

Abstract
The development of ANG II-dependent hypertension involves increased infiltration of macrophages (MΦ) and T cells into the kidney and the consequent elevation of intrarenal cytokines including IL-6, which facilitates the progression of hypertension and associated kidney injury. Intrarenal renin-angiotensin system (RAS) activation, including proximal tubular angiotensinogen (AGT) stimulation, has also been regarded as a cardinal mechanism contributing to these diseases. However, the interaction between immune cells and intrarenal RAS activation has not been fully delineated. Therefore, the present study investigated whether ANG II-treated MΦ induce AGT upregulation in renal proximal tubular cells (PTCs). MΦ were treated with 0–10−6 M ANG II for up to 48 h. PTCs were incubated with the collected medium from MΦ. In ANG II-treated MΦ, IL-6 mRNA and protein levels were increased (1.86 ± 0.14, protein level, ratio to control); moreover, IL-6 levels were higher than TNF-α and IL-1β in culture medium isolated from ANG II-treated MΦ. Elevated AGT expression (1.69 ± 0.04, ratio to control) accompanied by phosphorylated STAT3 were observed in PTCs that received culture medium from ANG II-treated MΦ. The addition of a neutralizing IL-6 antibody to the collected medium attenuated phosphorylation of STAT3 and AGT augmentation in PTCs. Furthermore, a JAK2 inhibitor also suppressed STAT3 phosphorylation and AGT augmentation in PTCs. These results demonstrate that ANG II-induced IL-6 elevation in MΦ enhances activation of the JAK-STAT pathway and consequent AGT upregulation in PTCs, suggesting involvement of an immune response in driving intrarenal RAS activity.
Keywords: interleukin-6, angiotensin II, angiotensinogen, macrophage, renin-angiotensin system
hypertension accounts for approximately one-fourth of all heart failure, with 60% of these cases attributable to hypertension in the elderly (46). Since the intrarenal renin-angiotensin system (RAS) plays cardinal roles in controlling blood pressure and regulating electrolyte and body fluid homeostasis (27, 28), an activated RAS in the kidney is a major risk factor contributing to the development of hypertension and kidney injury. A key process contributing to the elevation of intrarenal ANG II is augmentation of intrarenal angiotensinogen (AGT) (28), which is primarily produced in renal proximal tubular cells (PTCs) (11, 16, 41). In particular, renal proximal tubule-specific overexpression of AGT amplifies intrarenal ANG II levels and causes the development of hypertension and kidney injury (36, 48). Thus, elucidating the mechanisms that regulate intrarenal AGT is essential for the development of novel strategies to prevent and treat hypertension and RAS-associated tissue injury.
ANG II promotes AGT augmentation in the kidney as well as in the heart, liver, and adipose tissue (3, 7, 14, 16, 21, 23, 43). Thus, the intrarenal amplification of AGT by ANG II has been regarded as a pivotal mechanism facilitating the progression of hypertension. However, some in vitro studies using cultured human PTCs have failed to show AGT upregulation by direct treatment with ANG II (37, 38), suggesting that a mediator is required for the intrarenal AGT augmentation observed in ANG II-dependent hypertension (7, 16).
Chronic ANG II elevation induces increased immune cell infiltration in the kidneys, contributing to increases in intrarenal cytokine levels. In particular, IL-6 is produced in high quantities by activated macrophages (MΦ) driving inflammatory processes (8, 12, 15). Previous studies have shown that both plasma and intrarenal IL-6 levels are markedly elevated in chronic ANG II-infused animals (34, 35, 51), and, in IL-6 knockout (KO) mice, the severity of ANG II-induced hypertension and activation of STAT3 activity are reduced (2, 20). Moreover, IL-6 KO improves the development of albuminuria and renal fibrosis in ANG II-induced hypertension (24, 52). These results suggest that IL-6 and its signaling pathway are involved in the development of ANG II-induced hypertension and kidney injury.
IL-6 stimulates the secretion (30) and expression of hepatic AGT by activating the JAK-STAT pathway (13, 33). Notably, IL-6 and STAT3 activation are also required for ANG II-induced AGT augmentation in PTCs (37). Together, these findings provide a firm basis for our hypothesis that elevated IL-6 production by activated immune cells, especially MΦ, leads to intrarenal AGT augmentation via activation of the JAK-STAT pathway ultimately facilitating intrarenal RAS activation during the development of ANG II-dependent hypertension. However, in ANG II-dependent hypertensive animal models, ANG II is responsible for stimulating the production of multiple pathogenic factors, including cytokines, hormones, oxidative stress, mechanical stress, and immune cells other than MΦ. Moreover, immunosuppression broadly and nonselectively alters immune cell compositions and the consequent pathophysiological status, causing difficulty in distinguishing the exact nature of the roles that activated MΦ and IL-6 play in intrarenal AGT regulation. Therefore, in an in vitro setting using cultured MΦ and PTCs, we aimed to determine the role of IL-6 in proximal tubular AGT augmentation that occurs in ANG II-dependent hypertension.
MATERIALS AND METHODS
Cell culture.
Commercially available rat MΦ (male, NR8383, American Type Culture Collection) and immortalized rat PTCs (male) (10) were used in this study. Since immortalized PTCs proliferated over monolayer levels during tested treatments of MΦ (up to 48 h), coculture systems by transwell plates were not used, and, therefore, a sequential culture system was used in the present study. MΦ were cultured in RPMI-1640 medium (Invitrogen, Carlsbad, CA) supplemented with 10% heat-inactivated FCS (Invitrogen). MΦ were plated at a density of 1 × 105 cells/well in 12-well plates. Before stimulation, cells were serum starved for 24 h. MΦ were treated with 0–10−6 M ANG II (Phoenix Pharmaceuticals, Phoenix, AZ) for up to 48 h. Thereafter, culture media from MΦ was collected and filtered using 0.22-μm filters to remove floating MΦ from the collected media. PTCs were subsequently treated with the collected medium for 24 h. MΦ that were not treated with ANG II (0 μM ANG II) and PTCs that did not receive culture medium from MΦ served as control groups. To test the direct effects of ANG II on AGT regulation in PTCs, PTCs were incubated with 100 nM ANG II for 24 h.
