

Abstract
Glycoconjugates, glycans, carbohydrates, and sugars: these terms encompass a class of biomolecules that are diverse in both form and function ranging from free oligosaccharides, glycoproteins and proteoglycans, to glycolipids that make up a complex glycan-code that impacts normal physiology and disease. Recent data suggests that one mechanism by which glycoconjugates impact physiology is through the regulation of the process of autophagy. Autophagy is a degradative pathway necessary for differentiation, organism development, and the maintenance of cell and tissue homeostasis. In this review, we will highlight what is known about the regulation of autophagy by glycoconjugates focusing on signaling mechanisms from the extracellular surface and the regulatory roles of intracellular glycans. Glycan signaling from the extracellular matrix converges on “master” regulators of autophagy including AMPK and mTORC1, thus impacting their localization, activity and/or expression. Within the intracellular milieu, gangliosides are constituents of the autophagosome membrane, a subset of proteins composing the autophagic machinery are regulated by glycosylation, and oligosaccharide exposure in the cytosol triggers an autophagic response. The examples discussed provide some mechanistic insights into glycan regulation of autophagy and reveals areas for future investigation.
Keywords: post-translational modification, autophagy, glycosylation, glycoconjugates, lectin, extracellular surface, signaling, glycosaminoglycan, proteoglycan, hyaluronan, O-GlcNAc
Graphical abstract
Autophagy, a Central Pathway in Biology
Macroautophagy, herein referred to as autophagy is a key pathway in physiology and pathology governing diverse processes including development, innate immunity, cellular stress responses, and cell death [1–11]. Autophagy is a catabolic process necessary for the constitutive turnover of long-lived and damaged macromolecules as well as organelles. The process involves the capture and sequestration of bulk cytoplasm in double-membrane structures termed autophagosomes. The autophagosomes ultimately fuse with the lysosome to form autophagolysosomes (also known as autolysosomes). Lysosomal fusion makes the autophagosome cargo accessible to degradative enzymes leading to the generation of metabolites for cellular recycling [12, 13].
In addition to basal autophagic flux, autophagy is upregulated in response to diverse stimuli including low nutritional state, oxidative stress and microbial insult [1, 2, 8, 14–16]. These divergent signaling pathways converge on a set of core machinery, the autophagy related genes (Atg, yeast; ATG, human). Extensive genetic screening and proteomic analyses have identified the 38 ATG proteins controlling different steps along the autophagy pathway ([4, 17–19]; Table 1). These proteins cooperate as molecular machines that have distinct roles in induction, vesicle nucleation, autophagosome expansion and maturation, and fusion ([20–22]; Figure 1).
Table 1.
Proteins Involved in Regulating Autophagy*
| Yeast Name | Mammalian Names | Roles | Step |
|---|---|---|---|
| Atg1 | ULK1/2 | Kinase required for the induction of vesicle formation, often considered the first committed step of autophagy | Induction |
| Atg2 | ATG2a, ATG2b | Forms a complex with ATG18 and plays a role in recycling ATG9 | Recycling |
| Atg3 | ATG3 | An E2-like enzyme which catalyzes the lipidation of LC3and LC3-like proteins | Expansion |
| Atg4 | ATG4B | A cysteine protease critical for LC3 processing and activation | Expansion |
| Atg5 | ATG5 | Conjugates with ATG12 and forms an E3-like ligase important for the lipidation of LC3 | Expansion |
| Atg6 | Beclin 1 | Forms the VPS34 PI3-kinase complex which is critical for nucleation | Nucleation |
| Atg7 | ATG7 | An E1-like enzyme that regulates LC3 lipidation and the ATG12-ATG5/ATG3 interaction | Expansion |
| Atg8 | LC3 | Ubiquitin-like protein that conjugates with phosphatidylethanolamine and is critical for the formation of the autophagosome | Expansion |
| Atg9 | ATG9 | Transmembrane protein which cycles between Golgi, endosomes and PAS; interacts with the ATG2-WIPI1 complex | Nucleation/Expansion |
| Atg10 | ATG10 | An E2-like enzyme which conjugates ATG5 and ATG12 | Expansion |
| Atg12 | ATG12 | Conjugates with ATG5 and ATG3 | Expansion |
| Atg14 | ATG14 | Component of VPS34 PI3K complex | Nucleation |
| Atg16 | ATG16L | Forms the ATG16L-ATG12-ATG5 complex | Expansion |
| Atg17 | - | Component of Atg1/ULK1 complex: Atg1-Atg13-Atg17-Atg29-Atg31 in yeast. The function equivalent FIP200 is part of the ULK1/2-ATG13-FIP200-ATG101 complex in mammals | Nucleation |
| Atg18 | WIPI1 and other homologs | WD domain phosphoinositide interacting protein; Functions upstream of the ATG12-AT* G5-ATG16L1 complex and LC3, and downstream of the ULK1 and PI3-kinase complexes | Nucleation |
| Atg27 | - | Effector of Vps34 | Nucleation |
| Atg29 | - | Component of the Atg1 complex; Functions at the PAS | Nucleation |
| Other Key proteins | |||
| - | FIP200, RB1CC1 | FIP200 (FAK family kinase-interacting protein of 200 kDa) or RB1CC1 (RB1-inducible coiled-coil 1) interacts with ULK1 and is required for autophagosome biogenesis. | Nucleation |
| - | p62/SQSTM1 | Autophagy receptor that with ubiquitinated cargo and LC3 and is degraded by the lysosome | Cargo selection |
| - | SNAP29 | SNAP29 is a SNARE involved in regulating fusion of the autophagosome and lysosomal membranes | Fusion |
| Snf1 | AMPK | A serine/threonine protein kinase which promotes autophagy by phosphorylating and activating ULK1, and phosphorylating and inhibiting mTOR | Induction |
| Tor1, Tor2 | mTOR | A serine/threonine protein kinase which phosphorylates and inhibits ULK1 | Induction |
| Vps34 | PIK3C3, VPS34 | Phosphatidylinositol 3-kinase required for autophagy, as part of the autophagy-specific VPS34 PI3-kinase complex | Nucleation |
| Sec15 | Exoc6 | Exocyst complex component 6: Component of the exocyst complex involved in the docking of exocytic vesicles with fusion sites on the plasma membrane | Expansion |
This is not a comprehensive list of proteins that control autophagy but represents several of the mammalian regulatory proteins that are referenced in the main body of this chapter. Note: Atg is the yeast nomenclature, whereas ATG is the human and mouse nomenclature.
Figure 1. Autophagy.

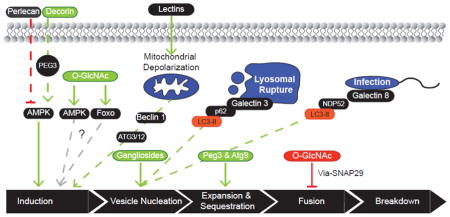
Upper Panel: Autophagy can be broken down into 5 key steps: 1) Induction; 2) Vesicle nucleation; 3) Expansion and Maturation; 4) Fusion with the Lysosome; and 5) Degradation. Critical regulatory events for each step are shown below. Lower Panel: Pro- and anti-autophagy signals converge on ULK1 (Atg1 in yeast), which performs the first committed step in autophagy. Activation of the ULK1 complex leads to the recruitment of the Beclin 1-VPS34 complex at the PAS (phagophore assembly site). Maturation of the phagophore is promoted by lipids contained in ATG9 vesicles. Expansion and sequestration requires two ubiquitin conjugating systems, which result in the lipidation of LC3 and its targeting to immature autophagosomes (phagophores). Autophagosomes fuse with the lysosome, in a manner dependent on SNAP29.The key proteins involved in regulating each step are shown, and are in light grey unless glycosylated or regulated by a glycan (lower panel). Proteins that are O-GlcNAc modified are indicated in black, whereas other types of glycosylation are highlighted in dark grey. The leading edge is highlighted by purple cicle.
The Atg1 complex in yeast regulates autophagy induction [21]. This complex functions as a central hub that receives activating or inhibitory signals; activation of the Atg1 complex is considered the first committed step in autophagy. In mammals, this complex contains the Atg1 homolog ULK1 (Unc-51-like kinase 1). The PI3K-AKT-mTORC1 and LKB1-AMPK pathways represent two of the many signaling platforms that control ULK1 activity [21, 22]. Nucleation of the isolation membrane (phagophore) is controlled by the class III phosphatidylinositol 3 kinase complex, Beclin 1-VPS34 (Vacuolar protein sorting-associated protein 34), which can be inhibited by 3-MA (3-methyladenine, Figure 1; [21, 22]). The concentrated levels of PI3P (phosphatidylinositol-3-phosphate) recruit effectors to the PAS (phagophore assembly site) that are needed to generate the autophagosomal membrane [23, 24]. During expansion, two ubiquitin-like conjugation systems cooperate to promote the covalent attachment of PE (phosphatidylethanolamine) to LC3 (microtubule-associated protein 1 light chain 3 β), referred to as the conversion of LC3-I to LC3-II (Figure 1). Lipidated LC3 associates with the phagophore membrane contributing to its expansion and maturation [21, 22]. LC3-II also contributes to cargo selection by the autophagosome which is achieved via autophagy receptors such as p62/sequestosome-1 [15, 25]. Complete autophagosomes are directed to lysosomes by the cytoskeleton [26, 27] and fusion is directed by the combined actions of Rab7a (Ras-related protein Rab-7a) GTPase, SNARE (soluble NSF [N-ethyl-maleimide-sensitive fusion protein] attachment protein receptor), and HOPS (Homotypic fusion and protein sorting) complexes [27–34].
Transcriptional control of the core machinery and PTMs (post-translational modifications) of the ATG proteins facilitate the dynamic regulation of autophagy. To date several types of PTMs have been characterized including lipidation of LC3 [35], phosphorylation of ULK1 by AMPK (AMP-activated protein kinase) and mTORC1 (mammalian target of rapamycin complex 1) [36], as well as Beclin 1 phosphorylation [37] and acetylation of ATG5, 7 and 12 [38]. The covalent modification of transcription factors and upstream signaling proteins also modulate the autophagic pathway [39, 40]. Recent studies highlight the importance of glycoconjugates in the regulation of autophagy (Figure 2), including glycosylation of the core machinery and glycoconjugates in the extracellular environment. In turn, autophagy has been implicated in the regulation of glycoconjugate homeostasis or turnover [41]. In this review, we focus on the role of glycoconjugates in regulating mammalian autophagy.
Figure 2. Common Glycoconjugates.

The location and constitution of key glycoconjugates are shown in the schematic on the left, including the glycan code (Adapted from Essentials in Glycobiology). Definitions of key “glycan” terms used throughout the text are shown on the right.
Glycoconjugates Regulate Autophagosome Membrane Dynamics
The mechanics of autophagosome formation have been a long-standing area of interest, particularly regarding the source of the autophagosome membrane. An early study employed lectin cytochemistry in conjunction with electron microscopy to characterize the source of the phagophore (immature autophagosomes), mature autophagosomes, and autolysosomes [42]. The lectins used in this study included ConA (concanavalin A), WGA (Wheat Germ Agglutinin) and RCA-120 (Ricinus Communis Agglutinin 120) that bind α-mannose residues, terminal GlcNAc (N-acetyl-Glucosamine) and Sialic acid residues, and terminal β-galactose residues, respectively. Lectin distribution proved heterogeneous in the immature autophagosomes, with Con A, WGA and RCA-120 binding focused on the leading edge of the phagophore (Figure 1). Mature autophagosomes exhibited lectin binding restricted to a narrow area, which is suggested to mark the site of closure of the leading edges of the phagophore. Unsurprisingly, the autolysosomes exhibit a more homogeneous lectin binding, likely due to the thick internal glycocalyx of the lysosome. The heterogeneity of lectin binding in the growing and mature autophagosome has two implications: First, Con A, WGA and RCA-120 binding are indicative of glycans generated in the ER (endoplasmic reticulum) and Golgi (Golgi apparatus), suggesting that the Golgi is one of the membrane sources utilized in the growth and maturation of the autophagosome; Second, the discrete localization of glycoproteins/glycolipids at the leading edges of the growing phagophore suggests that membrane lipid delivery involves a transient association between membrane glycoproteins/glycolipids and the growing phagophore. If the association of glycoproteins/glycolipids with the growing phagophore is static in nature, lectin staining of the mature autophagosome should have been more homogeneous.
A series of elegant studies favor the omegasome, a subdomain of the ER, as the nucleation site of the phagophore [43–45]. This is followed by growth and maturation of the phagophore/autophagosome utilizing lipids from the plasma membrane [46], ER-Golgi Intermediate Compartment (ERGIC) [47], Golgi [48] and ATG9 containing vesicles ([14, 24, 49–51]; Figure 1). Collectively, these studies implicate membrane-associated glycoconjugates in autophagosome maturation. ATG9, a transmembrane protein required for phagophore/autophagosome expansion [24], localizes to the TGN (Trans Golgi Network) and late endosomes and is N-glycosylated at asparagine 99 (N99) [14]. Autophagy activation mobilizes ATG9 via Golgi fission, resulting in trafficking of ATG9 containing vesicles to the PAS for membrane lipid delivery [52]. Perhaps the lectin cytochemistry conducted by Yamamoto et al. (1990; [42]) detected the transient association of ATG9 vesicles with the growing phagophore/autophagosomes. While knockout and knockdown experiments reveal the necessity of ATG9 for phagophore/autophagosome biogenesis [14], the role of ATG9 N-glycosylation has yet to be determined. Given the established role of N-glycosylation in protein folding/trafficking [53], one can postulate that ATG9 N-glycosylation functions in establishing the ATG9 reservoirs in the Golgi and late endosomes. The rapid redistribution of ATG9 vesicles upon autophagy induction likely reflects changes in the reservoir sorting/retention signal. Characterizing the N-glycan from Golgi retained ATG9 and the cytoplasmic ATG9 bearing vesicles will be useful in determining whether glycan editing is involved in the relocating ATG9 from the Golgi to the PAS.
