−-derived Mφ (BMlin−-Mφ), iPSCs, and CD45.1iPSC-derived Mφ (iPSC-Mφ) were incubated with OD660:0,1 heat-inactivated baker's yeast labeled with 10 mg/ml Kongo red in RPMI-based culture medium. At indicated time points, the number of yeasts per cells was counted using a binocular microscope.
Chemokine Release Assay
Mφ/well (105) were seeded overnight in tissue-culture-treated 24-well plates and stimulated with 1 μg/ml LPS (Sigma-Aldrich). Chemokine concentration was measured after 0, 24, and 48 hr using the Chemokines 6plex FlowCytomix Multiplex Kit (BMS821FF, eBioscience).
PCR on Genomic DNA
Genomic DNA (gDNA) was isolated from tissues or iPSCs using the Mammalian Genomic DNA Miniprep Kit (Sigma-Aldrich) according to the manufacturer's instructions. Detection of the neomycin resistance gene was performed using 100 μg of gDNA and primers (5′-GCGTTGGCTACCCGTGATAT-3′; 3′-GGTCTCCCAGACAAGCTTGAACC-5′) as described previously (Nishinakamura et al., 1995).
Clonogenic Progenitor Assay
Hematopoietic CD41+ progenitor cells obtained from iPSC differentiation (8 days after start of EB culture) were seeded at a concentration of 20,000–50,000/ml in basic and complete methylcellulose medium (HSC006 with no cytokines added and HSC007 containing IL-6, erythropoietin, SCF, and IL-3, respectively; R&D Systems). HSC007 was additionally supplemented with 20 ng/ml human G-CSF and 20 ng/ml murine M-CSF, while HSC006 was supplemented with 50 ng/ml murine GM-CSF (all Peprotech). Cells were grown for 7–10 days and colonies composed of more than 50 cells were counted.
ELISA
Concentration of GM-CSF was evaluated by ELISA following the manufacturer's instructions (Mouse GM-CSF ELISA Ready-SET-Go!, Affymetrix eBioscience).
Statistics
All graphs were created and statistical analysis performed with Prism V6 (GraphPad). One-way or two-way ANOVA testing was applied. Significance is indicated with asterisks (∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001), and specific tests applied are mentioned in each figure legend.
Author Contributions
A.M. designed experiments, performed experiments, analyzed the data, and wrote the manuscript; J.K. performed experiments, analyzed data, and wrote the manuscript; T.S., S.G., M.P.K., C.H., and A.K. performed experiments and analyzed data; S.B. performed experiments and wrote the manuscript; M.A. wrote the manuscript; A.S. provided reprogramming vectors; B.T., G.H., T.M., and N.L. conceived the project and provided financial support; A.M., T.M., and N.L. wrote the manuscript.
Acknowledgments
The authors thank M. Ballmaier and C. Struckmann (Cell Sorting Facility, Hannover Medical School) for scientific support and D. Lüttge and T. Buchegger for experimental and technical support. In addition, we would like to thank Elena Lopez-Rodriguez for performing immunofluorescence staining of lung sections. This work was supported by grants from the Else Kröner-Fresenius-Stiftung, the Deutsche Forschungsgemeinschaft (Cluster of Excellence REBIRTH, Exc62/1, grants MO 886/6-1, LA 3680/2-1, and the SFB738), the Bundesministerium für Bildung und Forschung (BMBF, project PidNet), and the German Center for Lung Research and Hannover Medical School internal programs (Hochschulinterne Leistungsförderung [HiLF] and Young Academy).
Published: July 21, 2016
Footnotes
Supplemental Information includes five figures and can be found with this article online at http://dx.doi.org/10.1016/j.stemcr.2016.06.011.
