

Abstract
Aims
Cardiac aging is associated with progressive structural changes and functional impairment, such as left ventricular hypertrophy, fibrosis and diastolic dysfunction. Aging also increases myocardial activity of endothelin-1 (ET-1), a multifunctional peptide with growth-promoting and pro-fibrotic activity. Because the G protein-coupled estrogen receptor (GPER) regulates vascular responsiveness to ET-1, we investigated whether GPER also plays a role in the regulation of the cardiac endothelin system with aging.
Main methods
Young (4 month-old) and aged (24 month-old) wild-type and Gper-deficient (Gper-/-) mice were studied. Gene expression levels of prepro-ET-1, endothelin converting enzymes ECE-1 and ECE-2, and endothelin ETA and ETB receptors were determined by qPCR in left ventricular myocardium.
Key findings
Aging markedly increased steady-state mRNA expression levels of ECE-1, ECE-2, ETA and ETB receptors (each p<0.001 vs. young mice). Deletion of Gper inhibited the age-dependent increase in ECE-2 and ETB receptor mRNA levels (57% and 40% reduction, respectively, each p<0.01 vs. wild-type mice), whereas gene expression of prepro-ET-1, ECE-1, or the ETA receptor was unaffected in Gper-/- mice.
Significance
We identified a novel regulatory mechanism through which the endogenous Gper facilitates the age-dependent increase in myocardial expression of ECE-2 and the ETB receptor, which is compatible with an activating role of GPER for the cardiac endothelin system with aging. Targeting GPER signaling by selective antagonists may therefore be considered a new therapeutic approach to reduce age-dependent increased ET-1 activity and the associated development of left ventricular hypertrophy, fibrosis and heart failure.
Keywords: Aging, Endothelin Converting Enzyme, Endothelin Receptor, Estrogen Receptor, ET-1, GPR30, Heart, Myocardium
Introduction
With the anticipated aging of the world's population, current estimates predict a marked increase in the prevalence of heart failure and the associated cost of care, necessitating already more hospitalizations of older patients than any other medical condition in the Western civilization [1]. Indeed, the elderly account for more than 90% of patients with heart failure [2]. Cardiac aging in humans is associated with progressive structural changes and functional impairment, such as left ventricular hypertrophy, fibrosis and impaired diastolic function, changes that are recapitulated in experimental animals [3,4].
In rodents, age-dependent cardiac hypertrophy and fibrosis have been associated with increased myocardial expression of the multifunctional peptide endothelin-1 (ET-1) [5-7], which can induce cardiomyocyte growth and collagen synthesis in cardiac fibroblasts [8,9]. Similarly, aging is associated with increased cardiac expression of endothelin ETA and ETB receptors [6,10], whereas cardiomyocyte hypertrophy and myocardial fibrosis are attenuated in aged mice with cardiomyocyte-specific deletion of the ETA receptor gene [6]. Furthermore, treatment with ETA and dual ETA/ETB receptor antagonists improves cardiac function and survival in animal models of acute and chronic heart failure [11-13]. In symptomatic patients with advanced heart failure, myocardial ET-1 peptide levels are also increased, which may be related to elevated expression of endothelin converting enzyme-1 (ECE-1) that catalyzes proteolytic cleavage of the precursor peptide big-ET-1 to form ET-1 [14]. Together, these findings suggest that cardiac aging activates the local endothelin system, yet the underlying mechanisms are still unclear.
Although myocardial function in adulthood and during aging critically depends on the function of G protein-coupled receptors (GPCRs) such as ETA and ETB receptors [15], much less is known about the orphan GPCR GPR30 that shows strong expression in the heart [16-18]. GPR30 was later identified to bind and induce rapid signaling in response to estrogen [19,20], which led to its designation as G protein-coupled estrogen receptor (GPER) [21]. However, chronic Gper-dependent effects in the absence of circulating ovarian estrogens have also been reported [22-24]. Activation of GPER using its selective agonist G-1 ameliorates cardiac function, hypertrophy, and fibrosis in animals with hypertensive cardiomyopathy or congestive heart failure [25-29], and reduces infarct size and improves cardiac remodeling after experimentally induced myocardial ischemia and reperfusion injury [30-33]. Whether GPER function plays a role in cardiac aging is unknown.
