

Abstract
Cerebellar ataxias represent a spectrum of disorders which are, however, linked by common symptoms of motor incoordination and typically associated with deficiency in Purkinje cell firing activity and, often, degeneration. Cerebellar ataxias currently lack a curative agent. The endocannabinoid (eCB) system includes eCB compounds and their associated metabolic enzymes, together with cannabinoid receptors, predominantly the cannabinoid CB1 receptor (CB1R) in the cerebellum; activation of this system in the cerebellar cortex is associated with deficits in motor coordination characteristic of ataxia, effects which can be prevented by CB1R antagonists. Of further interest are various findings that CB1R deficits may also induce a progressive ataxic phenotype. Together these studies suggest that motor coordination is reliant on maintaining the correct balance in eCB system signalling. Recent work also demonstrates deficient cannabinoid signalling in the mouse ‘ducky2J’ model of ataxia. In light of these points, the potential mechanisms whereby cannabinoids may modulate the eCB system to ameliorate dysfunction associated with cerebellar ataxias are considered.
Abbreviations
- 2‐AG
2‐arachidonylglycerol
- CBD
cannabidiol
- CBDV
cannabidivarin
- CF
climbing fibre
- eCB
endocannabinoid
- GPCR
G protein‐coupled receptor
- IN
interneuron
- LTD
long term depression
- MAGL
monoacylglycerol lipase
- PC
Purkinje cell
- pCB
phytocannabinoid
- PF
parallel fibre
- SCA
spinocerebellar ataxia
- Δ9‐THC
tetrahydrocannabinol
- VDCC
voltage‐dependent Ca2+ channel
Cerebellar ataxias are a diverse group of disorders lacking a therapeutic agent
Cerebellar ataxias comprise a group of progressive neurological diseases associated with deficits in motor coordination and are typically associated with dysfunction and/or degeneration of Purkinje cells (PCs), the sole efferent output of the cerebellar cortex. There are a range of acquired ataxias and different hereditary forms of the disease (Klockgether, 2011). Thus, ataxia can be acquired from, amongst others, traumatic head injury, bacterial infection (meningitis or encephalitis), viral infection (chickenpox or measles), disruption of blood flow (stroke or transient ischaemic attack, haemorrhage), CNS disease (cerebral palsy or multiple sclerosis), sustained long‐term alcohol misuse, underactive thyroid gland and cancer autoimmune conditions (lupus), and can also be iatrogenic. Hereditary ataxias may be autosomal‐dominant diseases, including forms of spinocerebellar ataxia (SCA), several of which are associated with polyglutamine repeats in the dysfunctional protein; for example: ataxin 1 in SCA1; ataxin 2 in SCA2; Cacna1a encoding the voltage‐dependent Ca2+ channel (VDCC) CaV2.1 subunit in SCA6 (also in episodic ataxia 2). There are also autosomal‐recessive diseases such as Friedreich's ataxia and ataxia telangiectasia associated with deficits in, respectively, the mitochondrial protein frataxin and a serine/threonine protein kinase termed ataxia telangiectasia mutated protein (Klockgether, 2011). Despite this range of causes and implicated proteins, deleterious effects are largely limited to the cerebellar cortex and are typically associated with cerebellar dysfunction and/or degeneration and are manifest as motor incoordination. This commonality of symptoms offers hope for providing treatment options; however, at present there is no known cure for cerebellar ataxia. There are treatments to ameliorate associated symptoms. For example, vitamin E and anti‐oxidants, such as co‐enzyme Q10 and its synthetic analogue idebenone, have been suggested to have some benefit, largely in Friedreich ataxia. However, as yet, such agents lack proven efficacy in controlled clinical trials (Cooper et al. 2008; Lynch et al. 2010), although some improvement in comparison to controls was seen in cross‐over trials, suggesting that patients with vitamin E‐deficient and co‐enzyme Q10‐deficient ataxia may receive some benefit (Cooper et al. 2008). In addition, administration of thyrotropin‐releasing hormone (TRH) was reported to ameliorate cerebellar ataxia in rolling Nagoya mice (Shibusawa et al. 2008) and the TRH analogue taltirelin is approved to improve motor performance in ataxic patients in Japan.
The elucidation of function of proteins associated with inherited ataxias within the cerebellar cortex may also lead to future therapeutic advances relevant across different forms of ataxia. Amongst target proteins, the CaV2.1 (α1A) VDCC represents a widely studied protein. CaV2.1 subunits are highly expressed in the cerebellum (Westenbroek et al. 1995; Kulik et al. 2004). In particular, CaV2.1 is expressed postsynaptically in PCs (which led to the designation of these subunits as carriers of P‐type Ca2+ current (Mintz et al. 1992)), at presynaptic terminals of inhibitory interneurons (INs) arising from basket and stellate cells, and of excitatory, parallel fibres (PFs) and climbing fibres (CFs); such inputs regulate PC, and thus cerebellar cortex, output activity (Regehr & Mintz, 1994; Mintz et al. 1995; Stephens et al. 2001; Lonchamp et al. 2009). Several mouse CaV2.1 mutants display ataxia (Pietrobon, 2010; Rajakulendran et al. 2012). Correspondingly, genetic deletion of CaV2.1 is associated with a clear ataxic behavioural phenotype (Jun et al. 1999). Moreover, conditional PC‐specific CaV2.1 knock‐down was shown to be sufficient to induce impaired synaptic transmission and ataxia (Mark et al. 2011; Todorov et al. 2012), the former study termed their mice ‘purky’. Cell‐specific work was extended to excitatory inputs into PCs, where it was shown that selective CaV2.1 knockdown in PFs (arising from mossy fibre inputs) in so‐called ‘quirky’ mice, also gave rise to an ataxic phenotype (Maejima et al. 2013). Of further interest here, is that mutations which increase CaV2.1 current also give impaired synaptic transmission and irregular PC firing (a cerebellar epitome predicted to lead to motor incoordination) (Gao et al. 2012); these reports suggest that correct VDCC activity must be maintained for PC firing fidelity. To add to purky and quirky, we also have ‘ducky’ mice (Barclay et al. 2001). Ducky, and the related ducky2J (du2J) strain, have mutations predicted to lead to deficits in α2δ‐2 auxiliary VDCC subunit protein, which is expressed at high levels in normal cerebella (Cole et al. 2005) and is associated predominantly with CaV2.1 in the cerebellum (Barclay et al. 2001). In different ducky strains, the ataxic phenotype is associated with a reduction in postsynaptic PC whole‐cell Ca2+ current (Brodbeck et al. 2002; Donato et al. 2006), together with irregular PC firing (Donato et al. 2006; Walter et al. 2006; Wang et al. 2013). Thus, several potential therapeutic targets have been suggested, largely confined to protein associated with inherited ataxias; however, as discussed above, as yet we have no cure for ataxia. The remainder of this review will focus on the potential to target the endocannabinoid (eCB) system to ameliorate cerebellar ataxia and, in particular, eCB compounds and their associated metabolic enzymes and G protein‐coupled CB1R, one of the most ubiquitously expressed proteins in the mammalian cerebellum, and a protein which also modulates CaV2.1 activity at the presynapse.