Supplemental Information
References
- Ackermann M., Lachmann N., Hartung S., Eggenschwiler R., Pfaff N., Happle C., Mucci A., Göhring G., Niemann H., Hansen G. Promoter and lineage independent anti-silencing activity of the A2 ubiquitous chromatin opening element for optimized human pluripotent stem cell-based gene therapy. Biomaterials. 2014;35:1531–1542. doi: 10.1016/j.biomaterials.2013.11.024. [DOI] [PubMed] [Google Scholar]
- Biffi A., Aubourg P., Cartier N. Gene therapy for leukodystrophies. Hum. Mol. Genet. 2011;20:R42–R53. doi: 10.1093/hmg/ddr142. [DOI] [PubMed] [Google Scholar]
- Blackwell T.S., Hipps A.N., Yamamoto Y., Han W., Barham W.J., Ostrowski M.C., Yull F.E., Prince L.S. NF-κB signaling in fetal lung macrophages disrupts airway morphogenesis. J. Immunol. 2011;187:2740–2747. doi: 10.4049/jimmunol.1101495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennig S., Lachmann N., Buchegger T., Hetzel M., Schambach A., Moritz T. Chemoprotection of murine hematopoietic cells by combined gene transfer of cytidine deaminase (CDD) and multidrug resistance 1 gene (MDR1) J. Exp. Clin. Cancer Res. 2015;34:148. doi: 10.1186/s13046-015-0260-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruscia E.M., Zhang P.-X., Ferreira E., Caputo C., Emerson J.W., Tuck D., Krause D.S., Egan M.E. Macrophages directly contribute to the exaggerated inflammatory response in cystic fibrosis transmembrane conductance regulator-/- mice. Am. J. Respir. Cell Mol. Biol. 2009;40:295–304. doi: 10.1165/rcmb.2008-0170OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartier N., Lewis C.-A., Zhang R., Rossi F.M.V. The role of microglia in human disease: therapeutic tool or target? Acta Neuropathol. 2014;128:363–380. doi: 10.1007/s00401-014-1330-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies L.C., Jenkins S.J., Allen J.E., Taylor P.R. Tissue-resident macrophages. Nat. Immunol. 2013;14:986–995. doi: 10.1038/ni.2705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di A., Brown M.E., Deriy L.V., Li C., Szeto F.L., Chen Y., Huang P., Tong J., Naren A.P., Bindokas V. CFTR regulates phagosome acidification in macrophages and alters bactericidal activity. Nat. Cell Biol. 2006;8:933–944. doi: 10.1038/ncb1456. [DOI] [PubMed] [Google Scholar]
- Feng Y.-H., Mao H. Expression and preliminary functional analysis of Siglec-F on mouse macrophages. J. Zhejiang Univ. Sci. B. 2012;13:386–394. doi: 10.1631/jzus.B1100218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandre-Babbe S., Paluru P., Aribeana C., Chou S.T., Bresolin S., Lu L., Sullivan S.K., Tasian S.K., Weng J., Favre H. Patient-derived induced pluripotent stem cells recapitulate hematopoietic abnormalities of juvenile myelomonocytic leukemia. Blood. 2013;121:4925–4929. doi: 10.1182/blood-2013-01-478412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez Perdiguero E., Klapproth K., Schulz C., Busch K., Azzoni E., Crozet L., Garner H., Trouillet C., de Bruijn M.F., Geissmann F., Rodewald H.-R. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature. 2015;518:547–551. doi: 10.1038/nature13989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen G., Hercus T.R., McClure B.J., Stomski F.C., Dottore M., Powell J., Ramshaw H., Woodcock J.M., Xu Y., Guthridge M. The structure of the GM-CSF receptor complex reveals a distinct mode of cytokine receptor activation. Cell. 2008;134:496–507. doi: 10.1016/j.cell.2008.05.053. [DOI] [PubMed] [Google Scholar]
- Happle C., Lachmann N., Skuljec J., Wetzke M., Ackermann M., Brennig S., Mucci A., Jirmo A.C., Groos S., Mirenska A. Pulmonary transplantation of macrophage progenitors as effective and long-lasting therapy for hereditary pulmonary alveolar proteinosis. Sci. Transl. Med. 2014;6:250ra113. doi: 10.1126/scitranslmed.3009750. [DOI] [PubMed] [Google Scholar]
