

Abstract
Short Structural Variants (SSVs) are short genomic variants (<50bp) other than SNPs. It has been suggested that SSVs contribute to many human complex traits. However, high-throughput analysis of SSVs presents numerous technical challenges. In order to facilitate the discovery and assessment of SSVs, we have developed a prototype bioinformatics tool, “SSV evaluation system”, which is a searchable, annotated database of SSVs in the human genome, with associated customizable scoring software that is used to evaluate and prioritize SSVs that are most likely to have significant biological effects and impact on disease risk. This new bioinformatics tool is a component in a larger strategy that we have been using to discover potentially important SSVs within candidate genomic regions that have been identified in genome wide association studies, with the goal to prioritize potential functional/causal SSVs and focus the follow-up experiments on a relatively small list of strong candidate SSVs. We describe our strategy and discuss how we have used the SSV evaluation system to discover candidate causal variants related to complex neurodegenerative diseases. We present the SSV evaluation system as a powerful tool to guide genetic investigations aiming to uncover SSVs that underlie human complex diseases including neurodegenerative diseases in aging.
Keywords: Structural variants, STR, causal variants, human complex diseases, bioinformatics tool
INTRODUCTION
Structural Variants (SVs) are genomic variants other than SNPs, and include deletions, insertions, microsatellites or simple sequence repeats/short tandem repeats (SSRs/STRs), copy number variation (CNV), block substitutions, and inversions (Frazer, et al., 2009). Our interests focus on Short SVs (hereafter SSVs), a category that is defined here as variants other than single nucleotide variation, that encompass short sequence (usually with size change less than 50 bp), and include short deletions, short insertions, insertion/deletions (In-del), mixed (cluster that contains multiple classes), multiple nucleotide polymorphism (MNP, the alleles remain the same length), and microsatellites or simple sequence repeats/short tandem repeats (SSRs/STRs).
Recently, there has been increased support for the idea that SSVs, as well as SNPs, may be responsible for many human complex traits (Mirkin, 2007; Pearson, et al., 2005; Willems, et al., 2014). Remarkably, a very recent study based on the analysis of an integrated SV map of 2504 human genomes showed that SVs are enriched on haplotypes identified by genome wide association studies (GWAS) (Sudmant, et al., 2015).
While the set of SSVs may contain many of the causal variants (CVs) responsible for human traits, the analysis of SSVs – particularly the class of repeat variations (such as short and long tandem repeats) – presents numerous technical challenges to the investigator. Many current next generation sequencing (NGS) technologies will not accurately detect polymorphic repetitive sequences, because of short reads and difficulties in assembling repeating elements into contigs. Other approaches that allow phased sequencing, such as PacBio or cloning coupled with traditional Sanger sequencing, are not feasible and/or very expensive to carry out at the whole-genome level. Therefore, we were motivated to develop a bioinformatics tool that would allow human geneticists to narrow down and efficiently focus the genotyping and sequencing efforts on a smaller list of SSVs that are predicted to have a high biological impact, and may be causal variants for the traits under investigation.
Our research strategy expands on the concept that changes, even subtle, in expression levels of normal (wild-type) proteins in the brain can lead to neurodegenerative diseases of the aging brain. Our major goal is to uncover functional variants that modulate expression regulation and lead to subtle changes in expression level of specific genes that play a role in pathways related to neurodegeneration. It has been suggested that many SSVs control gene expression, and so our current focus is on SSVs that are within putative regulatory regions, and their role in late onset neurodegenerative diseases. A new study identified >2000 expression STRs (eSTRs) in the human genome and found that eSTRs contribute to ~10-15% of the cis heritable variation in gene expression attributed to common variants, and hence provided further support for our focus on SSVs in the context of expression changes as an underlying mechanism for late onset neurodegenerative diseases (Gymrek, et al., 2016).
Herein we propose an evaluation system to guide genetic investigations exploring the role of SSVs in human complex diseases, particularly neurodegenerative diseases of aging. Our general strategy, outlined in Fig. 1, is to use available GWAS data to define candidate genomic regions, then to use the SSV evaluation system to prioritize particular SSVs within these regions, and finally in future studies to conduct laboratory testing to analyze the high-priority SSVs using experiments that include case-control samples to evaluate an association with a particular brain disease of interest coupled with functional studies to uncover the plausible mechanisms of action of the candidate SSV. This paper describes the stage of this general strategy in which we developed an in silico tool, the SSV evaluation system, for prioritization of candidate SSVs.
Fig. 1. The strategic flowchart of using the SSV evaluation system to discover causal genetic variants.

