

Abstract
Immune checkpoint inhibitors (CPIs), monoclonal antibodies that target inhibitory receptors expressed on T cells, represent an emerging class of immunotherapy used in treating solid organ and hematologic malignancies. We describe the clinical and histologic features of 13 patients with CPI-induced acute kidney injury (AKI) who underwent kidney biopsy. Median time from initiation of a CPI to AKI was 91 (range, 21 to 245) days. Pyuria was present in 8 patients, and the median urine protein to creatinine ratio was 0.48 (range, 0.12 to 0.98) g/g. An extra-renal immune-related adverse event occurred prior to the onset of AKI in 7 patients. Median peak serum creatinine was 4.5 (interquartile range, 3.6-7.3) mg/dl with 4 patients requiring hemodialysis. The prevalent pathologic lesion was acute tubulointerstitial nephritis in 12 patients, with 3 having granulomatous features, and one thrombotic microangiopathy. Among the 12 patients with acute tubulointerstitial nephritis, 10 received treatment with glucocorticoids, resulting in complete or partial improvement in renal function in 2 and 7 patients, respectively. However, the two patients with acute tubulointerstitial nephritis not given glucocorticoids had no improvement in renal function. Thus, CPI-induced AKI is a new entity that presents with clinical and histologic features similar to other causes of drug-induced acute tubulointerstitial nephritis, though with a longer latency period. Glucocorticoids appear to be a potentially effective treatment strategy. Hence, AKI due to CPIs may be caused by a unique mechanism of action linked to reprogramming of the immune system, leading to loss of tolerance.
Keywords: acute kidney injury, ipilimumab, nivolumab, pembrolizumab
Introduction
The recent emergence of immune checkpoint inhibitors (CPIs), a novel type of immunotherapy, represents a substantial advance in oncology. CPIs are monoclonal antibodies that target inhibitory receptors expressed on T cells, other immune cells, and tumor cells. These receptors include cytotoxic T lymphocyte-associated antigen 4 (CTLA-4), programmed death 1 protein (PD-1), and programmed death-ligand 1 (PD-L1) (1). CTLA-4 prevents T cell activation by outcompeting CD28 for its ligand, B7, thereby inhibiting T cell co-stimulation (2), whereas PD-1 downregulates effector T cell function by engaging its two ligands, PD-L1 and PD-L2 (3). Thus, by inhibiting CTLA-4 and PD-1/PD-L1, CPIs enhance tumor-directed immune responses and have been leveraged as novel therapeutic agents for solid and hematologic malignancies (1).
The efficacy of one CTLA-4 antagonist, ipilimumab, and two PD-1 antagonists, nivolumab and pembrolizumab, has been well established in the treatment of advanced melanoma (4-6) and non-small cell lung cancer (7, 8), and more recent data support their efficacy in patients with renal-cell carcinoma (9), Hodgkin's lymphoma (10), and many other malignancies. CPIs are known to cause a unique spectrum of side effects termed immune-related adverse events (IRAEs). The most common IRAEs include rash, colitis, hepatitis, and hypophysitis (1). Sparse case reports have described acute kidney injury (AKI) related to CPIs (11-13). Here, we present the largest series to date of CPI-induced AKI, with a focus on clinical features, pathology, and response to treatment.
Results
Baseline Characteristics
We identified 13 patients from 7 academic medical centers across the United States with CPI-induced AKI who underwent a kidney biopsy. Baseline characteristics are summarized in Table 1. All patients were Caucasian except for patient 10, who was Hispanic. Chronic kidney disease, defined as an eGFR<60 ml/min/1.73m2, was present in 4 patients. Dipstick proteinuria was negative in 7 patients, trace in one patient, and not available in 5 patients.
Table 1. Baseline Characteristics.
Estimated glomerular filtration rate (eGFR, ml/min) was calculated using the CKD-EPI equation (32).
| Pt | Age/ Gender | Malignancy | SCr / eGFR | Proteinuria (dipstick) | Comorbidities | Checkpoint Inhibitor Regimen | Cumulative Dose |
|---|---|---|---|---|---|---|---|
| 1 | 70M | Melanoma | 0.9 / 86 | NA | Asthma, osteoarthritis, basal and squamous cell carcinoma | Ipi 3mg/kg × 1 | Ipi 3mg/kg |
| 2 | 64M | Melanoma | 1.3 / 58 | Neg | CKD, hypertension, and BPH | Ipi 3mg/kg + Nivo 1 mg/kg q 6 weeks × 2 | Ipi 6mg/kg Nivo 2 mg/kg |
| 3 | 74M | Melanoma | 1.1 / 66 | Neg | Hypertension | Ipi 3mg/kg + Nivo 0.3mg/kg q 3 weeks × 2, followed by Ipi 3mg/kg × 1 (7.5 weeks later) | Ipi 9mg/kg Nivo 0.6mg/kg |
| 4 | 62F | Melanoma | 0.7 / 92 | Trace | CHF and atrial fibrillation | Ipi 10mg/kg q 3 weeks × 3 | Ipi 30mg/kg |
| 5 | 71F | NSCLC | 0.6 / 92 | NA | Hypothyroidism and history of pulmonary embolism | Ipi 3mg/kg q 12 weeks × 3 + Nivo 3mg/kg q 2 weeks × 14 | Ipi 9mg/kg Nivo 42mg/kg |
| 6 | 64M | Pancreatic Cancer | 0.8 / 78 | Neg | Adrenal insufficiency and hypothyroidism | Ipi 10mg/kg q 3 weeks × 4, followed by Ipi 10mg/kg × 1 (12 weeks later) | Ipi 50mg/kg |
| 7 | 71M | Melanoma | 1.0 / 57 | Neg | CKD and hypothyroidism | Nivo 0.1mg/kg q 2 weeks × 4, followed by Nivo 1mg/kg q 2 weeks × 12 | Nivo 12.4mg/kg |
| 8 | 58M | Melanoma | 0.5 / 107 | NA | Hypertension | Ipi 3mg/kg q 3 weeks × 8 | Ipi 24mg/kg |
| 9 | 75M | Melanoma | 0.9 / 63 | NA | BPH | Ipi 3mg/kg + Nivo 1mg/kg q 3 weeks × 2 | Ipi 6mg/kg Nivo 2mg/Kg |
| 10 | 32F | Hodgkin's Lymphoma | 1.0 / 74 | Neg | BMT for Hodgkins Lymphoma | Ipi 10mg/kg q 3 weeks × 4 | Ipi 40mg/kg |
| 11 | 73F | Melanoma | 1.0 / 56 | Neg | Hypertension | Ipi 3mg/kg q 3 weeks × 3 | Ipi 9mg/kg |
| 12 | 66M | Bladder Carcinoma | 1.5 / 48 | 1+ | CKD, hypertension, and hay fever | Pembro 2 mg/kg × 1 | Pembro 2mg/kg |
| 13 | 41F | Melanoma | 0.9/ 80 | Neg | Asthma and migraine headaches | Pembro 2mg/kg q 3 weeks × 10 | Pembro 20mg/kg |
Abbreviations: BMT, bone marrow transplant; BPH, Benign Prostatic Hyperplasia; CHF, Congestive Heart Failure; Ipi, ipilimumab; NA, not available; Nivo, nivolumab; NSCLC, non-small-cell lung carcinoma; Pembro, pembrolizumab; SCr, serum creatinine (mg/dl).
