

Abstract
A better understanding of the cellular physiological role that plasma membrane lipids, fatty acids and sterols play in various cellular systems may yield more insight into how cellular and whole organ function is altered during the ageing process. Membrane lipid rafts (MLRs) within the plasma membrane of most cells serve as key organizers of intracellular signalling and tethering points of cytoskeletal components. MLRs are plasmalemmal microdomains enriched in sphingolipids, cholesterol and scaffolding proteins; they serve as a platform for signal transduction, cytoskeletal organization and vesicular trafficking. Within MLRs are the scaffolding and cholesterol binding proteins named caveolin (Cav). Cavs not only organize a multitude of receptors including neurotransmitter receptors (NMDA and AMPA receptors), signalling proteins that regulate the production of cAMP (G protein‐coupled receptors, adenylyl cyclases, phosphodiesterases (PDEs)), and receptor tyrosine kinases involved in growth (Trk), but also interact with components that modulate actin and tubulin cytoskeletal dynamics (e.g. RhoGTPases and actin binding proteins). MLRs are essential for the regulation of the physiology of organs such as the brain, and age‐related loss of cholesterol from the plasma membrane leads to loss of MLRs, decreased presynaptic vesicle fusion, and changes in neurotransmitter release, all of which contribute to different forms of neurodegeneration. Thus, MLRs provide an active membrane domain that tethers and reorganizes the cytoskeletal machinery necessary for membrane and cellular repair, and genetic interventions that restore MLRs to normal cellular levels may be exploited as potential therapeutic means to reverse the ageing and neurodegenerative processes.
Abbreviations
- AD
Alzheimer's disease
- AMPAR
α‐amino‐3‐hydroxy‐5‐methyl‐4‐isoxazolepropionic acid receptor
- BBB
blood–brain barrier
- BDNF
brain derived neurotrophic factor
- Cav‐1
caveolin‐1
- ERK1/2
extracellular response kinase
- GPCR
G protein‐coupled receptor
- LDL
low‐density lipoprotein
- LTD
long‐term depression
- LTP
long‐term potentiation
- MLR
membrane lipid raft
- NMDAR
N‐methyl‐d‐aspartate receptor
- PD
Parkinson's disease
- Trk
tyrosine receptor kinase
Introduction
Genomics and proteomics have significantly advanced our basic understanding of biological functions and of disease processes. By contrast, research on lipids, fatty acids and sterols, which are key components of plasma membranes, has not received similar attention. Membranes are self‐assembled lipid complexes that provide the structural foundation for cells and serve as a source of energy that contributes to cell function and physiology. Membrane lipids indirectly control a great variety of biological functions through their regulation of enzymes, receptors, neurotransmitter clustering and activity (as already discussed previously), and therefore necessitate much more focus in the post‐genomic era. Membrane lipids provide order within a disordered environment and serve to encapsulate and thus protect enzymes and genes from the external environment. Alterations in membrane lipids occur as we age, and these changes have major consequences for brain structure, function and behaviour.
Ageing and decreased neuroplasticity
The population over 65 in the United States (U.S.) will increase to ∼87 million by 2050 (Leal & Yassa, 2013) with age alone being the greatest risk factor for developing Alzheimer's disease (AD) and other forms of neurodegeneration such as Parkinson's disease (PD) and depression. After AD, PD is the second most common neurodegenerative disease in individuals over 65 (Gresack et al. 2010; Paul et al. 2015). Individuals with AD and PD exhibit severe deficits in cognitive and motor function, sleep disorders, and deficits in attention, short‐term memory and executive function. In some cases, patients with PD also manifest β‐amyloid aggregates commonly found in AD (Mandal et al. 2006; Kalaitzakis et al. 2008). Depression, which is associated with co‐morbid medical illness and suicide, is a major problem among younger individuals and is also prevalent in the elderly population. With no prevention or treatment, individuals suffering from age‐related neuropathologies could reach 11–16 million in the U.S. by 2050; healthcare costs for AD alone were estimated at $200 billion in the U.S. in 2012, and with PD it is difficult to truly measure the financial burden on both families and the healthcare system. In addition to age‐related AD and PD, depression among the elderly has been estimated to increase health care costs by 50% accompanied by an increase in outpatient costs by 43–52% in these individuals compared to non‐depressed patients (Aziz & Steffens, 2013). Therefore, due to the increased ageing population and associated neurodegenerative illnesses, there is a great demand for therapeutic interventions to reverse age‐related neurological decline.