CopII vesicle proteins have long been implicated in autophagosome biogenesis [54]. Most recently, the Schekman lab have utilized an in vitro cell free LC3 lipidation assay to demonstrate that the ERGIC is a competent membrane source to generate LC3 lipidated vesicles under normal and starvation conditions [47, 55]. Members of the Sec protein family that coat CopII vesicles localize to the ERGIC membrane fraction [56, 57]. Previous investigations revealed that CopII proteins are modified by O-GlcNAc (O-linked β-N-acetylglucosamine), including Sec24p [58, 59]. In particular, the deglycosylation of Sec24p is associated with a block in ER-to-Golgi traffic. As the ERGIC is a membrane source for phagophore/autophagosome biogenesis, O-GlcNAcylation of Sec24p and other CopII proteins may promote autophagy via the maintenance of membrane flux between the ER and Golgi. Given that O-GlcNAc cycles on and off Sec24p, the O-GlcNAc levels of CopII proteins may respond to pro-autophagic stimuli to fine tune ERGIC membrane flux. However, mapping the O-GlcNAc sites and mutational studies would lend validity to O-GlcNAc’s role in mediating membrane availability for autophagosome formation.
Gangliosides (a sub-type of glycolipid) are important structural players in autophagosome biogenesis. These data are not surprising given that gangliosides function in organizing biological membranes, including the generation of curvature in membrane microenvironments [60]. In assessing the role of lipid rafts in autophagic flux, Matarrese et al. (2014; [61]) demonstrated that the ganglioside GD3 colocalizes with LC3 in response to classic autophagic stimuli. GD3 also colocalizes with PI3P, which recruits ATG18 and other components of the core machinery [61] during the nucleation stage. Inhibiting sphingolipid biosynthesis with FB1 (fumonisin B1), or knocking down ST8SIA1 (ST8 α-N-acetyl-neuraminide α-2, 8-sialyltransferase), to suppress GD3 synthesis reduced autophagosome levels [61]. FB1 autophagy inhibition was reversed with the addition of exogenous GD3 to the media, confirming the relevance of ganglioside synthesis in autophagosome membrane dynamics [61]. These data, in combination with other studies demonstrating enhanced autophagic flux associated with the administration of exogenous ganglioside mixture (GM1, GD1a, GD1b, GT1b) [62, 63], merit further investigation into the mechanisms involved in ganglioside regulation of autophagic flux.
Lectin – Ligand Interactions Transmit Pro-Autophagic Stimuli
Many studies in the field of glycobiology indicate that diverse facets of biology are driven by glycan codes. One method employed in translating these glycan-codes is through the interaction of glycans and motif specific carbohydrate binding proteins or lectins. A prime example of lectin signaling is seen in N-linked glycan biosynthesis. Glycoprotein quality control in the ER utilizes the lectin binding-cycles of calnexin and calreticulin to oligosaccharides to sample the folding of newly synthesized proteins [64]. Subsequently, transit from the ER to the Golgi is controlled by LMAN1/ERGIC-53, which recognizes the high mannose oligosaccharide Man8GlcNAc2 for cargo selection [53, 65]. Several studies provide strong evidence of the pro-autophagic potential of extracellular and intracellular lectins. The binding of specific cell surface receptors by exogenous lectins triggers autophagy that can manifest as antiproliferative activity. Additionally, transmembrane lectin receptors and intracellular lectins function in pattern recognition of damage signals derived from invading pathogens or organelle rupture. In these instances, lectins are implicated in the cytoprotective roles of autophagy.
Plant Lectins as Inducers of Autophagy
Several studies highlight the therapeutic potential of plant lectins in treating cancer and microbial infections [66]. The administration of lectins, including mistletoe lectin, ricin, and Con A, triggers cell death in different cancers by activating apoptotic and/or autophagic signaling. Con A binds high mannose oligosaccharides on cell surface receptors triggering clathrin-mediated endocytosis [67–69]. Several studies demonstrate that Con A accumulates in the mitochondria. This mitochondrial localization is associated with membrane depolarization, a known pro-autophagic stimulus [8, 15, 16]. Consistent with these observations, Con A treatment of HeLa cells results in increased expression of Beclin 1 and LC3-II, as well as autophagosome accumulation [70]. These changes in the expression of autophagy markers are correlated with increased cell death, suggesting that Con A-induced autophagy leads to cell death. In support of this model, the authors demonstrate that the autophagy inhibitor 3-MA reduces cell death induced by Con A. The mechanism of activation involves a reduction of PI3K/AKT/mTOR signaling, a pathway known to suppress autophagy [70] [71]. Con A treatment also enhanced pro-autophagic signaling through the MEK/ERK pathway [72]. In an alternative cancer model, U87 glioblastoma cells, Con A treatment reduces cell viability and also induces autophagy albeit by an alternative mechanism [72, 73]. Here, ConA induced expression of BNIP3 (BCL2/adenovirus E1B 19kDa interacting protein 3), a mitochondrial BH3 only protein, and the autophagy related proteins ATG3, ATG12, and ATG16L1 [74]. While BNIP3 overexpression is known to induce autophagy, the precise mechanism is a matter of some debate [75]. Some reports indicate that BNIP3 promotes mitochondrial membrane permeability/depolarization to initiate autophagy, while others indicate that BNIP3 autophagy occurs independently of mitochondrial membrane permeabilization [76].
PCL (Polygonatum cyrtonema lectin) is a mannose binding lectin that is cytotoxic in human melanoma (A375) and murine fibrosarcoma cells [77, 78]. PCL-induced cell death involves the upregulation of both autophagy and apoptosis. PCL activates different signaling pathways in these cancer cell types. A375 cells exposed to PCL accumulate ROS (reactive oxygen species), while reducing glutathione concentrations [77]. Similar to HeLa cells exposed to Con A, Beclin 1 expression was enhanced with PCL treatment. In accordance with other reports, the oxidative burst was associated with activation of p38 MAPK (p38 mitogen activated protein) kinase, a stress responsive kinase, and p53 [77]. Applying the p38 MAPK inhibitor SB 203580, or the p53 inhibitor pifithrin α, reduced PCL mediated autophagy and Beclin 1 expression. Although it was not assessed in PCL-induced autophagy, there is evidence that p38 MAPK signaling mediates Beclin 1 phosphorylation, a necessary step in starvation-induced autophagy [77]. In murine fibrosarcoma cells PCL activates autophagy by suppressing signaling from the Ras-Raf and PI3K-AKT pathways [78].
Galectins, Xenophagy, and Autophagy
The Galectins are a family of β-galactoside binding lectins that have intracellular roles in addition to their extracellular functions [79]. In comparison to the extracellular environment there is less glycan diversity in the cytoplasm where the major form of glycosylation is the O-GlcNAc modification (Figure 2). As such, the accumulation of complex oligosaccharides in the cytoplasm makes for an efficient danger or damage signal. The exposed glycans on damaged organelles are bound by galectins 3, 8 or 9 in the cytosol [80–82]. These cytosolic galectins also accumulate on pathogen-loaded vesicles [80, 82]. The downstream effects of galectin-damage detection include the targeting of organelles and microbes for autophagic degradation [25, 80]. Galectin-8 expression was shown to limit Salmonella typhimurium growth in HeLa cells [80]. The autophagy cargo receptor NDP52 (Nuclear dot protein 52) binds ubiquitin-coated S. typhimurium and recruits LC3 to target the bacteria for autophagy [25, 80]. Thurston et al. (2012; [80]) demonstrated that galectin-8 binds NDP52 via immunoprecipitation and confocal microscopy. Downregulating galectin-8 expression greatly reduced the number of NDP52 positive and LC3 positive S. typhimurium compared to control experiments [80]. Based on these data it seems that galectin-8 functions upstream in antimicrobial autophagy (xenophagy), recruiting NDP52 and LC3 to target the microbes to autophagosomes. The literature indicates that another ubiquitin binding protein, autophagy receptor, p62/sequestosome-1, is needed for efficient autophagy of S. typhimurium [83, 84]. The fact that p62/sequestosome-1 and NDP52 localize to discrete microdomains around S. typhimurium suggests that a signal independent of galectin-8 controls p62/sequestosome-1 localization.
Galectin-3 has been reported to regulate the turnover of damaged endosomes by autophagy. Calcium phosphate precipitates (CPP), used in mammalian transfections, causes transient autophagy activation that is though to result from damage to endosomes [85]. Using MβCD (methyl-β-cyclodextrin) and dynasore to inhibit clathrin mediated endocytosis, Chen and co-workers (2014; [85]) showed that CPP-induced autophagy relies on the endocytic pathway. These data agree with previous studies that describe the dependence of CPP transfection on endocytosis [86]. As complex glycans typically face the lumen of organelles (lysosomes, ER, Golgi), cytosolic galectins can only access glycans from damaged/lysed organelles. The authors used mCherry-galectin-3 to demonstrate that CPP endocytosis lysed endosomal vesicles. The generation of galectin-3 coated vesicles was suppressed with MβCD. Galectin-3 also colocalized with LC3 and p62/sequestosome-1 at vesicles loaded with CPP-DNA complexes [85]. These data suggests that galectin-3 recruits p62/sequestosome-1 and LC3 in a similar fashion to galectin-8 recruitment of NDP52 and LC3 to bacteria. In support of this concept, galectin-3 RNAi suppressed the formation of p62/sequestosome-1 and LC3 positive puncta induced by CPP [85].
Recent studies highlight the role of galectin-3 in the selective encapsulation of damaged lysosomes by autophagosomes [81]. Using LLOMe (Leu-Leu methyl ester hydrobromide) and silicon dioxide treatments to induce lysosomal membrane damage in cell culture, Maejima et al. (2013; [87]) clearly show specific detection of lysosomal membrane permeabilization by galectin-3 and the selective enclosure of compromised lysosomes in autophagosomes. In addition to the localization of LC3 at damaged lysosomes, the authors demonstrate that ULK1, WIPI1 (WD repeat domain phosphoinositide-interacting protein), ATG5 and p62/sequestosome-1 are also associated with lysosomes in response to damage induction [87]. Live cell imaging using fluorescent fusion proteins demonstrated that galectin-3 lysosomal association precedes ATG5 recruitment to damaged lysosomes. These data indicate that autophagy is induced by lysosomal damage and implies that galectin-3 transduces the damage signal. The damaged lysosomes appeared to be eliminated in an autophagy-dependent fashion as galectin-3 puncta decreased over time after LLOMe washout in control cells, but, were retained in cells expressing an inactive ATG4B mutant and ATG7−/− MEFs [81].
Dectin and LC3 Associated Phagocytosis
Transmembrane mammalian lectins are involved in promoting autophagy in response to immune challenges [88, 89]. In contrast to using conventional xenophagy to sequester pathogens within autophagosomes for lysosomal degradation, immune cells have been shown to recruit components of the autophagic pathway for an unusual form of phagocytosis that does not involve the formation of autophagosomes. This “LC3-associated phagocytosis” (LAP) produces a fungicidal effect by two methods: Firstly, LAP contributes to the maturation of the phagocytic vacuole and subsequent elimination of ingested pathogens; Secondly, LAP is implicated in antigen presentation. This unconventional phagocytosis begins with the detection of pathogens by pattern recognition receptors, including the C-type lectins, which transmit signals mediating pathogen ingestion and eventual pathogen killing.
The C-type lectins represent one of the largest classes of mammalian lectins. Many of the receptors on immune cells contain C-type lectins that distinguish endogenous glycans from foreign or xenoglycans [89]. The sugar binding specificity of C-type lectin carbohydrate recognition domains is diverse; some family members bind high mannose oligosaccharides [90], whereas others prefer galactose containing glycans [90] or sialylated glycans [90]. The C-type lectin receptors also employ diverse strategies in transmitting signals to the cytoplasm. The cytoplasmic domain found on C-type lectin receptors include the ITAM (immunoreceptor tyrosine-based activation motif) and ITAM-like domains [88]. The ITAMs are subject to tyrosine phosphorylation, which facilitates the recruitment of SH2 (Src homology type 2 domain) containing effectors [88]. SH3 (Src homology type 3 domains) and endocytosis motifs (YXXϕ) [88] are some of the other domains that have been characterized on C-type lectin receptors.
Dectin 1 is a C-type lectin receptor found on macrophages, dendritic cells, and neutrophils that activates LAP in response to fungal challenge [91–95]. The Dectin 1 carbohydrate recognition domain binds to fungal β-glucan, essentially a glucose polymer found in the cell walls of pathogenic bacteria and fungi [92]. Recognition of this PAMP (pathogen associated molecular pattern) initiates phagocytosis as well as respiratory burst and inflammation for the purpose of eliminating the pathogen [91, 92, 94]. The offending pathogen is internalized in a single-membraned phagocytic vacuole or phagosome (distinct from an autophagosome), which undergoes a series of phenotypic changes to gain the necessary degradative potential. The phagosome maturation sequence involves acidification, accumulation of ROS, and sequential fusion with intracellular compartments ultimately fusing with the lysosome. Though phagocytosis and autophagy are distinct processes, several components of the autophagy pathway are commandeered in the course Dectin 1-mediated LAP without inducing autophagic flux [93, 95–98].
LC3 is considered the canonical marker for autophagosomes. Ma et al. (2012; [93]) provide concrete evidence of rapid LC3 lipidation (≤ 10 minutes) and recruitment to phagocytic vacuoles when macrophages and dendritic cells are challenged with live or heat inactivated yeast and β-glucan particles. Beclin 1 and p62/sequestosome-1 were also localized at phagocytic vacuoles during Dectin 1-induced phagocytosis [95]. Clearly the ubiquitin-like reactions needed to produce LC3-II must be functional in LAP, as suppressing ATG5 expression blocked the ability of Dectin 1 to recruit LC3 to the phagocytic vacuole [93, 99]. We can assume that lipidation of LC3 allows it to associate with the membrane of the phagocytic vacuole. Beclin 1 recruitment may be involved in delivering VPS34 to the phagosome as significant levels of PI3P accumulates on the membrane following phagosome sealing [100]. PI3P enrichment at the phagosome was found to be critical for fusing with the lysosome as suppression of VPS34 activity arrested phagosomes in an immature state[100].
As autophagosome biogenesis is not observed during Dectin 1 mediated-LAP the signaling mechanism probably bypasses assembling the PAS. Interestingly LAP activation by other pattern recognition receptors occurs independently of the ULK1 complex [101, 102]. Perhaps the lack of ULK1 signaling precludes the formation of a functional PAS, resulting in the absence of autophagosome biogenesis. Taken together, the data suggests that a unique signaling paradigm must be at play in order to activate and recruit elements of the autophagic machinery without generating autophagosomes. The initial steps of LAP activation involve phosphorylation of the Dectin 1 ITAM-like (hemITAM) domain by Src kinases which recruits and activates Syk (Spleen tyrosine Kinase) [93]. In turn, Syk activates NADPH oxidase generating ROS in the phagosome [92]. ROS production is required to recruit LC3 to the phagosome, in addition to pathogen killing [93]. It is unclear how phagosomal ROS leads to the generation of LC3-II and association at the phagosome. However, the mechanism(s) of Dectin 1 cytokine induction offers viable avenues to uncover the downstream signaling involved in LAP [88, 91]. Many of the signaling proteins implicated in Dectin 1 cytokine induction, including JNK (c-Jun N-terminal kinase), p38 MAPK, TAK1 (Transforming growth factor-beta-activated kinase 1) and NFκB (nuclear factor kappa-light-chain-enhancer of activated B cells), are known regulators of autophagy [88]. Further work is also needed to determine how the autophagic components translocate to the phagocytic vacuole, as well as the interactions maintaining phagosomal interaction.