By focusing on genomic regions that are identified by GWAS and then prioritizing the SSVs in those ranges using the SSV evaluation system, the total number of regions to be tested in the laboratory can be reduced to a small number of likely candidate variants.
METHODS
Organization of the SSV evaluation system
Our prototype SSV evaluation system consists of an annotated, genome-wide list of SSVs (dbSSV), and a search and scoring program (SSV Search) that generates custom SSV Reports for a given query. Fig. 2 shows the data sources and organization of the main components of the system. The left side of this figure shows the public domain sources of the various data tracks, and the arrows indicate the processing of those tracks to make component databases and the final databases. This overall structure is easily expandable, permitting the future addition of new tracks within existing categories and the addition of entirely new categories.
Fig. 2. The organization and structure of the SSV evaluation system.

The boxes on the left indicate the public domain sources of raw data used in the system. Arrows indicate a transformation process. The various data sources were downloaded, reformatted, and assembled into the main database, dbSSV. The SSV Search program performs custom searches of dbSSV and writes SSV reports.
The SSV evaluation system is publicly accessible and can be downloaded in the following link at the Polymorphic DNA Technologies FTP site: http://polymorphicdna.tdl.com/download/user1 (username, "user1"; password, "rna389").
Database content and related fields
The current version of the dbSSV database contains information on approximately 4 million known SSVs and an additional 2 million SSRs found in the human genome. The position of each SSV is mapped to the human reference sequence version GRCh37/hg19. The known SSVs were extracted from the public database dbSNP (build 142) choosing essentially all variants that are not described as “simple” (i.e. SNP). The set of SSRs was created by a scan of the entire Human Reference Sequence, searching for simple repeats of any nucleotide unit of length ranging from 1 to 50 nucleotides. Data describing the location and definition of the SSV, variability indicators, repeat context, gene context, transcription factor and microRNA binding sites, other regulatory markers, conservation, position within a linkage disequilibrium (LD) block, GWAS signals, and tissue-specific regulatory signals are registered in the underlying database files. A list of the data sources for each of these fields is given in Supp. Table S1. Descriptions of the derivations of each field are given in Supp. Table S2.
Search and Scoring Software
The database searches and scoring are performed by “SSV Search”, which is an Excel file with a Visual Basic macro. This file includes a “Browser” worksheet that serves as the main user interface (Supp. Figure S1). The program finds all SSVs and related fields in dbSSV for the specified ranges, performs scoring of each SSV based upon all data fields, and creates a separate SSV Report file containing all related data fields and component scores. The report contains a worksheet named “Full Report” that lists all fields and partial scores. In addition, a separate worksheet called “Brief Report” lists a smaller number of data fields and only the major category scores and their associated total scores.
The algorithm: scoring parameters and considerations
The scoring algorithm creates numerical scores for 24 different properties of each SSV. For all numerical data and for some qualitative descriptions, we have assigned weighting factors or look-up values that can be used to produce partial scores for each property. It should be noted that the values we have assigned to these weighting factors are arbitrary and represent our knowledgeable prediction of the weight that describes the relative importance of each factor. The scoring parameters are contained in a separate “Scoring Rules” worksheet of the SSV Search file (Supp. Figure S2). Below, we briefly describe the significance of each data type and the scoring strategy:
Size Variability – We include fields for the number of known alleles of the SSV (from dbSNP), the known size range of the SSV region (calculated from dbSNP), and an SSR Slippage Index (calculated from a custom model of polymerase slippage). These parameters are all indictors of the extent to which an SSV may alter the size of a genomic region. Our scoring method assigns larger significance to data values that are related to larger size changes.
Repeat Context – The database includes descriptive fields from the “RepeatMasker” genome-wide track. These descriptions indicate regions that contain repeated elements, and such regions are known to be hotspots for mutation and for large size variations. Our scoring method gives higher values for “simple repeats”, “low complexity”, and “LTR”, since such regions are known to be sites of some causal variants. This category also employs a “clustering index”, which adds an additional score to pairs of SSVs that are in close proximity, since such adjacent SSVs may act synergistically.
Gene Context – For the site of each SSV, we determine whether it is within a gene and if so, whether it is in a coding exon, a 5’UTR, a 3’UTR, an intron, or a promoter region. We assign higher scores to coding regions, UTRs, and promoters because these are most likely to affect gene expression and/or peptide sequence. We also assign higher scores to larger introns since these may contain sequences with regulatory function.