Ageing, independent of AD and PD pathology, is associated with a decline in certain cognitive abilities such as mental speed, executive function, episodic memory, working memory, short‐term recollection, speed of processing new information, and spatial memory (Remy et al. 2004; Fjell et al. 2014). These behavioural deficits are due to losses in synaptic contacts, changes in neuronal morphology, reduced capacity to evoke neuroplasticity and dendritic branching (Henley & Wilkinson, 2013), and reductions in cortical (pre‐frontal, parietal, temporal and entorhinal) and hippocampal volume (Fjell et al. 2009, 2013, 2014). It has been proposed that certain areas of the brain that are more plastic throughout life (e.g. prefrontal cortex, limbic system) are in fact most vulnerable to age (Mesulam, 1999). The increased demand for life‐long plasticity makes these dynamic regions more susceptible to lesions and accumulation of pathology throughout life and is well summarized as ‘the inevitable manifestations of a failure to keep up with the increasingly more burdensome work of plasticity’ (Mesulam, 1999).
On a cellular level, neurons exhibit changes in plasma membrane biophysical properties (e.g. decreased cholesterol, gangliosides and phosphoinositides) (Ledesma et al. 2012), and impaired cholesterol synthesis and lipoprotein transport (Martin et al. 2010), all of which contribute to alterations in the molecular composition of synaptic membranes (Henley & Wilkinson, 2013). Additionally, age is accompanied by reduced membrane‐associated synaptic proteins (Jiang et al. 2010) and diminished presynaptic vesicle exocytosis and subsequent neurotransmitter release, the latter due to plasmalemmal cholesterol depletion or redistribution and subsequently altered membrane recruitment of SNARE complexes (Ledesma et al. 2012). Evidence also exists for age‐related deficits in cAMP‐mediated signalling, impairment in synaptic plasticity, and decreases in hippocampal long‐term potentiation (LTP) (Titus et al. 2013 a). The age‐related changes in plasmalemmal lipid composition and concomitant reduction in functional synaptic components (both pre‐ and postsynaptic) may contribute to the incapacity of the aged brain to evoke structural and functional plasticity (Wood et al. 2011; Ledesma et al. 2012; Morrison & Baxter, 2014). Although some therapeutic approaches attempt to regenerate the neurodegenerative or injured brain through delivery of exogenous pro‐growth stimuli (such as neurotrophins; Conte et al. 2008), the relative lack of efficacy of these approaches may be due in part to the reduction of cell surface receptors and their downstream effector molecules from the appropriate plasma membrane microdomains, thereby limiting transduction of extracellular signals. Genetic interventions that restore normal neuronal plasma membrane biophysical properties that facilitate pro‐growth signalling may enhance functional plasticity and reverse age‐related behavioural decline. Thus, therapies that activate essential molecular ‘switches’, which provide the structural maintenance of the brain, may allow for functional adaptations to the changing environment (i.e. synaptic plasticity).