Cannabinoid signalling and its potential links to cerebellar ataxia
Cannabinoids represent a diverse number of compounds, including (i) eCBs, for example, the lipid mediator 2‐arachidonylglycerol (2‐AG), which is the major eCB in the cerebellum (Szabo et al. 2006); (ii) plant‐derived phytocannabinoids (pCBs), for example, the major herbal Cannabis constituents tetrahydrocannabinol (Δ9‐THC) and cannabidiol (CBD) (Hill et al. 2012 a); and (iii) exogenous synthetic agents, namely CB1R agonists, for example, WIN 55,212‐2, an aminoalkylindole derivative, and CP 55940, which is structurally related to tetrahydrocannabinol, and CB1R antagonists/inverse agonists, for example, rimonabant (Pertwee et al. 2010), and newer allosteric modulators, for example, Org27569 (Price et al. 2005) and PSNCBAM‐1 (Horswill et al. 2007).
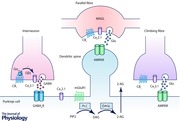
Within the CNS, cannabinoids predominantly activate CB1Rs, which represent the most widespread G protein‐coupled receptor (GPCR) in the mammalian cerebellum (Herkenham et al. 1991; Tsou et al. 1997). CB1R expression is reported to be very low at PC cell bodies; rather, expression is high at excitatory PF inputs into PCs, reportedly with a perisynaptic over extrasynaptic and synaptic localization, with lower CB1R expression at CF inputs onto PC dendritic shafts (Kawamura et al. 2006; see Abstract Figure). CB1Rs are expressed at higher levels on presynaptic terminals of inhibitory INs, predominantly basket cells, but also stellate cells, which form a specialized region surrounding the PC axon initial segment known as the pinceau (the French word for paintbrush) (Tsou et al. 1997; Kawamura et al. 2006; Rodríguez‐Cueto et al. 2014). Presynaptic CB1Rs are activated by retrograde ‘on demand’ release of 2‐AG from postsynaptic PCs. The major effect of presynaptic CB1R activation is a suppression of neurotransmitter release, whereby activation of presynaptic CB1Rs inhibits action potential‐evoked and spontaneous inhibitory postsynaptic currents (IPSCs) at IN–PC synapses (see Fig. 1) or excitatory postsynaptic currents (EPSCs) at PF–PC and CF–PC synapses (Takahashi & Linden, 2000; Szabo et al. 2004; Kano et al. 2009). We have also used multi‐electrode array recording to demonstrate that CB1R ligand‐induced changes to cerebellar cortex network activity are mediated, at least in part, via effects on inhibitory synaptic transmission (Ma et al. 2008). CB1R activation has been widely associated with a number of different forms of short‐ and long‐term synaptic plasticities which modulate cerebellar learning (Kano et al. 2009; Ohno‐Shosaku & Kano, 2014). Thus, 2‐AG release mediates the short‐term suppression of inhibitory GABA release from IN terminals (depolarization‐induced suppression of inhibition) or excitatory glutamate release (depolarization‐induced suppression of excitation) (Szabo et al. 2006; Tanimura et al. 2009). Seminal work by Ito (1989) linked long term depression (LTD) by associative stimulation of PF and CF inputs to PCs, to motor learning in the cerebellum. It is also known that the metabotropic glutamate receptor 1 (mGluR1) pathway is critically involved in cerebellar development and LTD (Aiba et al. 1994), and it was further proposed that CF inputs into individual PCs were required for normal motor coordination (Chen et al. 1995). More recent work has established that cerebellar LTD is a postsynaptic phenomenon requiring 2‐AG release from PCs and activation of presynaptic CB1Rs at PF–PC synapses (Safo & Regehr, 2005). Here, mGluR1 activation drives 2‐AG release (Kano et al. 2009; see Abstract Figure). Thus, CB1Rs have a privileged position in the function and control of overall output of the cerebellar cortex and, as such, represent good potential targets to modulate dysfunctional signalling associated with cerebellar ataxias.
Figure 1. Presynaptic CB1R modulation of inhibitory transmission at IN–PC synapses is deficient in ataxic ducky2J mice .
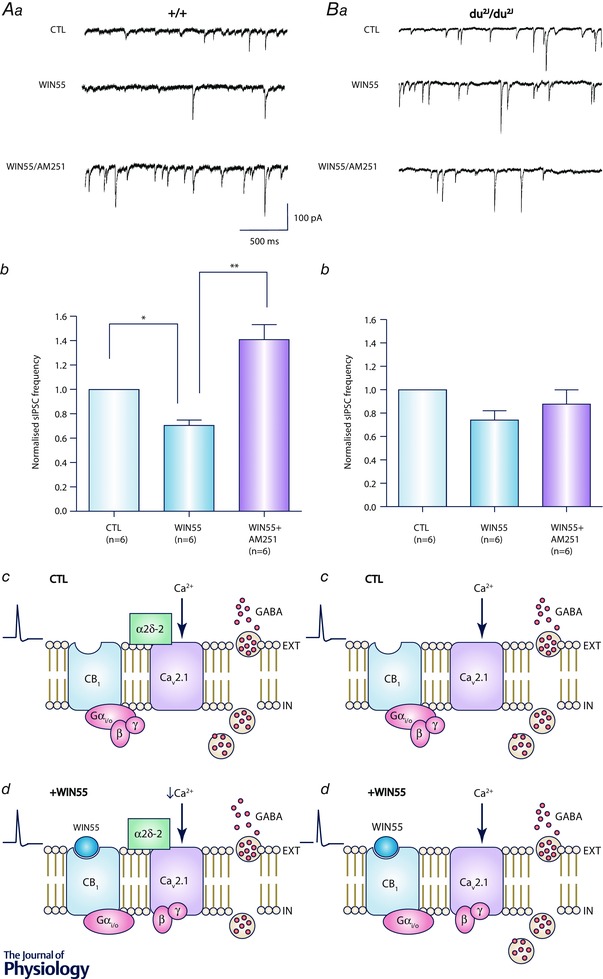
Aa and Ba, representative spontaneous inhibitory postsynaptic current (sIPSC) traces from +/+ (Aa) and du2J/du2J (Ba) PCs showing effect of WIN55 (5 μm), and also subsequent application of AM251 (2 μm). Ab and Bb, summary bar graphs showing that WIN55 significantly reduced, and AM251 significantly increased, normalized sIPSC frequency in +/+ (Ab), but was without effect in du2J/du2J (Bb), conditions. * P < 0.05; ** P < 0.01; repeated measurement one‐way ANOVA followed by Tukey's honest significant difference test. Ac and Bc, summary diagrams for +/+ (Ac) and du2J/du2J (Bc) conditions. Ad, in wild‐type conditions, CB1R activation (i.e. +WIN55) causes the release of Gβγ subunit from CB1R and subsequent inhibitory coupling of Gβγ to Cav2.1 at the presynapse to inhibit the action potential‐evoked GABA release seen in control (CTL). Bd, by contrast, in du2J/du2J conditions, CB1R activation (i.e. +WIN55) has no effect on the GABA release seen in control (CTL). AM251 effects were also absent (see Wang et al. 2013). Thus, we propose that at synapses lacking α2δ‐2 subunits (which associate predominantly with CaV2.1 in the cerebellum; Barclay et al. 2001), normal CB1R modulation of CaV2.1 is lacking. This deficit may relate to incorrect control of synaptic release by α2δ subunits (Hoppa et al. 2012); alternatively, it is possibly that lack of α2δ subunits may cause changes to CB1R‐mediated G protein inhibition of CaV2.1.