Given our previous observation that ET-1-mediated vasoconstriction is attenuated by G-1 [34] and potentiated in Gper-deficient male mice (e.g. in the absence of ovarian estrogen production) [23], we hypothesized that GPER may play a regulatory role in the cardiac endothelin system with age. We therefore set out to study steady-state gene expression of ET-1, as well as ECE-1, ECE-2, ETA and ETB receptors in left ventricular myocardium of young and aged GPER-deficient (Gper-/-) and wild-type male mice.
Materials and Methods
Materials
All materials were from Sigma-Aldrich (St. Louis, MO, USA) unless stated otherwise.
Animals
Male Gper-/- mice (Proctor & Gamble, Cincinnati, OH, USA, provided by Jan S. Rosenbaum) were generated and backcrossed onto the C57BL/6 background as described [24]. Gper-/- and wild-type littermates (Harlan Laboratories, Indianapolis, IN, USA) were housed at the Animal Resource Facility of the University of New Mexico Health Sciences Center with unlimited access to water and a rodent diet devoid of alfalfa or soybean meal to minimize the occurrence of natural phytoestrogens (Teklad 2020SX, Harlan Laboratories, Madison, WI, USA). Animals were maintained under controlled temperature of 22-23 °C on a 12h light-dark cycle. At 4 or 24 months of age, mice were sacrificed by intraperitoneal injection of sodium pentobarbital (2.2 mg/g body weight). The apex of the left ventricle was collected, immediately snap-frozen in liquid nitrogen and stored at -80 °C until further analysis. All procedures were approved by and carried out in accordance with institutional policies and the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Isolation and reverse transcription of myocardial mRNA
Frozen left ventricular myocardium (20 mg) was disrupted and homogenized using a rotor-stator homogenizer, and total RNA was extracted using the silica-based RNeasy Fibrous Tissue Mini Kit (Qiagen, Valencia, CA, USA). RNA was reverse-transcribed using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Carlsbad, CA, USA).
Quantitative real-time polymerase chain reaction (qPCR)
SYBR Green-based detection of amplified gene-specific cDNA fragments was performed on a 7500 FAST real-time PCR system (Applied Biosystems). The sets of primers used are given in Table 1. Relative gene expression was determined using the 2-∆CT method [35] with GAPDH serving as house-keeping control.
Table 1. Primer sets used for qPCR.
| Gene (GenBank ID) | Forward Primer | Reverse Primer |
|---|---|---|
| prepro-ET-1 (U35233.1) | 5′-AAC TCA GGG CCC AAA GTA CC-3′ | 5′-TTT GCA ACA CGA AAA GAT GC-3′ |
| ECE-1 (NM_199307.2) | 5′-GCC TAC CGG GCG TAC CAG AAC-3′ | 5′-GGT GTG CGG ACA GAG CAC CAG-3′ |
| ECE-2 (AF396699) | 5′-CCC GTG AAC GCT TAC TAC CTT-3′ | 5′-GGT CAT CAA AGG CAT GTG TCA-3′ |
| ETA receptor (BC008277) | 5′-GAA GGA CTG GTG GCT CTT TG-3′ | 5′-CTT CTC GAC GCT GTT TGA GG-3′ |
| ETB receptor (BC026553) | 5′-CGG TAT GCA GAT TGC TTT GA-3′ | 5′-AC CTG TGT GGA TTG CTC TG-3′ |
| eNOS (NM_008713) | 5′-AGA GCC TGC AAT TAC TAC CA-3′ | 5′-GTG GAT TTG CTG CTC TGT AG-3′ |
| GAPDH (NM_008084) | 5′-TTC ACC ACC ATG GAG AAG GC-3′ | 5′-GGC ATG GAC TGT GGT CAT GA-3′ |
Statistical analyses
Data was analyzed using two-way analysis of variance (ANOVA) followed by Bonferroni's post-hoc test for multiple comparisons (Prism version 5.0 for Macintosh, GraphPad Software, San Diego, CA, USA). Values are expressed as mean±s.e.m.; n equals the number of animals used. Statistical significance was accepted at a p value <0.05.