Dectin 1 phagocytosis is also involved in antigen presentation in dendritic cells and protein secretion in macrophages [93, 98]. Supporting this model, when BDMC cells are challenged with ovalbumin overexpressing Saccharomyces cerevisiae they successfully present antigen to OTII T-cells, resulting in the activation of the T-cells [103]. Because major MHC complexes (major histocompatibility complexes) are loaded with antigenic peptides at the phagosome, the authors investigated the role of LC3 in MHC-II antigen presentation. LC3 deficient dendritic (BDMC) cells proved to be very inefficient in recruiting MHC-II to phagosomes [93].
Intracellular Glycosylation: a Transduction Mechanism in Autophagy Signaling Pathways
The O-GlcNAc modification is well positioned to regulate the process of autophagy, modifying some 3000 cellular proteins and being implicated in regulating cellular processes such as transcription, translation, protein degradation and signal transduction [104]. The regulation of these pathways has been shown to be responsive to signals that also induce autophagy, such as nutritional deprivation [104–107], cell cycle and development [108, 109], and cellular stress [59, 110–112]. As proposed earlier, O-GlcNAc signaling could regulate autophagy via modification of CopII coat proteins. Though this model is yet to be demonstrated, several studies have provided evidence of autophagy regulation by O-GlcNAc signaling [113–117]. An early example of this was reported in a Caenorhabditis elegans model of neurodegenerative disease [117]. Null alleles (ogt-1; less O-GlcNAc) of the C. elegans OGT (O-GlcNAc transferase), the enzyme which adds the O-GlcNAc moiety to serine and threonine residues, alleviated neuronal death in the tauopathy model of frontotemporal dementia with parkinsonism chromosome 17 (FTDP-17). Whereas, the null allele for OGA (O-GlcNAcase, oga-1; elevated O-GlcNAc), did not worsen the thrashing defect. Expression of tau V337M, the causative lesion in FTDP-17, was suppressed in the ogt-1 and oga-1 null animals compared to control animals. These data demonstrate that O-GlcNAc cycling modulates either protein synthesis or turnover [117]. Analysis of autophagy in the ogt-1 and oga-1 mutants revealed higher total expression of the C. elegans LC3 ortholog, lgg-1, under basal conditions and increased expression of the lipidated form with nutrient deprivation compared to wild type controls [117]. OGT knockdown in Drosophilla melanogaster was also linked to enhanced stress-induced autophagy [116]. Wang et al. (2012) reported enhanced Daf-16f transcript levels, a FoxO (forkhead Box O) transcription factor, in the ogt-1 null animals and suppression of Daf-16a expression in the oga-1 null animals [117]. Interestingly, the FoxO transcription factors have been previously shown to promote autophagy [118, 119]. This differential regulation of Daf-16 isoforms (Daf-16f versus Daf-16a) in the ogt-1 and oga-1 null animals suggests that O-GlcNAc signaling regulates autophagy through multiple mechanisms.
Supporting direct regulation of key steps in the autophagic pathway by O-GlcNAc, a subset of the autophagy core machinery and other regulatory proteins are O-GlcNAc-modified [106, 113, 115, 116, 120, 121]. The core protein Beclin 1 is reported to be O-GlcNAc modified [115], as well as autophagy modulators such as the α and γ subunits of the AMPK heterotrimer [121], Bcl-2 [115], the forkhead family of transcription factors FoxO1/3/4 [118, 119, 122, 123], and SNAP29 (synaptosomal–associated protein 29) [113]. O-GlcNAcylation increases the catalytic and transcriptional activity of AMPK and FoxO1 respectively [106, 121], both of which are positive regulators of autophagy. However, O-GlcNAcylation of the FoxO transcription factors and AMPK subunits were not made in the context of autophagy regulation, as such there is a wealth of knowledge to be gained in determining whether the O-GlcNAcylation of these targets mediate pro-autophagic or anti-autophagic responses.
Akin to other forms of intracellular vesicle trafficking, autophagy utilizes Rab GTPases and the SNARE proteins to mediate autophagosome – lysosome fusion to produce the autolysosome [30–34, 124]. SNAP29 is an integral component of this fusion machinery [30, 124]. Guo et al. (2014; [113]) demonstrated that SNAP29 is O-GlcNAc modified at 4 residues: S2; S61, T130 and S153. Mutation of all four residues to alanine enhanced the association of SNAP29 with its cognate SNARE complex partners, syntaxin 17 (stx17) and VAMP8 (vesicle-associated membrane protein 8), promoting autophagic flux [113]. The O-GlcNAcylation status of SNAP29 proved to be differentially regulated based on the nutritional state, as glucose starvation downregulated SNAP29 O-GlcNAc levels [113]. The authors also observed increased autophagosome formation in the C. elegans ogt-1 mutant and also in OGT knockdown in mammalian cells, suggesting that autophagosome formation/nucleation was also affected in addition to the fusion steps. Given that LC3 accumulates in both the C. elegans oga-1 and ogt-1 strains [117] further investigation will be needed to determine if the autophagy initiation complex is directly subject to O-GlcNAc cycling/regulation or whether OGT and OGA deletion or knockdown elicits activating upstream signals that converge on the pathway. Both the mammalian OGT and OGA are essential genes, the depletion of which is result in cell death [125–128]. OGT in particular has been implicated in maintaining the integrity of the mitochondrial membrane potential [129], the disruption of which can activate autophagy [15, 76, 130].
Autophagy Regulation by the Extracellular Matrix (ECM): “Outside-In” Signaling by Glycoconjugates
Cell – ECM interactions have long been known to drive cellular behavior as demonstrated in development, wound healing and regenerative medicine, and cancer metastasis and angiogenesis [131]. Current reports highlight the role that cell–matrix interactions play in regulating autophagy [132–147]. The regulatory signals emanating from the extracellular space originate from diverse binding interactions including receptor engagement by soluble ligands, as well as classic cell-matrix adhesion signaling. On an interesting note, many of the soluble glycoprotein ligands activate autophagy. In contrast, glycoconjugates of the basement membrane, which attach cells the ECM, appear to suppress autophagy. In many of the cases discussed below the role of the glycan component has not been defined. However, deletion of the glycans is likely to suppress the expression of the protein scaffold, either due to changes in protein folding or trafficking, altering the regulation of autophagy.
Soluble Glycoproteins and Proteoglycan – Pro-autophagic ligands
Many of the secreted glycoproteins are not primarily employed in maintaining the structural integrity of tissues, but, instead regulate cellular functions including tissue remodeling via receptor-mediated signal transduction [148, 149]. The abundance of these ligands is dynamically regulated in response to different biochemical cues [150, 151]. Only recently has the pro-autophagic capacity of these ligands been recognized. Included in the list of secreted ligands are decorin, Tsp1 (thrombospondin-1) and lacritin. Decorin and Tsp1 are highly expressed during development, presumably because of their role in regulating and remodeling the fibrillar network, but, are also induced in response to tissue injury [152, 153]. Both glycoproteins elicit anticancer activity, which involves the upregulation of autophagy [135, 143]. Less is known regarding the regulation of lacritin expression. However, a lacritin deficiency is associated with several models of corneal disease. Cytoprotection in these models can be achieved by the administration of exogenous lacritin that results in the upregulation of autophagy. In addition to these soluble ligands, bioactive fragments of basement membrane glycoproteins have been implicated implicated in triggering autophagy [138, 140]. Endostatin and endorepellin are endogenous ligands derived from the proteolytic cleavage of collagen XVIII and perlecan, respectively [154–156]. Endostatin and endorepellin are released into response to diverse forms of cell stress or injury [157, 158]. Similar to decorin and Tsp1, these bioactive fragments have been shown to inhibit endothelial cell proliferation and migration in addition to upregulating autophagy [139, 159]. In experimental cancer models, Endostatin and endorepellin also proved proficient at inhibiting tumor angiogenesis and growth indicating their potential therapeutic value [160].
It is perhaps not surprising that many glycoprotein ligands are pro-autophagic as autophagy is generally implicated in development and tissue homeostasis/remodeling that are targets of these ligands. Autophagy induction by these glycoproteins is achieved via different signal mechanisms including transcriptional activation of ATG proteins [132, 140], functional activation or inhibition of master regulatory proteins AMPK and mTOR [133] or promotion of unique protein-protein interactions between transcription factors and ATG proteins in the cytosol [123]. More detailed descriptions of the signaling elicited by specific glycoprotein ligands are provided in the following paragraphs.
Decorin
Decorin, the founding member of the class of small leucine rich proteoglycans (SLRP; Figure 2), is modified by chondroitin and dermatan sulfate as well as N-linked oligosaccharides that regulate secretion [161]. Decorin may induce autophagy across all cell types as endothelial cells and breast carcinoma cells [133, 135] exhibit enhanced autophagy with the administration of exogenous decorin. Moreover, decorin null mice exhibited an impaired autophagic response to nutrient deprivation in cardiac tissue [134]. [134]. More significantly, the “classic” autophagic stimuli (nutrient deprivation and serum starvation) and the mTOR inhibitor, Torin 1, induce decorin mRNA and protein levels [134] in tissue and plasma which could potentially lead to rapid autophagy induction across multiple tissues.
In endothelial cells, decorin triggers autophagy upon binding and activation of the tyrosine receptor kinase VEGFR2 (vascular endothelial growth factor receptor-2). VEGFR2 activation induces expression of Peg 3 (paternally expressed gene 3) [132, 133, 135], a Krueppel C2H2 transcription factor [162], that increases Beclin 1 and LC3 mRNA levels. Peg3 also induces formation of the Beclin 1-VPS34 nucleation complex and binds this complex in the cytosol. The cytoplasmic sequestration of Peg3 by the Beclin 1-VPS34 complex may be a feedback mechanism finetuning Peg3 transcriptional activity. This cryptic interaction may also represent a novel function of Peg3. For example, Peg3 association may hasten the recruitment of necessary effectors to the nucleation complex or enhance the lipid kinase activity of VPS34. Decorin also activates AMPK, lending credence to its pro-autophagic role [133]. The protein core, as well as the proteoglycan form, of decorin produces the autophagic response in endothelial cells under basal conditions indicating that the GAG (glycosaminoglycans) -chains are not essential for VEGFR2 binding. However, it is not known whether the GAG-chains tune the binding affinity or half-life of decorin.
In triple negative breast cancer cells, decorin engages the Met (hepatocyte growth factor) receptor and activates mitophagy by enhancing expression of the mitochondrial protein mitostatin [135]. RNA interference (RNAi) experiments clearly show that decorin-induced-mitophagy is mitostatin-dependent [135]. Mitostatin exhibits tumor suppressor functionality across diverse cancer cell types [163, 164] and overexpression induces mitochondrial fragmentation and perinuclear aggregation [164]. These phenomena, key steps in mitophagy activation, were observed upon decorin treatment [135]. Decorin/Met signaling promotes the binding of mitostatin mRNA by PGC-1α (peroxisome proliferator-activated receptor γ coactivator-1α). This interaction stabilizes the mRNA transcripts allowing for the enhanced mitostatin protein levels. Further work will be needed to uncover how PGC-1α selectively binds mitostatin mRNA and delineate how mitostatin causes mitochondrial depolarization and alters the balance of mitochondrial fission and fusion events.
Lacritin
The glycoprotein lacritin is a prosecretory mitogen that performs cytoprotective roles in ocular homeostasis [136, 165, 166]. Lacritin is one of the cognate ligands of the transmembrane proteoglycan syndecan-1, an established signaling coreceptor involved in modulating cell and tissue behavior [167]. The lacritin-syndecan-1 interaction is mediated through GAG-binding (heparan sulfate and chondroitin sulfate), but, also through contacts with the hydrophobic surface of the protein core of syndecan-1 [168, 169]. This hybrid binding surface is formed when heparanase partially deglycosylates the N-terminus of syndecan-1 to expose a hydrophobic surface [168, 169]. Wang et al. (2013; [123]) were able to demonstrate that complementary hydrophobic residues at the lacritin C-terminus are needed for functional binding in addition to the trimmed GAGs.
Regarding ocular homeostasis, lacritin transiently and rapidly accelerates autophagic flux to suppress simulated inflammation in transformed HCE-T (human corneal epithelial) cells [123]. Upon binding syndecan-1, lacritin promotes the rapid association of ac-FoxO1 (acetylated FoxO1) with ATG7 and the interaction of ac-FoxO3 (acetylated FoxO3) with ATG101 [123]. ATG101 functions in stabilizing the ULK1 initiation complex by protecting ATG13 from degradation [170]. This is a crucial interaction, as ATG13 recruits downstream effectors needed for autophagosome formation [170]. The molecular function(s) of the ATG101 – ac-FoxO3 interaction on autophagy are currently undefined. However, several attractive models exist: the ac-FoxO3 – ATG101 interaction may increase the binding affinity/kinetics between ATG101 and ATG13 or may form a high affinity-binding surface with ATG13 to enhance recruitment of ATG9 vesicles and LC3 for autophagosome formation. Alternatively ac-FoxO3 may interact with ULK1, the autophagy initiating kinase, to potentiate its kinase activity. The ac-FoxO1-ATG7 interaction was also observed during autophagy induction under serum starvation in human cancer cells [119], suggesting that this interaction may be a common step in autophagy activation. ATG7 functions as an E1-like activating enzyme for the ubiquitin-like reactions employed in autophagosome expansion [6, 17]. However, the effects of ac-FoxO1 association on ATG7 activity have yet to been determined.