Cytokine ELISA.
Cytokine levels of macrophage cell media were analyzed via sandwich ELISA using a commercially available kit (Signosis, Santa Clara, CA) after the manufacturer's instructions. Briefly, 100 μl of standard, control, or sample were added to the proper well, incubated for 1 h, and washed before the addition of 100 μl of biotin-labeled antibody. After 1 h, excess antibody was removed via a wash. Wells were then probed with 100 μl of a streptavidin-horseradish peroxidase conjugate for 45 min before being washed. Substrate (100 μl) was added to begin the colorimetric reaction. The reaction was allowed to proceed for <30 min before the addition of stop solution and reading of the optical density at 450 nm.
Quantitative real-time RT-PCR.
Quantitative real-time RT-PCR was performed to evaluate rat IL-6 and AGT mRNA levels using the TaqMan PCR system. For total RNA isolation, treated cells were washed with 3 ml PBS. PBS was aspirated, and total RNA was isolated from cells using the RNeasy kit (Qiagen). Subsequently, quantitative real-time RT-PCR was performed as previously described (17, 38). All samples were analyzed in triplicate, and the data were normalized based on expression levels of rat β-actin mRNA.
Western blot analysis.
IL-6 and AGT protein levels were determined by Western blot analyses. Phosphorylation levels of STAT3 and expression levels of STAT3 were also detected by Western blot analyses to elucidate participation of these transcription factors in AGT regulation. The Western blots were performed as previously described (40). After treatment, cells were harvested with 80 μl lysis buffer containing 1% Triton X-100, 150 mmol/l NaCl, 1 mmol/l EDTA, 1% Nonidet P-40, 1 mmol/l Na3VO4, and 0.25% protease inhibitor cocktail (Sigma, St. Louis, MO). Lysates were sonicated three times for 10 s each and centrifuged at 13,000 rpm at 4°C for 30 min. Total protein concentration of the supernatant was quantified using a Micro BCA Protein Assay Kit (Pierce, Rockford, IL). Then, 20 μg of total protein were applied to a precast NuPAGE 4–12% gel (Invitrogen). The separated proteins were transferred to a nitrocellulose membrane (Bio-Rad, Hercules, CA). After incubation of the membrane with primary and secondary antibodies, detection and analysis of protein were performed using the Odyssey System (Li-Cor, Lincoln, NE). Data were normalized based on rat β-actin protein expression levels.
Immunofluorescence staining.
PTCs were cultured in four-well chambers (Lab-Tek). After treatment with culture medium of MΦ (CMM) or ANG II-treated CMM (ANG II-CMM), cells were rinsed with PBS and then fixed for 20 min with 4% paraformaldehyde. After 4 min of incubation with 0.2% Triton X-100, the blocking agent Image-iT FX signal enhancer (Invitrogen) was added to the chambers. Cells were incubated with an antibody against STAT3 for 3 h. After being washed with PBS, cells were incubated with an Alexa fluor 594-labeled secondary antibody. ProLong Gold antifade reagent with 4′,6-diamidino-2-phenylindole (Invitrogen) was used as a nuclear stain and a mount reagent. Localization of the stained proteins was observed and photographed under a fluorescence microscope (Olympus BX51, Olympus Optical).
Transfection of suppressor of cytokine signaling 3.
Rat suppressor of cytokine signaling (SOCS)3 cDNA was cloned by standard RT-PCR from rat kidney RNA isolates. The sense primer (5′-CTCCGTGCGCCATGGTCA-3′) and antisense primer (5′-GAGCATCATACTGGTCCAGGAACTC-3′) were designed based on the rat SOCS3 complete coding sequence (National Center for Biotechnology Information no. AF075383.1) and used in PCR. The amplified SOCS3 cDNA by PCR was subcloned into the mammalian expression vector pcDNA3.1/CT-GFP-TOPO (Invitrogen) in both sense and antisense orientations. pcDNA3.1/CT-GFP-TOPO plasmids containing SOCS3 cDNA in sense and antisense orientations were transfected into PTCs with Lipofectamine reagents.
Statistical analysis.
Data are expressed as means ± SE. Data were analyzed using a Student's t-test or one-way ANOVA followed by a post hoc Bonferroni/Dunn multiple-comparison test. P values of <0.05 were considered statistically significant.
RESULTS
Regulation of extracellular IL-6, TNF-α, and IL-1β levels by ANG II in MΦ.
The effect of ANG II on changes in extracellular IL-6, TNF-α, and IL-1β levels, which can serve as pathogenic factors in RAS-associated kidney injury (29, 35, 50, 52), in MΦ was investigated using a cytokine ELISA. In culture media from untreated MΦ, IL-6 was present in greater amounts than TNF-α and IL-1β (IL-6: 0.52 ± 0.07 ng/ml, TNF-α: 0.14 ± 0.004 ng/ml, and IL-1β: 0.20 ± 0.03 ng/ml, n = 4; Fig. 1). IL-6 levels were elevated after 1 μM ANG II treatment for 48 h (0.97 ± 0.08 ng/ml). Although TNF-α and IL-1β levels in CMM were also increased by ANG II, IL-6 levels were most abundant among the tested cytokines.
Fig. 1.
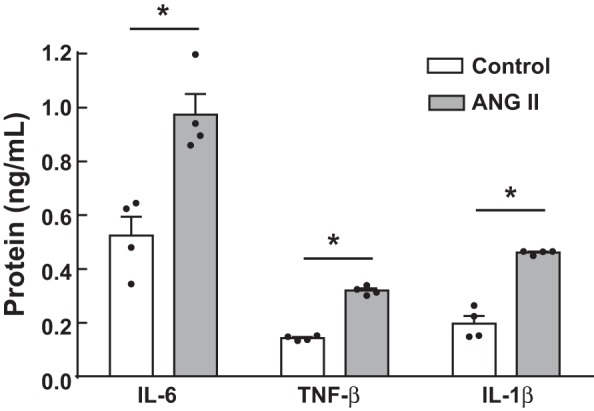
Regulation of extracellular IL-6, TNF-α, and IL-1β levels by ANG II in macrophages (MΦ). MΦ were treated with 1 μM ANG II for 48 h. After the treatment, culture media were collected, and cytokine levels were measured by ELISA. Data are expressed as means ± SE. *Significant difference compared with each control group (P < 0.05).