Results
Aging upregulates myocardial ECE and ET receptor gene expression
To study whether aging affects the cardiac endothelin system, gene expression levels of its individual components were quantified in left ventricular myocardium of young (4 month-old) and aged (24 month-old) wild-type mice. Aging was associated with a marked up-regulation of the ECEs and ET receptors in left ventricular myocardium: mRNA levels of ECE-1, ECE-2, ETA and ETB receptors were 4-fold to 12-fold higher compared to young mice (all p<0.001, n=6-7, Figures 1 and 2), whereas prepro-ET-1 expression was unaffected by aging. Furthermore, gene expression levels of ECE-1 were 19-fold and 6-fold higher than ECE-2 in left ventricular myocardium of young and aged mice, respectively (each n=6-7, p<0.001, Figure 1). In contrast, mRNA levels of ETA and ETB receptors were similar within each age group (each n=6-7, p=n.s., Figure 2).
Figure 1.
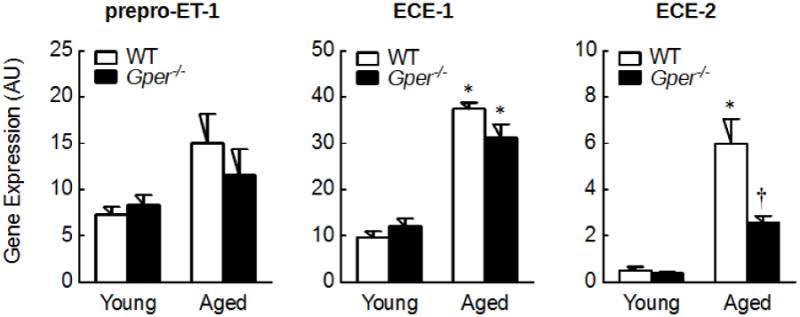
Gene expression levels of prepro-ET-1 and endothelin converting enzymes ECE-1 and ECE-2 in left ventricular myocardium of young (4 month-old) and aged (24 month-old) wild-type (WT) and Gper-deficient (Gper-/-) mice. All data (n=5-7) are mean±s.e.m.; *P<0.001 vs. young mice, †p<0.001 vs. wild-type control. AU, arbitrary units.
Figure 2.

Gene expression levels of endothelin ETA and ETB receptors in left ventricular myocardium of young (4 month-old) and aged (24 month-old) wild-type (WT) and Gper-deficient (Gper-/-) mice. All data (n=5-7) are mean±s.e.m.; *P<0.01 vs. young mice, †p<0.01 vs. wild-type control. AU, arbitrary units.
GPER mediates the age-dependent upregulation of ECE-2 and ETB receptor gene expression
To determine whether endogenous GPER affects the upregulation of the cardiac endothelin system with aging, left ventricular myocardium of Gper-/- mice was analyzed. In young mice, myocardial gene expression of prepro-ET-1, ECE-1, ECE-2, ETA or ETB receptors was unaffected by deletion of Gper (each n=5-7, p=n.s. vs. wild-type control, Figures 1 and 2). In contrast, the increase in ECE-2 mRNA level with aging was markedly reduced in myocardium lacking Gper (57% reduction, n=6, p<0.001 vs. wild-type control, Figure 1). Similarly, deletion of Gper significantly inhibited the age-dependent upregulation of myocardial ETB receptor gene expression (40% reduction, n=6, p<0.01 vs. wild-type control, Figure 2).
Given that activation of GPER induces production of nitric oxide (NO) by endothelial NO synthase (eNOS) [22,24,34], and since the cardiac eNOS and endothelin systems closely interact in the pathogenesis of cardiac dysfunction, hypertrophy and fibrosis [36,37], we next determined myocardial eNOS gene expression in wild-type and Gper-/- mice. Surprisingly, eNOS mRNA levels were neither affected by aging nor by Gper deletion (n=5-7, p=n.s., Figure 3).
Figure 3.

Gene expression levels of endothelial NO synthase (eNOS) in left ventricular myocardium of young (4 month-old) and aged (24 month-old) wild-type (WT) and Gper-deficient (Gper-/-) mice. All data (n=5-7) are mean±s.e.m. AU, arbitrary units.
Taken together, the presence of Gper in aged mice is required to facilitate the upregulation of specific components of the cardiac endothelin system with age, including ECE-2 and the ETB receptor.