The most common malignancy in our cohort was melanoma (9 out of 13 patients). Other malignancies included non-small cell lung cancer (n=1), pancreatic cancer (n=1), Hodgkin's Lymphoma (n=1), and bladder carcinoma (n=1). The treatment regimen varied widely across the cohort. Patients received ipilimumab alone (n=6), ipilimumab in combination with nivolumab (n=4), nivolumab alone (n=1), and pembrolizumab alone (n=2).
Concomitant medications
Concomitant medications are shown in Supplemental Table 1. In three cases, a medication associated with AIN was introduced within four weeks prior to AKI onset: patient 1 began pantoprazole 24 days prior to AKI; patient 8 began ibuprofen 19 days prior to AKI; and patient 12 began taking a three-day course of ciprofloxacin 20 days prior to AKI.
Clinical Features of CPI-induced AKI
The clinical features of AKI following treatment with a CPI are summarized in Table 2. The interval from initiation of a CPI to AKI ranged from 21 to 245 days (median [IQR], 91 [60 to 183] days), and the interval from the last CPI dose to AKI ranged from 7 to 63 days (median [IQR], 21 [18-49] days). Pyuria, defined as > 5 white blood cells per high power field (hpf), was present in 8 out of 13 patients. Hematuria, defined as > 2 red blood cells per hpf, was present in 3 out of 13 patients. Eosinophilia occurred in one patient. Complement levels (C3 and C4) were normal in all 8 patients who were tested.
Table 2. Clinical Features of CPI-induced AKI.
| Pt | Urine Sediment¶ | Proteinuria (Dipstick / UPCR) | Day of AKI† | #Days since last dose of CPI | Eos | HTN‡ | Oliguria* | Kidney Size (cm) | Peak SCr (mg/dl) | Requirement for RRT | IRAEs |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 5-10 WBCs** 2 RBCs |
1+ / 0.6 | 54 | 54 | No | No | No | R 12.8 L 13.8 |
6.2 | No | Hypophysitis |
| 2 | 2-3 WBCs 3-5 RBCs |
Trace / NA | 91 | 49 | No | No | No | R 12.2 L 13.2 |
4.1 | No | Thyroiditis; Ileitis |
| 3 | 5-10 WBCs 0 RBCs 0-2 WBC casts |
Trace / NA | 69 | 14 | No | No | No | R 11.6 L 12.6 |
9.7 | 3 HD treatments starting on day 130 | Hepatitis |
| 4 | 16-34 WBCs | NA / NA | 70 | 28 | NA | No | No | R 13.0 L 13.0 |
3.6 | No | None |
| 5 | 5 WBCs** 1 RBC |
Neg / 0.26 | 245 | 63 | No | No | No | R 13.2 L 13.0 |
2.9 | No | Hypophysitis; Thyroiditis |
| 6 | 0 WBC 0 RBC |
Neg / 0.74 | 183 | 36 | No | Yes | Yes | R 10.9 L 13.5 |
11.7 | HD dependent starting on day 183 | Hypophysitis;§ Colitis |
| 7 | 0 WBC** 0 RBC |
Neg / NA | 224 | 14 | No | No | No | R 11.8 L 12.2 |
3.8 | No | Sicca syndrome with sialadenitis on lip biopsy; Colitis |
| 8 | 6-9 WBCs 0-3 RBCs |
1+ / 0.98 | 154 | 7 | No | No | Yes | R 12.8 L 11.8 |
5.6 | HD dependent starting on day 210 | None |
| 9 | 9 WBCs** 8 RBCs WBC casts |
2+ / 0.12 | 42 | 21 | No | Yes | No | R 12.4 L 13.0 |
7.3 | No | Rash; Colitis |
| 10 | 3 WBCs** 3 RBCs WBC casts |
1+ / 0.73 | 120 | 57 | No | No | No | R 8.0 L 10.0 |
2.9 | No | None |
| 11 | 50-100 WBCs 0-2 RBCs |
1+ / 0.18 | 60 | 18 | 14.7% | No | No | R 10.2 L 10.0 |
4.5 | No | None |
| 12 | 20-50 WBCs 0-2 RBCs |
1+ / NA | 21 | 21 | No | No | No | NA | 13.3 | 3 HD treatments starting on day 21 | None |
| 13 | 11-20 WBCs 0 RBCs |
Neg/ 0.36 | 231 | 21 | No | No | No | R 10.7 L 11.9 |
2.5 | No | Iritis, Colitis |
|
| |||||||||||
| Median | 0.48 | 91 | 21 | R 12.0, L 12.8 |
4.5 | ||||||
| IQR | 0.24-0.73 | 60-183 | 18-49 | R 10.9-12.8 L 11.9-13.1 |
3.6-7.3 | ||||||
Number of cells are reported per high-powered field, and number of casts are reported per low-powered field.