GWAS – dbSSV currently contains GWAS “signals” for 19 different disease traits. To get these signals, we first take the logarithm of the negative logarithm of the p-values of each GWAS-SNP to get a signal value at each SNP site. This “log of the log” calculation is done to reduce the range of the GWAS signals from hundreds of orders of magnitude to just a few orders of magnitude. We then use the known local recombination rates to compute a continuous GWAS signal over the chromosome locations flanking each GWAS-SNP, with that signal dropping off with accumulated loss of association because of recombination. Each SSV is assigned this calculated GWAS signal value and scores are assigned in proportion to signal strength. This parameter assigns higher scores to SSVs that are closer to a strongly associated GWAS SNP.
- Regulation – We have selected tracks available from several data sources: NIH Roadmap Epigenomics project, ENCyclopedia Of DNA Elements (ENCODE), Biobase, TargetScan.
- 5.1. NIH Roadmap Epigenomics project: We have chosen six tissue-specific regulatory signals, which are the four histone modifications H3K4me1, H3K4me3, H3K9ac, and H3K27ac, and also tracks for DNase I hypersensitivity and for methylation. The current version of the SSV evaluation system includes these tracks for four brain tissues [brain hippocampus middle (BHM), brain inferior temporal lobe (BITL), brain mid frontal lobe (BMFL), and brain substantia nigra (BSN)]. These epigenetic signals reflect the extent to which genomic regions are active in various gene expression processes. For scoring, we have initially scaled the weighting factors for each of the six signals in order to get a range of partial scores of 0 to 5 for each signal.
- 5.2. ENCODE: We have chosen two tracks, Genome Segmentation and Transcription Factor ChIPs. (i) Genome Segmentation represents multivariate genome-segmentation results based on ENCODE data from 6 cell-lines (Consortium, 2012) and using machine learning techniques. The genome was automatically segmented into disjoint segments and each segment belongs to one of seven specific genomic "states" (region): Promoter (including transcription start site,TSS), Promoter-flanked, Enhancer, Weak Enhancer, Transcribed, Repressed, and CTCF enriched element. Scores were assigned with higher values for Enhancer and CTCF enriched regions since these regions are more involved in regulation. (ii) Transcription Factor ChIP track represents a comprehensive set of human transcription factor binding sites based on ChIP-seq experiments generated by all production groups in the ENCODE Consortium (Consortium, 2012). The report includes the names of the transcription factors (TFs), and scores for their signal strength using a linear scoring factor.
- 5.3. Biobase and TargetScan: We have chosen two matrix databases, for TF binding sites and for microRNA derived from in silico analyses. (i) Conserved Transcription Factor Binding Sites (TFBS) contains the location and score of transcription factor binding sites conserved in the human/mouse/rat alignment. In this track, a binding site is considered conserved across the alignment if its score meets the threshold score for its binding matrix in all 3 species. The score and threshold are computed with the Transfac Matrix Database (v7.0) created by Biobase. The scoring factor used for this track is the same as that used for the Transcription Factor ChIP track. (ii) TargetScan represents conserved mammalian microRNA regulatory target sites for conserved microRNA families in the 3' UTR regions of Refseq Genes, as predicted by TargetScanHuman (v7.0) (Agarwal, et al., 2015). These sites are typically only 8 base-pairs in length, but we have extended the range in which to apply their signal to larger window sizes of either 60, 400, order 2000 bp. For scoring, we multiply these signals by a linear scoring factor.
Conservation – The SSV evaluation system includes tracks for both primate and mammalian conservation, and we have calculated “smoothened” tracks averaged over 25, 75, 125, or 225 base-pairs. The 25 base-pair averaged signal is named the “small window” track and the user can choose one of the other averaged tracks as the “long window” track. The system allows the user to score these in various ways, but our preferred choice is to subtract the short-window mammalian signal from the long-window mammalian signal and score that difference. This strategy provides higher scores to regions that are conserved over a larger region (long window) but have been subject to variation in the immediate region (short window).
The SSV evaluation system has been frequently updated and we are currently pursuing ongoing efforts to include additional datasets from the NIH Roadmap Epigenomics project, the ENCODE integrative data, and the new build of dbSNP (dbSNP144), with the goal of ensuring that the SSV evaluation system is continuously up to date and includes new regulatory and annotation tracks as they become publicly available.