Discussion
This study identifies endogenous GPER as an age-dependent stimulatory regulator of myocardial ECE-2 and ETB receptor gene expression in male mice. In the presence of GPER, increased ECE-2 expression is likely to contribute to augmented local synthesis of ET-1. Thus, GPER may facilitate the activation of the cardiac endothelin system with aging [5-7,10]. Furthermore, GPER-dependent regulation of ETB receptor expression suggests functional cross-talk between the two GPCRs. Since increased activity of the cardiac endothelin system has been implicated in the progression of heart failure with aging [5-7,10], inhibiting GPER may provide a new approach to reduce myocardial ECE-2 and ETB receptor expression and thus increased ET-1 activity.
Elevated circulating levels and myocardial expression of ET-1 have been associated with age-dependent cardiac hypertrophy and fibrosis [5-7], and can be observed in animals and humans with heart failure [11-14]. In the present study, we found an age-dependent increase in myocardial ECE-1, ECE-2, as well as ETA and ETB receptor gene expression, while prepro-ET-1 mRNA levels were unaffected by aging. However, given that ET-1 is formed locally through a 39-amino acid intermediate, big-ET-1, which undergoes subsequent proteolytic cleavage by ECE-1 and ECE-2 [14,38], enhanced conversion of precursor peptides by ECE may be relevant for elevated ET-1 levels in cardiac aging or age-related cardiovascular disease. Among the ECE identified, the membrane bound metalloproteinase ECE-1 was found to display increased expression levels in the myocardium of patients with advanced heart failure [14], and inhibition of ECE-1 activity or expression reduces in vitro hypertrophy of cardiomyocytes stimulated by the adrenergic agonist phenylephrine [39]. ECE-2, which similar to GPER [19] is expressed intracellularly in the Golgi network [38], plays a role in the embryonic development of cardiac outflow structures, and may be functionally involved in cardiac ET-1 formation similar to ECE-1 [40]. Although the role of ECE-2 in cardiac aging is unclear and its expression level is substantially lower compared to ECE-1, we observed a substantial age-dependent increase in ECE-2 mRNA levels that largely depended on the presence of Gper. Thus, GPER-driven ECE-2 expression may potentiate age-dependent increases in cardiac ET-1 production. In addition, it is conceivable that local ET-1 activity would be further enhanced by increased expression of ETA and/or ETB receptors with aging [6,10], which have been implicated in age-dependent pathologies such as podocyte injury and glomerulosclerosis. Indeed, ETA receptor blockade for several weeks in part reverses structural and functional renal aging [41,42]. Given that ETB receptor mRNA expression is also stimulated by the presence of GPER as shown in the present study, this may further underscore a role of the endogenous GPER for increased activity of the cardiac endothelin system with aging.
The role of GPER in the physiological aging process of the heart is unknown. Previous studies have shown impaired vascular function related to GPER in blood vessels from aged female rats [43], while metabolic effects on glucose tolerance and inflammation become evident in male Gper-/- mice with age [44]. In line with these findings, the present study now also provides evidence of a novel role for GPER in cardiac aging. This may not be surprising given the high cardiac expression of GPER [16-18] and its emerging protective role in heart failure models upon activation by the selective ligand G-1 [25-32]. To the contrary, the observation that GPER expression is required to increase certain components of the myocardial endothelin system was unexpected, even more so since we have previously observed that deletion of Gper increases vasoconstrictor responses to ET-1 [23], whereas G-1 inhibits ET-1-dependent contractions of coronary arteries [34]. However, these GPER-mediated effects on vascular reactivity to ET-1 have been observed in blood vessels from healthy young animals as opposed to the present findings regarding the cardiac endothelin system in mice at advanced age, which has recently been considered a “diseaselike state” [3]. It may therefore be possible that GPER – contrary to its effects under healthy conditions – contributes in part to the increased activity of myocardial ET-1 with cardiac aging, as recently also observed for ET-1-stimulated migration of certain cancer cells [45]. Furthermore, activation of GPER potently induces eNOS-derived NO formation [22,24,34], an effect that may explain the inhibition of ET-1-dependent contractions in blood vessels. On the other hand, despite the strong interaction between the cardiac eNOS and endothelin systems [36,37], we found that neither Gper deletion nor aging affected myocardial eNOS expression, indicating that GPER – at least on the gene expression level – regulates the cardiac endothelin but not the eNOS system with aging.