Thrombospondin-1
The anti-angiogenic glycoprotein Tsp1 (thrombospondin-1) induces autophagosome accumulation in cancer cell lines expressing mutant H-Ras [143]. While Tsp1 can bind several receptors [148], a decapeptide derived from the CD47 binding region of Tsp1 selectively reduced tumor outgrowth in mice and increased cell death in B6ras cells. However, this phenotype was not observed in the non-tumorigenic MDFB6 cell line [143]. The induction of cell death by Tsp1 did not exhibit an apoptotic profile, but, significantly increased the number of autophagosomes [143]. Downstream effectors of the Tsp1/CD47 axis include BNIP3, a known autophagy inducer [171]. While the mechanisms underlying autophagy activation in B6ras cells are undefined, in immortalized Jurkat T lymphocytes and T cells derived from mice and human donors, BNIP3 expression and mitochondrial localization is induced by CD47 activation [171]. While the use of Tsp1 peptides suggests that glycosylation is dispensable for autophagy activation, Tsp1 glycosylation would be needed for the extracellular transit of the full-length glycoprotein [53, 172, 173] and there is evidence that receptor GAGs mediate Tsp1 binding in T cells [174]. Given the diverse interactome of Tsp1 [175] and the reported CD47-independent activities of the Tsp1 (4NIK) peptide [176], knockdown experiments targeting Tsp1 binding partners should be utilized in validating the how exogenous Tsp1 regulates autophagy.
In contrast to the work in cancer models, the depletion of either CD47 or Tsp1 leads to increased autophagic flux which is associated with protection against radiation in endothelial cells, Jurkat T-cells and mice [177, 178]. Suppressing CD47 signaling greatly increased gene expression of Beclin 1, ATG5 and ATG7 in response to ionizing radiation compared to wild type [177]. Basal LC3-II levels were also enhanced in CD47 deficient cells [177]. These results, in addition to the reduced p62/sequestosome-1 expression, indicate that suppressing Tsp1/CD47 signaling enhances autophagic flux under basal and radiation stress conditions.
Endorepellin
As previously mentioned, endorepellin is an endogenously processed C-terminal fragment of perlecan [155, 157]. This region of perlecan contains three LG (laminin-like globular) repeats and is proteolytically cleaved by Cathepsin L to liberate endorepellin [155]. Endorepellin can be further processed by the BMP-1/Tolloid family of metalloproteases to a bioacive 26kDa fragment [157]. Endorepellin possesses both angiostatic [157, 179] and pro-autophagic functionality [140]. Similar to decorin, endorepellin – induced autophagy involves binding VEGFR2 [140]. This interaction occurs between LG1 and LG2 of endorepellin and the immunoglobulin-like repeats Ig3–5 of the VEGFR2 ectodomain [179]. Though endorepellin is glycosylated, the roles that the glycans play in binding VEGFR2 are unknown. In endothelial cells, endorepellin treatment induces Peg3 expression a necessary event to upregulate Beclin 1 and LC3 mRNA and protein expression. All three proteins, along with VPS34, were found to colocalize at the autophagosomes [140]. The crucial trigger to initiate autophagy may be to the suppression of mTOR signaling on endorepellin treatment as described in an earlier study [159].
Endostatin
Akin to perlecan, endogenous proteolytic processing of collagen XVIII, a basement membrane heparan sulfate proteoglycan, generates the C-terminal fragment endostatin that has anti-angiogenic capacity [154, 167]. In endothelial cells (EAhy926), endostatin treatment induces the accumulation of autophagosomes in 6 hours, however prolonged exposure (24 hours) enhanced autophagosome accumulation and induced ~90% cell death [138]. Similar to cancer cells treated with Tsp1, endostatin-induced cell death is due primarily to the autophagy pathway [138]. Experiments in HUVEC (human umbilical vein endothelial cells) uncovered several mechanistic details of endostatin-induced autophagy. Endostatin binds the α5β1 integrin heterotrimer (fibronectin receptor), increasing Beclin 1 expression while suppressing expression of Bcl-2, Bcl-xL and β-catenin. Two possible mechanisms underlie these observations: Firstly, the dissociation of Beclin 1 and Bcl-2 promotes autophagy, which suggests that altering the Beclin 1:Bcl-2 ratio is sufficient for endostatin to switch on autophagy [180, 181]. Secondly, endostatin treatment relieves repression of the autophagy pathway by down regulating Wnt/β-catenin, a known negative regulator [182].
Kringle 5
An endogenous inhibitor of angiogenesis, Kringle 5 (K5), is generated by cleavage of the plasma glycoprotein plasminogen [141]. In HUVEC cells, recombinant K5 induces both apoptosis and autophagy. K5 treatment increased Beclin 1 expression within 8 hours and increased autophagosome accumulation could be clearly seen at 4 and 8 hours with a plateau at 24 hours. The activation profiles for caspases 7 and 3 revealed enhanced activation at later time points (≥8 hours), indicating that K5-induced autophagy precedes apoptosis activation. Although K5 treatment for 24 hours caused mitochondrial depolarization, a common trigger for both pathways [15, 16, 75, 76, 130], autophagy is not required for K5-induced apoptosis as Beclin 1 RNAi suppressed K5-induced autophagy while increasing caspase activation [141]. These data suggest that K5-induced autophagy is pro-survival. This autophagic response appears to be specific for endothelial cells, as ovarian cancer cells and human foreskin fibroblasts did not exhibit autophagy induction with K5 treatment for 24 hours [141].
Basement membrane glycoproteins – Adhesive signaling maintains baseline autophagy
Adhesion to the extracellular matrix provides biochemical cues needed for tissue homeostasis, cell proliferation and development. Several studies indicate that loss of ECM adhesion results in the manifestation of metabolic and oxidative stress [183–185] which can culminate in an apoptotic response referred to as anoikis [186]. As series of investigations using mammary epithelial cells cultured under adherent or suspension conditions demonstrate significant induction of autophagic flux in matrix detached cells as a pro-survival program [184, 185, 187]. These studies have provided a comprehensive analysis of the intracellular signaling mediating detachment-induced autophagy: ROS induction activates protein kinase R like endoplasmic reticulum kinase (PERK) which then activates AMPK. AMPK inhibits mTORC1 leading to an induction in autophagy[184, 185]. More recent studies have assessed individual basement membrane glycoproteins involved in adhesion regulation of autophagy.
Perlecan
Perlecan is expressed across a broad range of tissues and is a functional component of the basement membrane [188]. Perlecan plays critical roles in tissue development and homeostasis, as well as angiogenesis [149, 188, 189]. Ning et al. (2015; [137]) investigated the role that perlecan plays in regulating autophagy in skeletal muscle. Perlecan depletion is lethal, and this can be overcome by re-expression of transgenic perlecan in cartilage [137]. Perlecan deficiency was correlated with increased tenotomy-induced soleus atrophy and autophagosome accumulation compared to control animals [137]. These data suggested that perlecan suppresses autophagy. The perlecan deficient mice exhibited greater catabolic potential under basal conditions than control animals; p62/sequestosome-1 levels were significantly reduced in perlecan deficient animals [137]. Consistent with a pro-autophagic paradigm, LC3-II levels and AMPK signaling were enhanced (pro-autophagic) whereas inhibitory mTORC1 signaling was suppressed [137].
The receptor or signaling platform responsible for transmitting perlecan’s anti-autophagy signal is yet to be identified. However, pervious studies have identified functional interactions that mediate perlecan’s bioactivity. The protein core of perlecan contains conserved motifs that bind ligands including progranulin and hedgehog, but, also interacts directly with receptors (VEGFR1/2, α2β1 integrin, α-dystroglycan) [188–190]. Perlecan also bind cytokines and growth factors, including FGF10 (fibroblast growth factor 10) and IL2 (interleukin 2) [188–190], through its HS (heparan sulfate) chains. These binding interactions can have coreceptor function to enhance receptor-ligand interactions [149, 188, 189]. Perlecan’s HS chains can also act as a ligand storage sink controlling sequestration and release of ligands such as PDGF (platelet derived growth factor) [191]. As such, Perlecan’s HS specified bioactivities can be suppressed by the action of heparanase that degrades HS [189]. On an interesting note, exogenous heparanase and heparanase overexpression increase autophagy by downregulating signaling from the negative regulator mTORC1 [192]. While the colocalization of intracellular heparanase, autophagosome and lysosomes correlates with enhanced autophagy, one cannot rule out the possibility that the degradation of extracellular HS chains and other HSPG (heparan sulfate proteoglycans) could liberate ligands for autophagy activation.
Laminin α2
A component of the heterotrimeric basement membrane protein laminin 211, laminin α2, is required for proper muscle development and functionality [193, 194]. Muscle integrity is mediated through laminin binding of cell surface receptors including integrins and dystroglycan [193, 194]. The C-terminus of laminin α2 is primarily responsible for these binding interactions. Congenital muscular dystrophy type 1A (MDC1A) is caused by mutations in laminin α2 resulting in complete or partial deficiency [194]. The muscle wasting observed in MDC1A is associated with increased degradation by the proteasome and autophagy pathways [142, 194]. The mRNA and protein levels of several autophagy related proteins were upregulated in laminin α2 deficient mice including LC3, Beclin 1 and VPS34 [142]. LC3 expression was also upregulated in muscle biopsies of patients with MDC1A. Using 3-MA, the authors suppressed autophagy and returned gene expression of autophagy related proteins to wild type levels in laminin α2-deficient muscle. In addition, 3-MA reduced fibrosis in quadricep muscle and muscle wet weight while muscle fiber diameter returned to wild type levels [142]. These results clearly indicate that constitutively enhanced autophagy is deleterious. Laminin α2 is reported to be a glycoprotein, though the function(s) of these glycans and their impact on autophagy are currently undefined.
Intracellular Hyaluronan triggers Autophagy
HA (Hyaluronan) is unique among the glycosaminoglycans as it not typically covalently linked to a protein core and is unsulfated [195, 196]. The repeating unit, a disaccharide of GluA (glucuronic acid) and GlcNAc, can generate linear chains of varying molecular weights [197]. The heterogeneity of HA matrices are due to the complex interplay of HAS (hyaluronan synthase) enzymes that generate the polymer and hyaluronidases that catabolize HA. Hyaluronan metabolism is also dictated by the nucleotide-sugar reservoir [198, 199]. High and low molecular-weight HA mediate disparate physiological effects via differential receptor binding that activates diverse signaling pathways [200].
Unlike the Golgi-dependent reactions typically utilized in glycosaminoglycan biosynthesis, HA synthesis is carried out at the plasma membrane by the HAS enzymes [201, 202]. Although the plasma membrane is the final destination for this family of transmembrane proteins, several studies have established the existence of intracellular pools of these enzymes that are thought to facilitate rapid localization to the plasma membrane [201, 202]. Normally HAS enzyme isoforms are inactive until inserted into the plasma membrane [201, 203]. However, animal models of diabetic nephropathy and quiescent mesangial cells stimulated to grow in high glucose (25.5mM) media exhibit formation of intracellular HA chains, enhanced extracellular HA content, and marked immunohistochemical staining for LC3 [144, 147]. This intracellular HA most likely reflects activation of the organellar HAS enzyme reservoirs culminating in ER stress and autophagy induction. Activation of the intracellular HAS enzymes may be a result of increased lipogenic flux in the ER, which was observed in the glomeruli of diabetic rats and mesangial cells grown in hyperglycemic conditions [147]. Supporting these data, treatment of rat mesangial cells grown in hyperglycemic conditions with heparin suppressed lipid accumulation, LC3 expression and the formation of autophagosomes [147]. Another mechanism which may result in activation of intracellular HAS enzymes, is signaling though cyclin D3 and C/EBPα (CCAAT/enhancer-binding protein α). The expression of cyclin D3 and C/EBPα, and subsequent formation of the cyclin D3-CDK4-C/EBPα complex, is induced by high glucose treatment (24h) [144]. This complex colocalizes with intracellular LC3 and HA, suggesting that HAS enzymes may be directly phosphorylated and activated by CDK4 (cyclin-dependent kinase 4) [144]. Indeed, RNAi suppression of cyclin D3 under hyperglycemia reduced total hyaluronan content and also reduced C/EBPα expression [144]. C/EBPα also upregulates lipogenesis which may also trigger HAS activation as discussed above [204].
The overexpression of the intracellular Hyaluronan Binding Proteins or hyaldherins in fibroblast and cancer cell lines have vastly different effects on the autophagy pathway. Overexpressed HABP1 (Hyaluronan Binding Protein-1) was previously shown to accumulate in mitochondria raising the ROS levels [205]. Saha et al (2013; [145]) confirmed that Beclin 1 and LC3 and expression were upregulated in fibroblasts constitutively overexpressing HABP1 (F-HABP07 cells) suggesting the induction of autophagy. Use of the inhibitor bafilomycin confirmed that HABP1 overexpression enhanced autophagic flux. In agreement with other studies, excess ROS reduced the levels of HA [145]. The addition of exogenous HA reversed autophagosome accumulation and ROS levels in F-HABP07 cells. This may be due to the reported antioxidant property of HA [145]. In contrast to these studies, HABP1 expression in the human liver cancer line HepG2 enhances HA levels and expression of the HA receptor CD44, but, suppresses expression of the autophagy marker LC3 under basal conditions and nutrient deprivation [146]. Inhibition of HA synthesis with 4-MU (4-methylumbelliferone) increased ROS levels as well as increased Beclin 1 expression and basal autophagy [146]. A potential caveat of 4-MU use is that it depletes the cellular pool of UDP-GluA (UDP-Glucuronic acid) in addition to suppressing HAS2 and HAS3 gene expression [198]. Depletion of the nucleotide sugar pool could cause ER stress that would trigger autophagy.
Autophagy Regulates Glycoconjugate Turnover
The release of unconjugated glycan moieties in the cytosol occurs as a result of the hydrolysis of lipid-linked oligosaccharides, the donors for N-glycan biosynthesis and a subset of O-glycan biosynthesis [206–208]. This pool of free sugars can be augmented during the unfolded protein response in which glycoproteins that fail to fold properly are retrotranslocated from the ER and are targeted to the proteasome for degradation [208]. A cytoplasmic peptide:N-glycanase removes the N-linked glycans on the unfolded proteins [208]. Recent studies indicate that autophagy is involved in the elimination of these free glycans. Suppressing ATG5 and ATG9a expression in MEFs (mouse embryonic fibroblasts) impairs basal and stressed-induced autophagy [2, 209], resulting in the accumulation of cytoplasmic sialyloligosaccharides over time [41]. In wild type MEFs 51% of the free sialyloligosaccharides are found in the lysosomal fraction and 45% in the cytosolic fraction. In contrast, when autophagy is suppressed by deletion of ATG5, only 25% of free sialyloligosaccarides are targeted to the lysosomal fraction. Using an ATG5 “tet-off ” system [210] the authors promoted sialyloligosaccharide build-up. Releasing ATG5 suppression (basal autophagy) did not significantly reduce free sialyloligosaccharides, however 6 hours of starvation induced autophagy drastically reduced the levels of cytosolic sialyloligosaccharides and high-mannose type glycans [41]. These data demonstrate that free oligosaccharides accumulated in the “fed” state and that basal and induced autophagy participate in eliminating free oligosaccharides.