Augmentation of intracellular IL-6 levels in ANG II-treated MΦ.
Further analyses for IL-6 regulation by ANG II in MΦ were performed using several concentrations of ANG II at early (6 h) and late (48 h) time points. After 48 h of treatment, IL-6 mRNA levels and intracellular protein levels were augmented by 10−8 M (mRNA: 1.67 ± 0.11-fold and protein: 2.05 ± 0.09-fold, ratio to each control) and 10−6 M (mRNA: 2.18 ± 0.24-fold and protein: 3.43 ± 0.20-fold, ratio to each control) ANG II (n = 4; Fig. 2, A and B) compared with untreated (0 μM ANG II) MΦ. A lower concentration of ANG II (10−10 M) induced minor increases in IL-6 mRNA and intracellular protein upregulation, but they were not statistically significant. Whereas elevation of IL-6 mRNA was observed in ANG II-treated MΦ at 48 h, no change occurred after 6 h of treatment (Fig. 2C). Therefore, treatment of MΦ with ANG II for 48 h was used throughout the remainder of our study.
Fig. 2.
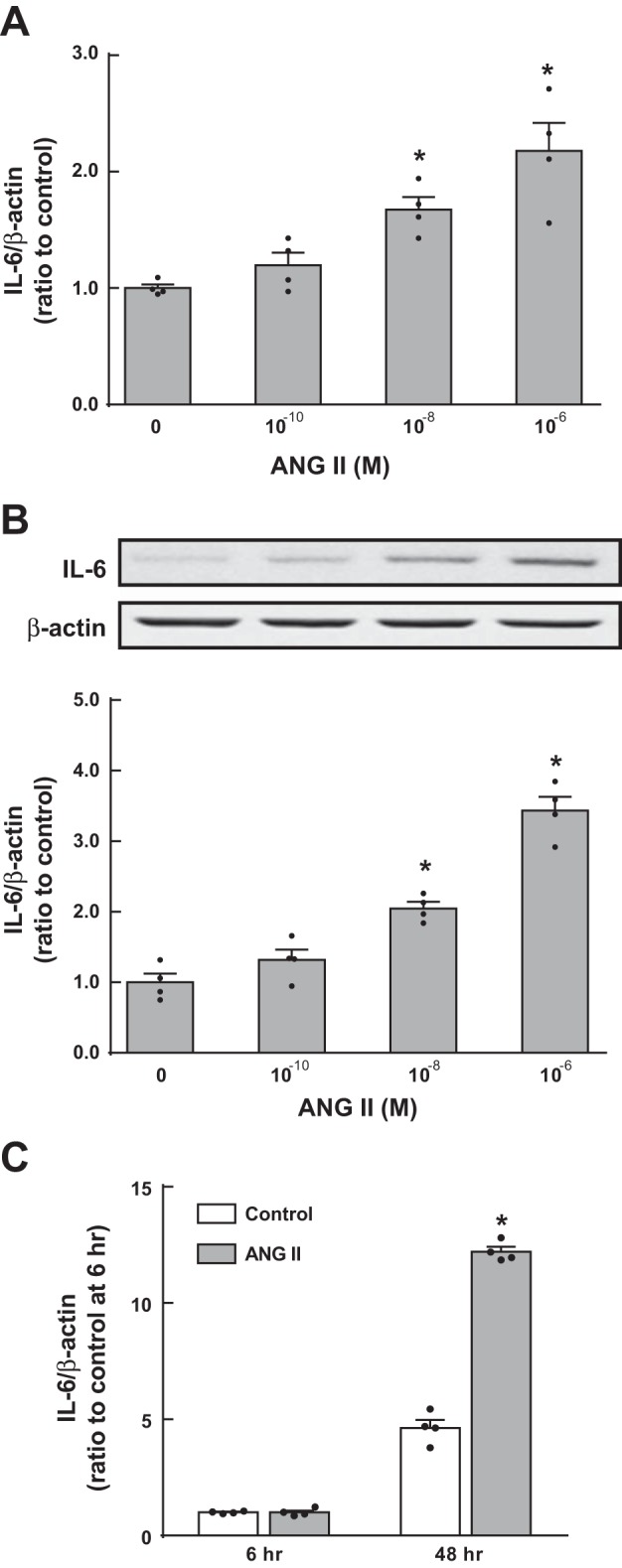
Augmentation of intracellular IL-6 levels in ANG II-treated MΦ. MΦ were treated with 0–1 μM ANG II for 6 or 48 h. After 48 h of treatment, IL-6 mRNA (A) and protein (B) levels were determined by quantitative real-time RT-PCR and Western blot analysis, respectively. The effect of ANG II on IL-6 mRNA expression was compared at 6 and 48 h (C). Data are expressed as means ± SE. *Significant difference compared with each control group consisting of untreated (0 μM ANG II) MΦ (P < 0.05).
Involvement of ANG II type 1 receptors in ANG II-induced IL-6 augmentation in MΦ.
First, we tested if ANG II alters ANG II type 1 receptor (AT1R) expression levels in MΦ. Since specificities of commercial anti-AT1R antibodies are controversial (9), AT1aR mRNA levels were measured by quantitative real-time RT-PCR and protein levels by Western blot analysis. No changes in AT1R levels after ANG II treatment were observed in these analyses (n = 4; Fig. 3, A and B). MΦ pretreated for 1 h with 1 μM olmesartan (Sankyo) (37), an AT1R blocker, showed significant attenuation of IL-6 augmentation after ANG II treatment (n = 4; Fig. 3C), suggesting that AT1Rs mediate ANG II-induced IL-6 upregulation in MΦ.