Interestingly, despite low endogenous estrogen production, studies in mice have previously established that endogenous GPER is also active in the cardiovascular system of males [22,23] and ovariectomized females [24], e.g. in the absence of ovarian estrogen production. The present study extends these previous findings demonstrating that deletion of the Gper gene in male mice is sufficient to regulate the expression of components of the cardiac endothelin system with aging. Such effects may be explained by a certain level of intrinsic activity generally attributed to GPCRs [46], or by localized myocardial conversion of androgen precursors into estrogens catalyzed by the enzyme aromatase, which facilitates sufficient localized estrogen biosynthesis that might activate signaling of GPER and/or the “classical” estrogen receptors, ERα and ERβ [47].
Conclusion
The present study presents evidence for a novel regulatory mechanism that is required to stimulate the age-dependent increase in myocardial expression of ECE-2 and ETB receptors through the presence of the Gper gene. This endogenous effect of GPER will likely increase the overall activity of the cardiac endothelin system with aging. Aged mice closely mimic changes of cardiac aging in humans, including left-ventricular hypertrophy, fibrosis, and diastolic dysfunction, which has been associated with increased activity of ET-1 [3-7,10], yet the extent to which GPER-mediated, age-dependent upregulation of ECE-2 and ETB expression translates into structural or functional cardiac changes remains to be clarified. Given these GPER-mediated effects, targeting GPER signaling by selective antagonists may be considered a new therapeutic approach to reduce increased activity of the cardiac endothelin system with aging and the development of heart failure associated with it [3-7,10].
Acknowledgments
We thank Dr. Chelin Hu and Daniel F. Cimino for expert technical assistance. This study was supported by the National Institutes of Health (NIH R01 CA127731 & CA163890 to E.R.P.), Dedicated Health Research Funds from the University of New Mexico School of Medicine allocated to the Signature Program in Cardiovascular and Metabolic Diseases (to E.R.P.), and the Swiss National Science Foundation (grants 135874 & 141501 to M.R.M. and grants 108258 & 122504 to M.B.). N.C.F. was supported by NIH training grant HL07736.
Footnotes
Financial Disclosure and Competing Interests Statement: M.R.M., G.S., M.B., and E.R.P. are inventors on U.S. patent applications for therapeutic use of compounds targeting GPER. E.R.P. is an inventor on U.S. patent Nos. 7,875,721 and 8,487,100 for GPER-selective ligands and imaging agents.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Heidenreich PA, Albert NM, Allen LA, Bluemke DA, Butler J, Fonarow GC, et al. Forecasting the impact of heart failure in the United States: a policy statement from the American Heart Association. Circ Heart Fail. 2013;6(3):606–19. doi: 10.1161/HHF.0b013e318291329a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Blaha MJ, et al. Heart disease and stroke statistics--2014 update: a report from the American Heart Association. Circulation. 2014;129(3):e28–e292. doi: 10.1161/01.cir.0000441139.02102.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lakatta EG. So! What's aging? Is cardiovascular aging a disease? J Mol Cell Cardiol. 2015;83:1–13. doi: 10.1016/j.yjmcc.2015.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boyle AJ, Shih H, Hwang J, Ye J, Lee B, Zhang Y, et al. Cardiomyopathy of aging in the mammalian heart is characterized by myocardial hypertrophy, fibrosis and a predisposition towards cardiomyocyte apoptosis and autophagy. Exp Gerontol. 2011;46(7):549–59. doi: 10.1016/j.exger.2011.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Iemitsu M, Miyauchi T, Maeda S, Tanabe T, Irukayama-Tomobe Y, Goto K, et al. Effects of aging and subsequent exercise training on gene expression of endothelin-1 in rat heart. Clin Sci (Lond) 2002;103(Suppl 48):152S–7S. doi: 10.1042/CS103S152S. [DOI] [PubMed] [Google Scholar]