RESULTS AND DISCUSSION
Genome-wide association studies (GWAS) and related genome-based approaches have resulted in the identification of extensive lists of SNPs associated with human complex traits and diseases, but the precise causal genetic variants and the molecular mechanisms underlying those genetic associations remain largely unknown. To date, GWAS using SNP platforms have been the primary genetic analytical approach to study human complex diseases and expression traits (eQTL), while the analysis of SSVs has been underrepresented in such studies. Recently, there has been increased support for the idea that SSVs may contribute to variation in gene expression in humans and also contribute to many human complex traits (Gymrek, et al., 2016; Mirkin, 2007; Pearson, et al., 2005; Willems, et al., 2014). Herein, we present a tool that supports and guides human genetic studies that aim to understand the contribution of SSVs to complex neurodegenerative disorders in aging brains including Alzheimer, Parkinson and related disorders.
Short SVs (SSVs) are thought to affect phenotype by altering the regulation of gene transcription (Akai, et al., 1999; Chiba-Falek and Nussbaum, 2001; Okladnova, et al., 1998; Peters, et al., 1999; Searle and Blackwell, 1999; Shimajiri, et al., 1999), splicing (Hefferon, et al., 2004), and translation, and it is by these mechanisms that SSVs may play a role in the etiology of human diseases, including complex disorders. Furthermore, recent studies proposed a potential mechanism whereby SSRs affect transcription. These studies showed that certain repetitive DNA sequences, when present in the flanking regions of specific transcription factor (TF) binding sites, can have a magnitude effect on the intensity of TF-DNA binding, through a mechanism we termed “non-consensus binding” (Afek, et al., 2015; Afek, et al., 2014). Notably, a recent study based upon the analysis of an integrated SV map of 2504 human genomes showed that SVs are enriched on haplotypes identified by GWAS and exhibit enrichment for expression quantitative trait loci (Sudmant, et al., 2015). Furthermore, another new analysis showed that STRs in the human genome extensively contribute to variation in gene expression and that these eSTRs are enriched in genomic regions associated with clinically relevant phenotypes (Gymrek, et al., 2016).
There are several public resources that contain extensive information on SVs, mainly focused on large SV. dbVar (http://www.ncbi.nlm.nih.gov/dbvar) is NCBI's database of genomic structural variation (Sayers, et al., 2012): insertions, deletions, duplications, inversions, multinucleotide substitutions, mobile element insertions, translocations, and complex chromosomal rearrangements. Copy number variants (CNV) are extensively described in this database and the database includes data from all species. In addition to information about the variant region, the database also contains information on clinical assertions and genotypes. Extensive browsing, search and data download facilities are provided; the database can also be used with the Variation Viewer (http://www.ncbi.nlm.nih.gov/variation/view/). The ENSEMBL genome browser (http://useast.ensembl.org) contains extensive annotation of SVs. A highly curated catalog of structural variation in healthy control human samples is available from the Database of Genomic Variants (16) (http://dgv.tcag.ca/dgv/app/home). These public resources offer an extensive catalog of structural variation in the human genome. For studies that are focused on larger (> 50bp) SVs, these resources provide data for bioinformatics analysis of these classes of genetic variation and serve as a starting point for next generation sequencing studies for genetic association studies.
Generally, variants smaller than 50bp are submitted to dbSNP instead of dbVar. dbVar also contains the structural variation data from the 1000 genomes project (63,000 variant regions with over 6 million calls from 2,504 subjects) (Sudmant, et al., 2015). The level of annotation for these smaller SVs is more limited than for the SVs that are >50bp. Our dbSSV database integrates data from a variety of sources to provide more extensive annotation of these variants. The SSV evaluation system focuses on the subclass of short SVs and offers a scoring system to prioritize SSVs that are likely to be regulatory and/or causal in relation to human traits in health and disease. The scoring system is an innovative, empirical approach to prioritizing SSVs for genotyping. The scoring system is customizable by the individual investigator enabling a priori knowledge to be included in the evaluation. Although several bioinformatics approaches have been described to localize causal variants based on linkage disequilibrium (Bochdanovits, et al., 2013; Zhu, et al., 2012), the specific aims of the SSV evaluation system is to focus on SSVs that are likely to be associated with gene regulation for follow up laboratory studies using approaches such as nCounter (Nanostring) to measure gene expression from samples that define the trait of interest. Similar to the CAVIAR-Gene (Causal Variants Identification in Associated Regions) approach (Hormozdiari, et al., 2015), the SSV evaluation system is designed to operate across large LD regions of the genome with the aim of more precisely localizing the variants and genes that are causal for complex traits such as Alzheimer disease in order to facilitate fine mapping studies. Both approaches also use GWAS data as a first step for localization of causal or informative variants. CAVIAR focuses on SNPs and partitions the SNPs at a locus into genes in order to identify causal genes and notably can correct for population structure. The SSV evaluation system is designed to find specific, highly informative SSVs and does not aggregate variants at the gene level, although associated genes are listed in the annotation. Information on gene context, regulation and conservation is included in the scoring algorithm in order to maximize the probability of finding variants that are associated with gene regulation.