Fig. 3.

Involvement of ANG II type 1 receptors (AT1Rs) in ANG II-induced IL-6 augmentation in MΦ. MΦ were treated with 1 μM ANG II for 48 h. After the treatment, AT1R mRNA (A) and protein (B) levels were determined by quantitative real-time RT-PCR and Western blot analysis, respectively. Furthermore, MΦ were pretreated with olmesartan (C) to test if AT1Rs mediate ANG II-induced IL-6 upregulation. Data are expressed as means ± SE. *Significant difference compared with each control group (P < 0.05); #significant difference compared with the ANG II-treated group (P < 0.05).
Stimulating effects of CMM on AGT expression in PTCs.
In PTCs, AGT mRNA expression levels were 1.41 ± 0.07-fold increased by CMM (n = 4; Fig. 4A) compared with AGT mRNA levels in untreated PTCs (control). Augmentation of AGT mRNA expression was further enhanced when PTCs received ANG II-CMM (2.39 ± 0.05-fold, ratio to control). In contrast, direct treatment of PTCs with 100 nM ANG II did not alter AGT mRNA (0.95 ± 0.13, ratio to control, n = 6) or protein (0.96 ± 0.11, ratio to control, n = 6) levels under the experimental conditions. An IL-6-neutralizing antibody (R&D Systems) added to collected ANG II-CMM significantly suppressed the elevation of AGT mRNA expression, indicating that IL-6 mediates AGT augmentation in PTCs treated with ANG II-CMM. However, AGT expression in PTCs treated with the anti-IL-6 antibody remained slightly higher than control levels (Fig. 4A) even when a higher concentration of the antibody was used. Regulation of AGT protein levels corresponded closely to changes in mRNA levels (Fig. 4B).
Fig. 4.
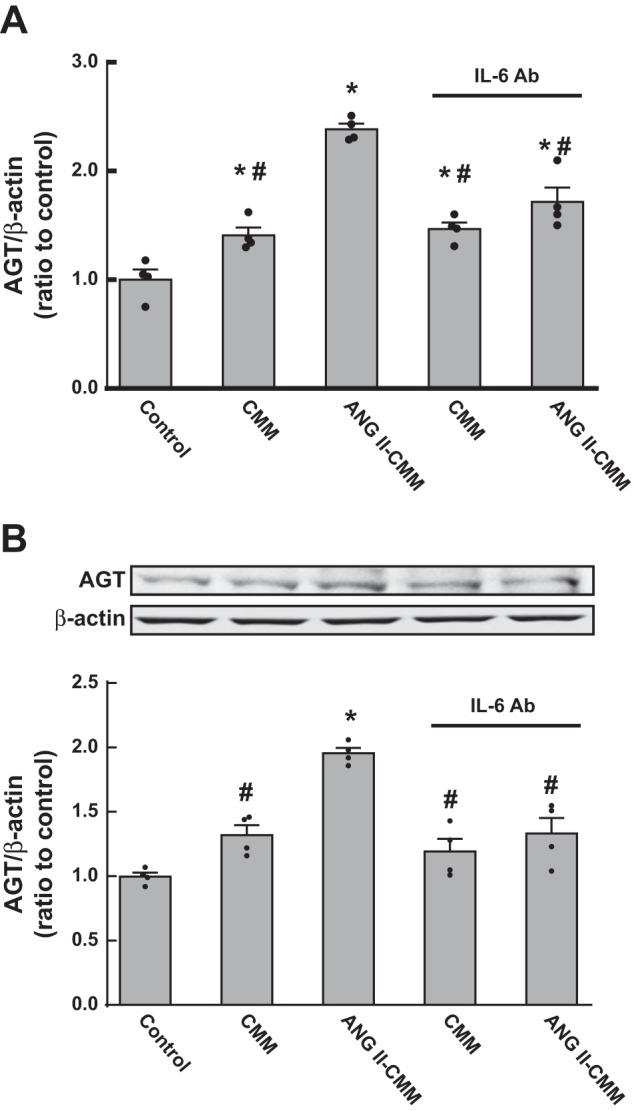
Stimulating effects of conditioned medium from activated MΦ (CMM) on angiotensinogen (AGT) expression in proximal tubular cells (PTCs). PTCs were treated with CMM or ANG II-treated CMM (ANG II-CMM) for 24 h. Thereafter, AGT mRNA (A) and protein (B) levels were evaluated by quantitative real-time RT-PCR and Western blot analysis, respectively. A neutralizing IL-6 antibody (Ab) was used to test if IL-6 mediates CMM-induced AGT augmentation in PTCs. Data are expressed as means ± SE. *Significant difference compared with each control group consisting of untreated PTCs (P < 0.05); #significant difference compared with the ANG II-CMM-treated group (P < 0.05).
Activation of STAT3 by ANG II-CMM in PTCs.
Since IL-6 has been characterized as a strong stimulator of the JAK-STAT pathway, STAT3 activity was evaluated in PTCs. CMM slightly increased phosphorylation levels of STAT3 in PTC (1.43 ± 0.09-fold, ratio to control, n = 4; Fig. 5A). ANG II-CMM treatment induced further phosphorylation in PTCs (2.76 ± 0.23-fold), which was prevented by the IL-6-neutralizing antibody. Immunocytochemistry demonstrated translocalization of STAT3 into nuclei after incubation with ANG II-CMM (Fig. 5B).
Fig. 5.

Activation of STAT3 by ANG II-CMM in PTCs. PTCs were treated with CMM or ANG II-CMM for 24 h. STAT3 phosphorylation levels (A) and translocation of STAT3 to the nucleus (B) were evaluated by Western blot analysis and immunocytochemistry, respectively. pSTAT3, phosphorylated STAT3; tSTAT3, total STAT3. Data are expressed as means ± SE. *Significant difference compared with each control consisting of untreated PTCs (P < 0.05); #significant difference compared with the ANG II-CMM-treated group (P < 0.05).