Number of days between the first dose of a CPI and AKI onset.
Refers to new or worsened hypertension only.
Oliguria was defined as urine output <500 ml/day.
Denotes independent review of sediment by nephrologist.
Manifestations included hypogonadism, hypothyroidism, and adrenal insufficiency.
Abbreviations: CPI, checkpoint inhibitor; Eos, eosinophilia; HD, hemodialysis; HTN, hypertension; IQR, interquartile range; IRAEs, immune-related adverse events; NA, not available; RRT, renal replacement therapy; SCr, serum creatinine; UPCR, urine protein:creatinine ratio (g/g).
New or worsened hypertension occurred in two patients. Patient 6, who had a baseline blood pressure (BP) of 115/70, had a BP of 170/95 when he first developed AKI. By the following day, his BP had fallen to 140/90 spontaneously. Patient 9, who had a baseline BP of 145/80, had an initial BP of 218/95 when he first developed AKI. He required triple anti-hypertensive therapy with amlodipine, furosemide, and labetalol to control his BP.
The median peak serum creatinine (SCr) during AKI was 4.5 (IQR, 3.6-7.3) mg/dL. Two patients had oliguric AKI. Among the 8 patients whose proteinuria was quantified, the urine protein:creatinine ratio ranged from 0.12 to 0.98 g/g. At least one extra-renal IRAE was documented prior to AKI onset in 7 patients and concurrently with AKI in one additional patient. The most common IRAEs were hypophysitis and colitis (n=3 each). Two patients developed AKI while on a glucocorticoid taper for an extra-renal IRAE.
Pathology
Representative renal pathology images are shown in Figure 1 (larger versions are available in Supplemental Figure 1A-F) and the detailed findings are described in Supplemental Table 2. Acute tubulointerstitial nephritis (AIN) was the primary pathologic lesion in 12 out of 13 patients (Figure 1A). The infiltrates were composed predominantly of lymphocytes, with varying degrees of plasma cells and eosinophils (Figure 1B). Further characterization of the lymphocyte infiltrate in three patients (3, 7, and 9) with CD3 and CD20 staining revealed a predominance of CD3+ T lymphocytes in all three patients. Additional staining performed in patient 9 showed the CD3+ T lymphocytes were predominantly CD4+ (Figure 1C; CD3, CD8 and CD20 stains shown in Supplemental Figure 1C). Granulomatous features were present in 3 out of 12 AIN cases (Figure 1D). Immunofluorescence typically yielded only background staining for C3 along vessel walls with absence of tubular basement membrane and glomerular staining (Supplemental Table 2). Electron microscopy was notable for mild-to-moderate foot process effacement and the absence of electron dense deposits, with the exception of patient 10, who had subepithelial and intramembranous deposits (Supplemental Table 2). Patient 10 had a negative ANA titer, no evidence of infection, and no other evident explanation for the deposits. In patient 8, the primary lesion was acute thrombotic microangiopathy (TMA) (Figures 1E and 1F) with no evidence of AIN.
Figure 1. Representative images of CPI-induced AKI.
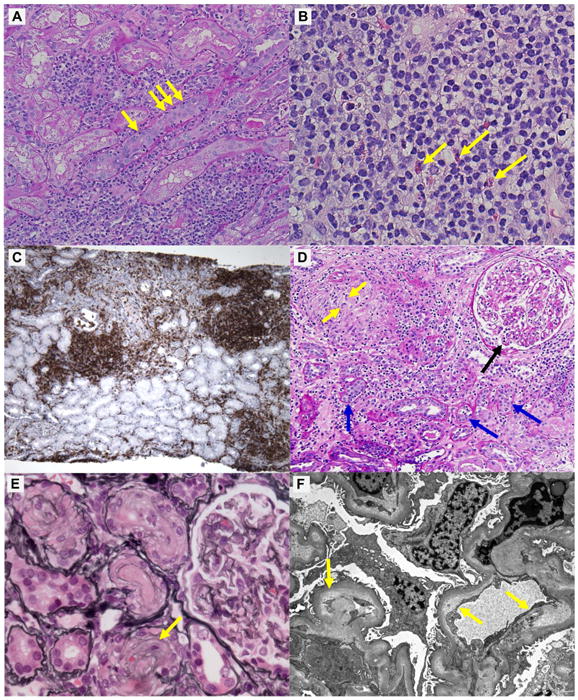
Panels A-C, core needle-biopsy specimens from patient 9, show “typical” features of AIN; panel D, from patient 2, shows granulomatous AIN; panels E and F, from patient 8, show acute thrombotic microangiopathy. A) Periodic acid-Schiff (PAS) stain shows diffuse interstitial inflammation and focal severe tubulitis with infiltrating lymphocytes (arrows, ×200; scale bar, 50μm). B) Hematoxylin and eosin stain shows diffuse interstitial infiltrates predominantly composed of lymphocytes, with several eosinophils (arrows, ×400; scale bar, 25μm). C) Immunohistochemistry reveals the lymphocytic infiltrates in the interstitium to be predominantly CD4+ T cells (×40; scale bar, 100μm). D) PAS stain shows a non-caseating granuloma with multi-nucleated giant cells (yellow arrows), severe interstitial inflammation and tubulitis (blue arrows), and severe glomerulitis (black arrow, ×200; scale bar, 50μm). E) Silver stain shows diffusely wrinkled glomerular basement membranes and “onion-skin” lesion of small arteries (arrow, ×200; scale bar, 50μm). F) Electron microscopy shows swollen endothellium and subintimal widening filled with electron-lucent “fluffy” material (arrows, ×1400; scale bar, 4μm). Larger versions of these images are shown in Supplemental Figure 1.