Synaptic plasticity and age‐related deficits
Genetic profiling studies have elucidated more specifically the underlying mechanisms that lead to alterations in synaptic plasticity in the aged brain (Conte et al. 2008; Neumann et al. 2010). Important components essential to normal synaptic plasticity are plasma membrane receptors that regulate calcium flux and homeostasis (e.g. N‐methyl‐d‐aspartate receptors (NMDARs) and α‐amino‐3‐hydroxy‐5‐methyl‐4‐isoxazolepropionic acid receptors (AMPARs)); these receptors are decreased in the aged brain (Head et al. 2010). Others have shown that calcium channels, intraneuronal calcium buffering capacity, and receptors involved in calcium entry are decreased with age and render the brain more vulnerable to excitotoxic stress (Stiess & Bradke, 2011; Breceda & Dromerick, 2013; Phillips et al. 2014). This dysregulation of calcium handling and increased vulnerability to excitotoxicity could explain why ischaemic tolerance is absent in the aged brain (Lowery & Van Vactor, 2009). In addition to decreases in synaptic glutamate receptors, their downstream kinases (i.e. calcium–calmodulin kinases) (Dent et al. 2011), G protein‐coupled receptors (GPCRs), adenylyl cyclases, and neurotrophin signalling are also significantly downregulated in the aged brain (Conte et al. 2008; Hall & Lalli, 2010; Stiess & Bradke, 2011; Head et al. 2014). These age‐related deficits in functional synaptic signalling components dampen the regenerative capacity of the aged brain to re‐organize its neuronal circuitry. Although some therapeutic approaches attempt to regenerate the neurodegenerative or injured brain through delivery of exogenous pro‐growth stimuli such as brain derived neurotrophic factor (BDNF) (Dent & Gertler, 2003; Bassani & Passafaro, 2012), the ineffectiveness of these approaches is likely to be due to the absence of the coupling of membrane receptors to their downstream effector molecules via key scaffolding proteins, an essential bridge for facilitating proper signal transduction. What therefore appears to be required for neuronal regeneration in the aged brain is not only receptor expression but also expression of downstream signalling molecules scaffolded in close proximity to these plasma membrane receptors in order to achieve functional signal transduction (Hall & Lalli, 2010).
Cholesterol in the brain
Cholesterol is found in all animal cellular membranes and is essential for plasma membrane structure and fluidity. Although the brain comprises only 2–5% of total body mass, it contains 20–25% of the body's cholesterol (Russell et al. 2009). The amount of cellular cholesterol is proportional to membrane surface area. The brain contains approximately 15 mg g−1 of tissue, followed by adrenal glands (9 mg g−1), which use it for steroid hormone synthesis, and the lung (6 mg g−1), which uses it to increase alveolar surface area for facilitating gas exchange. To better understand the physiological significance of cholesterol, one needs to comprehensively dissect the influence of its biologically active oxidized forms, i.e. oxysterols (Lutjohann et al. 1996; Lund et al. 2003; Xie et al. 2003; Heverin et al. 2005, 2015; Martin et al. 2014).
Cellular cholesterol can be obtained by receptor‐mediated endocytosis of lipoproteins or synthesized in the endoplasmic reticulum and distributed to cellular membranes by way of vesicular and non‐vesicular transport (Martin et al. 2014). The latter involves a biosynthetic pathway comprising 20 enzymes. Several of these enzymes have received much attention due to their role in cardiovascular disease and neuroprotection (Liscum et al. 1983; Brown & Goldstein, 1997; Peri & Serio, 2008). 3‐Hydroxy‐3‐methyl‐glutaryl (HMG)‐CoA reductase is the rate‐controlling enzyme that converts 3‐methyl‐glutaryl‐CoA into mevalonate (Liscum et al. 1983), which is subsequently converted into cholesterol and other isoprenoids. Another enzyme, 3‐β‐hydroxysterol‐∆‐24‐reductase, which is encoded by the seladin‐1 gene, catalyses the synthesis of cholesterol from desmosterol, the final enzymatic step in the cholesterol biosynthetic pathway (Peri & Serio, 2008). To facilitate brain cholesterol elimination, the enzyme cholesterol 24‐hydroxylase converts cholesterol back into 24S‐hydroxycholesterol (24S‐OHC) (Ohyama et al. 2006), and in humans efflux of 24S‐OHC accounts for as much as 5–7 mg of cholesterol removal from the brain each day (Lutjohann et al. 1996; Bjorkhem et al. 1998). Cholesterol 24‐hydroxylase is extremely stable in the adult brain and evidence indicates that oxidative stress increases its transcriptional activity. Studies that genetically disrupted the CYP46A1 gene, which encodes cholesterol 24‐hydroxylase, demonstrated a 65% reduction of net sterol flux from the brain; however, no biochemical or phenotypic alterations