- 6.Ceylan-Isik AF, Dong M, Zhang Y, Dong F, Turdi S, Nair S, et al. Cardiomyocyte-specific deletion of endothelin receptor A rescues aging-associated cardiac hypertrophy and contractile dysfunction: role of autophagy. Basic Res Cardiol. 2013;108(2):335. doi: 10.1007/s00395-013-0335-3. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 7.Wang X, Guo Z, Ding Z, Khaidakov M, Lin J, Xu Z, et al. Endothelin-1 upregulation mediates aging-related cardiac fibrosis. J Mol Cell Cardiol. 2015;80:101–9. doi: 10.1016/j.yjmcc.2015.01.001. [DOI] [PubMed] [Google Scholar]
- 8.Guarda E, Katwa LC, Myers PR, Tyagi SC, Weber KT. Effects of endothelins on collagen turnover in cardiac fibroblasts. Cardiovasc Res. 1993;27(12):2130–4. doi: 10.1093/cvr/27.12.2130. [DOI] [PubMed] [Google Scholar]
- 9.Ito H, Hirata Y, Adachi S, Tanaka M, Tsujino M, Koike A, et al. Endothelin-1 is an autocrine/paracrine factor in the mechanism of angiotensin II-induced hypertrophy in cultured rat cardiomyocytes. J Clin Invest. 1993;92(1):398–403. doi: 10.1172/JCI116579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ishihata A, Katano Y. Role of angiotensin II and endothelin-1 receptors in aging-related functional changes in rat cardiovascular system. Ann N Y Acad Sci. 2006;1067:173–81. doi: 10.1196/annals.1354.021. [DOI] [PubMed] [Google Scholar]
- 11.Hocher B, George I, Rebstock J, Bauch A, Schwarz A, Neumayer HH, et al. Endothelin system-dependent cardiac remodeling in renovascular hypertension. Hypertension. 1999;33(3):816–22. doi: 10.1161/01.hyp.33.3.816. [DOI] [PubMed] [Google Scholar]
- 12.Mulder P, Richard V, Bouchart F, Derumeaux G, Munter K, Thuillez C. Selective ETA receptor blockade prevents left ventricular remodeling and deterioration of cardiac function in experimental heart failure. Cardiovasc Res. 1998;39(3):600–8. doi: 10.1016/s0008-6363(98)00159-x. [DOI] [PubMed] [Google Scholar]
- 13.Sakai S, Miyauchi T, Kobayashi M, Yamaguchi I, Goto K, Sugishita Y. Inhibition of myocardial endothelin pathway improves long-term survival in heart failure. Nature. 1996;384(6607):353–5. doi: 10.1038/384353a0. [DOI] [PubMed] [Google Scholar]
- 14.Morawietz H, Goettsch W, Szibor M, Barton M, Shaw S, Hakim K, et al. Increased expression of endothelin-converting enzyme-1 in failing human myocardium. Clin Sci (Lond) 2002;103(Suppl 48):237S–40S. doi: 10.1042/CS103S237S. [DOI] [PubMed] [Google Scholar]
- 15.Violin JD, Soergel DG, Boerrigter G, Burnett JC, Jr, Lark MW. GPCR biased ligands as novel heart failure therapeutics. Trends Cardiovasc Med. 2013;23(7):242–9. doi: 10.1016/j.tcm.2013.01.002. [DOI] [PubMed] [Google Scholar]
- 16.Feng Y, Gregor P. Cloning of a novel member of the G protein-coupled receptor family related to peptide receptors. Biochem Biophys Res Commun. 1997;231(3):651–4. doi: 10.1006/bbrc.1997.6161. [DOI] [PubMed] [Google Scholar]
- 17.Kvingedal AM, Smeland EB. A novel putative G-protein-coupled receptor expressed in lung, heart and lymphoid tissue. FEBS Lett. 1997;407(1):59–62. doi: 10.1016/s0014-5793(97)00278-0. [DOI] [PubMed] [Google Scholar]
- 18.Owman C, Blay P, Nilsson C, Lolait SJ. Cloning of human cDNA encoding a novel heptahelix receptor expressed in Burkitt's lymphoma and widely distributed in brain and peripheral tissues. Biochem Biophys Res Commun. 1996;228(2):285–92. doi: 10.1006/bbrc.1996.1654. [DOI] [PubMed] [Google Scholar]