Treatment and Response
Treatment regimens are shown in Table 3. The implicated CPI was discontinued in all patients because of AKI and other IRAEs, except for patient 10, who was continued on scheduled therapy. Glucocorticoids were administered to 10 of the 12 patients with AIN, as well as patient 8, who had TMA. Six patients were treated with intravenous solumedrol or hydrocortisone followed by oral prednisone. An additional five patients were treated with oral prednisone alone.
Table 3. Treatment and Outcomes.
| Pt | Treatment | Renal Function | Re-treated with CPI? | IRAEs with re-treatment? | AKI with re-treatment? |
|---|---|---|---|---|---|
| 1 | Pred 60mg daily, tapered off over 3 months | Partial recovery | Pembro | No | No |
| 2 | Pred 60mg daily, tapered off over 6 weeks | Complete recovery | No | NA | NA |
| 3 | Episode 1: Pred 60mg daily, tapered off over 2 weeks; Episode 2: hydrocortisone 100mg IV q12 hours × 1 day, then prednisone 60mg daily, tapered to 10 mg daily over 3 months and continued at 10mg daily for an additional 3 months | Partial recovery | No | NA | NA |
| 4 | Conservative management | No recovery | No | NA | NA |
| 5 | Solumedrol 35mg IV daily × 4 days, then Pred 80mg daily, tapered off over 8 weeks | Partial recovery | No | NA | NA |
| 6 | Solumedrol 500mg IV daily × 3 days, 250mg IV daily × 3 days, then pred 80mg daily, tapered off over 4 weeks | No recovery | No | NA | NA |
| 7 | Pred 70mg BID, tapered over 3 weeks to 10mg daily, tapered off over 9 weeks | Partial recovery | Ipi | Colitis | No |
| 8 | Pred 60mg daily, tapered off over 2 weeks | No recovery | No | NA | NA |
| 9 | Solumedrol 500mg IV daily × 3 days, then Pred 60mg daily; 2 weeks later Pred increased to 80mg BID and MMF 1g BID added | Partial recovery | No | NA | NA |
| 10 | Conservative management | No recovery | Ipi | No | No |
| 11 | Pred 60mg daily × 4 weeks, then tapered off over 8 weeks | Partial recovery | No | NA | NA |
| 12 | Solumedrol 500mg IV daily × 3 days, then Pred 40mg daily, tapered off over 4 weeks | Partial recovery | No | NA | NA |
| 13 | Solumedrol 250mg × 1, then Pred 40 mg daily, tapered off over 3 months | Complete recovery | No | NA | NA |
Abbreviations: CPI, checkpoint inhibitor; IRAEs, immune-related adverse events; MMF, mycophenolate mofetil; NA, not applicable; Pembro, pembrolizumab; Pred, prednisone.
Response to treatment is shown in Figure 2. Among the 10 patients with AIN treated with glucocorticoids, 9 had complete (n=2) or partial (n=7) recovery of renal function. Patient 8, who had TMA, did not recover renal function with glucocorticoids. The renal function of patient 3 initially improved with a 2-week course of prednisone, but 6 weeks later the patient developed a recurrent episode of AKI that responded partially to a prolonged prednisone taper (Figure 2). Patient 9 had an initial response to solumedrol followed by prednisone, but two weeks later developed worsening renal function necessitating an increase in prednisone dose and addition of mycophenolate mofetil. Hemodialysis was required in 4 patients: two patients (3 and 12) required dialysis only transiently, whereas the other two patients (6 and 8) remained dialysis-dependent.
Figure 2. Time course of events and response to treatment.
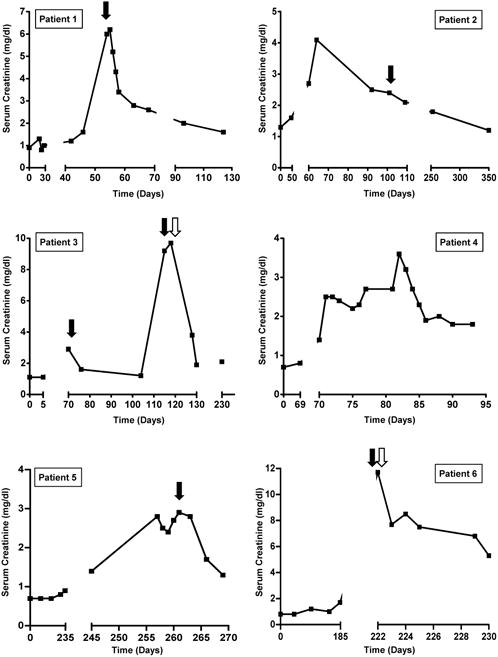
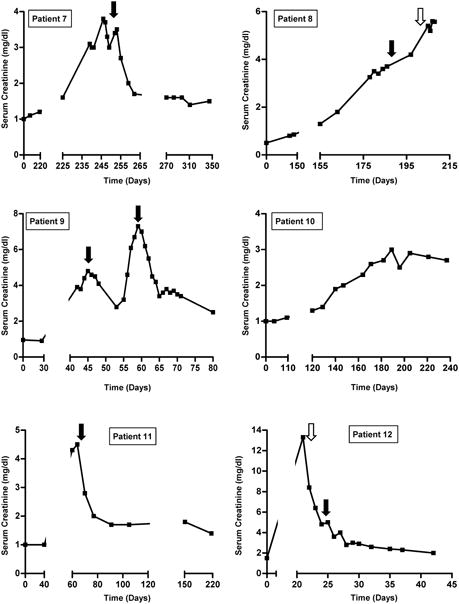
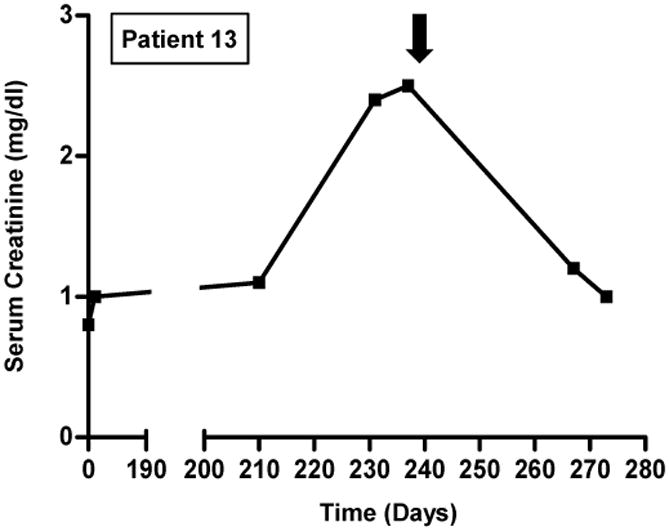
Solid black arrows indicate initiation of steroids (dosing regimens are provided in Table 3). Open arrows indicate initiation of hemodialysis (patients 3 and 12 received three sessions of hemodialysis and subsequently recovered; patients 6 and 8 remained dialysis-dependent). Day 0 refers to CPI initiation.