were observed between these genetically modified mice versus their wild‐type littermates (Lund et al. 2003; Xie et al. 2003). Another manner through which cholesterol may be eliminated from the brain is its conversion to the metabolite 27‐hydroxycholesterol (27OHC) by sterol 27‐hydroxylase (Heverin et al. 2005), albeit to a much lesser degree than conversion of 24S‐OHC; 27OHC makes up 5–10% of total brain oxysterols compared to 24S‐OHC (Lutjohann et al. 1996; Heverin et al. 2005). Moreover, evidence now shows that 27OHC is in fact neurotoxic (Heverin et al. 2015), elevated in patients with the Swedish mutation for amyloid precursor protein, and may contribute to the neurodegenerative progression of AD based on its ability to be taken up by the brain from the circulation independent of BBB damage (Shafaati et al. 2011; Heverin et al. 2015). Therefore, a better understanding of cholesterol metabolism and homeostasis in the brain may shed light on the aetiology that contributes to age‐related alterations in neuronal membrane cholesterol, neuronal function and neuroplasticity as well as the contribution of cholesterol and cholesterol metabolites to certain neurodegenerative diseases (Bjorkhem et al. 2010; Maioli et al. 2013).
Although the brain contains close to 400 miles of blood vessels (Begley & Brightman, 2003), the blood–brain barrier (BBB) prevents the plasma lipoproteins from entering the brain, thus forcing the brain to primarily depend upon biosynthesis of its own cholesterol (Jurevics & Morell, 1995; Osono et al. 1995; Jurevics et al. 1997; Turley et al. 1998). Within the brain two pools of cholesterol exist: 70% within the myelin of white matter that ensheaths axons, and 30% within the plasmalemmal and subcellular membranes of neurons and glia that compose the grey matter. Evidence shows that the rate of cholesterol synthesis in the brain is highest during development and following injury, and the predominant source of cholesterol in the brain is provided by glia after development (Vance & Hayashi, 2010). Extracellular uptake by neurons of cholesterol occurs when apo E‐containing lipoproteins are internalized by low‐density lipoprotein (LDL) receptors (e.g. LDLR, LRP1, vLDLR and apo ER2). Apoproteins are taken up by the axons and transported retrogradely from the axon terminal to the neuronal cell body (also termed soma). While LDLR is more highly expressed in glia, LRP1 is found predominantly in neurons. Interestingly vLDLR is specifically expressed in neuronal growth cones and is presumed to facilitate membrane expansion. Glia secrete apolipoproteins (E and J), with apo E being the primary subtype; apo E is synthesized by a few types of hippocampal and cortical neurons as well. Apo E exists in three isoforms: E2, E3 and E4; apo E3 carries 2‐ to ‐3‐fold more cholesterol than E4, the latter isoform of which is associated with a 12‐fold higher risk of developing AD (Vance & Hayashi, 2010). After injury, apo E synthesis in glia increases 150‐fold, and apo E deficiency renders neurons more vulnerable to ischaemic insult. Thus apo E containing lipoproteins serve to deliver cholesterol to neurons for axonal growth, repair and synaptogenesis.
Cholesterol is important for neuronal membrane integrity and neuronal physiology (i.e. synaptic signalling and plasticity) during development and throughout adulthood. Therefore, it should come as no surprise that alterations in the cholesterol synthesis, transport, and uptake have been implicated in the pathogenesis of several neurodegenerative diseases (Martin et al. 2014), yet the mechanisms that underlie these alterations have yet to be defined. Cholesterol contributes to neuronal membrane integrity; supports membrane protein clustering and function, cell morphology, and intercellular communication; and facilitates high‐fidelity signal transduction. Imbalances in cholesterol homeostasis in the brain can contribute to neurodegenerative diseases such as AD and PD. Additional age‐related impairments in glial‐derived cholesterol biosynthesis and transport, or uptake of cholesterol by neurons in the brain may adversely affect development, plasticity and synaptic circuitry, and contribute to the inability of the aged brain to sprout new neurites in order to respond to an ever changing environment (Bulloj et al. 2008; Cecchi et al. 2008; Peri & Serio, 2008; Vanmierlo et al. 2009). Because the brain is heavily dependent upon endogenous intracellular cholesterol biosynthesis rather than circulating plasma lipoproteins, understanding how cholesterol is properly utilized to form new neuronal membranes necessary for maintaining proper signalling, growth and expansion may permit us to restore high‐order brain function in the aged and neurodegenerative brain through genetic interventions that restore plasmalemmal cholesterol.