- Hashimoto D., Chow A., Noizat C., Teo P., Beasley M.B., Leboeuf M., Becker C.D., See P., Price J., Lucas D. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity. 2013;38:792–804. doi: 10.1016/j.immuni.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirata S., Takayama N., Jono-Ohnishi R., Endo H., Nakamura S., Dohda T., Nishi M., Hamazaki Y., Ishii E.-I., Kaneko S. Congenital amegakaryocytic thrombocytopenia iPS cells exhibit defective MPL-mediated signaling. J. Clin. Invest. 2013;123:3802–3814. doi: 10.1172/JCI64721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodge S., Hodge G., Scicchitano R., Reynolds P.N., Holmes M. Alveolar macrophages from subjects with chronic obstructive pulmonary disease are deficient in their ability to phagocytose apoptotic airway epithelial cells. Immunol. Cell Biol. 2003;81:289–296. doi: 10.1046/j.1440-1711.2003.t01-1-01170.x. [DOI] [PubMed] [Google Scholar]
- Hussell T., Bell T.J. Alveolar macrophages: plasticity in a tissue-specific context. Nat. Rev. Immunol. 2014;14:81–93. doi: 10.1038/nri3600. [DOI] [PubMed] [Google Scholar]
- Jiang Y., Cowley S.A., Siler U., Melguizo D., Tilgner K., Browne C., Dewilton A., Przyborski S., Saretzki G., James W.S. Derivation and functional analysis of patient-specific induced pluripotent stem cells as an in vitro model of chronic granulomatous disease. Stem Cells. 2012;30:599–611. doi: 10.1002/stem.1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleff V., Sorg U.R., Bury C., Suzuki T., Rattmann I., Jerabek-Willemsen M., Poremba C., Flasshove M., Opalka B., Trapnell B. Gene therapy of βc-deficient pulmonary alveolar proteinosis (βc-PAP): studies in a murine in vivo model. Mol. Ther. 2008;16:757–764. doi: 10.1038/mt.2008.7. [DOI] [PubMed] [Google Scholar]
- Kovtunovych G., Ghosh M.C., Ollivierre W., Weitzel R.P., Eckhaus M.A., Tisdale J.F., Yachie A., Rouault T.A. Wild-type macrophages reverse disease in heme oxygenase 1-deficient mice. Blood. 2014;124:1522–1530. doi: 10.1182/blood-2014-02-554162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuehle J., Turan S., Cantz T., Hoffmann D., Suerth J.D., Maetzig T., Zychlinski D., Klein C., Steinemann D., Baum C. Modified lentiviral LTRs allow Flp recombinase-mediated cassette exchange and in vivo tracing of “factor-free” induced pluripotent stem cells. Mol. Ther. 2014;22:919–928. doi: 10.1038/mt.2014.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lachmann N., Happle C., Ackermann M., Lüttge D., Wetzke M., Merkert S., Hetzel M., Kensah G., Jara-Avaca M., Mucci A. Gene correction of human induced pluripotent stem cells repairs the cellular phenotype in pulmonary alveolar proteinosis. Am. J. Respir. Crit. Care Med. 2014;189:167–182. doi: 10.1164/rccm.201306-1012OC. [DOI] [PubMed] [Google Scholar]
- Lachmann N., Czarnecki K., Brennig S., Phaltane R., Heise M., Heinz N., Kempf H., Dilloo D., Kaever V., Schambach A. Deoxycytidine-kinase knockdown as a novel myeloprotective strategy in the context of fludarabine, cytarabine or cladribine therapy. Leukemia. 2015;29:2266–2269. doi: 10.1038/leu.2015.108. [DOI] [PubMed] [Google Scholar]
- Lavin Y., Winter D., Blecher-Gonen R., David E., Keren-Shaul H., Merad M., Jung S., Amit I. Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell. 2014;159:1312–1326. doi: 10.1016/j.cell.2014.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang G., Zhang Y. Genetic and epigenetic variations in iPSCs: potential causes and implications for application. Cell Stem Cell. 2013;13:149–159. doi: 10.1016/j.stem.2013.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litvack M.L., Wigle T.J., Lee J., Wang J., Ackerley C., Grunebaum E., Post M. Alveolar-like stem cell-derived Myb(neg) macrophages promote recovery and survival in airway disease. Am. J. Respir. Crit. Care Med. 2016;193:1219–1229. doi: 10.1164/rccm.201509-1838OC. [DOI] [PubMed] [Google Scholar]