- 19.Revankar CM, Cimino DF, Sklar LA, Arterburn JB, Prossnitz ER. A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science. 2005;307(5715):1625–30. doi: 10.1126/science.1106943. [DOI] [PubMed] [Google Scholar]
- 20.Thomas P, Pang Y, Filardo EJ, Dong J. Identity of an estrogen membrane receptor coupled to a G protein in human breast cancer cells. Endocrinology. 2005;146(2):624–32. doi: 10.1210/en.2004-1064. [DOI] [PubMed] [Google Scholar]
- 21.Prossnitz ER, Barton M. The G-protein-coupled estrogen receptor GPER in health and disease. Nat Rev Endocrinol. 2011;7(12):715–26. doi: 10.1038/nrendo.2011.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Meyer MR, Amann K, Field AS, Hu C, Hathaway HJ, Kanagy NL, et al. Deletion of G protein-coupled estrogen receptor increases endothelial vasoconstriction. Hypertension. 2012;59(2):507–12. doi: 10.1161/HYPERTENSIONAHA.111.184606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meyer MR, Field AS, Kanagy NL, Barton M, Prossnitz ER. GPER regulates endothelin-dependent vascular tone and intracellular calcium. Life Sci. 2012;91:623–7. doi: 10.1016/j.lfs.2012.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Meyer MR, Fredette NC, Howard TA, Hu C, Ramesh C, Daniel C, et al. G protein-coupled estrogen receptor protects from atherosclerosis. Sci Reports. 2014;4:7564. doi: 10.1038/srep07564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jessup JA, Lindsey SH, Wang H, Chappell MC, Groban L. Attenuation of salt-induced cardiac remodeling and diastolic dysfunction by the GPER agonist G-1 in female mRen2.Lewis rats. PLoS One. 2010;5(11):e15433. doi: 10.1371/journal.pone.0015433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang H, Jessup JA, Lin MS, Chagas C, Lindsey SH, Groban L. Activation of GPR30 attenuates diastolic dysfunction and left ventricle remodelling in oophorectomized mRen2.Lewis rats. Cardiovasc Res. 2012;94(1):96–104. doi: 10.1093/cvr/cvs090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.De Francesco EM, Angelone T, Pasqua T, Pupo M, Cerra MC, Maggiolini M. GPER mediates cardiotropic effects in spontaneously hypertensive rat hearts. PLoS One. 2013;8(8):e69322. doi: 10.1371/journal.pone.0069322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lenhart PM, Broselid S, Barrick CJ, Leeb-Lundberg LM, Caron KM. G-protein-coupled receptor 30 interacts with receptor activity-modifying protein 3 and confers sex-dependent cardioprotection. J Mol Endocrinol. 2013;51(1):191–202. doi: 10.1530/JME-13-0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kang S, Liu Y, Sun D, Zhou C, Liu A, Xu C, et al. Chronic activation of the G protein-coupled receptor 30 with agonist G-1 attenuates heart failure. PLoS One. 2012;7(10):e48185. doi: 10.1371/journal.pone.0048185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Deschamps AM, Murphy E. Activation of a novel estrogen receptor, GPER, is cardioprotective in male and female rats. Am J Physiol Heart Circ Physiol. 2009;297(5):H1806–13. doi: 10.1152/ajpheart.00283.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bopassa JC, Eghbali M, Toro L, Stefani E. A novel estrogen receptor GPER inhibits mitochondria permeability transition pore opening and protects the heart against ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol. 2010;298(1):H16–23. doi: 10.1152/ajpheart.00588.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee TM, Lin SZ, Chang NC. Both GPER and membrane oestrogen receptor-alpha activation protect ventricular remodelling in 17beta oestradiol-treated ovariectomized infarcted rats. J Cell Mol Med. 2014;18(12):2454–65. doi: 10.1111/jcmm.12430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kabir ME, Singh H, Lu R, Olde B, Leeb-Lundberg LM, Bopassa JC. G protein-coupled estrogen receptor 1 mediates acute estrogen-induced cardioprotection via MEK/ERK/GSK-3beta pathway after ischemia/reperfusion. PLoS One. 2015;10(9):e0135988. doi: 10.1371/journal.pone.0135988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Meyer MR, Baretella O, Prossnitz ER, Barton M. Dilation of epicardial coronary arteries by the G protein-coupled estrogen receptor agonists G-1 and ICI 182,780. Pharmacology. 