Two patients (4 and 10) did not receive immunosuppression for their AKI; these patients did not recover renal function, though patient 10's renal function stabilized at a SCr of 2.7 mg/dL despite continuation of ipilimumab. Patients 1 and 7 were re-challenged with a CPI following improvement of AKI after treatment with prednisone. Upon re-challenge, neither developed AKI (Table 3).
Estimated Incidence of CPI-associated AKI
To estimate the incidence of CPI-associated AKI, we analyzed data from all published phase II and III clinical trials that included at least 100 patients treated with CPIs and that provided data on renal outcomes (Supplemental Figure 2). The estimated incidence of CPI-associated AKI, stratified by CPI regimen, is summarized in Figure 3 (data from individual clinical trials is shown in Supplemental Table 3). In a combined analysis of 3,695 patients treated with a CPI, the overall incidence of AKI was 2.2%, and the incidence of grade III or IV AKI, defined as an increase in SCr > 3-fold above baseline, an increase in SCr to a level >4.0 mg/dl, or the need for renal replacement therapy, was 0.6%. AKI occurred more frequently in patients who received combination therapy with ipilimumab and nivolumab (4.9%) than in patients who received monotherapy with ipilimumab (2.0%), nivolumab (1.9%), or pembrolizumab (1.4%) (P<0.01 for all comparisons).
Figure 3. Estimated incidence of CPI-associated AKI.
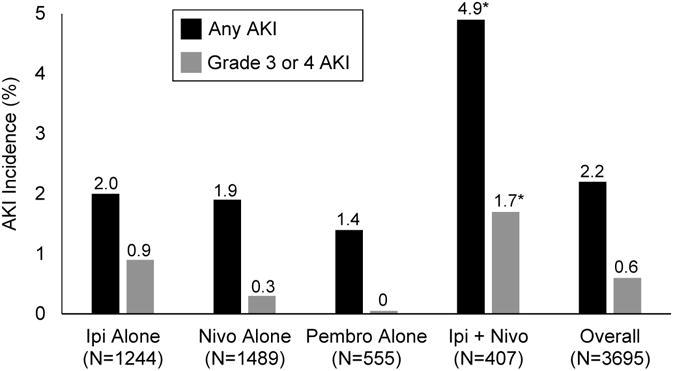
Abbreviations: Ipi, ipilimumab; Nivo, nivolumab; Pembro, pembrolizumab. *P<0.01 for each of the following comparisons: Ipi+Nivo compared to Ipi alone, Nivo alone, and Pembro alone.
Discussion
CPI-induced AKI is an emerging entity that is being observed with increasing frequency as the use of CPIs in oncology continues to expand. Thus, it is likely that nephrologists will be increasingly charged with diagnosing and managing AKI that may be related to these agents. To date, four biopsy-confirmed reports of CPI-induced AKI have described granulomatous AIN (n=3) and lupus nephritis (n=1) (11-13). In other reports, no kidney biopsy was performed (14, 15). Additionally, a recent abstract described biopsy-proven AIN in two patients receiving pembrolizumab for non-small cell lung cancer (Mae et al., TH-P01051, presented at the American Society of Nephrology Kidney Week in San Diego, 2015). Here, we describe the clinical features, pathology, and response to treatment in 13 patients with CPI-induced AKI, 12 of whom had AIN. Granulomatous features were present only in a subset of patients with CPI-induced AIN, and thus appear to be non-specific.
We found that the clinical and histologic features of CPI-induced AIN mirror other etiologies of AIN. Patients often present with pyuria and sub-nephrotic range proteinuria (16, 17). Similar to other causes of AIN, eosinophilia, rash, and fever are absent in the majority of cases and are poor predictors of the disease (18). In our cohort, 7 of 12 patients with CPI-induced AIN had an extra-renal IRAE prior to AKI onset, and an additional patient had an extra-renal IRAE concurrently with AKI. Thus, the presence of current or prior extra-renal IRAEs should raise suspicion of CPI-induced AIN in a patient with AKI.
A notable feature of CPI-induced AKI is the variable time course from CPI exposure to AKI. Time from initiation of a CPI to the diagnosis of CPI-induced AKI ranged from 21 to 245 days. Furthermore, CPI-induced AKI was diagnosed up to 63 days after the last CPI dose. The delayed onset of AKI following exposure to CPIs is consistent with several reported cases of hypophysitis that occurred greater than two months following the last CPI dose (19) and in line with an immune-related etiology.