We are currently using the SSV evaluation system in functional genomic studies of complex neurodegenerative diseases. As an illustration of the use of the system and the overall research strategy, a prior study suggested that clusterin (CLU; MIM# 185430) and membrane spanning four domain subfamiliy A, member 4A (MS4A4A; MIM# 606547) expression-associated- SNPs (eSNPs) might explain the late-onset Alzheimer Disease (LOAD) risk association at these loci (Allen, et al., 2012). We annotated and scored SSVs within 1Mb regions of CLU and MS4A4A loci; the distribution of the total impact scores and the top scoring SSVs (total impact score≥30) are presented in Fig. 3a and 3b, respectively. Note that the distribution is skewed towards lower impact scores (median of 24). The relatively few SSVs in the part of the distribution with higher scores effectively prioritizes the variants for likelihood of a functional impact based on the criteria that define the database and the weightings in the scoring equation. The shape of the distribution shown in Fig. 3a has been observed for multiple experiments on different regions of the genome for genetic association studies for Alzheimer Disease, obesity and Amyotrophic Lateral Sclerosis (ALS) (Supp. Figure S3a-c). These high priority SSVs are subject to ongoing follow up laboratory investigations to assess their associations with LOAD and gene expression in brain, demonstrating the potential of the SSV evaluation system to guide follow up studies on GWAS discoveries.
Fig. 3. SSV evaluation analysis of the late onset Alzheimer disease (LOAD)-GWA genes: CLU and MS4A4A.
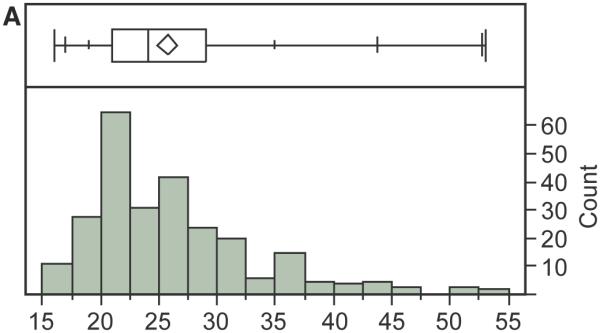

CLU and MS4A4A genes and 1Mb regions surrounding these loci were analyzed using the SSV evaluation system (version 4.1). The search was performed using the GWAS signal for Alzheimer disease, the regulatory signals selected for “Brain Hippocampus Middle” tissue, and default scoring. The specific derivation of each data item (e.g. column) and the scoring formula are provided in the Supporting Information. (a) The distribution of total potential impact scores. The quantile box plot is a simple, graphical depiction of the quantiles of the distribution: the 25% and 75% quartiles are defined by the rectangle with the median as the line in the middle; the diamond shows the mean and 95% confidence interval for the mean; the short, horizontal lines on either side of the rectangle define quantiles: 0.5%, 2.5%, 10%, 90%, 97.5% and 99.5%. (b) Top-scoring SSVs for potential impact on LOAD in 1Mb regions surrounding CLU and MS4A4A. This is a section of the “Brief” SSV Report, an Excel file output result of a SSV evaluation system search for CLU and MS4A4A genes regions.
To further demonstrate that the SSV evaluation system may serve as a powerful tool in the design of human genetic studies, below we discuss several published examples of implementation of the SSV evaluation system in our research program of genetic studies of Lewy body and Alzheimer spectrum disorders from our research program:
(1) SNCA-CT rich haplotype
We have used the SSV evaluation system to analyze the SSVs in the alpha-synuclein gene (SNCA; MIM# 163890) with the goal to identify candidate causal variants for synucleinopathies, in particular for Lewy body (LB) variant of Alzheimer disease (LBV/AD). The SSV evaluation system provided high scores (representative Total Impact Score=42) in a CT-rich, low-complexity region of intron 4 of SNCA. We cloned and sequenced this region in case (LBV/AD) and control (AD) subjects and identified four distinct haplotypes within this region, with specific haplotype-conferred risk to develop LB pathology in AD patients (Lutz, et al., 2015). We further demonstrated that the risk haplotype was significantly associated with elevated levels of SNCA-mRNA in human brain tissues relevant to the LB pathology and suggested that the CT-rich site acts as an enhancer element of SNCA transcription (Lutz, et al., 2015). Experiments using induced pluripotent stem cells (iPSCs) and genome-editing technologies to validate and modulate the regulatory effect of the candidate causal/functional SSVs are underway.