In cell lines and native neurons, CB1R activation causes pertussis toxin‐sensitive inhibition of CaV2 family VDCCs, and can also activate inwardly rectifier K+ channels (Mackie & Hille, 1992; Twitchell et al. 1997; Guo & Ikeda, 2004). It is likely that CB1Rs couple to presynaptic CaV2.1 (P/Q‐type) VDCCs at IN–PC synapses to reduce action potential evoked GABA release (Forti et al. 2000; Stephens et al. 2001; Lonchamp et al. 2009) and to CaV2.1 (and to a lesser extent CaV2.2 and CaV2.3) at PF–PC synapses to reduce action potential‐evoked glutamate release (Brown et al. 2004); these effects are most likely mediated by direct binding of G protein Gβγ subunits to VDCCs (Abstract Figure and Fig. 1).
CB1R agonists also cause clear reductions in frequency of ‘miniature’ IPSCs at IN–PC synapses (Takahashi & Linden, 2000; Yamasaki et al. 2006; Ma et al. 2008), consistent with an inhibition of action potential‐independent GABA release. These effects are proposed to occur downstream of actions on voltage‐dependent ion channels and are also consistent with direct effects on the synaptic release machinery, and also may be mediated by Gβγ subunits (Stephens, 2009). By contrast, CB1R agonist effects on miniature EPSCs at PF–PC synapses were only apparent when extracellular Ca2+ levels were increased (Yamasaki et al. 2006). Moreover, CB1R antagonists/inverse agonists such as AM251, rimonabant and the pCB Δ9‐tetrahydrocannabivarin all increase inhibitory GABA release at IN–PC synapses (Ma et al. 2008). Such effects are consistent with the presence of a strong, modulatable eCB tone in the cerebellum (Kreitzer et al. 2002; Galante & Diana, 2004), which provides further opportunity for therapeutic intervention in cerebellar ataxia.
Importantly, activation of presynaptic CB1Rs by synthetic cannabinoids and eCBs has been shown to promote cerebellar dysfunction, causing severe motor incoordination and modelling cerebellar ataxia (Lichtman et al. 1998; DeSanty & Dar, 2001; Patel & Hillard, 2001); in these studies pre‐treatment with a CB1R antagonist or CB1R antisense prevented the induction of an ataxic phenotype. Such data suggest that CB1R antagonism may be useful in the pathogenic situation. In comparison to administration of CB1R ligands, data with CB1R knock‐out mice are somewhat more equivocal. Thus, young/mature CB1R deficient mice are reported not to exhibit clear motor discoordination or changes to gait (Steiner et al. 1999; Kishimoto & Kano, 2006); however, deficits in motor function were reported in mature and older mice, in comparison to unaffected younger mice, in rotarod tests (Bilkei‐Gorzo et al. 2005). One interpretation of these studies is that a progressive ataxic pathogenesis may be associated with long‐term loss of CB1R. One common feature of CB1R knock‐out mice, chronic marijuana users or animals administered CB1R agonists is a reported deficit in delay eyeblink conditioning, a cerebellar‐dependent, motor learning process (Kishimoto & Kano, 2006; Skosnik et al. 2008; Steinmetz & Freeman, 2010). These data are consistent with CB1R controlling discrete motor function. Kishimoto & Kano (2006) also report that pharmacological block of CB1R had no effect on motor function in wild‐type mice; however, it is also important to point out that CB1R deficiency and/or lack of effect of CB1R antagonism in a non‐pathogenic situation does not preclude a role in disease; for example, whilst SR141617A (rimonabant) reversed CB1R‐induced dysfunction, it had no effects itself on motor incoordination in non‐ataxic animals (Lichtman et al. 1998; DeSanty & Dar, 2001). Together, these data suggest that CB1Rs modulate cerebellar circuitry in ataxic disease, potentially with a progressive onset of effect. Therefore, targeting CB1Rs may be beneficial in modulating motor incoordination in cerebellar ataxia, as discussed more fully below.
An ataxic mouse model has deficient CB1R signalling
Whilst the role of ion channels (in particular, CaV2.1) has been broadly studied in animal models of ataxia, there has been much less work on the presynaptic receptors that modulate neurotransmitter release and the postsynaptic receptors responsible for onward signalling in such models. A study in CaV2.1 mutant tottering mice by Zhou and co‐workers reported that presynaptic inhibition mediated by GABAB or α2‐adrenoceptor GPCRs was enhanced at excitatory PF–PC synapses, although this may be as consequence of a switch to a reliance on CaV2.2 (N‐type) channels for transmitter release (Zhou et al. 2003). Tottering mice also had a reduction in GABAA receptor expression, with specific deficits in granule cells (Kaja et al. 2007). We have shown that ataxic du2J mutant mice exhibit increased irregularity of PC and, to a lesser extent, granule cell firing in multi‐electrode array recordings from cerebellar brain slices (Wang et al. 2013). Of note, clear effects on PC firing regularity in du2J/du2J mice were not seen in heterozygous +/du2J mice, and the latter also lacked a clear behavioural ataxic phenotype. Importantly, the CB1R‐mediated inhibition at IN–PC synapses seen in litter‐matched controls was completely absent in both +/du2J and du2J/du2J mice. These data demonstrate that ataxic α2δ‐2‐deficient mice have aberrant presynaptic CB1R‐mediated signalling. The question arises as to whether deficiency in CB1R‐mediated signalling is involved in ataxia pathogenesis or whether it occurs as a result of the disease. It appears that, in this model, both alleles need to be affected in order for an ataxic phenotype to be seen (Wang et al. 2013), and thus progressive deficits may be associated with du2J mice. We saw no changes in PC firing regularity in response to CB1R ligands in wild‐type or du2J mice, consistent with a lack of postsynaptic CB1R effects in this model. We propose that such deficits occur due to compromised Ca2+ channel activity consequent to reduced presynaptic α2δ‐2 expression in du2J mice (Fig. 1). In this regard, α2δ‐2 subunits have been shown to be essential not only for Ca2+ channel trafficking (Dolphin, 2012), but also for synaptic function, the latter by increasing transmitter release probability and also protecting release from inhibitory effects of intracellular Ca2+ chelators (Hoppa et al. 2012).