It is well established that the ageing brain presents functional deficits. Whether these age‐related deficits are based upon a failure to evoke de novo sprouting or due to neurodevelopmental issues earlier in life is a matter of debate (Crutcher, 2002). The ageing nervous system consists of neurons that are in essence as old as the individual itself, and therefore what determines an ‘aged’ neuron may simply be how well it developed earlier in life. Because growth cones are essential for neurite guidance and maturation during early life neurodevelopment and synaptogenesis, understanding the membrane biology of growth cones and how this contributes to growth cone function does ultimately have consequences for age‐related functional changes. Conventional thought is that growth cones are only associated with neuritogenesis during neuronal development; however, growth cones are in fact necessary for de novo neuronal sprouting in the adult CNS that occurs after injury (e.g. ischaemia or trauma) or in the aged brain (Mishra et al. 2011; Tong et al. 2011). Evidence shows that neurite outgrowth and normal growth cone morphology is altered in neurodegenerative conditions (Kao et al. 2010). Because growth cones serve to guide neuronal processes during the neurite outgrowth and axonal regeneration, any alterations to growth cone function (i.e. reduced membrane cholesterol and membrane lipid raft (MLR)‐associated pro‐growth receptors) may dampen the ability of the aged brain to re‐evoke plasticity in response to a constant changing environment. The next section will describe how MLRs contribute to growth cone biology and function. A better understanding of MLR and growth cone membrane biology and how alterations to MLR and growth cone function can ultimately contribute to aged‐related functional changes to the brain may yield potential genetic targets for the purpose of attenuating age‐related functional decline.
Membrane lipid rafts: regulators of signal transduction, cytoskeletal tethering and cellular polarity
MLRs are discrete plasmalemmal microdomains enriched in cellular cholesterol, glycosphingolipids (i.e. gangliosides) and a variety of signalling and scaffolding proteins (Head et al. 2014). Within neurons, MLRs are essential for pro‐growth signalling (e.g. BDNF/TrkB activation), and synapse development, stabilization and maintenance (Head et al. 2014). Moreover, caveolin‐1 (Cav‐1), a cholesterol binding and resident protein of MLRs, organizes and targets synaptic components of the neurotransmitter and neurotrophic receptor signalling pathways to MLRs (Head et al. 2008, 2011). Specifically, these synaptic signalling components localize to MLRs in growth cones and, when activated, converge upon formation of cAMP, a second messenger molecule that has an important role in synaptic plasticity (Head et al. 2014). Because MLRs have been detected at synapses (Suzuki, 2002; Gil et al. 2006; Allen et al. 2007; Wasser & Kavalali, 2009; Mailman et al. 2011), inhibition of cholesterol biosynthesis and subsequent loss of these microdomains can greatly impact pre‐ and postsynaptic function leading to cognitive deficits (Schilling et al. 2014). Previous work from our group demonstrated that Cav‐1 and synaptic components essential for neurotransmitter and neurotrophin signalling (e.g. NMDAR, AMPAR, TrkB) decrease in the aged brain, specifically in synaptosomes and MLRs (Head et al. 2010).