Implications of the activated JAK-STAT pathway in AGT augmentation.
STAT3 phosphorylation and AGT expression were facilitated by CMM and ANG II-CMM (n = 4 in each experiment; Fig. 6, A and B) as shown above. Pretreatment of PTCs with 1 μM AG-490, a JAK2 inhibitor, diminished STAT3 phosphorylation induced by both CMM and ANG II-CMM (Fig. 6A) to control levels (or slightly lower than the control). AGT augmentation by CMM and ANG II-CMM was also attenuated to the control levels (Fig. 6B), although suppression of AGT levels by an IL-6-neutralizing antibody was still greater than the control (Fig. 4). Overexpression of SOCS3-GFP (Fig. 6C), an endogenous suppressor of the JAK-STAT pathway, prevented AGT upregulation in PTCs treated with CMM and ANG II-CMM, reinforcing JAK-STAT signaling as a critical mediator in AGT expression.
Fig. 6.

Implications of the activated JAK-STAT pathway in AGT augmentation. AG-490 (A and B) and suppressor of cytokine signaling (SOCS)3 overexpression (C) were used to investigate the involvement of the activated JAK-STAT pathway in CMM-induced AGT augmentation in PTCs. Data are expressed as means ± SE. *Significant difference compared with each control group consisting of untreated PTCs (P < 0.05); #significant difference compared with the ANG II-CMM-treated group (P < 0.05).
DISCUSSION
Since intrarenal ANG II is elevated in many forms of hypertension, the renal RAS is acknowledged as a key target for clinical and biochemical studies (27). Importantly, animal studies have demonstrated that elevated ANG II stimulates renal cortical AGT production, amplifying intrarenal RAS activity during the development of ANG II-dependent hypertension (7, 16). Recent studies have reported that chronic ANG II infusion in mice concomitantly increases AGT and IL-6 levels in renal cortical tubules (28, 35) and that IL-6 is required to augment AGT expression in cultured PTCs (37). The elevation of IL-6 levels in ANG II-dependent hypertension and stimulating effects of IL-6 on AGT expression have been demonstrated in other tissues (6, 13, 33, 34), suggesting the importance of elevated IL-6 in local RAS activation and the development of hypertension. This view is further supported by previous studies that revealed gene deletion of IL-6 in mice abrogates ANG II-induced hypertension (2, 20). During the development of hypertension mediated by elevated ANG II, infiltration of MΦ, which are a primary source of IL-6, and T cells is enhanced in the kidney (32, 53) as well as the vascular endothelium (6). These MΦ and T cells have been shown to play crucial roles in accelerating the progression of hypertension and associated tissue damages including kidney injury (25, 42, 44, 45, 49).
Previously, conflicting studies describing the role that ANG II plays in IL-6 expression in MΦ have been presented. In one study (8), it was shown that 1 μM ANG II enhances IL-6 production at 6 h in mouse MΦ, whereas other work (22) using the same dose of ANG II demonstrated an inhibitory effect of lipopolysaccharide-induced IL-6 augmentation in mouse MΦ. Thus, the activating effect of ANG II on IL-6 expression in MΦ was controversial. In the present study, ANG II increased IL-6 mRNA and protein levels in cultured rat MΦ as well as levels of the cytokine secreted in CMM. While TNF-α and IL-1β concentrations were increased in ANG II-treated CMM, IL-6 levels were significantly elevated, suggesting that IL-6 is one of the core proinflammatory cytokines in AGT regulation produced by ANG II-activated MΦ. In the present study, basal levels of IL-6 in the control group were elevated at 48 h compared with 6 h. The augmentation could be due to a number of things, including autocrine and paracrine signaling induced by secreted factors from these MΦ during the 48-h incubation. Although there is no doubt that TNF-α and IL-1β contribute to the development of kidney injury, including renal inflammation, these cytokines have been reported to suppress AGT in PTCs (38, 40). Accordingly, our present study highlights the significant impact of IL-6 on AGT upregulation. In these experiments, a high dose of ANG II (>10 nM) and a long-term treatment duration were required to induce significant IL-6 augmentation in MΦ, findings in agreement with a previous study (8). This effect might be due to the use of only a single treatment of cells at the beginning of the experiment. Furthermore, the high dose of ANG II did not change AT1R levels in MΦ, which also corresponds with a previous finding that only lower doses of ANG II increase AT1R levels in MΦ. Experiments using olmesartan in the present study confirmed that AT1Rs mediate the ANG II-induced IL-6 upregulation observed in MΦ. This may explain how AT1R blockage inhibits intrarenal AGT elevation in ANG II-infused rats (18) and spontaneously hypertensive rats (17), whereas direct treatment of cultured PTCs with ANG II does not alter AGT expression levels, as shown in a previous study (37) and the present study.
CMM collected from MΦ slightly but significantly increased AGT mRNA and protein expression in PTCs. CMM collected from ANG II-treated MΦ markedly enhanced the AGT augmentation. Importantly, the enhanced AGT upregulation was attenuated by an IL-6-neutralizing antibody, indicating that the elevated IL-6 in CMM mediates stimulation of AGT expression in PTCs. The AGT augmentation by CMM and ANG II-treated CMM was not lowered to the control levels even after higher concentrations of the IL-6 antibody were used. This may be due to incomplete blockade of the receptor by the antibody, in which case the decrease in AGT after its addition would not fully represent the total effect of IL-6. It is also possible that besides IL-6, MΦ may produce another factor(s) that stimulates AGT expression in PTCs. For example, previous studies have demonstrated ROS as well as interferon-γ as factors that also contribute to intrarenal AGT augmentation (26, 39). Therefore, MΦ activated by ANG II might stimulate AGT expression in PTCs via complex mechanisms in which elevated IL-6 is the primary contributor.