Concluding Remarks
Glycoconjugates play key roles in regulating many facets of biology, including development and differentiation, immunity, inflammation, and angiogenesis. Dysregulation of the regulatory pathways that control glycoconjugate biosynthesis or deconvolute glycan codes underly an array of diseases. Newly added to the large catalog of biological activities controlled by glycoconjugates is autophagy, a gatekeeper of cell survival. As detailed in the preceding sections a diverse assortment of glyconjugates exert a stimulatory or inhibitory effect on autophagy in a broad range of physiologically relvant models. However, the examples we have referenced are only a small portion of the full repertoire of mammalian glyconjugates. To define a more complete map of the role of glycans in the regulation of autophagy and their impact on physiology numerous studies are required. While these studies run the gamut of glycobiology, there are several critical approaches that are summarized below.
Many of the steps of autophagy include proteins which can be O-GlcNAc-modified, and several studies have implicated O-GlcNAc in regulating both the induction of autophagy and key steps in the autophagic process. These data lead to some obvious questions: First, which other autophagy proteins are modified by O-GlcNAc; Second, how do signals which induce autophagy impact O-GlcNAcylation of these proteins; Third, how does O-GlcNAc regulate these proteins and the processes they are involved in. Critical to understanding the role of O-GlcNAc in autophagy are site-mapping studies enabling researchers to mutate key residues and to develop site-specific antibodies which will facilitate the application of this information to other physiological models in which autophagy is involved. Such studies should be extended to N- and O-linked glycoproteins. As complete disruption of glycoconjugates biosynthetic pathways are likely to impact protein folding and thus protein expression, the identification of the glycan structures and the sites of modification are critical. Studying these glycoconjugates in cells, such as the Lec mutant CHO cells [211], in which the latter steps in glycoconjuagtes biosynthesis have been disrupted may provide critical information about the roles of protein-glycan interactions in the regulation of autophagy as well as identifying the key structures that are critical to autophagy. A similar approach would deconvolute the role that the glycosaminoglycan components of proteoglycans play in the regulation of autophagy. Perhaps the most intruging role of glyconjugates in autophagy is their role as PAMPs. Identifying the PAMP receptors will be challenging. One tool that should facilitate the identification of these receptors are UV cross-linkable sugars [212, 213]. Here unnatural sugars modified by diazirines are salvaged into the nucleotide sugar biosynthetic pathway and are ultimately added to proteins. The diazirine can be cross-linked to nearby proteins, enabling the identification of protein complexes and glycan receptors. Such a technique will also be invaluable to identifying the role of ER/Golgi derived proteins in the induction of autophagy and the generation of the autophagosome membranes. Undoubtedly, these and other studies focused on glycans will provide insight into the regulation of autophagy and the impact that glycan-mediated autophagy has on physiological processes and disease pathogenesis, as well as providing novel targets for therapeutic intervention.
Research Highlights.
Diverse glycoconjugates are implicated in the regulation of the autophagy pathway
Proteoglycans and glycoproteins signal the “master regulators” of autophagy
Autophagy regulatory proteins and core machinery are targets of glycosylation
Lectins bind extracellular and intracellular danger signals initiating autophagy
Work is required to understand the mechanisms by which glycans regulate autophagy
Abbreviations
- 3-MA
3-methyladenine
- 4-MU
4-methylumbelliferone
- AMPK
AMP-activated kinase
- ATG
Autophagy related
- BMDC
Bone marrow derived dendritic cells
- C/EBPα
CCAAT/enhancer-binding protein α
- CDK4
Cyclin-dependent kinase 4
- Con A
Concanavalin A
- CPP
calcium phosphate precipitate
- CS
Chondroitin sulfate
- DPI
Diphenyleneiodonium
- ECM
Extracellular Matrix
- ER
Endoplasmic reticulum
- ERGIC
ER-Golgi Intermediate Compartment
- FB1
Fumonisin B1
- FGF10
Fibroblast growth factor 10
- FoxO
Forkhead Box O transcription factor
- FTDP-17
Frontotemporal dementia with parkinsonism chromosome 17
- GAG
Glycosaminoglycan
- GluA
Glucuronic acid
- Golgi
Golgi Apparatus
- HA
Hyaluronan
- HABP1
Hyaluronan binding protein-1
- HAS
Hyaluronan synthase
- HCE-T
Transformed human corneal epithelial cells
- HS
Heparan sulfate
- HSPG
Heparan sulfate proteoglycans
- HUVEC
Human umbilical vein endothelial cells
- ITAM
Immunoreceptor tyrosine-based activation motif
- JNK
c-Jun N-terminal kinase
- K5
Kringle 5
- LAP
LC3-associated phagocytosis
- LC3
Microtubule-associated protein 1 light chain 3
- LG
Laminin-like globular repeat
- LKB1
Liver kinase B1
- LLOMe
Leu-Leu methyl ester hydrobromide
- MDC1A
Congenital muscular dystrophy type 1A
- MEF
Mouse embryonic fibroblasts
- MHC- II
Major histocompatiblity complex- II
- mtDNA
Mitochondrial DNA
- mTORC1
Mammalian target of rapamycin complex 1
- MβCD
Methyl-β-cyclodextrin
- NDP52
Nuclear dot protein 52
- O-GlcNAc
O-linked β-N-acetylglucosamine
- OGA
O-GlcNAcase
- OGT
O-GlcNAc transferase
- p38 MAPK
p38 mitogen activated protein kinase
- PAMP
Pathogen associated molecular pattern
- PAS
Phagophore assembly site
- PCL
Polygonatum cyrtonema lectin
- PDGF
Platelet derived growth factor
- Peg3
Paternally expressed gene 3
- PI3P
Phosphatidylinositol-3-phosphate
- PERK
Protein kinase R like endoplasmic reticulum kinase
- Rab7a
Ras-related protein Rab-7a GTPase
- RCA-120
Ricinus Communis Agglutinin 120
- RNAi
RNA interference
- ROS
Reactive oxygen species
- SNAP29
Synaptosomal–associated protein 29
- SNARE
Soluble NSF [N-ethyl-maleimide-sensitive fusion protein] attachment protein receptor
- stx17
Syntaxin 17
- Syk
Spleen tyrosine kinase
- TAK1
Transforming growth factor-beta-activated kinase 1
- Tsp1
Thrombospondin-1
- ULK1
Unc-51-like kinase 1
- VAMP8
Vesicle-associated membrane protein 8
- VEGFR
Vascular endothelial growth factor receptor
- VPS34
Vacuolar protein sorting-associated protein 34
- WGA
Wheat germ agglutinin
- WIPI1
WD repeat domain phosphoinositide-interacting protein
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Nakagawa I, et al. Autophagy defends cells against invading group A Streptococcus. Science. 2004;306(5698):1037–40. doi: 10.1126/science.1103966. [DOI] [PubMed] [Google Scholar]
- 2.Kuma A, et al. The role of autophagy during the early neonatal starvation period. Nature. 2004;432(7020):1032–6. doi: 10.1038/nature03029. [DOI] [PubMed] [Google Scholar]
- 3.Wei Y, et al. The stress-responsive kinases MAPKAPK2/MAPKAPK3 activate starvation-induced autophagy through Beclin 1 phosphorylation. Elife. 2015;4:e05289. doi: 10.7554/eLife.05289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Harding TM, et al. Isolation and characterization of yeast mutants in the cytoplasm to vacuole protein targeting pathway. J Cell Biol. 1995;131(3):591–602. doi: 10.1083/jcb.131.3.591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Berry DL, Baehrecke EH. Growth arrest and autophagy are required for salivary gland cell degradation in Drosophila. Cell. 2007;131(6):1137–48. doi: 10.1016/j.cell.2007.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Juhász G, et al. Atg7-dependent autophagy promotes neuronal health, stress tolerance, and longevity but is dispensable for metamorphosis in Drosophila. Genes Dev. 2007;21(23):3061–6. doi: 10.1101/gad.1600707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim Y, Sun H. Functional genomic approach to identify novel genes involved in the regulation of oxidative stress resistance and animal lifespan. Aging Cell. 2007;6(4):489–503. doi: 10.1111/j.1474-9726.2007.00302.x. [DOI] [PubMed] [Google Scholar]
- 8.Zhang Y, et al. The role of autophagy in mitochondria maintenance: characterization of mitochondrial functions in autophagy-deficient S. cerevisiae strains. Autophagy. 2007;3(4):337–46. doi: 10.4161/auto.4127. [DOI] [PubMed] [Google Scholar]
- 9.Komatsu M, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441(7095):880–4. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- 10.Hara T, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441(7095):885–9. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- 11.Degenhardt K, et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell. 2006;10(1):51–64. doi: 10.1016/j.ccr.2006.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Onodera J, Ohsumi Y. Autophagy is required for maintenance of amino acid levels and protein synthesis under nitrogen starvation. J Biol Chem. 2005;2005(280):36. doi: 10.1074/jbc.M506736200. [DOI] [PubMed] [Google Scholar]
- 13.Lum JJ, et al. Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell. 2005;120(2):237–48. doi: 10.1016/j.cell.2004.11.046. [DOI] [PubMed] [Google Scholar]
- 14.Young AR, et al. Starvation and ULK1-dependent cycling of mammalian Atg9 between the TGN and endosomes. J Cell Sci. 2006;119(Pt 18):3888–900. doi: 10.1242/jcs.03172. [DOI] [PubMed] [Google Scholar]
- 15.Okatsu K, et al. p62/SQSTM1 cooperates with Parkin for perinuclear clustering of depolarized mitochondria. Genes Cells. 2010;15(8):887–900. doi: 10.1111/j.1365-2443.2010.01426.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vives-Bauza C, et al. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc Natl Acad Sci U S A. 2010;107(1):378–83. doi: 10.1073/pnas.0911187107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mizushima N, et al. A protein conjugation system essential for autophagy. Nature. 1998;395(6700):395–8. doi: 10.1038/26506. [DOI] [PubMed] [Google Scholar]
- 18.Itakura E, Mizushima N. Characterization of autophagosome formation site by a hierarchical analysis of mammalian Atg proteins. Autophagy. 2010;6(6):764–76. doi: 10.4161/auto.6.6.12709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dengjel J, et al. Identification of autophagosome-associated proteins and regulators by quantitative proteomic analysis and genetic screens. Mol Cell Proteomics. 2012;11(3):M111.014035. doi: 10.1074/mcp.M111.014035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Amaya C, Fader CM, Colombo MI. Autophagy and proteins involved in vesicular trafficking. FEBS Lett. 2015;589(22):3343–53. doi: 10.1016/j.febslet.2015.09.021. [DOI] [PubMed] [Google Scholar]
- 21.Feng Y, et al. The machinery of macroautophagy. Cell Res. 2014;24(1):24–41. doi: 10.1038/cr.2013.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang Z, Klionsky DJ. Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol. 2010;22(2):124–31. doi: 10.1016/j.ceb.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lamb CA, Yoshimori T, Tooze SA. The autophagosome: origins unknown, biogenesis complex. Nat Rev Mol Cell Biol. 2013;14(12):759–74. doi: 10.1038/nrm3696. [DOI] [PubMed] [Google Scholar]
- 24.Yamamoto H, et al. Atg9 vesicles are an important membrane source during early steps of autophagosome formation. J Cell Biol. 2012;198(2):219–33. doi: 10.1083/jcb.201202061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li S, et al. Sterical hindrance promotes selectivity of the autophagy cargo receptor NDP52 for the danger receptor galectin-8 in antibacterial autophagy. Sci Signal. 2013 Feb 5;6(261):ra9. doi: 10.1126/scisignal.2003730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Olsvik HL, et al. FYCO1 Contains a C-terminally Extended, LC3A/B-preferring LC3-interacting Region (LIR) Motif Required for Efficient Maturation of Autophagosomes during Basal Autophagy. J Biol Chem. 2015;290(49):29361–74. doi: 10.1074/jbc.M115.686915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pankiv S, et al. FYCO1 is a Rab7 effector that binds to LC3 and PI3P to mediate microtubule plus end-directed vesicle transport. J Cell Biol. 2010;188(2):253–69. doi: 10.1083/jcb.200907015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McEwan DG, et al. PLEKHM1 regulates autophagosome-lysosome fusion through HOPS complex and LC3/GABARAP proteins. Mol Cell. 2015;57(1):39–54. doi: 10.1016/j.molcel.2014.11.006. [DOI] [PubMed] [Google Scholar]
- 29.Jiang P, et al. The HOPS complex mediates autophagosome–lysosome fusion through interaction with syntaxin 17. Mol Biol Cell. 2014;25(8):1327–37. doi: 10.1091/mbc.E13-08-0447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Morelli E, et al. Multiple functions of the SNARE protein Snap29 in autophagy, endocytic, and exocytic trafficking during epithelial formation in Drosophila. Autophagy. 2014;10(12):2251–68. doi: 10.4161/15548627.2014.981913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Itakura E, Kishi-Itakura C, Mizushima N. The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell. 2012;151(6):1256–69. doi: 10.1016/j.cell.2012.11.001. [DOI] [PubMed] [Google Scholar]
- 32.Renna M, et al. Autophagic substrate clearance requires activity of the syntaxin-5 SNARE complex. J Cell Sci. 2011;124(Pt 3):469–82. doi: 10.1242/jcs.076489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jäger S, et al. Role for Rab7 in maturation of late autophagic vacuoles. J Cell Sci. 2004;117(Pt 20):4837–48. doi: 10.1242/jcs.01370. [DOI] [PubMed] [Google Scholar]
- 34.Gutierrez MG, et al. Rab7 is required for the normal progression of the autophagic pathway in mammalian cells. J Cell Sci. 2004;117(Pt 13):2687–97. doi: 10.1242/jcs.01114. [DOI] [PubMed] [Google Scholar]
- 35.Ichimura Y, et al. A ubiquitin-like system mediates protein lipidation. Nature. 2000;408(6811):488–92. doi: 10.1038/35044114. [DOI] [PubMed] [Google Scholar]