- Ma Y.D., Lugus J.J., Park C., Choi K. Differentiation of mouse embryonic stem cells into blood. Curr. Protoc. Stem Cell Biol. 2008;Chapter 1:Unit 1F.4. doi: 10.1002/9780470151808.sc01f04s6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma N., Liao B., Zhang H., Wang L., Shan Y., Xue Y., Huang K., Chen S., Zhou X., Chen Y. Transcription activator-like effector nuclease (TALEN)-mediated gene correction in integration-free -thalassemia induced pluripotent stem cells. J. Biol. Chem. 2013;288:34671–34679. doi: 10.1074/jbc.M113.496174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malur A., Baker A.D., McCoy A.J., Wells G., Barna B.P., Kavuru M.S., Malur A.G., Thomassen M.J. Restoration of PPARγ reverses lipid accumulation in alveolar macrophages of GM-CSF knockout mice. Am. J. Physiol. Lung Cell Mol. Physiol. 2011;300:L73–L80. doi: 10.1152/ajplung.00128.2010. [DOI] [PubMed] [Google Scholar]
- Menon T., Firth A.L., Scripture-Adams D.D., Galic Z., Qualls S.J., Gilmore W.B., Ke E., Singer O., Anderson L.S., Bornzin A.R. Lymphoid regeneration from gene-corrected SCID-X1 subject-derived iPSCs. Cell Stem Cell. 2015;16:367–372. doi: 10.1016/j.stem.2015.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neri T., Muggeo S., Paulis M., Caldana M.E., Crisafulli L., Strina D., Focarelli M.L., Faggioli F., Recordati C., Scaramuzza S. Targeted gene correction in osteopetrotic-induced pluripotent stem cells for the generation of functional Osteoclasts. Stem Cell Rep. 2015;5:558–568. doi: 10.1016/j.stemcr.2015.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishinakamura R., Nakayama N., Hirabayashi Y., Inoue T., Aud D., McNeil T., Azuma S., Yoshida S., Toyoda Y., Arai K. Mice deficient for the IL-3/GM-CSF/IL-5 beta c receptor exhibit lung pathology and impaired immune response, while beta IL3 receptor-deficient mice are normal. Immunity. 1995;2:211–222. doi: 10.1016/1074-7613(95)90046-2. [DOI] [PubMed] [Google Scholar]
- Okamura K., Watanabe T., Onishi T., Watanabe H., Fujii E., Mori K., Matsuda J. Successful allogeneic unrelated bone marrow transplantation using reduced-intensity conditioning for the treatment of X-linked adrenoleukodystrophy in a one-yr-old boy. Pediatr. Transplant. 2009;13:130–133. doi: 10.1111/j.1399-3046.2008.00962.x. [DOI] [PubMed] [Google Scholar]
- Park I.H., Arora N., Huo H., Maherali N., Ahfeldt T., Shimamura A., Lensch M.W., Cowan C., Hochedlinger K., Daley G.Q. Disease-specific induced pluripotent stem cells. Cell. 2008;134:877–886. doi: 10.1016/j.cell.2008.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaff N., Lachmann N., Kohlscheen S., Sgodda M., Araúzo-Bravo M.J., Greber B., Kues W., Glage S., Baum C., Niemann H. Efficient hematopoietic redifferentiation of induced pluripotent stem cells derived from primitive murine bone marrow cells. Stem Cells Dev. 2012;21:689–701. doi: 10.1089/scd.2011.0010. [DOI] [PubMed] [Google Scholar]
- Rybtsov S., Sobiesiak M., Taoudi S., Souilhol C., Senserrich J., Liakhovitskaia A., Ivanovs A., Frampton J., Zhao S., Medvinsky A. Hierarchical organization and early hematopoietic specification of the developing HSC lineage in the AGM region. J. Exp. Med. 2011;208:1305–1315. doi: 10.1084/jem.20102419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senju S., Haruta M., Matsunaga Y., Fukushima S., Ikeda T., Takahashi K., Okita K., Yamanaka S., Nishimura Y. Characterization of dendritic cells and macrophages generated by directed differentiation from mouse induced pluripotent stem cells. Stem Cells. 2009;27:1021–1031. doi: 10.1002/stem.33. [DOI] [PubMed] [Google Scholar]
- Sgambato J.A., Park T.S., Miller D., Panicker L.M., Sidransky E., Lun Y., Awad O., Bentzen S.M., Zambidis E.T., Feldman R.A. Gaucher disease-induced pluripotent stem cells display decreased erythroid potential and aberrant myelopoiesis. Stem Cells Transl. Med. 2015;4:878–886. doi: 10.5966/sctm.2014-0213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata Y., Berclaz P.Y., Chroneos Z.C., Yoshida M., Whitsett J.A., Trapnell B.C. GM-CSF regulates alveolar macrophage differentiation and innate immunity in the lung through PU.1. Immunity. 2001;15:557–567. doi: 10.1016/s1074-7613(01)00218-7. [DOI] [PubMed] [Google Scholar]