(2) SNCA-Rep1
Previously, we also discovered the Rep1 element, a SSV cluster associated with increased PD-risk, that regulates SNCA transcription in human brain (Linnertz, et al., 2009). We confirmed these findings using luciferase reporter assay (Chiba-Falek and Nussbaum, 2001; Chiba-Falek, et al., 2003) and a humanized mouse model (Cronin, et al., 2009). Retrospectively, the Rep1 site was evaluated and had a high score (Total Impact Score=31) relative to the complete list of SSRs within SNCA genomic regions including +/−50kb flanking regions in SSV evaluation system was confirmed.
(3) TOMM40-‘523’
Using phylogenetic analyses based on phased sequence data, we identified a variable intronic poly-T, in the TOMM40 (MIM# 608061) gene that is associated with risk for LOAD and age of onset (Roses, et al.). We demonstrated using AD-affected and normal brain tissues and a luciferase reporter system that this highly-variable poly-T site regulates the transcript levels of both TOMM40 and its neighboring gene, APOE (Linnertz, et al., 2014). Retrospectively, this particular polyT site (rs10524523) was shown to have a very high score (Total Impact Score=46) in the analysis of this genomic region by the SSV evaluation system.
In addition we also validated the utility of our tool for the prioritization of regulatory/causal variants using examples from studies of ALS and frontal temporal dementia (FTD) by other groups. A massive hexanucleotide (GGGGCC) repeat expansion mutation in the C9orf72 (MIM# 614260) gene has been linked to the majority of familial ALS, familial-FTD, some sporadic forms of FTD, and mixed ALS-FTD cases (DeJesus-Hernandez, et al., 2011; Haeusler, et al., 2014; Renton, et al., 2011). This repeat is positioned in the non-coding region of C9orf72 and it has been postulated to have a regulatory function. We analyzed the C9orf72 gene and flanking regions using our SSV evaluation system and showed that this structural variant generated a high score (Total Impact Score=52) relative to the landscape of the C9orf72 region.
We also assessed the SSV evaluation system for its scoring of “negative SSVs”, that is variants from our laboratory studies that showed no evidence to have neither a functional effect nor impact on disease risk. Towards this goal, we analyzed two SSVs, a ‘TTAG’ deletion and poly(GT) SSR positioned within SNCA locus that previously showed no association with LB in AD cases (n=214, p=0.50, p= 0.12, respectively) and no effects on SNCA-mRNA levels (our unpublished data). These SSVs produced Total Impact Scores of 26 and 19, respectively, which is below the cut off score we suggested here for the top priority SSVs. This evaluation strengthens the evidence for the utility and the potential of our new tool to effectively and specifically prioritize only strong candidate SSVs.
In conclusion, the SSV evaluation system leverages on existing SSVs databases and other relevant genomic datasets and offers an integrated, customizable scoring system to rank SSVs that are more likely to be regulatory and/or causal in relation to human traits in health and disease. This screening and prioritization strategy enables us to focus our re-sequencing efforts on highly informative SSVs that are likely to be functional and potentially causal. The SSV evaluation system has proven to be an efficient, effective tool facilitating the wet/dry cycle of experimentation necessary to understand the genetic underpinnings of disease. Thus, our new developed SSV evaluation system is a powerful tool to guide genetic investigations aiming to uncover SSVs that underlie human complex diseases including idiopathic neurodegenerative disease in aging.
Supplementary Material
ACKNOWLEDGMENTS
This work was funded in part by the National Institutes of Health/National Institute of Neurological Disorders and Stroke (NIH/NINDS) [R01 NS085011 to O.C.], the National Institute on Aging (NIA) [R01AG040370 to AR], the Blue Wine Group Charitable Fund [M.W.L. and O.C,] and Cabernet Pharmaceuticals, Inc.[R.S. and A.D.R.].
Footnotes
Conflict of Interest
Dr. Robert Saul is the chief executive officer of Polymorphic DNA Technologies, Inc.