The heterogeneity of the time course and delayed response are suggestive of a mechanism distinct from typical drug-induced AIN. Indeed, CTLA-4 knockout mice develop a systemic immune hyperactivation syndrome characterized by diffuse lymphocytic tissue invasion and death at 3-4 weeks of age (20). Likewise, PD-1 knockout mice develop interstitial nephritis and glomerulonephritis (3, 21). Similar findings were observed in mice treated with an anti-PD-1 monoclonal antibody (22). Thus, CPI-induced AIN may be due to “re-programming” of the immune system, leading to loss of tolerance against endogenous kidney antigens, as opposed to the delayed-type hypersensitivity response characteristic of other drugs (23). This unique mechanism may explain the long latency period observed in some cases. However, an alternative plausible mechanism is that CPIs reduce tolerance to concomitant medications known to cause AIN.
The efficacy of glucocorticoids in the treatment of AIN remains controversial due to conflicting observational studies and the absence of a randomized trial (16, 17). In our cohort, 9 out of 10 patients with CPI-induced AIN treated with glucocorticoids had complete or partial recovery of renal function, whereas neither of the two patients managed conservatively recovered spontaneously. Although we acknowledge the limited sample size and observational nature of our study, these data suggest that glucocorticoids are beneficial in CPI-induced AIN, though optimal dosing regimens and treatment duration remain unknown. Of note, the two patients with AIN who achieved a complete response (2 and 13) had absent or minimal fibrosis appreciated on biopsy. Conversely, patients with AIN who were treated with glucocorticoids and did not achieve a complete response had mild (n=2), moderate (n=5), or uninterpretable (n=1; due to severe inflammation and edema) interstitial fibrosis. These findings suggest that as with other forms of AIN, preservation of kidney function may be best achieved by early recognition of injury and prompt treatment.
An important consideration is whether patients who develop CPI-associated AKI can continue CPI therapy. In our series, the one patient who was continued on therapy had relatively stable renal function. This suggests a mechanism distinct from typical drug-induced AIN, where continued exposure to the offending medication would be expected to lead to a more rapid deterioration in renal function. Rather, this patient may have had an insidious autoimmune process more akin to Sjӧgren's syndrome, where the decline in renal function is often slow (24). In general, experts recommend permanent discontinuation of a CPI if it results in grade III or IV toxicity (25). For renal toxicity, this corresponds to a tripling of SCr or the need for renal replacement therapy. However, like all treatment decisions, balancing the potential oncologic benefits of continued treatment with CPIs against the risk of recurrent AKI must be carefully weighed. Interestingly, there is limited data that nivolumab may be well tolerated among patients who suffer a grade III or IV IRAE from ipilimumab, though the number of patients with AIN was small (26).
Whether patients who suffer IRAEs have a more potent anti-tumor response as a consequence of enhanced immune activation remains unknown. The largest study examining this hypothesis was a retrospective analysis of 298 patients with metastatic melanoma treated with ipilimumab. In this study, 19% of patients developed an IRAE requiring immunosuppressive treatment, and these patients had the same overall survival and time to treatment failure as the patients who did not develop an IRAE (27). It is possible that immunosuppression and withholding of the CPI mitigated any oncologic benefit obtained from a more vigorous state of immune activation. Nonetheless, this study underscores that in the setting of a severe IRAE, immunosuppressive therapy should not be withheld for fear of cancer progression.
Interestingly, one patient in our series developed TMA that temporally correlated with CPI therapy. Metastatic cancer alone has been associated with TMA leading to AKI (28), although not typically with melanoma. Nonetheless, mechanisms of CPI-induced TMA are unclear, and additional cases are needed before TMA can be reliably ascribed to CPIs.
We estimated the incidence of CPI-associated AKI by analyzing data from large clinical trials. In a pooled analysis of 3,695 patients treated with CPIs, we found the incidence of treatment-related AKI to be 2.2%. Additionally, we found that AKI was more common in patients receiving ipilimumab and nivolumab combination therapy compared to CPI monotherapy. These data suggest that while AKI is an important and often severe complication of CPI therapy, it is a relatively uncommon event. Moreover, the risk of AKI is likely outweighed by the benefits of CPI therapy in a patient population that often has few therapeutic options.
Our study has several limitations. First, by only including patients with biopsy-confirmed disease, our study is enriched with patients who had severe AKI, and thus the findings may not be generalizable to more mild cases of CPI-associated AKI that are not routinely biopsied. Second, while glucocorticoids appear to be beneficial, the small numbers and lack of a control group precludes firm conclusions about the efficacy of glucocorticoids in this setting. Third, renal biopsies were not centrally reviewed, which would have provided internal standardization, though the data presented are more reflective of real-world data that clinicians are charged with interpreting. Fourth, in some cases we cannot exclude the possibility that AIN was caused by a concomitant medication rather than a CPI. In most cases, however, concurrent medications associated with AIN were started many months or years prior to the onset of AKI, making a causal relationship less likely, though a synergistic effect between CPIs and concomitant medications is certainly possible. Finally, we estimated the incidence of CPI-associated AKI by analyzing AKI event rates in published clinical trials. This approach could have led to overestimation of the incidence of CPI-associated AKI, since some of the reported cases could have been due to etiologies unrelated to CPI therapy. Alternatively, it is also possible that mild cases of CPI-associated AKI were undetected or erroneously attributed to another etiology, thus resulting in underestimation of the true incidence of CPI-associated AKI.
Given the novelty of CPIs, much remains unknown about CPI-induced AKI. CPI-induced AKI appears to be relatively rare and, as with other IRAEs, responds to glucocorticoids in most cases. Further, glucocorticoids do not appear to negatively impact tumor-response or survival (27). In cases of suspected CPI-induced AKI, prompt and close collaboration between oncologists and nephrologists is encouraged. Further investigation is needed to determine the best approach to diagnosis and treatment of CPI-induced AKI.
Methods
All protocols were approved by our hospital's Institutional Review Board. Cases were obtained by contacting renal pathology and oncology departments at large academic medical centers across the United States to inquire about potential cases of AKI related to CPIs. A total of 20 centers were contacted, and 7 contributed cases to the current series. AKI was defined in accordance with criteria established by KDIGO (29), and was considered to be attributable to a CPI if AKI developed after initiation of a CPI and the treating nephrologist considered a CPI to be the most likely etiology of AKI.