There are few studies measuring CB1R expression in ataxic animals; we reported no clear changes in expression in the cerebellar cortex of +/du2J and du2J/du2J mice (Wang et al. 2013). In a recent study, post‐mortem cerebellar tissue from patients with SCAs, CB1R (and CB2R) expression was generally up‐regulated in PCs, and also in glial cells (Rodríguez‐Cueto et al. 2014 a). Of interest here was that CB1R expression was reported in PC soma and pinceau in SCA patients, but was confined largely to the pinceau in control patients. It is possible that upregulated postsynaptic CB1R expression may affect 2‐AG release in SCA patients; however, Rodriguez‐Cueto and co‐workers suggest that this CB1R expression is associated with degenerating PCs and may represent a marker for degeneration and/or a protective response against such degeneration. A further study in SCA patients reported an up‐regulation of eCB degradative fatty acid amide hydrolase and monoacylglycerol lipase (MAGL) enzymes (Rodríguez‐Cueto et al. 2014 b), proposed to lead to reduced eCB levels in disease. In these studies, a compensatory up‐regulation of cannabinoid receptor expression may occur as a consequence of reduced eCB levels; alternatively, it may be argued that eCBs are suppressed in order not to overactivate the system. Thus, it may also be possible to target eCB metabolizing enzymes for future therapeutic development. In this regard, potential avenues to increase 2‐AG include inhibition of MAGL, localized predominantly to the PF terminal (Tanimura et al. 2012), or activation of the biosynthetic enzyme diacylglycerol lipase‐α (DAGLα) localized predominantly to the base of postsynaptic dendritic spines (Yoshida et al. 2006) (see Abstract Figure). It is also clear that we will need to investigate potential changes to eCB signalling in different animal models of ataxia to inform development of the most useful therapeutic strategies and also to determine if any such changes represent useful markers for different forms of ataxia.
Do cannabinoids have therapeutic utility in cerebellar ataxia?
There are anecdotal reports that cannabis smokers can achieve symptom relief for several CNS disorders. Such evidence has fuelled the investigation of use of CB1R agonists as potential neuroprotective agents for a range of conditions including epilepsy, neurodegenerative diseases such as Alzheimer's, Parkinson's and Huntington's disease and appetitive disorders (Fernández‐Ruiz et al. 2011; Hill et al. 2012 a). Earlier evidence for ataxia is largely confined to two case studies which suggest that oral Δ9‐THC or marijuana improved motor coordination in some multiple sclerosis patients (Clifford, 1983; Meinck et al. 1989). At the clinical level, synthetic Δ9‐THC has been used in management of nausea, emesis and pain, and nabiximols (Sativex) (containing ∼1:1 Δ9‐THC:CBD) represents the first phytocannabinoid medicine, used as an oromucosal spray for pain and spasticity associated with multiple sclerosis (Hill et al. 2012 a); Sativex was also stated to delay onset of ataxia symptoms in the Medicines and Healthcare products Regulatory Agency (MHRA) Public Information Report UK/H/961/01/DC. Such reports contributed to fuelling a major review on the clinical effects of cannabinoids in ataxia associated with multiple sclerosis (Mills et al. 2007); although cannabinoids showed promise, it was concluded that better standardized measures of ataxia were needed to fully establish the utility of cannabis‐based medicines in ataxia. The role of cannabinoids in disease‐associated movement disorders and tremor has been further discussed more recently by Arjmand et al. (2015) and Kluger et al. (2015), who similarly concluded that further work on cannabinoids in different models of ataxia is warranted. In this regard, an interesting recent report suggests that a ‘Sativex‐like’ combination of Δ9‐THC and CBD, as well as the individual administration of Δ9‐THC or CBD, was able to improve motor deficits in a viral model of multiple sclerosis (Feliú et al. 2015). Clearly, studies which suggest CB1R activation may be useful in cerebellar ataxia contrast to preclinical data where CB1R agonists induce an ataxic phenotype (Lichtman et al. 1998; DeSanty & Dar, 2001; Patel & Hillard, 2001); however, these data do support the hypothesis that maintaining the correct balance in eCB system signalling is a major factor for proper control of motor coordination. This hypothesis is further supported by data from CaV2.1 mutants described above, where both decreases (Maejima et al. 2013) and increases in Ca2+ current (Gao et al. 2012) can produce an ataxia phenotype; moreover, deficits in delay eyeblink conditioning are reported for both CB1R agonists and CB1R antagonists/inverse agonists using the same experimental design (Steinmetz & Freeman, 2010). CB1R agonists have also been shown to possess functional selectively or ‘biased agonism’, whereby different ligands (including eCBs) preferentially activate different CB1R signalling pathways (Laprairie et al. 2014; Khajehali et al. 2015). Whilst, as argued above, it is likely that CB1Rs act predominantly on presynaptic CaV2.1 to reduce transmitter release in the cerebellar cortex, alternative signalling pathways include inhibition of cAMP or stimulation of phosphorylation of signal regulated kinases (Howlett et al. 2010). Thus, it could be argued that by using knowledge of biased agonism that we can target specific pathways associated with diseases, including cerebellar ataxia. Finally here, CB1R agonists such as Δ9‐THC have also been proposed to possess anti‐oxidant (Hampson et al. 1998) and anti‐inflammatory (although largely CB2R‐mediated) (Fernández‐Ruiz et al. 2011) properties; in common with other degenerative diseases, such properties may benefit the amelioration of cerebellar ataxia symptoms.
The demonstration that pre‐treatment with a CB1R antagonist prevents the induction of motor incoordination by CB1R agonists (DeSanty & Dar, 2001; Patel & Hillard, 2001) suggests that CB1R antagonists/inverse agonists may be protective in ataxia. The archetypal agent rimonabant was introduced as an anti‐obesity agent, but was withdrawn due to fears of increased suicidality and depression in patients (Nathan et al. 2011). Since then, therapeutic development of CB1R antagonists/inverse agonists has largely been put on hold. Interesting potential alternatives are CB1R negative allosteric antagonists, such as Org‐27569 and PSNCBAM‐1. These compounds have a somewhat unique pharmacological profile as they increase orthosteric agonist binding, but decrease agonist activity; more intriguingly, allosteric antagonism action is ligand‐dependent and also shows biased antagonism for different signalling pathways (Baillie et al. 2013). We have shown that such functional selectivity for PSNCBAM‐1 extends to effects on orthosteric ligands at IN–PC synapses in the cerebellar cortex (Wang et al. 2011); thus, PSNCBAM‐1 attenuated CP55940 agonist and AM251 antagonist effects, but had no clear effects against WIN 55,212‐2. Moreover, when applied alone, PSNCBAM‐1 was not associated with potentially deleterious effects on eCB tone, a concern associated with use of CB1R antagonists/inverse agonists such as rimonabant. These studies indicate that exogenous allosteric CB1R ligands have potential to fine tune eCB orthosteric agonist effects in a ligand‐ and/or cell signalling‐selective manner within the cerebellar cortex; moreover, biased antagonism effects may allow for further useful therapeutic development.