REFERENCES
- Afek A, Cohen H, Barber-Zucker S, Gordan R, Lukatsky DB. Nonconsensus Protein Binding to Repetitive DNA Sequence Elements Significantly Affects Eukaryotic Genomes. PLoS Comput Biol. 2015;11(8):e1004429. doi: 10.1371/journal.pcbi.1004429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afek A, Schipper JL, Horton J, Gordan R, Lukatsky DB. Protein-DNA binding in the absence of specific base-pair recognition. Proc Natl Acad Sci U S A. 2014;111(48):17140–5. doi: 10.1073/pnas.1410569111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal V, Bell GW, Nam JW, Bartel DP. Predicting effective microRNA target sites in mammalian mRNAs. Elife. 2015:4. doi: 10.7554/eLife.05005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akai J, Kimura A, Hata RI. Transcriptional regulation of the human type I collagen alpha2 (COL1A2) gene by the combination of two dinucleotide repeats. Gene. 1999;239(1):65–73. doi: 10.1016/s0378-1119(99)00380-7. [DOI] [PubMed] [Google Scholar]
- Allen M, Zou F, Chai HS, Younkin CS, Crook J, Pankratz VS, Carrasquillo MM, Rowley CN, Nair AA, Middha S, Maharjan S, Nguyen T, et al. Novel late-onset Alzheimer disease loci variants associate with brain gene expression. Neurology. 2012;79(3):221–8. doi: 10.1212/WNL.0b013e3182605801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bochdanovits Z, Simon-Sanchez J, Jonker M, Hoogendijk WJ, van der Vaart A, Heutink P. Accurate prediction of a minimal region around a genetic association signal that contains the causal variant. Eur J Hum Genet. 2013 doi: 10.1038/ejhg.2013.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiba-Falek O, Nussbaum RL. Effect of allelic variation at the NACP-Rep1 repeat upstream of the alpha-synuclein gene (SNCA) on transcription in a cell culture luciferase reporter system. Hum Mol Genet. 2001;10(26):3101–9. doi: 10.1093/hmg/10.26.3101. [DOI] [PubMed] [Google Scholar]
- Chiba-Falek O, Touchman JW, Nussbaum RL. Functional analysis of intra-allelic variation at NACP-Rep1 in the alpha-synuclein gene. Hum Genet. 2003;113(5):426–31. doi: 10.1007/s00439-003-1002-9. [DOI] [PubMed] [Google Scholar]
- Consortium EP. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489(7414):57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cronin KD, Ge D, Manninger P, Linnertz C, Rossoshek A, Orrison BM, Bernard DJ, El-Agnaf OM, Schlossmacher MG, Nussbaum RL, Chiba-Falek O. Expansion of the Parkinson disease-associated SNCA-Rep1 allele upregulates human alpha-synuclein in transgenic mouse brain. Hum Mol Genet. 2009;18(17):3274–85. doi: 10.1093/hmg/ddp265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, Nicholson AM, Finch NA, Flynn H, Adamson J, Kouri N, Wojtas A, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72(2):245–56. doi: 10.1016/j.neuron.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frazer KA, Murray SS, Schork NJ, Topol EJ. Human genetic variation and its contribution to complex traits. Nat Rev Genet. 2009;10(4):241–51. doi: 10.1038/nrg2554. [DOI] [PubMed] [Google Scholar]
- Gymrek M, Willems T, Guilmatre A, Zeng H, Markus B, Georgiev S, Daly MJ, Price AL, Pritchard JK, Sharp AJ, Erlich Y. Abundant contribution of short tandem repeats to gene expression variation in humans. Nat Genet. 2016;48(1):22–29. doi: 10.1038/ng.3461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haeusler AR, Donnelly CJ, Periz G, Simko EA, Shaw PG, Kim MS, Maragakis NJ, Troncoso JC, Pandey A, Sattler R, Rothstein JD, Wang J. C9orf72 nucleotide repeat structures initiate molecular cascades of disease. Nature. 2014;507(7491):195–200. doi: 10.1038/nature13124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hefferon TW, Groman JD, Yurk CE, Cutting GR. A variable dinucleotide repeat in the CFTR gene contributes to phenotype diversity by forming RNA secondary structures that alter splicing. Proc Natl Acad Sci U S A. 2004;101(10):3504–9. doi: 10.1073/pnas.0400182101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hormozdiari F, Kichaev G, Yang WY, Pasaniuc B, Eskin E. Identification of causal genes for complex traits. Bioinformatics. 