Data Collection
The following de-identified patient data were obtained for each case: age, gender, race, type of malignancy, CPI regimen and dosing schedule, SCr trend before, during, and after AKI, urine sediment, renal ultrasound, presence of peripheral eosinophilia, complement levels, pathology reports with images, treatment for AKI including need for dialysis, tumor response, and change in renal function if the patient was re-treated with a CPI. The urine sediments presented were obtained from the hospital laboratory. In cases where the consulting nephrologist independently reviewed the sediment (n=5), the presence of additional findings (e.g., casts, crystals, dysmorphic red blood cells) were incorporated into the results. The degree of interstitial fibrosis on biopsy specimens was expressed as follows: mild (6-25%), moderate (25-50%), and severe (> 50%).
Definitions of complete and partial recovery
Complete recovery of renal function following AKI was defined as a return of SCr to <0.35 mg/dl above the baseline value (30), while partial recovery was defined as a return of SCr to >0.35 mg/dl but less than twice the baseline value. Patients who transitioned from dialysis-dependence to dialysis-independence were considered to have complete recovery if their steady-state SCr returned to <0.35 mg/dl above the baseline, and were considered to have partial recovery if their steady-state SCr returned to >0.35 mg/dl.
Estimated incidence of CPI-associated AKI
To estimate the incidence of CPI-associated AKI, we analyzed all published phase II and III clinical trials in which at least 100 patients were treated with CPI therapy. On February 20, 2016, we conducted a search on pubmed.org for clinical trials using the following keywords: “ipilimumab”; “nivolumab”; and “pembrolizumab.” The search yielded 115 studies, of which 23 were phase II or III and included at least 100 patients treated with CPI therapy. Among these, 11 studies reported data on renal outcomes (Supplemental Figure 2). We collected data on the incidence of all AKI events and grade III or IV AKI events. Events were graded according to the Common Terminology Criteria for Adverse Events Version 4.0, whereby AKI severity is graded as follows: grade 1, an absolute increase in SCr > 0.3 mg/dL within 48 hours or a relative increase in SCr 1.5 to 2.0 fold above baseline; grade 2, an increase in SCr of 2-3 fold above baseline; grade 3, an increase in SCr > 3 fold above baseline or an increase to a SCr > 4.0 mg/dL; and grade 4, requirement for renal replacement therapy (31).
Statistical Analysis
We used the chi-square test to compare the proportion of patients who developed AKI across different CPI treatment strategies. All comparisons are two-tailed, with P<0.05 considered significant.
Supplementary Material
Acknowledgments
We thank Dr. Vivette D. D'Agati, from the Department of Renal Pathology at Columbia University, Dr. Rex Neal Smith, from the Department of Renal Pathology at Massachusetts General Hospital, and Dr. Surya Seshan, from the Department of Pathology at Weill Cornell Medical College, for their invaluable assistance.
Sources of support: DEL is supported by K23DK106448 from the National Institute of Diabetes and Digestive Kidney Diseases.
Disclosures/Funding Sources: DEL is supported by K23DK106448 from the National Institute of Diabetes and Digestive Kidney Diseases. JRB received grant support from Bristol-Myers Squibb (BMS) and consulted for Merck. DTL received research funding from BMS and Merck and speaking honorarium from Merck. IGG served on an advisory committee for Otsuka and has stock ownership in Pfizer. JW served as a consultant and has received research support from BMS, Merck, Medimmune, and Genentech. EJP consulted for BMS, Merck, Amgen, and Castle Biosciences. PAO received research support to the institution from BMS and Merck and consulted for BMS, Amgen, and Alexion. FSH received research support from BMS and consulted for Merck and Genentech.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Postow MA, Callahan MK, Wolchok JD. Immune Checkpoint Blockade in Cancer Therapy. J Clin Oncol. 2015;33:1974–1982. doi: 10.1200/JCO.2014.59.4358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Krummel MF, Allison JP. CTLA-4 engagement inhibits IL-2 accumulation and cell cycle progression upon activation of resting T cells. J Exp Med. 1996;183:2533–2540. doi: 10.1084/jem.183.6.2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zha Y, Blank C, Gajewski TF. Negative regulation of T-cell function by PD-1. Crit Rev Immunol. 2004;24:229–237. doi: 10.1615/critrevimmunol.v24.i4.10. [DOI] [PubMed] [Google Scholar]
- 4.Ribas A, Puzanov I, Dummer R, et al. Pembrolizumab versus investigator-choice chemotherapy for ipilimumab-refractory melanoma (KEYNOTE-002): a randomised, controlled, phase 2 trial. Lancet Oncol. 2015;16:908–918. doi: 10.1016/S1470-2045(15)00083-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hodi FS, O'Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711–723. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Postow MA, Chesney J, Pavlick AC, et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N Engl J Med. 2015;372:2006–2017. doi: 10.1056/NEJMoa1414428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lynch TJ, Bondarenko I, Luft A, et al. Ipilimumab in combination with paclitaxel and carboplatin as first-line treatment in stage IIIB/IV non-small-cell lung cancer: results from a randomized, double-blind, multicenter phase II study. J Clin Oncol. 2012;30:2046–2054. doi: 10.1200/JCO.2011.38.4032. [DOI] [PubMed] [Google Scholar]