This review has focused on cannabinoids as agents acting on the eCB system in the cerebellum; moreover, the modulation of CB1Rs has been highlighted. It may transpire that, for exogenous compounds, targeting alternative modes of action offers improved therapeutic potential in diseases such as ataxia. The last few years have seen increasing calls for the use of medical marijuana to treat a range of disorders; of course, use of marijuana is intimately associated with psychoactive effects of the CB1R partial agonist Δ9‐THC. Therapeutically, a more attractive option may be use of non‐psychoactive pCBs. Thus, CBD and cannabidivarin (CBDV) have reported utility in epilepsy and, potentially, other CNS disorders (Hill et al. 2012 b, 2013; Devinsky et al. 2014). The demonstration that Sativex can improve motor activity in multiple sclerosis (Feliú et al. 2015) is consistent with beneficial effects of a CB1R activator (Δ9‐THC) in combination with CBD as a potential ameliorating agent for unwanted Δ9‐THC effects (McPartland et al. 2015); for example, CBD alone was also effective in improving motor deficits, potentially via an action on peroxisome proliferator‐activated receptor γ (PPARγ) receptors (Feliú et al. 2015). In this regard, CBD has recently been awarded orphan drug status for the severe childhood epilepsy Dravet syndrome and is currently progressing well through clinical trials. CBD and CBDV have only low affinity at CB1Rs and CBD has been proposed, amongst other possibilities, to act at alternative GPCRs or at transient receptor potential ion channels or, possibly, to augment eCB tone via effects on metabolic enzymes (Hill et al. 2012 a; McPartland et al. 2015). A recent study has proposed that CBD, as well as having CB1R‐independent actions, may also act as a CB1R negative allosteric antagonist (Laprairie et al. 2015); therefore, CBD may share useful properties of this class of agents discussed above. It is also of interest that the hypophagic effects of the allosteric antagonist Org27569 have been suggested to occur independently of CB1Rs (Ding et al. 2014; Gamage et al. 2014). Thus, the use of cannabinoids with CB1R‐independent and/or allosteric actions should also be considered.
In the future, it will be of interest in particular to test agents such as Sativex, and perhaps CBD as an individual compound, in cerebellar ataxia. There are a number of general points to consider, including whether deficits in CB1R‐mediated signalling are hallmark characteristics of different forms of ataxia, how best to target such deficits and whether aberrant cannabinergic signalling represents a useful biomarker for early or asymptomatic cerebellar ataxia. The answer to such questions will go some way to determining if modulation of the eCB system has therapeutic utility in cerebellar ataxia.
Additional information
Competing interests
The author has no conflict of interest.
Funding
Work in my group included here was supported by The Wellcome Trust and an Ataxia UK Postgraduate Fellowship awarded to Xiaowei Wang, who also received a University of Reading Postgraduate Research Studentship Award and was co‐supervised by Prof. Benjamin Whalley. I also acknowledge the support of GW Pharmaceuticals in supplying material and associated grant funding during some of this work.
Biography
Gary Stephens held postdoctoral and Fellowship positions with Wyeth Research, Imperial College and University College London before moving to University of Reading in 2005, where he is now Professor of Pharmacology and Director of Pharmacology and Therapeutics. His research interests are in the modulation of presynaptic calcium channels and G protein‐coupled receptors, with a particular interest in cerebellar function.

This review was presented at the symposium “Mechanisms of cerebellar ataxias and neurodegeneration”, which took place at Ageing and Degeneration: A Physiological Perspective in Edinburgh, UK, 10–11 April 2015.
References
- Aiba A, Kano M, Chen C, Stanton ME, Fox GD, Herrup K, Zwingman TA & Tonegawa S (1994). Deficient cerebellar long‐term depression and impaired motor learning in mGluR1 mutant mice. Cell 79, 377–388. [PubMed] [Google Scholar]
- Arjmand S, Vaziri Z, Behzadi M, Abbassian H, Stephens GJ & Shabani M (2015). Cannabinoids and tremor induced by motor‐related disorders: friend or foe? Neurotherapeutics 12, 778–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baillie GL, Horswill JG, Anavi‐Goffer S, Reggio PH, Bolognini D, Abood ME, McAllister S, Strange PG, Stephens GJ, Pertwee RG & Ross RA (2013). CB1 receptor allosteric modulators display both agonist and signaling pathway specificity. Mol Pharmacol 83, 322–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barclay J, Balaguero N, Mione M, Ackerman SL, Letts VA, Brodbeck J, Canti C, Meir A, Page KM, Kusumi K, Perez‐Reyes E, Lander ES, Frankel WN, Gardiner RM, Dolphin AC & Rees M (2001). Ducky mouse phenotype of epilepsy and ataxia is associated with mutations in the Cacna2d2 gene and decreased calcium channel current in cerebellar PCs. J Neurosci 21, 6095–6104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilkei‐Gorzo A, Racz I, Valverde O, Otto M, Michel K, Sastre M & Zimmer A (2005). Early age‐related cognitive impairment in mice lacking cannabinoid CB1 receptors. Proc Natl Acad Sci USA 102, 15670–15675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodbeck J, Davies A, Courtney JM, Meir A, Balaguero N, Canti C, Moss FJ, Page KM, Pratt WS, Hunt SP, Barclay J, Rees M & Dolphin AC (2002). The ducky mutation in Cacna2d2 results in altered Purkinje cell morphology and is associated with the expression of a truncated α2δ‐2 protein with abnormal function. J Biol Chem 277, 7684–7693. [DOI] [PubMed] [Google Scholar]
- Brown SP, Safo PK & Regehr WG (2004). Endocannabinoids inhibit transmission at granule cell to Purkinje cell synapses by modulating three types of presynaptic calcium channels. J Neurosci 24, 5623–5631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Kano M, Abeliovich A, Chen L, Bao S, Kim JJ, Hashimoto K, Thompson RF & Tonegawa S (1995). Impaired motor coordination correlates with persistent multiple climbing fiber innervation in PKCγ mutant mice. Cell 83, 1233–1242. [DOI] [PubMed] [Google Scholar]