- 36.Kim J, et al. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13(2):132–41. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Russell RC, et al. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat Cell Biol. 2013;15(7):741–50. doi: 10.1038/ncb2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee IH, Finkel T. Regulation of autophagy by the p300 acetyltransferase. J Biol Chem. 2009;284(10):6322–8. doi: 10.1074/jbc.M807135200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sarkar S, et al. Complex inhibitory effects of nitric oxide on autophagy. Mol Cell. 2011;43(1):19–32. doi: 10.1016/j.molcel.2011.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Settembre C, et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. 2012;31(5):1095–108. doi: 10.1038/emboj.2012.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Seino J, et al. Basal autophagy is required for the efficient catabolism of sialyloligosaccharides. J Biol Chem. 2013;288(37):26898–907. doi: 10.1074/jbc.M113.464503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yamamoto A, Masaki R, Tashiro Y. Characterization of the isolation membranes and the limiting membranes of autophagosomes in rat hepatocytes by lectin cytochemistry. J Histochem Cytochem. 1990;38(4):573–80. doi: 10.1177/38.4.2319125. [DOI] [PubMed] [Google Scholar]
- 43.Axe EL, et al. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J Cell Biol. 2008;182(4):685–701. doi: 10.1083/jcb.200803137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ylä-Anttila P, et al. 3D tomography reveals connections between the phagophore and endoplasmic reticulum. Autophagy. 2009 Nov;5(8):1180–5. doi: 10.4161/auto.5.8.10274. Epub 2009 Nov 8., 2009. 5(8): p. 1180–5. [DOI] [PubMed] [Google Scholar]
- 45.Hayashi-Nishino M, et al. A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nat Cell Biol. 2009;11(12):1433–7. doi: 10.1038/ncb1991. [DOI] [PubMed] [Google Scholar]
- 46.Ravikumar B, et al. Plasma membrane contributes to the formation of pre-autophagosomal structures. Nat Cell Biol. 2010;12(8):747–57. doi: 10.1038/ncb2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ge L, et al. The ER-Golgi intermediate compartment is a key membrane source for the LC3 lipidation step of autophagosome biogenesis. Elife. 2013;2:e00947. doi: 10.7554/eLife.00947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.van der Vaart A, Griffith J, Reggiori F. Exit from the Golgi is required for the expansion of the autophagosomal phagophore in yeast Saccharomyces cerevisiae. Mol Biol Cell. 2010;21(13):2270–84. doi: 10.1091/mbc.E09-04-0345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Reggiori F, et al. Atg9 cycles between mitochondria and the pre-autophagosomal structure in yeasts. Autophagy. 2005;1(2):101–9. doi: 10.4161/auto.1.2.1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Suzuki SW, et al. Atg13 HORMA domain recruits Atg9 vesicles during autophagosome formation. Proc Natl Acad Sci U S A. 2015;112(11):3350–5. doi: 10.1073/pnas.1421092112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.He C, et al. Self-interaction is critical for Atg9 transport and function at the phagophore assembly site during autophagy. Mol Biol Cell. 2008;19(12):5506–16. doi: 10.1091/mbc.E08-05-0544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Takahashi Y, et al. Bif-1 regulates Atg9 trafficking by mediating the fission of Golgi membranes during autophagy. Autophagy. 2011;7(1) doi: 10.4161/auto.7.1.14015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Appenzeller C, et al. The lectin ERGIC-53 is a cargo transport receptor for glycoproteins. Nat Cell Biol. 1999;1(6):330–4. doi: 10.1038/14020. [DOI] [PubMed] [Google Scholar]
- 54.Ishihara N, et al. Autophagosome requires specific early Sec proteins for its formation and NSF/SNARE for vacuolar fusion. Mol Biol Cell. 2001;12(11):3690–702. doi: 10.1091/mbc.12.11.3690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ge L, Schekman R. The ER-Golgi intermediate compartment feeds the phagophore membrane. Autophagy. 2014;10(1):170–2. doi: 10.4161/auto.26787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ge L, Zhang M, Schekman R. Phosphatidylinositol 3-kinase and COPII generate LC3 lipidation vesicles from the ER-Golgi intermediate compartment. Elife. 2014;3:e04135. doi: 10.7554/eLife.04135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Barlowe C, et al. COPII: A membrane coat formed by Sec proteins that drive vesicle budding from the endoplasmic reticulum. Cell. 1994;77(6):895–907. doi: 10.1016/0092-8674(94)90138-4. [DOI] [PubMed] [Google Scholar]
- 58.Dudognon P, et al. Regulation of a COPII component by cytosolic O-glycosylation during mitosis. FEBS Lett. 2004;561(1–3):44–50. doi: 10.1016/S0014-5793(04)00109-7. [DOI] [PubMed] [Google Scholar]
- 59.Zachara NE, et al. The dynamic stress-induced “O-GlcNAc-ome” highlights functions for O-GlcNAc in regulating DNA damage/repair and other cellular pathways. Amino Acids. 2011;40(3):793–808. doi: 10.1007/s00726-010-0695-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Akiyoshi K, et al. Induction of neuron-like tubes and liposome networks by cooperative effect of gangliosides and phospholipids. FEBS Lett. 2003;534(1–3):33–8. doi: 10.1016/s0014-5793(02)03743-2. [DOI] [PubMed] [Google Scholar]
- 61.Matarrese P, et al. Evidence for the involvement of GD3 ganglioside in autophagosome formation and maturation. Autophagy. 2014;10(5):750–65. doi: 10.4161/auto.27959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hwang J, et al. Gangliosides induce autophagic cell death in astrocytes. Br J Pharmacol. 2010;159(3):586–603. doi: 10.1111/j.1476-5381.2009.00563.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hwang J, et al. NF-κB as a common signaling pathway in ganglioside-induced autophagic cell death and activation of astrocytes. J Neuroimmunol. 2010;226(1–2):66–72. doi: 10.1016/j.jneuroim.2010.05.037. [DOI] [PubMed] [Google Scholar]
- 64.Caramelo JJ, Parodi AJ. Getting in and out from calnexin/calreticulin cycles. J Biol Chem. 2008;283(16):10221–5. doi: 10.1074/jbc.R700048200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Itin C, et al. ERGIC-53 is a functional mannose-selective and calcium-dependent human homologue of leguminous lectins. Mol Biol Cell. 1996;7(3):483–93. doi: 10.1091/mbc.7.3.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jiang QL, et al. Plant lectins, from ancient sugar-binding proteins to emerging anti-cancer drugs in apoptosis and autophagy. Cell Prolif. 2015;48(1):17–28. doi: 10.1111/cpr.12155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bhattacharyya L, et al. Concanavalin A interactions with asparagine-linked glycopeptides. Bivalency of high mannose and bisected hybrid type glycopeptides. J Biol Chem. 1987;262(3):1288–93. [PubMed] [Google Scholar]
- 68.Albertini DF, Anderson E. Microtubule and microfilament rearrangements during capping of concanavalin A receptors on cultured ovarian granulosa cells. J Cell Biol. 1977;73(1):111–27. doi: 10.1083/jcb.73.1.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Callaghan TM, et al. Modulation of the binding and endocytosis of concanavalin A by guinea pig keratinocytes: reversible antagonistic effects of cholesterol and phospholipid-liposomes. J Invest Dermatol. 1990;94(1):58–64. doi: 10.1111/1523-1747.ep12873359. [DOI] [PubMed] [Google Scholar]
- 70.Roy B, et al. Role of PI3K/Akt/mTOR and MEK/ERK pathway in Concanavalin A induced autophagy in HeLa cells. Chem Biol Interact. 2014;210:96–102. doi: 10.1016/j.cbi.2014.01.003. [DOI] [PubMed] [Google Scholar]
- 71.Wu YT, et al. Activation of the PI3K-Akt-mTOR signaling pathway promotes necrotic cell death via suppression of autophagy. Autophagy. 2009;5(6):824–34. doi: 10.4161/auto.9099. [DOI] [PubMed] [Google Scholar]
- 72.Pratt J, Annabi B. Induction of autophagy biomarker BNIP3 requires a JAK2/STAT3 and MT1-MMP signaling interplay in Concanavalin-A-activated U87 glioblastoma cells. Cell Signal. 2014;26(5):917–24. doi: 10.1016/j.cellsig.2014.01.012. [DOI] [PubMed] [Google Scholar]
- 73.Belkaid A, et al. Necrosis induction in glioblastoma cells reveals a new “bioswitch” function for the MT1-MMP/G6PT signaling axis in proMMP-2 activation versus cell death decision. Neoplasia. 2007;9(4):332–40. doi: 10.1593/neo.07142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pratt J, Roy R, Annabi B. Concanavalin-A-induced autophagy biomarkers requires membrane type-1 matrix metalloproteinase intracellular signaling in glioblastoma cells. Glycobiology. 2012;22(9):1245–55. doi: 10.1093/glycob/cws093. [DOI] [PubMed] [Google Scholar]
- 75.Vande Velde C, et al. BNIP3 and genetic control of necrosis-like cell death through the mitochondrial permeability transition pore. Mol Cell Biol. 2000;20(15):5454–68. doi: 10.1128/mcb.20.15.5454-5468.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rikka S, et al. Bnip3 impairs mitochondrial bioenergetics and stimulates mitochondrial turnover. Cell Death Differ. 2011;18(4):721–31. doi: 10.1038/cdd.2010.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Liu B, et al. Polygonatum cyrtonema lectin induces apoptosis and autophagy in human melanoma A375 cells through a mitochondria-mediated ROS-p38-p53 pathway. Cancer Lett. 2009;275(1):54–60. doi: 10.1016/j.canlet.2008.09.042. [DOI] [PubMed] [Google Scholar]
- 78.Liu B, et al. Polygonatum cyrtonema lectin induces murine fibrosarcoma L929 cell apoptosis and autophagy via blocking Ras-Raf and PI3K-Akt signaling pathways. Biochimie. 2010;92(12):1934–8. doi: 10.1016/j.biochi.2010.08.009. [DOI] [PubMed] [Google Scholar]
- 79.Arthur CM, et al. Evolving mechanistic insights into galectin functions. Methods Mol Biol. 2015;1207:1–35. doi: 10.1007/978-1-4939-1396-1_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Thurston TL, et al. Galectin 8 targets damaged vesicles for autophagy to defend cells against bacterial invasion. Nature. 2012;482(7385):414–8. doi: 10.1038/nature10744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Aits S, et al. Sensitive detection of lysosomal membrane permeabilization by lysosomal galectin puncta assay. Autophagy. 2015;11(8):1408–24. doi: 10.1080/15548627.2015.1063871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Paz I, et al. Galectin-3, a marker for vacuole lysis by invasive pathogens. Cell Microbiol. 2010;12(4):530–44. doi: 10.1111/j.1462-5822.2009.01415.x. [DOI] [PubMed] [Google Scholar]
- 83.Cemma M, Kim PK, Brumell JH. The ubiquitin-binding adaptor proteins p62/SQSTM1 and NDP52 are recruited independently to bacteria-associated microdomains to target Salmonella to the autophagy pathway. Autophagy. 2011;7(3):341–5. doi: 10.4161/auto.7.3.14046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zheng YT, et al. The adaptor protein p62/SQSTM1 targets invading bacteria to the autophagy pathway. J Immunol. 2009;183(9):5909–16. doi: 10.4049/jimmunol.0900441. [DOI] [PubMed] [Google Scholar]
- 85.Chen X, et al. Autophagy induced by calcium phosphate precipitates targets damaged endosomes. J Biol Chem. 2014;289(16):11162–74. doi: 10.1074/jbc.M113.531855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Loyter A, et al. Mechanisms of DNA entry into mammalian cells. II. Phagocytosis of calcium phosphate DNA co-precipitate visualized by electron microscopy. Exp Cell Res. 1982;139(1):223–34. doi: 10.1016/0014-4827(82)90336-6. [DOI] [PubMed] [Google Scholar]
- 87.Maejima I, et al. Autophagy sequesters damaged lysosomes to control lysosomal biogenesis and kidney injury. EMBO J. 2013;32(17):2336–47. doi: 10.1038/emboj.2013.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Plato A, Willment JA, Brown GD. C-type lectin-like receptors of the dectin-1 cluster: ligands and signaling pathways. Int Rev Immunol. 2013;32(2):134–56. doi: 10.3109/08830185.2013.777065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lichtenstein RG, Rabinovich GA. Glycobiology of cell death: when glycans and lectins govern cell fate. Cell Death Differ. 2013;20(8):976–86. doi: 10.1038/cdd.2013.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Drickamer K, Taylor ME. Recent insights into structures and functions of C-type lectins in the immune system. Curr Opin Struct Biol. 2015;34:26–34. doi: 10.1016/j.sbi.2015.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Huang JH, et al. CR3 and Dectin-1 Collaborate in Macrophage Cytokine Response through Association on Lipid Rafts and Activation of Syk-JNK-AP-1 Pathway. PLoS Pathog. 2015;11(7):e1004985. doi: 10.1371/journal.ppat.1004985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Underhill DM, et al. Dectin-1 activates Syk tyrosine kinase in a dynamic subset of macrophages for reactive oxygen production. Blood. 2005;106(7):2543–50. doi: 10.1182/blood-2005-03-1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ma J, et al. Dectin-1-triggered recruitment of light chain 3 protein to phagosomes facilitates major histocompatibility complex class II presentation of fungal-derived antigens. J Biol Chem. 2012;287(41):34149–56. doi: 10.1074/jbc.M112.382812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Blanco-Menéndez N, et al. SHIP-1 Couples to the Dectin-1 hemITAM and Selectively Modulates Reactive Oxygen Species Production in Dendritic Cells in Response to Candida albicans. J Immunol. 2015;95(9):4466–78. doi: 10.4049/jimmunol.1402874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Tam JM, et al. Dectin-1-dependent LC3 recruitment to phagosomes enhances fungicidal activity in macrophages. J Infect Dis. 2014;210(11):1844–54. doi: 10.1093/infdis/jiu290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kyrmizi I, et al. Corticosteroids block autophagy protein recruitment in Aspergillus fumigatus phagosomes via targeting dectin-1/Syk kinase signaling. J Immunol. 2013;91(3):1287–99. doi: 10.4049/jimmunol.1300132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ma J, et al. Cutting edge: FYCO1 recruitment to dectin-1 phagosomes is accelerated by light chain 3 protein and regulates phagosome maturation and reactive oxygen production. J Immunol. 2014;192(4):1356–60. doi: 10.4049/jimmunol.1302835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Öhman T, et al. Dectin-1 pathway activates robust autophagy-dependent unconventional protein secretion in human macrophages. J Immunol. 2014;192(12):5952–62. doi: 10.4049/jimmunol.1303213. [DOI] [PubMed] [Google Scholar]