The contribution of activated JAK-STAT signaling to the development of kidney injury has been established using JAK inhibitors or SOCS overexpression (1, 31, 47). Moreover, IL-6 augments AGT expression via activation of the JAK-STAT pathway in hepatocytes and PTCs (33, 37). In the present study, STAT3 phosphorylation was induced by CMM, and further activation of STAT3 was observed in PTCs that received CMM obtained from ANG II-treated MΦ. An IL-6-neutralizing antibody and a JAK2 inhibitor (AG-490) prevented the phosphorylation of STAT3. CMM-induced AGT augmentation in PTCs was also attenuated by AG-490. Interestingly, AG-490 completely diminished AGT augmentation to control levels. Additionally, SOCSs have been identified as endogenous suppressors of the JAK-STAT pathway (19). In the present study, overexpression of SOCS3 in PTCs also resulted in partial suppression of AGT augmentation stimulated by CMM obtained from ANG II-treated MΦ. Taken together, these results indicate that activation of the JAK-STAT pathway mediates CMM-induced AGT augmentation.
In conclusion, the findings in the present study suggest sequential mechanisms underlying proximal tubular AGT upregulation that occur in ANG II-dependent hypertension. In this system, elevated IL-6 production in activated MΦ stimulates AGT expression in PTCs via the JAK-STAT pathway. This may explain why suppression of immune cell infiltration (5) through the administration of mycophenolate mofetil, an immunosuppressive drug, reduces intrarenal ANG II and the consequent increases in blood pressure (4). The findings provide a mechanistic rationale for targeting the MΦ-IL-6-AGT axis to treat hypertension and associated kidney injury.
GRANTS
This study was supported by National Institutes of Health (NIH) IDeA Program (COBRE) Grant P30-GM-103337, NIH Grant R01-DK-107694-01, and American Heart Association Greater Southeast Affiliate Summer 2013 Health Sciences Scholarship 14PRE19130002.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: R.O., H.P., K.M., and R.S. conception and design of research; R.O., H.P., K.M., and R.S. performed experiments; R.O., H.P., K.M., and R.S. analyzed data; R.O., H.P., K.M., and R.S. interpreted results of experiments; R.O., H.P., K.M., and R.S. prepared figures; R.O., H.P., K.M., and R.S. drafted manuscript; R.O., H.P., K.M., and R.S. edited and revised manuscript; R.O., H.P., K.M., and R.S. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors thank Dr. Julie R. Ingelfinger (Massachusetts General Hospital And Harvard Medical School) for providing established PTCs. The authors acknowledge the valuable advice of Dr. L. Gabriel Navar (Tulane University) and excellent technical assistance of Akemi Katsurada (Tulane University).
REFERENCES
- 1.Banes AK, Shaw S, Jenkins J, Redd H, Amiri F, Pollock DM, Marrero MB. Angiotensin II blockade prevents hyperglycemia-induced activation of JAK and STAT proteins in diabetic rat kidney glomeruli. Am J Physiol Renal Physiol 286: F653–F659, 2004. [DOI] [PubMed] [Google Scholar]
- 2.Brands MW, Banes-Berceli AK, Inscho EW, Al-Azawi H, Allen AJ, Labazi H. Interleukin 6 knockout prevents angiotensin II hypertension. Role of renal vasoconstriction and Janus kinase 2/signal transducer and activator of transcription 3 activation. Hypertension 56: 879–884, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brasier AR, Jamaluddin M, Han Y, Patterson C, Runge MS. Angiotensin II induces gene transcription through cell-type-dependent effects on the nuclear factor-κB (NF-κB) transcription factor. Mol Cell Biochem 212: 155–169, 2000. [PubMed] [Google Scholar]
- 4.Bravo Y, Quiroz Y, Ferrebuz A, Vaziri ND, Rodriguez-Iturbe B. Mycophenolate mofetil administration reduces renal inflammation, oxidative stress, and arterial pressure in rats with lead-induced hypertension. Am J Physiol Renal Physiol 293: F616–F623, 2007. [DOI] [PubMed] [Google Scholar]
- 5.Crowley SD, Frey CW, Gould SK, Griffiths R, Ruiz P, Burchette JL, Howell DN, Makhanova N, Yan M, Kim HS, Tharaux PL, Coffman TM. Stimulation of lymphocyte responses by angiotensin II promotes kidney injury in hypertension. Am J Physiol Renal Physiol 295: F515–F524, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gomolak JR, Didion SP. Angiotensin II-induced endothelial dysfunction is temporally linked with increases in interleukin-6 and vascular macrophage accumulation. Front Physiol 5: 396, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gonzalez-Villalobos RA, Seth DM, Satou R, Horton H, Ohashi N, Miyata K, Katsurada A, Tran DV, Kobori H, Navar LG. Intrarenal angiotensin II and angiotensinogen augmentation in chronic angiotensin II-infused mice. Am J Physiol Renal Physiol 295: F772–F779, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guo F, Chen XL, Wang F, Liang X, Sun YX, Wang YJ. Role of angiotensin II type 1 receptor in angiotensin II-induced cytokine production in macrophages. J Interf Cytok Res 31: 351–361, 2011. [DOI] [PubMed] [Google Scholar]
- 9.Herrera M, Sparks MA, Alfonso-Pecchio AR, Harrison-Bernard LM, Coffman TM. Lack of specificity of commercial antibodies leads to misidentification of angiotensin type 1 receptor protein. Hypertension 61: 253–258, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ingelfinger JR, Jung F, Diamant D, Haveran L, Lee E, Brem A, Tang SS. Rat proximal tubule cell line transformed with origin-defective SV40 DNA: autocrine ANG II feedback. Am J Physiol Renal Physiol 276: F218–F227, 1999. [DOI] [PubMed] [Google Scholar]