2010;86(1):58–64. doi: 10.1159/000315497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25(4):402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 36.Vignon-Zellweger N, Relle K, Kienlen E, Alter M, Seider P, Sharkovska J, et al. Endothelin-1 overexpression restores diastolic function in eNOS knockout mice. J Hypertens. 2011;29(5):961–70. doi: 10.1097/HJH.0b013e3283450770. [DOI] [PubMed] [Google Scholar]
- 37.Vignon-Zellweger N, Relle K, Rahnenfuhrer J, Schwab K, Hocher B, Theuring F. Endothelin-1 overexpression and endothelial nitric oxide synthase knock-out induce different pathological responses in the heart of male and female mice. Life Sci. 2014;118(2):219–25. doi: 10.1016/j.lfs.2013.12.003. [DOI] [PubMed] [Google Scholar]
- 38.Emoto N, Yanagisawa M. Endothelin-converting enzyme-2 is a membrane-bound, phosphoramidon-sensitive metalloprotease with acidic pH optimum. J Biol Chem. 1995;270(25):15262–8. doi: 10.1074/jbc.270.25.15262. [DOI] [PubMed] [Google Scholar]
- 39.Kaburagi S, Hasegawa K, Morimoto T, Araki M, Sawamura T, Masaki T, et al. The role of endothelin-converting enzyme-1 in the development of alpha1-adrenergic-stimulated hypertrophy in cultured neonatal rat cardiac myocytes. Circulation. 1999;99(2):292–8. doi: 10.1161/01.cir.99.2.292. [DOI] [PubMed] [Google Scholar]
- 40.Yanagisawa H, Hammer RE, Richardson JA, Emoto N, Williams SC, Takeda S, et al. Disruption of ECE-1 and ECE-2 reveals a role for endothelin-converting enzyme-2 in murine cardiac development. J Clin Invest. 2000;105(10):1373–82. doi: 10.1172/JCI7447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ortmann J, Amann K, Brandes RP, Kretzler M, Munter K, Parekh N, et al. Role of podocytes for reversal of glomerulosclerosis and proteinuria in the aging kidney after endothelin inhibition. Hypertension. 2004;44(6):974–81. doi: 10.1161/01.HYP.0000149249.09147.b4. [DOI] [PubMed] [Google Scholar]
- 42.Barton M. Reversal of proteinuric renal disease and the emerging role of endothelin. Nat Clin Pract Nephrol. 2008;4(9):490–501. doi: 10.1038/ncpneph0891. [DOI] [PubMed] [Google Scholar]
- 43.Lindsey SH, da Silva AS, Silva MS, Chappell MC. Reduced vasorelaxation to estradiol and G-1 in aged female and adult male rats is associated with GPR30 downregulation. Am J Physiol Endocrinol Metab. 2013;305(1):E113–8. doi: 10.1152/ajpendo.00649.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sharma G, Hu C, Brigman JL, Zhu G, Hathaway HJ, Prossnitz ER. GPER deficiency in male mice results in insulin resistance, dyslipidemia, and a proinflammatory state. Endocrinology. 2013;154(11):4136–45. doi: 10.1210/en.2013-1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bartella V, De Francesco EM, Perri MG, Curcio R, Dolce V, Maggiolini M, et al. The G protein estrogen receptor (GPER) is regulated by endothelin-1 mediated signaling in cancer cells. Cell Signal. 2016;28(2):61–71. doi: 10.1016/j.cellsig.2015.11.010. [DOI] [PubMed] [Google Scholar]
- 46.Bond RA, Ijzerman AP. Recent developments in constitutive receptor activity and inverse agonism, and their potential for GPCR drug discovery. Trends Pharmacol Sci. 2006;27(2):92–6. doi: 10.1016/j.tips.2005.12.007. [DOI] [PubMed] [Google Scholar]
- 47.Grohe C, Kahlert S, Lobbert K, Vetter H. Expression of oestrogen receptor alpha and beta in rat heart: role of local oestrogen synthesis. J Endocrinol. 1998;156(2):R1–7. doi: 10.1677/joe.0.156r001. [DOI] [PubMed] [Google Scholar]