- 99.Pyo JO, et al. Overexpression of Atg5 in mice activates autophagy and extends lifespan. Nat Commun. 2013;4:2300. doi: 10.1038/ncomms3300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Vieira OV, et al. Distinct roles of class I and class III phosphatidylinositol 3-kinases in phagosome formation and maturation. The Journal of cell biology. 2001;155(1):19–25. doi: 10.1083/jcb.200107069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Henault J, et al. Noncanonical autophagy is required for type I interferon secretion in response to DNA-immune complexes. Immunity. 2012;37(6):986–97. doi: 10.1016/j.immuni.2012.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Martinez J, et al. Microtubule-associated protein 1 light chain 3 alpha (LC3)-associated phagocytosis is required for the efficient clearance of dead cells. Proc Natl Acad Sci U S A. 2011;108(42):17396–401. doi: 10.1073/pnas.1113421108. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 103.Barnden MJ, et al. Defective TCR expression in transgenic mice constructed using cDNA-based alpha- and beta-chain genes under the control of heterologous regulatory elements. Immunol Cell Biol. 1998;76(1):34–40. doi: 10.1046/j.1440-1711.1998.00709.x. [DOI] [PubMed] [Google Scholar]
- 104.Hart GW. Three Decades of Research on O-GlcNAcylation - A Major Nutrient Sensor That Regulates Signaling, Transcription and Cellular Metabolism. Front Endocrinol (Lausanne) 2014;5:183. doi: 10.3389/fendo.2014.00183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Hu Y, et al. Increased enzymatic O-GlcNAcylation of mitochondrial proteins impairs mitochondrial function in cardiac myocytes exposed to high glucose. The Journal of biological chemistry. 2009;284(1):547–55. doi: 10.1074/jbc.M808518200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Housley MP, et al. O-GlcNAc regulates FoxO activation in response to glucose. J Biol Chem. 2008;283(24):16283–92. doi: 10.1074/jbc.M802240200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Aguilar H, et al. Role for high-glucose-induced protein O-GlcNAcylation in stimulating cardiac fibroblast collagen synthesis. Am J Physiol Cell Physiol. 2014;306(9):C794–804. doi: 10.1152/ajpcell.00251.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Slawson C, et al. Perturbations in O-linked beta-N-acetylglucosamine protein modification cause severe defects in mitotic progression and cytokinesis. The Journal of biological chemistry. 2005;280(38):32944–56. doi: 10.1074/jbc.M503396200. [DOI] [PubMed] [Google Scholar]
- 109.Zhang S, et al. Modification of histones by sugar beta-N-acetylglucosamine (GlcNAc) occurs on multiple residues, including histone H3 serine 10, and is cell cycle-regulated. The Journal of biological chemistry. 2011;286(43):37483–95. doi: 10.1074/jbc.M111.284885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ngoh GA, et al. O-GlcNAc signaling attenuates ER stress-induced cardiomyocyte death. American journal of physiology Heart and circulatory physiology. 2009;297(5):H1711–9. doi: 10.1152/ajpheart.00553.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Krishnamoorthy V, Donofrio AJ, Martin JL. O-GlcNAcylation of alphaB-crystallin regulates its stress-induced translocation and cytoprotection. Mol Cell Biochem. 2013;379(1–2):59–68. doi: 10.1007/s11010-013-1627-5. [DOI] [PubMed] [Google Scholar]
- 112.Kazemi Z, et al. O-linked beta-N-acetylglucosamine (O-GlcNAc) regulates stress-induced heat shock protein expression in a GSK-3beta-dependent manner. The Journal of biological chemistry. 2010;285(50):39096–107. doi: 10.1074/jbc.M110.131102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Guo B, et al. O-GlcNAc-modification of SNAP-29 regulates autophagosome maturation. Nat Cell Biol. 2014;16(12):1215–26. doi: 10.1038/ncb3066. [DOI] [PubMed] [Google Scholar]
- 114.Love DC, et al. Dynamic O-GlcNAc cycling at promoters of Caenorhabditis elegans genes regulating longevity, stress, and immunity. Proc Natl Acad Sci U S A. 2010;107(16):7413–8. doi: 10.1073/pnas.0911857107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Marsh SA, et al. Cardiac O-GlcNAcylation blunts autophagic signaling in the diabetic heart. Life Sci. 2013;92(11):648–56. doi: 10.1016/j.lfs.2012.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Park S, et al. O-GlcNAc modification is essential for the regulation of autophagy in Drosophila melanogaster. Cell Mol Life Sci. 2015;72(16):3173–83. doi: 10.1007/s00018-015-1889-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Wang P, et al. O-GlcNAc cycling mutants modulate proteotoxicity in Caenorhabditis elegans models of human neurodegenerative diseases. Proc Natl Acad Sci U S A. 2012;109(43):17669–74. doi: 10.1073/pnas.1205748109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.van der Vos KE, et al. Modulation of glutamine metabolism by the PI(3)K-PKB-FOXO network regulates autophagy. Nat Cell Biol. 2012;14(8):829–37. doi: 10.1038/ncb2536. [DOI] [PubMed] [Google Scholar]
- 119.Zhao Y, et al. Cytosolic FoxO1 is essential for the induction of autophagy and tumour suppressor activity. Nat Cell Biol. 2010;12(7):665–75. doi: 10.1038/ncb2069. [DOI] [PubMed] [Google Scholar]
- 120.Ho SR, et al. O-GlcNAcylation enhances FOXO4 transcriptional regulation in response to stress. FEBS Lett. 2010;584(1):49–54. doi: 10.1016/j.febslet.2009.11.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Bullen JW, et al. Cross-talk between two essential nutrient-sensitive enzymes: O-GlcNAc transferase (OGT) and AMP-activated protein kinase (AMPK) J Biol Chem. 2014;289(15):10592–606. doi: 10.1074/jbc.M113.523068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Housley MP, et al. A PGC-1alpha-O-GlcNAc transferase complex regulates FoxO transcription factor activity in response to glucose. J Biol Chem. 2009;284(8):5148–57. doi: 10.1074/jbc.M808890200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Wang N, et al. Lacritin rescues stressed epithelia via rapid forkhead box O3 (FOXO3)-associated autophagy that restores metabolism. J Biol Chem. 2013;288(25):18146–61. doi: 10.1074/jbc.M112.436584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Hohenstein AC, Roche PA. SNAP-29 is a promiscuous syntaxin-binding SNARE. Biochem Biophys Res Commun. 2001;285(2):167–71. doi: 10.1006/bbrc.2001.5141. [DOI] [PubMed] [Google Scholar]
- 125.Keembiyehetty C, et al. Conditional knock-out reveals a requirement for O-linked N-Acetylglucosaminase (O-GlcNAcase) in metabolic homeostasis. J Biol Chem. 2015;290(11):7097–113. doi: 10.1074/jbc.M114.617779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Yang YR, et al. O-GlcNAcase is essential for embryonic development and maintenance of genomic stability. Aging Cell. 2012;11(3):439–48. doi: 10.1111/j.1474-9726.2012.00801.x. [DOI] [PubMed] [Google Scholar]
- 127.O'Donnell N, et al. Ogt-dependent X-chromosome-linked protein glycosylation is a requisite modification in somatic cell function and embryo viability. Mol Cell Biol. 2004;24(4):1680–90. doi: 10.1128/MCB.24.4.1680-1690.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Shafi R, et al. The O-GlcNAc transferase gene resides on the X chromosome and is essential for embryonic stem cell viability and mouse ontogeny. Proc Natl Acad Sci U S A. 2000;97(11):5735–9. doi: 10.1073/pnas.100471497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Jones SP, et al. Cardioprotection by N-acetylglucosamine linkage to cellular proteins. Circulation. 2008;117(9):1172–82. doi: 10.1161/CIRCULATIONAHA.107.730515. [DOI] [PubMed] [Google Scholar]
- 130.Yamano K, Matsuda N, Tanaka K. The ubiquitin signal and autophagy: an orchestrated dance leading to mitochondrial degradation. EMBO Rep. 2016;17(3):300–316. doi: 10.15252/embr.201541486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Lu P, et al. Extracellular matrix degradation and remodeling in development and disease. Cold Spring Harb Perspect Biol. 2011;3(12) doi: 10.1101/cshperspect.a005058. p. pii: a005058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Buraschi S, et al. Decorin causes autophagy in endothelial cells via Peg3. Proc Natl Acad Sci U S A. 2013;110(28):E2582–91. doi: 10.1073/pnas.1305732110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Goyal A, et al. Decorin activates AMPK, an energy sensor kinase, to induce autophagy in endothelial cells. Matrix Biol. 2014;34:46–54. doi: 10.1016/j.matbio.2013.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Gubbiotti MA, et al. Decorin is an autophagy-inducible proteoglycan and is required for proper in vivo autophagy. Matrix Biol. 2015;48:14–25. doi: 10.1016/j.matbio.2015.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Neill T, et al. Decorin induces mitophagy in breast carcinoma cells via peroxisome proliferator-activated receptor γcoactivator-1α (PGC-1α) and mitostatin. J Biol Chem. 2014;289(8):4952–68. doi: 10.1074/jbc.M113.512566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.McKown RL, et al. A cleavage-potentiated fragment of tear lacritin is bactericidal. J Biol Chem. 2014;289(32):22172–82. doi: 10.1074/jbc.M114.570143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Ning L, et al. Perlecan inhibits autophagy to maintain muscle homeostasis in mouse soleus muscle. Matrix Biol. 2015;48:26–35. doi: 10.1016/j.matbio.2015.08.002. [DOI] [PubMed] [Google Scholar]
- 138.Nguyen TM, et al. Endostatin induces autophagy in endothelial cells by modulating Beclin 1 and beta-catenin levels. J Cell Mol Med. 2009;13(9B):3687–98. doi: 10.1111/j.1582-4934.2009.00722.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Xu X, et al. Endostar, a modified recombinant human endostatin, suppresses angiogenesis through inhibition of Wnt/β-catenin signaling pathway. PLoS One. 2014;9(9):e107463. doi: 10.1371/journal.pone.0107463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Poluzzi C, et al. Endorepellin evokes autophagy in endothelial cells. J Biol Chem. 2014;289(23):16114–28. doi: 10.1074/jbc.M114.556530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Nguyen TM, et al. Kringle 5 of human plasminogen, an angiogenesis inhibitor, induces both autophagy and apoptotic death in endothelial cells. Blood. 2007;109(11):4793–802. doi: 10.1182/blood-2006-11-059352. [DOI] [PubMed] [Google Scholar]
- 142.Carmignac V, et al. Autophagy is increased in laminin α2 chain-deficient muscle and its inhibition improves muscle morphology in a mouse model of MDC1A. Hum Mol Genet. 2011;20(24):4891–902. doi: 10.1093/hmg/ddr427. [DOI] [PubMed] [Google Scholar]
- 143.Kalas W, et al. Thrombospondin-1 receptor mediates autophagy of RAS-expressing cancer cells and triggers tumour growth inhibition. Anticancer Res. 2013;33(4):1429–38. [PubMed] [Google Scholar]
- 144.Ren J, Hascall VC, Wang A. Cyclin D3 mediates synthesis of a hyaluronan matrix that is adhesive for monocytes in mesangial cells stimulated to divide in hyperglycemic medium. J Biol Chem. 2009;284(24):16621–32. doi: 10.1074/jbc.M806430200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Saha P, et al. Autophagic vacuolation induced by excess ROS generation in HABP1/p32/gC1qR overexpressing fibroblasts and its reversal by polymeric hyaluronan. PLoS One. 2013;8(10):e78131. doi: 10.1371/journal.pone.0078131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Saha P, Ghosh I, Datta K. Increased hyaluronan levels in HABP1/p32/gC1qR overexpressing HepG2 cells inhibit autophagic vacuolation regulating tumor potency. PLoS One. 2014;9(7):e103208. doi: 10.1371/journal.pone.0103208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Wang A, et al. Heparin prevents intracellular hyaluronan synthesis and autophagy responses in hyperglycemic dividing mesangial cells and activates synthesis of an extensive extracellular monocyte-adhesive hyaluronan matrix after completing cell division. J Biol Chem. 2014;289(13):9418–29. doi: 10.1074/jbc.M113.541441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Murphy-Ullrich JE, Sage EH. Revisiting the matricellular concept. Matrix Biol. 2014;37:1–14. doi: 10.1016/j.matbio.2014.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Lord MS, et al. The role of vascular-derived perlecan in modulating cell adhesion, proliferation and growth factor signaling. Matrix Biol. 2014;35:112–22. doi: 10.1016/j.matbio.2014.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Iruela-Arispe ML, et al. Differential expression of thrombospondin 1, 2, and 3 during murine development. Dev Dyn. 1993;197(1):40–56. doi: 10.1002/aja.1001970105. [DOI] [PubMed] [Google Scholar]
- 151.Dupuis LE, Kern CB. Small leucine-rich proteoglycans exhibit unique spatiotemporal expression profiles during cardiac valve development. Developmental Dynamics. 2014;243(4):601–611. doi: 10.1002/dvdy.24100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Bornstein P, Agah A, Kyriakides TR. The role of thrombospondins 1 and 2 in the regulation of cell-matrix interactions, collagen fibril formation, and the response to injury. Int J Biochem Cell Biol. 2004;36(6):1115–25. doi: 10.1016/j.biocel.2004.01.012. [DOI] [PubMed] [Google Scholar]
- 153.Fiedler LR, et al. Decorin regulates endothelial cell motility on collagen I through activation of insulin-like growth factor I receptor and modulation of alpha2beta1 integrin activity. The Journal of biological chemistry. 2008;283(25):17406–15. doi: 10.1074/jbc.M710025200. [DOI] [PubMed] [Google Scholar]