- Clifford DB (1983). Tetrahydrocannabinol for tremor in multiple sclerosis. Ann Neurol 13, 669–671. [DOI] [PubMed] [Google Scholar]
- Cole RL, Lechner SM, Williams ME, Prodanovich P, Bleicher L, Varney MA & Gu G (2005). Differential distribution of voltage‐gated calcium channel alpha‐2 delta (α2δ) subunit mRNA‐containing cells in the rat central nervous system and the dorsal root ganglia. J Comp Neurol 491, 246–269. [DOI] [PubMed] [Google Scholar]
- Cooper JM, Korlipara LV, Hart PE, Bradley JL & Schapira AH (2008). Coenzyme Q10 and vitamin E deficiency in Friedreich's ataxia: predictor of efficacy of vitamin E and CoQ10 therapy. Eur J Neurol 15, 1371–1379. [DOI] [PubMed] [Google Scholar]
- DeSanty KP & Dar MS (2001). Cannabinoid‐induced motor incoordination through the cerebellar CB1 receptor in mice. Pharmacol Biochem Behav 69, 251–259. [DOI] [PubMed] [Google Scholar]
- Devinsky O, Cilio MR, Cross H, Fernandez‐Ruiz J, French J, Hill C, Katz R, Di Marzo V, Jutras‐Aswad D, Notcutt WG, Martinez‐Orgado J, Robson PJ, Rohrback BG, Thiele E, Whalley B & Friedman D (2014). Cannabidiol: Pharmacology and potential therapeutic role in epilepsy and other neuropsychiatric disorders. Epilepsia 55, 791–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Y, Qiu Y, Jing L, Thorn DA, Zhang Y & Li JX (2014). Behavioral effects of the cannabinoid CB1 receptor allosteric modulator ORG27569 in rats. Pharmacol Res Perspect 2, e00069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolphin AC (2012). Calcium channel auxiliary α2δ and β subunits: trafficking and one step beyond. Nat Rev Neurosci 13, 542–555. [DOI] [PubMed] [Google Scholar]
- Donato R, Page KM, Koch D, Nieto‐Rostro M, Foucault I, Davies A, Wilkinson T, Rees M, Edwards FA & Dolphin AC (2006). The ducky2J mutation in Cacna2d2 results in reduced spontaneous Purkinje cell activity and altered gene expression. J Neurosci 26, 12576–12586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feliú A, Moreno‐Martet M, Mecha M, Carrillo‐Salinas FJ, de Lago E, Fernández‐Ruiz J & Guaza C (2015). A Sativex®‐like combination of phytocannabinoids as a disease‐modifying therapy in a viral model of multiple sclerosis. Br J Pharmacol 172, 3579–3595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández‐Ruiz J, Moreno‐Martet M, Rodríguez‐Cueto C, Palomo‐Garo C, Gómez‐Cañas M, Valdeolivas S, Guaza C, Romero J, Guzmán M, Mechoulam R & Ramos JA (2011). Prospects for cannabinoid therapies in basal ganglia disorders. Br J Pharmacol 163, 1365–1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forti L, Pouzat C & Llano I (2000). Action potential‐evoked Ca2+ signals and calcium channels in axons of developing rat cerebellar interneurones. J Physiol 527, 33–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galante M & Diana MA (2004). Group I metabotropic glutamate receptors inhibit GABA release at interneuron‐Purkinje cell synapses through endocannabinoid production. J Neurosci 24, 4865–4874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamage TF, Ignatowska‐Jankowska BM, Wiley JL, Abdelrahman M, Trembleau L, Greig IR, Thakur GA, Tichkule R, Poklis J, Ross RA, Pertwee RG & Lichtman AH (2014). In‐vivo pharmacological evaluation of the CB1‐receptor allosteric modulator Org‐27569. Behav Pharmacol 25, 182–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Z, Todorov B, Barrett CF, van Dorp S, Ferrari MD, van den Maagdenberg AM, De Zeeuw CI & Hoebeek FE (2012). Cerebellar ataxia by enhanced CaV2.1 currents is alleviated by Ca2+‐dependent K+‐channel activators in Cacna1a(S218L) mutant mice. J Neurosci 32, 15533–15546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J & Ikeda SR (2004). Endocannabinoids modulate N‐type calcium channels and G‐protein‐coupled inwardly rectifying potassium channels via CB1 cannabinoid receptors heterologously expressed in mammalian neurons. Mol Pharmacol 65, 665–674. [DOI] [PubMed] [Google Scholar]
- Hampson AJ, Grimaldi M, Axelrod J & Wink D (1998). Cannabidiol and (‐)Δ9‐tetrahydrocannabinol are neuroprotective antioxidants. Proc Natl Acad Sci USA 95, 8268–8273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herkenham M, Groen BG, Lynn AB, De Costa BR & Richfield EK (1991). Neuronal localization of cannabinoid receptors and second messengers in mutant mouse cerebellum. Brain Res 552, 301–310. [DOI] [PubMed] [Google Scholar]
- Hill AJ, Mercier MS, Hill TD, Glyn SE, Jones NA, Yamasaki Y, Futamura T, Duncan M, Stott CG, Stephens GJ, Williams CM & Whalley BJ (2012. b). Cannabidivarin is anticonvulsant in mouse and rat in vitro and in seizure models. Br J Pharmacol 167, 1629–1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill AJ, Williams CM, Whalley BJ & Stephens GJ (2012. a). Phytocannabinoids as novel therapeutic agents in CNS disorders. Pharmacol Ther 133, 79–97. [DOI] [PubMed] [Google Scholar]
- Hill TD, Cascio MG, Romano B, Duncan M, Pertwee RG, Williams CM, Whalley BJ & Hill AJ (2013). Cannabidivarin‐rich cannabis extracts are anticonvulsant in mouse and rat via a CB1 receptor‐independent mechanism. Br J Pharmacol 170, 679–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoppa MB, Lana B, Margas W, Dolphin AC & Ryan TA (2012). α2δ expression sets presynaptic calcium channel abundance and release probability. Nature 486, 122–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horswill JG, Bali U, Shaaban S, Keily JF, Jeevaratnam P, Babbs AJ, Reynet C & Wong Kai In P (2007). PSNCBAM‐1, a novel allosteric antagonist at cannabinoid CB1 receptors with hypophagic effects in rats. Br J Pharmacol 152, 805–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howlett AC, Blume LC & Dalton GD (2010). CB1 cannabinoid receptors and their associated proteins. Curr Med Chem 17, 1382–1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito M (1989). Long‐term depression. Annu Rev Neurosci 12, 85–102. [DOI] [PubMed] [Google Scholar]
- Jun K, Piedras‐Rentería ES, Smith SM, Wheeler DB, Lee SB, Lee TG, Chin H, Adams ME, Scheller RH, Tsien RW & Shin HS (1999). Ablation of P/Q‐type Ca2+ channel currents, altered synaptic transmission, and progressive ataxia in mice lacking the α1A‐subunit. Proc Natl Acad Sci USA 96, 15245–15250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaja S, Hann V, Payne HL & Thompson CL (2007). Aberrant cerebellar granule cell‐specific GABAA receptor expression in the epileptic and ataxic mouse mutant, Tottering . Neuroscience 148, 115–125. [DOI] [PubMed] [Google Scholar]