2015;31(12):i206–13. doi: 10.1093/bioinformatics/btv240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linnertz C, Anderson L, Gottschalk W, Crenshaw D, Lutz MW, Allen J, Saith S, Mihovilovic M, Burke JR, Welsh-Bohmer KA, Roses AD, Chiba-Falek O. The cis-regulatory effect of an Alzheimer's disease-associated poly-T locus on expression of TOMM40 and apolipoprotein E genes. Alzheimers Dement. 2014;10(5):541–51. doi: 10.1016/j.jalz.2013.08.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linnertz C, Saucier L, Ge D, Cronin KD, Burke JR, Browndyke JN, Hulette CM, Welsh-Bohmer KA, Chiba-Falek O. Genetic regulation of alpha-synuclein mRNA expression in various human brain tissues. PLoS One. 2009;4(10):e7480. doi: 10.1371/journal.pone.0007480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutz MW, Saul R, Linnertz C, Glenn OC, Roses AD, Chiba-Falek O. A cytosine-thymine (CT)-rich haplotype in intron 4 of SNCA confers risk for Lewy body pathology in Alzheimer's disease and affects SNCA expression. Alzheimers Dement. 2015 doi: 10.1016/j.jalz.2015.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirkin SM. Expandable DNA repeats and human disease. Nature. 2007;447(7147):932–40. doi: 10.1038/nature05977. [DOI] [PubMed] [Google Scholar]
- Okladnova O, Syagailo YV, Tranitz M, Stober G, Riederer P, Mossner R, Lesch KP. A promoter-associated polymorphic repeat modulates PAX-6 expression in human brain. Biochem Biophys Res Commun. 1998;248(2):402–5. doi: 10.1006/bbrc.1998.8972. [DOI] [PubMed] [Google Scholar]
- Pearson CE, Nichol Edamura K, Cleary JD. Repeat instability: mechanisms of dynamic mutations. Nat Rev Genet. 2005;6(10):729–42. doi: 10.1038/nrg1689. [DOI] [PubMed] [Google Scholar]
- Peters DG, Kassam A, St Jean PL, Yonas H, Ferrell RE. Functional polymorphism in the matrix metalloproteinase-9 promoter as a potential risk factor for intracranial aneurysm. Stroke. 1999;30(12):2612–6. doi: 10.1161/01.str.30.12.2612. [DOI] [PubMed] [Google Scholar]
- Renton AE, Majounie E, Waite A, Simon-Sanchez J, Rollinson S, Gibbs JR, Schymick JC, Laaksovirta H, van Swieten JC, Myllykangas L, Kalimo H, Paetau A, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72(2):257–68. doi: 10.1016/j.neuron.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roses AD, Lutz MW, Amrine-Madsen H, Saunders AM, Crenshaw DG, Sundseth SS, Huentelman MJ, Welsh-Bohmer KA, Reiman EM. A TOMM40 variable-length polymorphism predicts the age of late-onset Alzheimer's disease. Pharmacogenomics J. 10(5):375–84. doi: 10.1038/tpj.2009.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayers EW, Barrett T, Benson DA, Bolton E, Bryant SH, Canese K, Chetvernin V, Church DM, Dicuccio M, Federhen S, Feolo M, Fingerman IM, et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2012;40:D13–25. doi: 10.1093/nar/gkr1184. Database issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Searle S, Blackwell JM. Evidence for a functional repeat polymorphism in the promoter of the human NRAMP1 gene that correlates with autoimmune versus infectious disease susceptibility. J Med Genet. 1999;36(4):295–9. [PMC free article] [PubMed] [Google Scholar]
- Shimajiri S, Arima N, Tanimoto A, Murata Y, Hamada T, Wang KY, Sasaguri Y. Shortened microsatellite d(CA)21 sequence down-regulates promoter activity of matrix metalloproteinase 9 gene. FEBS Lett. 1999;455(1-2):70–4. doi: 10.1016/s0014-5793(99)00863-7. [DOI] [PubMed] [Google Scholar]
- Sudmant PH, Rausch T, Gardner EJ, Handsaker RE, Abyzov A, Huddleston J, Zhang Y, Ye K, Jun G, Hsi-Yang Fritz M, Konkel MK, Malhotra A, et al. An integrated map of structural variation in 2,504 human genomes. Nature. 2015;526(7571):75–81. doi: 10.1038/nature15394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willems T, Gymrek M, Highnam G, Genomes Project C. Mittelman D, Erlich Y. The landscape of human STR variation. Genome Res. 2014;24(11):1894–904. doi: 10.1101/gr.177774.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Q, Ge D, Heinzen EL, Dickson SP, Urban TJ, Zhu M, Maia JM, He M, Zhao Q, Shianna KV, Goldstein DB. Prioritizing genetic variants for causality on the basis of preferential linkage disequilibrium. Am J Hum Genet. 2012;91(3):422–34. doi: 10.1016/j.ajhg.2012.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.