- 154.Felbor U, et al. Secreted cathepsin L generates endostatin from collagen XVIII. The EMBO journal. 2000;19(6):1187–94. doi: 10.1093/emboj/19.6.1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Cailhier JF, et al. Caspase-3 activation triggers extracellular cathepsin L release and endorepellin proteolysis. J Biol Chem. 2008;283(40):27220–9. doi: 10.1074/jbc.M801164200. [DOI] [PubMed] [Google Scholar]
- 156.Oda O, et al. Purification and characterization of perlecan fragment in urine of end-stage renal failure patients. Clin Chim Acta. 1996;255(2):119–32. doi: 10.1016/0009-8981(96)06395-4. [DOI] [PubMed] [Google Scholar]
- 157.Gonzalez EM, et al. BMP-1/Tolloid-like metalloproteases process endorepellin, the angiostatic C-terminal fragment of perlecan. J Biol Chem. 2005;280(8):7080–7. doi: 10.1074/jbc.M409841200. [DOI] [PubMed] [Google Scholar]
- 158.Lauten A, et al. Ischemia-reperfusion injury activates early extracellular matrix processing and expression of endostatin in the heart with differential effects of temperature. Basic Res Cardiol. 2009;104(5):559–69. doi: 10.1007/s00395-009-0013-7. [DOI] [PubMed] [Google Scholar]
- 159.Goyal A, et al. Endorepellin affects angiogenesis by antagonizing diverse vascular endothelial growth factor receptor 2 (VEGFR2)-evoked signaling pathways: transcriptional repression of hypoxia-inducible factor 1alpha and VEGFA and concurrent inhibition of nuclear factor of activated T cell 1 (NFAT1) activation. The Journal of biological chemistry. 2012;287(52):43543–56. doi: 10.1074/jbc.M112.401786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Poluzzi C, Iozzo RV, Schaefer L. Endostatin and endorepellin: A common route of action for similar angiostatic cancer avengers. Adv Drug Deliv Rev. 2016;97:156–73. doi: 10.1016/j.addr.2015.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Mann DM, et al. Analysis of glycosaminoglycan substitution in decorin by site-directed mutagenesis. J Biol Chem. 1990;265(9):5317–23. [PubMed] [Google Scholar]
- 162.Thiaville MM, et al. DNA-binding motif and target genes of the imprinted transcription factor PEG3. Gene. 2013;512(2):314–20. doi: 10.1016/j.gene.2012.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Fassan M, et al. Mitostatin is down-regulated in human prostate cancer and suppresses the invasive phenotype of prostate cancer cells. PLoS One. 2011;6(5):e19771. doi: 10.1371/journal.pone.0019771. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 164.Vecchione A, et al. MITOSTATIN, a putative tumor suppressor on chromosome 12q24.1, is downregulated in human bladder and breast cancer. Oncogene. 2009;28(2):257–69. doi: 10.1038/onc.2008.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Vantaku VR, et al. Lacritin Salvages Human Corneal Epithelial Cells from Lipopolysaccharide Induced Cell Death. Sci Rep. 2015;5:18362. doi: 10.1038/srep18362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Wang W, et al. Lacritin-mediated regeneration of the corneal epithelia by protein polymer nanoparticles. J Mater Chem B Mater Biol Med. 2014;2(46):8131–41. doi: 10.1039/C4TB00979G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Iozzo RV, SL Proteoglycan form and function: A comprehensive nomenclature of proteoglycans. Matrix Biol. 2015;42:11–55. doi: 10.1016/j.matbio.2015.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Ma P, et al. Heparanase deglycanation of syndecan-1 is required for binding of the epithelial-restricted prosecretory mitogen lacritin. J Cell Biol. 2006;174(7):1097–106. doi: 10.1083/jcb.200511134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Zhang Y, et al. Targeting of Heparanase-modified Syndecan-1 by Prosecretory Mitogen Lacritin Requires Conserved Core GAGAL plus Heparan and Chondroitin Sulfate as a Novel Hybrid Binding Site That Enhances Selectivity. J Biol Chem. 2013;288(17):12090–101. doi: 10.1074/jbc.M112.422717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Mercer CA, Kaliappan A, DPB A novel, human Atg13 binding protein, Atg101, interacts with ULK1 and is essential for macroautophagy. Autophagy. 2009 Jul;5(5):649–62. doi: 10.4161/auto.5.5.8249. Epub 2009 Jul 20., 2009 5(5)649–62. [DOI] [PubMed] [Google Scholar]
- 171.Lamy L, et al. CD47 and the 19 kDa interacting protein-3 (BNIP3) in T cell apoptosis. The Journal of biological chemistry. 2003;278(26):23915–21. doi: 10.1074/jbc.M301869200. [DOI] [PubMed] [Google Scholar]
- 172.Defaus S, et al. Mammalian protein glycosylation – structure versus function. The Analyst. 2014;139(12):2944. doi: 10.1039/c3an02245e. [DOI] [PubMed] [Google Scholar]
- 173.Hofsteenge J, et al. C-Mannosylation and O-Fucosylation of the Thrombospondin Type 1 Module. Journal of Biological Chemistry. 2000;276(9):6485–6498. doi: 10.1074/jbc.M008073200. [DOI] [PubMed] [Google Scholar]
- 174.Kaur S, et al. Heparan sulfate modification of the transmembrane receptor CD47 is necessary for inhibition of T cell receptor signaling by thrombospondin- 1. The Journal of biological chemistry. 2011;286(17):14991–5002. doi: 10.1074/jbc.M110.179663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Resovi A, et al. Current understanding of the thrombospondin-1 interactome. Matrix Biol. 2014;37:83–91. doi: 10.1016/j.matbio.2014.01.012. [DOI] [PubMed] [Google Scholar]
- 176.Leclair P, Lim CJ. CD47-independent effects mediated by the TSP-derived 4N1K peptide. PloS one. 2014;9(5):e98358. doi: 10.1371/journal.pone.0098358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Soto-Pantoja DR, et al. CD47 deficiency confers cell and tissue radioprotection by activation of autophagy. Autophagy. 2012;8(11):1628–42. doi: 10.4161/auto.21562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Soto-Pantoja DR, et al. Blockade of CD47 increases survival of mice exposed to lethal total body irradiation. Sci Rep. 2013;3:1038. doi: 10.1038/srep01038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Willis CD, et al. Endorepellin laminin-like globular 1/2 domains bind Ig3–5 of vascular endothelial growth factor (VEGF) receptor 2 and block pro-angiogenic signaling by VEGFA in endothelial cells. FEBS J. 2013;280(10):2271–84. doi: 10.1111/febs.12164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Pattingre S, et al. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122(6):927–39. doi: 10.1016/j.cell.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 181.Molejon MI, et al. The VMP1-Beclin 1 interaction regulates autophagy induction. Scientific reports. 2013;3:1055. doi: 10.1038/srep01055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Petherick KJ, et al. Autolysosomal β-catenin degradation regulates Wnt-autophagy-p62 crosstalk. EMBO J. 2013;32(13):1903–16. doi: 10.1038/emboj.2013.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Grassian AR, Coloff JL, Brugge JS. Extracellular matrix regulation of metabolism and implications for tumorigenesis. Cold Spring Harbor symposia on quantitative biology. 2011;76:313–24. doi: 10.1101/sqb.2011.76.010967. [DOI] [PubMed] [Google Scholar]
- 184.Avivar-Valderas A, et al. PERK integrates autophagy and oxidative stress responses to promote survival during extracellular matrix detachment. Molecular and cellular biology. 2011;31(17):3616–29. doi: 10.1128/MCB.05164-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Avivar-Valderas A, et al. Regulation of autophagy during ECM detachment is linked to a selective inhibition of mTORC1 by PERK. Oncogene. 2013;32(41):4932–40. doi: 10.1038/onc.2012.512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Gilmore AP. Anoikis. Cell death and differentiation. 2005;12(Suppl 2):1473–7. doi: 10.1038/sj.cdd.4401723. [DOI] [PubMed] [Google Scholar]
- 187.Fung C, et al. Induction of autophagy during extracellular matrix detachment promotes cell survival. Molecular biology of the cell. 2008;19(3):797–806. doi: 10.1091/mbc.E07-10-1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Whitelock JM, Melrose J, Iozzo RV. Diverse cell signaling events modulated by perlecan. Biochemistry. 2008;47(43):1174–11183. doi: 10.1021/bi8013938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Patel VN, et al. Heparanase cleavage of perlecan heparan sulfate modulates FGF10 activity during ex vivo submandibular gland branching morphogenesis. Development. 2007 Dec;134(23):4177–86. doi: 10.1242/dev.011171. Epub 2007 Oct 24., 2007, 134 (23), 4177–86. [DOI] [PubMed] [Google Scholar]
- 190.Friedrich MV, et al. Structural basis of glycosaminoglycan modification and of heterotypic interactions of perlecan domain V. J Mol Biol. 1999;294(1):259–70. doi: 10.1006/jmbi.1999.3259. [DOI] [PubMed] [Google Scholar]
- 191.Whitelock JM, Iozzo RV. Heparan sulfate: a complex polymer charged with biological activity. Chem Rev. 2005;105(7):2745–64. doi: 10.1021/cr010213m. [DOI] [PubMed] [Google Scholar]
- 192.Shteingauz A, et al. Heparanase Enhances Tumor Growth and Chemoresistance by Promoting Autophagy. Cancer Res. 2015;75(18):3946–57. doi: 10.1158/0008-5472.CAN-15-0037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Endo T. Glycobiology of alpha-dystroglycan and muscular dystrophy. Journal of biochemistry. 2015;157(1):1–12. doi: 10.1093/jb/mvu066. [DOI] [PubMed] [Google Scholar]
- 194.Carmignac V, Quéré R, Durbeej M. Proteasome inhibition improves the muscle of laminin α2 chain-deficient mice. Hum Mol Genet. 2011;20(3):541–52. doi: 10.1093/hmg/ddq499. [DOI] [PubMed] [Google Scholar]
- 195.Meyer K, Palmer JW. ON GLYCOPROTEINS: II. THE POLYSACCHARIDES OF VITREOUS HUMOR AND OF UMBILICAL CORD. J Biol Chem. 1936;114(3):689–703. [Google Scholar]
- 196.Meyer K, Palmer JW. THE POLYSACCHARIDE OF THE VITREOUS HUMOR. J Biol Chem. 1934;107:629–34. [Google Scholar]
- 197.Rothenhöfer M, et al. Qualitative and quantitative analysis of hyaluronan oligosaccharides with high performance thin layer chromatography using reagent-free derivatization on amino-modified silica and electrospray ionization-quadrupole time-of-flight mass spectrometry coupling on normal phase. J Chromatogr A. 2012;1248:169–77. doi: 10.1016/j.chroma.2012.05.057. [DOI] [PubMed] [Google Scholar]
- 198.Kultti A, et al. 4-Methylumbelliferone inhibits hyaluronan synthesis by depletion of cellular UDP-glucuronic acid and downregulation of hyaluronan synthase 2 and 3. Exp Cell Res. 2009;315(11):1914–23. doi: 10.1016/j.yexcr.2009.03.002. [DOI] [PubMed] [Google Scholar]
- 199.Rilla K, et al. Hyaluronan synthase 1 (HAS1) requires higher cellular UDP-GlcNAc concentration than HAS2 and HAS3. J Biol Chem. 2013;288(8):5973–83. doi: 10.1074/jbc.M112.443879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Stern R, Asari AA, Sugahara KN. Hyaluronan fragments: an information-rich system. Eur J Cell Biol. 2006;65(8):699–715. doi: 10.1016/j.ejcb.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 201.Rilla K, et al. Plasma membrane residence of hyaluronan synthase is coupled to its enzymatic activity. J Biol Chem. 2005;280(36):31890–7. doi: 10.1074/jbc.M504736200. [DOI] [PubMed] [Google Scholar]
- 202.Törrönen K, et al. Tissue distribution and subcellular localization of hyaluronan synthase isoenzymes. Histochem Cell Biol. 2014;141(1):17–31. doi: 10.1007/s00418-013-1143-4. [DOI] [PubMed] [Google Scholar]
- 203.Ontong P, et al. Effect of a cholesterol-rich lipid environment on the enzymatic activity of reconstituted hyaluronan synthase. Biochem Biophys Res Commun. 2014;442(2):666–71. doi: 10.1016/j.bbrc.2013.12.028. [DOI] [PubMed] [Google Scholar]
- 204.Pedersen TA, et al. Distinct C/EBPalpha motifs regulate lipogenic and gluconeogenic gene expression in vivo. EMBO J. 2007;26(4):1081–93. doi: 10.1038/sj.emboj.7601563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Chowdhury AR, Ghosh I, Datta K. Excessive reactive oxygen species induces apoptosis in fibroblasts: role of mitochondrially accumulated hyaluronic acid binding protein 1 (HABP1/p32/gC1qR) Experimental cell research. 2008;314(3):651–67. doi: 10.1016/j.yexcr.2007.10.033. [DOI] [PubMed] [Google Scholar]
- 206.Villers C, et al. Release of oligomannoside-type glycans as a marker of the degradation of newly synthesized glycoproteins. Biochem J. 1994;298(Pt 1):135–42. doi: 10.1042/bj2980135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Verbert A, Cacan R. Trafficking of oligomannosides released during N-glycosylation- a clearing mechanism of the rough endoplasmic reticulum. Biochim Biophys Acta. 1999;1473(1):137–46. doi: 10.1016/s0304-4165(99)00174-9. [DOI] [PubMed] [Google Scholar]
- 208.Suzuki T, Funakoshi Y. Free N-linked oligosaccharide chains: formation and degradation. Glycoconjugate journal. 2006;23(5–6):291–302. doi: 10.1007/s10719-006-6975-x. [DOI] [PubMed] [Google Scholar]
- 209.Orsi A, et al. Dynamic and transient interactions of Atg9 with autophagosomes, but not membrane integration, are required for autophagy. Mol Biol Cell. 2012;23(10):1860–73. doi: 10.1091/mbc.E11-09-0746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Hosokawa N, Hara Y, Mizushima N. Generation of cell lines with tetracycline-regulated autophagy and a role for autophagy in controlling cell size. FEBS letters. 2006;580(11):2623–9. doi: 10.1016/j.febslet.2006.04.008. [DOI] [PubMed] [Google Scholar]
- 211.Patnaik SK, Stanley P. Lectin-resistant CHO glycosylation mutants. Methods Enzymol. 2006;416:159–82. doi: 10.1016/S0076-6879(06)16011-5. [DOI] [PubMed] [Google Scholar]
- 212.Yu SH, et al. Metabolic labeling enables selective photocrosslinking of O-GlcNAc-modified proteins to their binding partners. Proc Natl Acad Sci U S A. 2012;109(13):4834–9. doi: 10.1073/pnas.1114356109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Bond MR, et al. Photocrosslinking of glycoconjugates using metabolically incorporated diazirine-containing sugars. Nat Protoc. 2009;4(7):1044–63. doi: 10.1038/nprot.2009.85. [DOI] [PubMed] [Google Scholar]