- Kano M, Ohno‐Shosaku T, Hashimotodani Y, Uchigashima M & Watanabe M (2009). Endocannabinoid‐mediated control of synaptic transmission. Physiol Rev 89, 309–380. [DOI] [PubMed] [Google Scholar]
- Kawamura Y, Fukaya M, Maejima T, Yoshida T, Miura E, Watanabe M, Ohno‐Shosaku T & Kano M (2006). CB1 is the major cannabinoid receptor at excitatory presynaptic site in the hippocampus and cerebellum. J Neurosci 26, 2991–3001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khajehali E, Malone DT, Glass M, Sexton PM, Christopoulos A & Leach K (2015). Biased agonism and biased allosteric modulation at the CB1 cannabinoid receptor. Mol Pharmacol 88, 368–379. [DOI] [PubMed] [Google Scholar]
- Kishimoto Y & Kano M (2006). Endogenous cannabinoid signaling through the CB1 receptor is essential for cerebellum‐dependent discrete motor learning. J Neurosci 26, 8829–8837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klockgether T (2011). Update on degenerative ataxias. Curr Opin Neurol 24, 339–345. [DOI] [PubMed] [Google Scholar]
- Kluger B, Triolo P, Jones W & Jankovic J (2015). The therapeutic potential of cannabinoids for movement disorders. Mov Disord 30, 313–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitzer AC, Carter AG & Regehr WG (2002). Inhibition of interneuron firing extends the spread of endocannabinoid signaling in the cerebellum. Neuron 34, 787–796. [DOI] [PubMed] [Google Scholar]
- Kulik A, Nakadate K, Hagiwara A, Fukazawa Y, Luján R, Saito H, Suzuki N, Futatsugi A, Mikoshiba K, Frotscher M & Shigemoto R (2004). Immunocytochemical localization of the α1A subunit of the P/Q‐type calcium channel in the rat cerebellum. Eur J Neurosci 19, 2169–2178. [DOI] [PubMed] [Google Scholar]
- Laprairie RB, Bagher AM, Kelly ME & Denovan‐Wright EM (2015). Cannabidiol is a negative allosteric modulator of the type 1 cannabinoid receptor. Br J Pharmacol 172, 4790–4805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laprairie RB, Bagher AM, Kelly ME, Dupré DJ & Denovan‐Wright EM (2014). Type 1 cannabinoid receptor ligands display functional selectivity in a cell culture model of striatal medium spiny projection neurons. J Biol Chem 289, 24845–24862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtman AH, Wiley JL, LaVecchia KL, Neviaser ST, Arthur DB, Wilson DM & Martin BR (1998). Effects of SR 141716A after acute or chronic cannabinoid administration in dogs. Eur J Pharmacol 357, 139–148. [DOI] [PubMed] [Google Scholar]
- Lonchamp E, Dupont JL, Doussau F, Shin HS, Poulain B & Bossu JL (2009). Deletion of CaV2.1(α1A) subunit of Ca2+‐channels impairs synaptic GABA and glutamate release in the mouse cerebellar cortex in cultured slices. Eur J Neurosci 30, 2293–2307. [DOI] [PubMed] [Google Scholar]
- Lynch DR, Perlman SL & Meier T (2010). A phase 3, double‐blind, placebo‐controlled trial of idebenone in Friedreich ataxia. Arch Neurol 67, 941–947. [DOI] [PubMed] [Google Scholar]
- Ma YL, Weston SE, Whalley BJ & Stephens GJ (2008). The phytocannabinoid Δ9‐tetrahydrocannabivarin modulates inhibitory neurotransmission in the cerebellum. Br J Pharmacol 154, 204–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackie K & Hille B (1992). Cannabinoids inhibit N‐type calcium channels in neuroblastoma‐glioma cells. Proc Natl Acad Sci USA 89, 3825–3829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maejima T, Wollenweber P, Teusner LU, Noebels JL, Herlitze S & Mark MD (2013). Postnatal loss of P/Q‐type channels confined to rhombic‐lip‐derived neurons alters synaptic transmission at the parallel fiber to purkinje cell synapse and replicates genomic Cacna1a mutation phenotype of ataxia and seizures in mice. J Neurosci 33, 5162–5174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mark MD, Maejima T, Kuckelsberg D, Yoo JW, Hyde RA, Shah V, Gutierrez D, Moreno RL, Kruse W, Noebels JL & Herlitze S (2011). Delayed postnatal loss of P/Q‐type calcium channels recapitulates the absence epilepsy, dyskinesia, and ataxia phenotypes of genomic Cacna1A mutations. J Neurosci 31, 4311–4326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPartland JM, Duncan M, Di Marzo V & Pertwee R (2015). Are cannabidiol and Δ9‐tetrahydrocannabivarin negative modulators of the endocannabinoid system? A systematic review. Br J Pharmacol 172, 737–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meinck HM, Schönle PW & Conrad B (1989). Effect of cannabinoids on spasticity and ataxia in multiple sclerosis. J Neurol 236, 120–122. [DOI] [PubMed] [Google Scholar]
- Mills RJ, Yap L & Young CA (2007). Treatment for ataxia in multiple sclerosis. Cochrane Database Syst Rev 1, CD005029. [DOI] [PubMed] [Google Scholar]
- Mintz IM, Sabatini BL & Regehr WG (1995). Calcium control of transmitter release at a cerebellar synapse. Neuron 15, 675–688. [DOI] [PubMed] [Google Scholar]
- Mintz IM, Venema VJ, Swiderek KM, Lee TD, Bean BP & Adams ME (1992). P‐type calcium channels blocked by the spider toxin omega‐Aga‐IVA. Nature 355, 827–829. [DOI] [PubMed] [Google Scholar]
- Nathan PJ, O'Neill BV, Napolitano A & Bullmore ET (2011). Neuropsychiatric adverse effects of centrally acting antiobesity drugs. CNS Neurosci Ther 17, 490–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno‐Shosaku T & Kano M (2014). Endocannabinoid‐mediated retrograde modulation of synaptic transmission. Curr Opin Neurobiol 29, 1–8. [DOI] [PubMed] [Google Scholar]
- Patel S & Hillard CJ (2001). Cannabinoid CB1 receptor agonists produce cerebellar dysfunction in mice. J Pharmacol Exp Ther 297, 629–637. [PubMed] [Google Scholar]
- Pertwee RG, Howlett AC, Abood ME, Alexander SP, Di Marzo V, Elphick MR, Greasley PJ, Hansen HS, Kunos G, Mackie K, Mechoulam R & Ross RA (2010). International Union of Basic and Clinical Pharmacology. LXXIX. Cannabinoid receptors and their ligands: beyond CB₁ and CB₂. Pharmacol Rev 62, 588–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietrobon D (2010). CaV2.1 channelopathies. Pflugers Arch 460, 375–393. [DOI] [PubMed] [Google Scholar]
- Price MR, Baillie GL, Thomas A, Stevenson LA, Easson M, Goodwin R, McLean A, McIntosh L, Goodwin G, Walker G, Westwood P, Marrs J, Thomson F, Cowley P, Christopoulos A, Pertwee RG & Ross RA (2005). Allosteric modulation of the cannabinoid CB1 receptor. Mol Pharmacol 68, 1484–1495. [DOI] [PubMed] [Google Scholar]