- 11.Ingelfinger JR, Zuo WM, Fon EA, Ellison KE, Dzau VJ. In situ hybridization evidence for angiotensinogen messenger RNA in the rat proximal tubule. An hypothesis for the intrarenal renin angiotensin system. J Clin Invest 85: 417–423, 1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Iwashita M, Sakoda H, Kushiyama A, Fujishiro M, Ohno H, Nakatsu Y, Fukushima T, Kumamoto S, Tsuchiya Y, Kikuchi T, Kurihara H, Akazawa H, Komuro I, Kamata H, Nishimura F, Asano T. Valsartan, independently of AT1 receptor or PPARγ, suppresses LPS-induced macrophage activation and improves insulin resistance in cocultured adipocytes. Am J Physiol Endocrinol Metab 302: E286–E296, 2012. [DOI] [PubMed] [Google Scholar]
- 13.Jain S, Li Y, Patil S, Kumar A. HNF-1α plays an important role in IL-6-induced expression of the human angiotensinogen gene. Am J Physiol Cell Physiol 293: C401–C410, 2007. [DOI] [PubMed] [Google Scholar]
- 14.Jamaluddin M, Meng T, Sun J, Boldogh I, Han Y, Brasier AR. Angiotensin II induces nuclear factor (NF)-κB1 isoforms to bind the angiotensinogen gene acute-phase response element: a stimulus-specific pathway for NF-κB activation. Mol Endocrinol 14: 99–113, 2000. [DOI] [PubMed] [Google Scholar]
- 15.Kleemann R, Zadelaar S, Kooistra T. Cytokines and atherosclerosis: a comprehensive review of studies in mice. Cardiovasc Res 79: 360–376, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kobori H, Harrison-Bernard LM, Navar LG. Expression of angiotensinogen mRNA and protein in angiotensin II-dependent hypertension. JASN 12: 431–439, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kobori H, Ozawa Y, Suzaki Y, Nishiyama A. Enhanced intrarenal angiotensinogen contributes to early renal injury in spontaneously hypertensive rats. JASN 16: 2073–2080, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kobori H, Prieto-Carrasquero MC, Ozawa Y, Navar LG. AT1 receptor mediated augmentation of intrarenal angiotensinogen in angiotensin II-dependent hypertension. Hypertension 43: 1126–1132, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Larsen L, Ropke C. Suppressors of cytokine signalling: SOCS. APMIS 110: 833–844, 2002. [DOI] [PubMed] [Google Scholar]
- 20.Lee DL, Sturgis LC, Labazi H, Osborne JB Jr, Fleming C, Pollock JS, Manhiani M, Imig JD, Brands MW. Angiotensin II hypertension is attenuated in interleukin-6 knockout mice. Am J Physiol Heart Circ Physiol 290: H935–H940, 2006. [DOI] [PubMed] [Google Scholar]
- 21.Li J, Brasier AR. Angiotensinogen gene activation by angiotensin II is mediated by the rel A (nuclear factor-κB p65) transcription factor: one mechanism for the renin angiotensin system positive feedback loop in hepatocytes. Mol Endocrinol 10: 252–264, 1996. [DOI] [PubMed] [Google Scholar]
- 22.Lo CJ, Lo EJ. Angiotensin II inhibits interleukin-6 mRNA expression of LPS-stimulated macrophages through down-regulating calcium signaling. J Surg Res 181: 287–292, 2013. [DOI] [PubMed] [Google Scholar]
- 23.Lu H, Boustany-Kari CM, Daugherty A, Cassis LA. Angiotensin II increases adipose angiotensinogen expression. Am J Physiol Endocrinol Metab 292: E1280–E1287, 2007. [DOI] [PubMed] [Google Scholar]
- 24.Manhiani MM, Quigley JE, Socha MJ, Motamed K, Imig JD. IL6 suppression provides renal protection independent of blood pressure in a murine model of salt-sensitive hypertension. Kidney Blood Press Res 30: 195–202, 2007. [DOI] [PubMed] [Google Scholar]
- 25.McMaster WG, Kirabo A, Madhur MS, Harrison DG. Inflammation, immunity, and hypertensive end-organ damage. Circ Res 116: 1022–1033, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miyata K, Ohashi N, Suzaki Y, Katsurada A, Kobori H. Sequential activation of the reactive oxygen species/angiotensinogen/renin-angiotensin system axis in renal injury of type 2 diabetic rats. Clin Exp Pharmacol Physiol 35: 922–927, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Navar LG, Harrison-Bernard LM, Nishiyama A, Kobori H. Regulation of intrarenal angiotensin II in hypertension. Hypertension 39: 316–322, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Navar LG, Prieto MC, Satou R, Kobori H. Intrarenal angiotensin II and its contribution to the genesis of chronic hypertension. Curr Opin Pharmacol 11: 180–186, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Navarro JF, Milena FJ, Mora C, Leon C, Garcia J. Renal pro-inflammatory cytokine gene expression in diabetic nephropathy: effect of angiotensin-converting enzyme inhibition and pentoxifylline administration. Am J Nephrol 26: 562–570, 2006. [DOI] [PubMed] [Google Scholar]
- 30.Ohtani R, Yayama K, Takano M, Itoh N, Okamoto H. Stimulation of angiotensinogen production in primary cultures of rat hepatocytes by glucocorticoid, cyclic adenosine 3′,5′-monophosphate, and interleukin-6. Endocrinology 130: 1331–1338, 1992. [DOI] [PubMed] [Google Scholar]
- 31.Ortiz-Munoz G, Lopez-Parra V, Lopez-Franco O, Fernandez-Vizarra P, Mallavia B, Flores C, Sanz A, Blanco J, Mezzano S, Ortiz A, Egido J, Gomez-Guerrero C. Suppressors of cytokine signaling abrogate diabetic nephropathy. JASN 21: 763–772, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ozawa Y, Kobori H, Suzaki Y, Navar LG. Sustained renal interstitial macrophage infiltration following chronic angiotensin II infusions. Am J Physiol Renal Physiol 292: F330–F339, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ray S, Boldogh I, Brasier AR. STAT3 NH2-terminal acetylation is activated by the hepatic acute-phase response and required for IL-6 induction of angiotensinogen. Gastroenterology 129: 1616–1632, 2005. [DOI] [PubMed] [Google Scholar]