- Suzuki T., Sakagami T., Rubin B.K., Nogee L.M., Wood R.E., Zimmerman S.L., Smolarek T., Dishop M.K., Wert S.E., Whitsett J.A. Familial pulmonary alveolar proteinosis caused by mutations in CSF2RA. J. Exp. Med. 2008;205:2703–2710. doi: 10.1084/jem.20080990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T., Maranda B., Sakagami T., Catellier P., Couture C.Y., Carey B.C., Chalk C., Trapnell B.C. Hereditary pulmonary alveolar proteinosis caused by recessive CSF2RB mutations. Eur. Respir. J. 2011;37:201–204. doi: 10.1183/09031936.00090610. [DOI] [PubMed] [Google Scholar]
- Suzuki T., Arumugam P., Sakagami T., Lachmann N., Chalk C., Sallese A., Abe S., Trapnell C., Carey B., Moritz T. Pulmonary macrophage transplantation therapy. Nature. 2014;514:450–454. doi: 10.1038/nature13807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T., Mayhew C., Sallese A., Chalk C., Carey B.C., Malik P., Wood R.E., Trapnell B.C. Use of induced pluripotent stem cells to recapitulate pulmonary alveolar proteinosis pathogenesis. Am. J. Respir. Crit. Care Med. 2014;189:183–193. doi: 10.1164/rccm.201306-1039OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabó P.E., Hübner K., Schöler H., Mann J.R. Allele-specific expression of imprinted genes in mouse migratory primordial germ cells. Mech. Dev. 2002;115:157–160. doi: 10.1016/s0925-4773(02)00087-4. [DOI] [PubMed] [Google Scholar]
- Takahashi K., Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- Takahashi K., Tanabe K., Ohnuki M., Narita M., Ichisaka T., Tomoda K., Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- Trapnell B.C., Whitsett J.A. GM-CSF regulates pulmonary surfactant homeostasis and alveolar macrophage-mediated innate host defense. Annu. Rev. Physiol. 2002;64:775–802. doi: 10.1146/annurev.physiol.64.090601.113847. [DOI] [PubMed] [Google Scholar]
- Trapnell B.C., Whitsett J.A., Nakata K. Pulmonary alveolar proteinosis. N. Engl. J. Med. 2003;349:2527–2539. doi: 10.1056/NEJMra023226. [DOI] [PubMed] [Google Scholar]
- Trapnell B.C., Carey B.C., Uchida K., Suzuki T. Pulmonary alveolar proteinosis, a primary immunodeficiency of impaired GM-CSF stimulation of macrophages. Curr. Opin. Immunol. 2009;21:514–521. doi: 10.1016/j.coi.2009.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tubsuwan A., Abed S., Deichmann A., Kardel M.D., Bartholomä C., Cheung A., Negre O., Kadri Z., Fucharoen S., von Kalle C. Parallel assessment of globin lentiviral transfer in induced pluripotent stem cells and adult hematopoietic stem cells derived from the same transplanted β-thalassemia patient. Stem Cells. 2013;31:1785–1794. doi: 10.1002/stem.1436. [DOI] [PubMed] [Google Scholar]
- Viana G.M., Buri M.V., Paredes-Gamero E.J., Martins A.M., D'Almeida V. Impaired hematopoiesis and disrupted monocyte/macrophage homeostasis in mucopolysaccharidosis type I mice. J. Cell Physiol. 2016;231:698–707. doi: 10.1002/jcp.25120. [DOI] [PubMed] [Google Scholar]
- Ye Z., Zhan H., Mali P., Dowey S., Williams D.M., Jang Y.-Y., Dang C.V., Spivak J.L., Moliterno A.R., Cheng L. Human-induced pluripotent stem cells from blood cells of healthy donors and patients with acquired blood disorders. Blood. 2009;114:5473–5480. doi: 10.1182/blood-2009-04-217406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanoni I., Ostuni R., Granucci F. Generation of mouse bone marrow-derived macrophages (BM-MFs) Protoc. Exch. 2009 [Google Scholar]
- Zhuang L., Pound J.D., Willems J.J.L.P., Taylor A.H., Forrester L.M., Gregory C.D. Pure populations of murine macrophages from cultured embryonic stem cells. Application to studies of chemotaxis and apoptotic cell clearance. J. Immunol. Methods. 2012;385:1–14. doi: 10.1016/j.jim.2012.06.008. [DOI] [PubMed] [Google Scholar]
- Zou J., Mali P., Huang X., Dowey S.N., Cheng L. Site-specific gene correction of a point mutation in human iPS cells derived from an adult patient with sickle cell disease. Blood. 2011;118:4599–4608. doi: 10.1182/blood-2011-02-335554. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.