- 8.Brahmer J, Reckamp KL, Baas P, et al. Nivolumab versus Docetaxel in Advanced Squamous-Cell Non-Small-Cell Lung Cancer. N Engl J Med. 2015;373:123–135. doi: 10.1056/NEJMoa1504627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Motzer RJ, Escudier B, McDermott DF, et al. Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. N Engl J Med. 2015;373:1803–1813. doi: 10.1056/NEJMoa1510665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ansell SM, Lesokhin AM, Borrello I, et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin's lymphoma. N Engl J Med. 2015;372:311–319. doi: 10.1056/NEJMoa1411087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Izzedine H, Gueutin V, Gharbi C, et al. Kidney injuries related to ipilimumab. Invest New Drugs. 2014;32:769–773. doi: 10.1007/s10637-014-0092-7. [DOI] [PubMed] [Google Scholar]
- 12.Fadel F, El Karoui K, Knebelmann B. Anti-CTLA4 antibody-induced lupus nephritis. N Engl J Med. 2009;361:211–212. doi: 10.1056/NEJMc0904283. [DOI] [PubMed] [Google Scholar]
- 13.Thajudeen B, Madhrira M, Bracamonte E, et al. Ipilimumab granulomatous interstitial nephritis. Am J Ther. 2015;22:e84–87. doi: 10.1097/MJT.0b013e3182a32ddc. [DOI] [PubMed] [Google Scholar]
- 14.Forde PM, Rock K, Wilson G, et al. Ipilimumab-induced immune-related renal failure--a case report. Anticancer Res. 2012;32:4607–4608. [PubMed] [Google Scholar]
- 15.Voskens CJ, Goldinger SM, Loquai C, et al. The price of tumor control: an analysis of rare side effects of anti-CTLA-4 therapy in metastatic melanoma from the ipilimumab network. PLoS One. 2013;8:e53745. doi: 10.1371/journal.pone.0053745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Clarkson MR, Giblin L, O'Connell FP, et al. Acute interstitial nephritis: clinical features and response to corticosteroid therapy. Nephrol Dial Transplant. 2004;19:2778–2783. doi: 10.1093/ndt/gfh485. [DOI] [PubMed] [Google Scholar]
- 17.Gonzalez E, Gutierrez E, Galeano C, et al. Early steroid treatment improves the recovery of renal function in patients with drug-induced acute interstitial nephritis. Kidney Int. 2008;73:940–946. doi: 10.1038/sj.ki.5002776. [DOI] [PubMed] [Google Scholar]
- 18.Muriithi AK, Leung N, Valeri AM, et al. Biopsy-proven acute interstitial nephritis, 1993-2011: a case series. Am J Kidney Dis. 2014;64:558–566. doi: 10.1053/j.ajkd.2014.04.027. [DOI] [PubMed] [Google Scholar]
- 19.Ryder M, Callahan M, Postow MA, et al. Endocrine-related adverse events following ipilimumab in patients with advanced melanoma: a comprehensive retrospective review from a single institution. Endocr Relat Cancer. 2014;21:371–381. doi: 10.1530/ERC-13-0499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tivol EA, Borriello F, Schweitzer AN, et al. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity. 1995;3:541–547. doi: 10.1016/1074-7613(95)90125-6. [DOI] [PubMed] [Google Scholar]
- 21.Nishimura H, Nose M, Hiai H, et al. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity. 1999;11:141–151. doi: 10.1016/s1074-7613(00)80089-8. [DOI] [PubMed] [Google Scholar]
- 22.Zheng G, Wang Y, Mahajan D, et al. The role of tubulointerstitial inflammation. Kidney Int Suppl. 2005:S96–100. doi: 10.1111/j.1523-1755.2005.09423.x. [DOI] [PubMed] [Google Scholar]
- 23.Spanou Z, Keller M, Britschgi M, et al. Involvement of drug-specific T cells in acute drug-induced interstitial nephritis. J Am Soc Nephrol. 2006;17:2919–2927. doi: 10.1681/ASN.2006050418. [DOI] [PubMed] [Google Scholar]
- 24.Maripuri S, Grande JP, Osborn TG, et al. Renal involvement in primary Sjogren's syndrome: a clinicopathologic study. Clin J Am Soc Nephrol. 2009;4:1423–1431. doi: 10.2215/CJN.00980209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Spain L, Diem S, Larkin J. Management of toxicities of immune checkpoint inhibitors. Cancer Treat Rev. 2016;44:51–60. doi: 10.1016/j.ctrv.2016.02.001. [DOI] [PubMed] [Google Scholar]
- 26.Weber J, Gibney G, Kudchadkar R, et al. Phase I/II Study of Metastatic Melanoma Patients Treated with Nivolumab Who Had Progressed after Ipilimumab. Cancer Immunol Res. 2016 doi: 10.1158/2326-6066.CIR-15-0193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Horvat TZ, Adel NG, Dang TO, et al. Immune-Related Adverse Events, Need for Systemic Immunosuppression, and Effects on Survival and Time to Treatment Failure in Patients With Melanoma Treated With Ipilimumab at Memorial Sloan Kettering Cancer Center. J Clin Oncol. 2015;33:3193–3198. doi: 10.1200/JCO.2015.60.8448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.George JN. Systemic malignancies as a cause of unexpected microangiopathic hemolytic anemia and thrombocytopenia. Oncology (Williston Park) 2011;25:908–914. [PubMed] [Google Scholar]
- 29.Kidney Disease; Improving Global Outcomes (KDIGO) Acute Kidney Injury Work Group. KDIGO clinical practice guideline for acute kidney injury. Kidney Int Suppl. 2012;2:1–138. [Google Scholar]
- 30.Dewitte A, Joannes-Boyau O, Sidobre C, et al. Kinetic eGFR and Novel AKI Biomarkers to Predict Renal Recovery. Clin J Am Soc Nephrol. 2015;10:1900–1910. doi: 10.2215/CJN.12651214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Common Terminology Criteria for Adverse Events (CTCAE) Version 4.0. National Institutes of Health, National Cancer Institute, US Department of Health and Human Services; 2009. [Google Scholar]
- 32.Levey AS, Stevens LA, Schmid CH, et al. A new equation to estimate glomerular filtration rate. Ann Intern Med. 2009;150:604–612. doi: 10.7326/0003-4819-150-9-200905050-00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.