- 34.Recinos A 3rd, LeJeune WS, Sun H, Lee CY, Tieu BC, Lu M, Hou T, Boldogh I, Tilton RG, Brasier AR. Angiotensin II induces IL-6 expression and the Jak-STAT3 pathway in aortic adventitia of LDL receptor-deficient mice. Atherosclerosis 194: 125–133, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ruiz-Ortega M, Ruperez M, Lorenzo O, Esteban V, Blanco J, Mezzano S, Egido J. Angiotensin II regulates the synthesis of proinflammatory cytokines and chemokines in the kidney. Kidney Int Suppl 2002: 12–22, 2002. [DOI] [PubMed] [Google Scholar]
- 36.Sachetelli S, Liu Q, Zhang SL, Liu F, Hsieh TJ, Brezniceanu ML, Guo DF, Filep JG, Ingelfinger JR, Sigmund CD, Hamet P, Chan JS. RAS blockade decreases blood pressure and proteinuria in transgenic mice overexpressing rat angiotensinogen gene in the kidney. Kidney Int 69: 1016–1023, 2006. [DOI] [PubMed] [Google Scholar]
- 37.Satou R, Gonzalez-Villalobos RA, Miyata K, Ohashi N, Katsurada A, Navar LG, Kobori H. Costimulation with angiotensin II and interleukin 6 augments angiotensinogen expression in cultured human renal proximal tubular cells. Am J Physiol Renal Physiol 295: F283–F289, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Satou R, Gonzalez-Villalobos RA, Miyata K, Ohashi N, Urushihara M, Acres OW, Navar LG, Kobori H. IL-6 augments angiotensinogen in primary cultured renal proximal tubular cells. Mol Cell Endocrinol 311: 24–31, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Satou R, Miyata K, Gonzalez-Villalobos RA, Ingelfinger JR, Navar LG, Kobori H. Interferon-γ biphasically regulates angiotensinogen expression via a JAK-STAT pathway and suppressor of cytokine signaling 1 (SOCS1) in renal proximal tubular cells. FASEB J 26: 1821–1830, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Satou R, Miyata K, Katsurada A, Navar LG, Kobori H. Tumor necrosis factor-α suppresses angiotensinogen expression through formation of a p50/p50 homodimer in human renal proximal tubular cells. Am J Physiol Cell Physiol 299: C750–C759, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Terada Y, Tomita K, Nonoguchi H, Marumo F. PCR localization of angiotensin II receptor and angiotensinogen mRNAs in rat kidney. Kidney Int 43: 1251–1259, 1993. [DOI] [PubMed] [Google Scholar]
- 42.Thang LV, Demel SL, Crawford R, Kaminski NE, Swain GM, Van Rooijen N, Galligan JJ. Macrophage depletion lowers blood pressure and restores sympathetic nerve α2-adrenergic receptor function in mesenteric arteries of DOCA-salt hypertensive rats. Am J Physiol Heart Circ Physiol 309: H1186–H1197, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.van Kats JP, Danser AH, van Meegen JR, Sassen LM, Verdouw PD, Schalekamp MA. Angiotensin production by the heart: a quantitative study in pigs with the use of radiolabeled angiotensin infusions. Circulation 98: 73–81, 1998. [DOI] [PubMed] [Google Scholar]
- 44.Wallace K, Novotny S, Heath J, Moseley J, Martin JN Jr, Owens MY, LaMarca B. Hypertension in response to CD4+ T cells from reduced uterine perfusion pregnant rats is associated with activation of the endothelin-1 system. Am J Physiol Regul Integr Comp Physiol 303: R144–R149, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xiao L, Kirabo A, Wu J, Saleh MA, Zhu L, Wang F, Takahashi T, Loperena R, Foss JD, Mernaugh RL, Chen W, Roberts J 2nd, Osborn JW, Itani HA, Harrison DG. Renal denervation prevents immune cell activation and renal inflammation in angiotensin II-induced hypertension. Circ Res 117: 547–557, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yamasaki N, Kitaoka H, Matsumura Y, Furuno T, Nishinaga M, Doi Y. Heart failure in the elderly. Intern Med 42: 383–388, 2003. [DOI] [PubMed] [Google Scholar]
- 47.Yang N, Luo M, Li R, Huang Y, Zhang R, Wu Q, Wang F, Li Y, Yu X. Blockage of JAK/STAT signalling attenuates renal ischaemia-reperfusion injury in rat. Nephrol Dial Transplant 23: 91–100, 2008. [DOI] [PubMed] [Google Scholar]
- 48.Ying J, Stuart D, Hillas E, Gociman BR, Ramkumar N, Lalouel JM, Kohan DE. Overexpression of mouse angiotensinogen in renal proximal tubule causes salt-sensitive hypertension in mice. Am J Hypertens 25: 684–689, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang J, Crowley SD. Role of T lymphocytes in hypertension. Curr Opin Pharmacol 21: 14–19, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang J, Patel MB, Griffiths R, Mao A, Song YS, Karlovich NS, Sparks MA, Jin H, Wu M, Lin EE, Crowley SD. Tumor necrosis factor-α produced in the kidney contributes to angiotensin II-dependent hypertension. Hypertension 64: 1275–1281, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang L, Du J, Hu Z, Han G, Delafontaine P, Garcia G, Mitch WE. IL-6 and serum amyloid A synergy mediates angiotensin II-induced muscle wasting. JASN 20: 604–612, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang W, Wang W, Yu H, Zhang Y, Dai Y, Ning C, Tao L, Sun H, Kellems RE, Blackburn MR, Xia Y. Interleukin 6 underlies angiotensin II-induced hypertension and chronic renal damage. Hypertension 59: 136–144, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zimmerman MA, Baban B, Tipton AJ, O'Connor PM, Sullivan JC. Chronic ANG II infusion induces sex-specific increases in renal T cells in Sprague-Dawley rats. Am J Physiol Renal Physiol 308: F706–F712, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]