- Rajakulendran S, Kaski D & Hanna MG (2012). Neuronal P/Q‐type calcium channel dysfunction in inherited disorders of the CNS. Nat Rev Neurol 8, 86–96. [DOI] [PubMed] [Google Scholar]
- Rodríguez‐Cueto C, Benito C, Fernández‐Ruiz J, Romero J, Hernández‐Gálvez M & Gómez‐Ruiz M (2014. a). Changes in CB1 and CB2 receptors in the post‐mortem cerebellum of humans affected by spinocerebellar ataxias. Br J Pharmacol 171, 1472–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez‐Cueto C, Benito C, Romero J, Hernández‐Gálvez M, Gómez‐Ruiz M & Fernández‐Ruiz J (2014. b). Endocannabinoid‐hydrolysing enzymes in the post‐mortem cerebellum of humans affected by hereditary autosomal dominant ataxias. Pathobiology 81, 149–159. [DOI] [PubMed] [Google Scholar]
- Regehr WG & Mintz IM (1994). Participation of multiple calcium channel types in transmission at single climbing fiber to Purkinje cell synapses. Neuron 12, 605–613. [DOI] [PubMed] [Google Scholar]
- Safo PK & Regehr WG (2005). Endocannabinoids control the induction of cerebellar LTD. Neuron 48, 647–659. [DOI] [PubMed] [Google Scholar]
- Shibusawa N, Hashimoto K & Yamada M (2008). Thyrotropin‐releasing hormone (TRH) in the cerebellum. Cerebellum 7, 84–95. [DOI] [PubMed] [Google Scholar]
- Skosnik PD, Edwards CR, O'Donnell BF, O'Donnell BF, Steffen A, Steinmetz JE & Hetrick WP (2008). Cannabis use disrupts eyeblink conditioning: evidence for cannabinoid modulation of cerebellar‐dependent learning. Neuropsychopharmacol 33, 1432–1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner H, Bonner TI, Zimmer AM, Kitai S & Zimmer A (1999). Altered gene expression in striatal projection neurons in CB1 cannabinoid receptor knockout mice. Proc Natl Acad Sci USA 96, 5786–5790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinmetz AB & Freeman JH (2010). Central cannabinoid receptors modulate acquisition of eyeblink conditioning. Learn Mem 17, 571–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens GJ (2009). G‐protein‐coupled‐receptor‐mediated presynaptic inhibition in the cerebellum. Trends Pharmacol Sci 30, 421–430. [DOI] [PubMed] [Google Scholar]
- Stephens GJ, Morris NP, Fyffe RE & Robertson B (2001). The CaV2.1/α1A (P/Q‐type) voltage‐dependent calcium channel mediates inhibitory neurotransmission onto mouse cerebellar Purkinje cells. Eur J Neurosci 13, 1902–1912. [DOI] [PubMed] [Google Scholar]
- Szabo B, Than M, Thorn D & Wallmichrath I (2004). Analysis of the effects of cannabinoids on synaptic transmission between basket and Purkinje cells in the cerebellar cortex of the rat. J Pharmacol Exp Ther 310, 915–925. [DOI] [PubMed] [Google Scholar]
- Szabo B, Urbanski MJ, Bisogno T, Di Marzo V, Mendiguren A, Baer WU & Freiman I (2006). Depolarization‐induced retrograde synaptic inhibition in the mouse cerebellar cortex is mediated by 2‐arachidonoylglycerol. J Physiol 577, 263–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi KA & Linden DJ (2000). Cannabinoid receptor modulation of synapses received by cerebellar Purkinje cells. J Neurophysiol 83, 1167–1180. [DOI] [PubMed] [Google Scholar]
- Tanimura A, Kawata S, Hashimoto K & Kano M (2009). Not glutamate but endocannabinoids mediate retrograde suppression of cerebellar parallel fiber to Purkinje cell synaptic transmission in young adult rodents. Neuropharmacol 57, 157–163. [DOI] [PubMed] [Google Scholar]
- Tanimura A, Uchigashima M, Yamazaki M, Uesaka N, Mikuni T, Abe M, Hashimoto K, Watanabe M, Sakimura K & Kano M (2012). Synapse type‐independent degradation of the endocannabinoid 2‐arachidonoylglycerol after retrograde synaptic suppression. Proc Natl Acad Sci USA 109, 12195–12200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todorov B, Kros L, Shyti R, Plak P, Haasdijk ED, Raike RS, Frants RR, Hess EJ, Hoebeek FE, De Zeeuw CI & van den Maagdenberg AM (2012). Purkinje cell‐specific ablation of CaV2.1 channels is sufficient to cause cerebellar ataxia in mice. Cerebellum 11, 246–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsou K, Brown S, Sañudo‐Peña MC, Mackie K & Walker JM (1997). Immunohistochemical distribution of cannabinoid CB1 receptors in the rat central nervous system. Neuroscience 83, 393–411. [DOI] [PubMed] [Google Scholar]
- Twitchell W, Brown S & Mackie K (1997). Cannabinoids inhibit N‐ and P/Q‐type calcium channels in cultured rat hippocampal neurons J Neurophysiol 78, 43–50. [DOI] [PubMed] [Google Scholar]
- Walter JT, Alviña K, Womack MD, Chevez C & Khodakhah K (2006). Decreases in the precision of Purkinje cell pacemaking cause cerebellar dysfunction and ataxia. Nat Neurosci 9, 389–397. [DOI] [PubMed] [Google Scholar]
- Wang X, Horswill JG, Whalley BJ & Stephens GJ (2011). Effects of the allosteric antagonist PSNCBAM‐1 on CB1 receptor modulation in the cerebellum. Mol Pharmacol 79, 758–767. [DOI] [PubMed] [Google Scholar]
- Wang X, Whalley BJ & Stephens GJ (2013). The du2J mouse model of ataxia and absence epilepsy has deficient cannabinoid CB1 receptor‐mediated signalling. J Physiol 591, 3919–3933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westenbroek RE, Sakurai T, Elliott EM, Hell JW, Starr TV, Snutch TP & Catterall WA (1995). Immunochemical identification and subcellular distribution of the alpha 1A subunits of brain calcium channels. J Neurosci 15, 6403–6418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamasaki M, Hashimoto K & Kano M (2006). Miniature synaptic events elicited by presynaptic Ca2+ rise are selectively suppressed by cannabinoid receptor activation in cerebellar Purkinje cells. J Neurosci 26, 86–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida T, Fukaya M, Uchigashima M, Miura E, Kamiya H, Kano M & Watanabe M (2006). Localization of diacylglycerol lipase‐alpha around postsynaptic spine suggests close proximity between production site of an endocannabinoid, 2‐arachidonoyl‐glycerol, and presynaptic cannabinoid CB1 receptor. J Neurosci 26, 4740–4751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou YD, Turner TJ & Dunlap K (2003). Enhanced G protein‐dependent modulation of excitatory synaptic transmission in the cerebellum of the Ca2+ channel‐mutant mouse, tottering . J Physiol 547, 497–507. [DOI] [PMC free article] [PubMed] [Google Scholar]