The mechanical basis of axonal and dendritic growth is cytoskeletal dynamics, wherein rearrangements of actin and microtubules create enhanced motility in growth cones. Neurite outgrowth (or neuritogenesis) occurs in three steps: protrusion, engorgement and consolidation (Lowery & Van Vactor, 2009; Stiess & Bradke, 2011). In the protrusion stage, actin polymerization (localized in the growth cone itself) increases, causing filopodia and lamellipodia to extend. At the same time, some actin filaments are pulled backwards by retrograde flow, condensing in filamentous (F) actin ‘Arc’, which prevents microtubules (localized in the neurite shaft) from invading. In the engorgement stage, actin filaments dissociate and reorient at the interface between the neurite shaft and growth cone, allowing microtubule invasion. In consolidation, actin polymerization is suppressed while microtubules are stabilized in the newly invaded region, extending the neurite shaft, which eventually matures into an axon or dendrite. For these morphological changes to occur, the plasma membrane needs to establish a polarized signalling platform that tethers and regulates cytoskeletal components and transduces extracellular stimuli (e.g. growth and repellant cues). MLRs provide an essential plasma membrane platform that establishes cellular polarity by compartmentalizing pro‐growth signalling components (i.e. TrK receptors) while at the same time tethering cytoskeletal proteins (Kamiguchi, 2006; Head et al. 2013) critical for neuritogenesis. MLRs are located at the leading edge of neuronal growth cones (Kamiguchi, 2006), and loss or disruption of MLRs from the leading edge results in growth cone collapse and inhibition of neuritogenesis (Niethammer et al. 2002).
Axonal growth and guidance is crucial for the development of functional neuronal networks. The growth cone at the tip of the axon is essential for axonal growth, pathfinding and proper targeting (Dent et al. 2011). While the growth cone is responsive to both attraction and repulsion cues, the ‘steering’ of the growing axon via the growth cone is mediated by dynamic changes within the underlying actin cytoskeleton (Hall & Lalli, 2010). Collapse of the growth cone, induced by actin dysregulation, leads to stunted axonal growth, loss of connectivity and the development of aberrant connections; as such, growth cone collapse causes dysfunction in neuronal networks (Dent & Gertler, 2003; Dent et al. 2011). Because MLRs serve to tether and directly influence cytoskeletal component dynamics within growth cones (Bassani & Passafaro, 2012; Head et al. 2013), the regulation and balance of actin dynamics can determine the fate of key neurodevelopmental events, such as growth cone collapse and guidance. Therefore, interventions that promote or enhance MLR formation within the plasma membrane of aged neurons may evoke structural and functional synaptic plasticity and potentially reverse behavioural decline.
Cholesterol, sphingolipids and gangliosides: synaptic transmission and receptor function
Understanding age‐related changes to the biochemistry of the brain is fundamental to reversing behavioural deficits. The decline in brain function with age is thought to occur through the accumulation of toxic products from normal oxidative metabolism, cardiovascular disease or changes in the normal body homeostasis. However, the aged brain contains few dead neurons, suggesting that ageing may in part be caused by other factors such as alterations in the cellular membrane biophysical and biochemical properties. Recent evidence points to the contribution of changes in membrane cholesterol and lipid composition of membranes, which can directly affect neurotransmitter and neurotrophin release from presynaptic membranes ultimately influencing cognitive and motor function.
In 1958, the first evidence was obtained that membrane lipid composition of the brain changes with age (Burger & Seidel, 1958; Rouser & Yamamoto, 1968). More recent evidence has confirmed that age‐related lipid alterations occur in human and rodent brains, as well as cultured neurons (Ledesma et al. 2012). Much evidence over the years has demonstrated that reduced cholesterol occurs specifically in the human hippocampus and synaptosomes from aged rodents (Soderberg et al. 1990; Yamamoto et al. 2008; Sodero et al. 2011). Moreover, it has been reported that increased cholesterol catabolism occurs with age (Lutjohann et al. 1996; Bjorkhem & Diczfalusy, 2004; Thelen et al. 2006). Interestingly, in addition to decreases in membrane cholesterol with age, the real problem may lie in the redistribution of cholesterol from the cytofacial to the exofacial leaflet of the plasma membrane (Igbavboa et al. 1996; Wood et al. 2011) (Fig. 1). These hippocampal changes in cholesterol are believed to be due to decreases in cholesterol synthesis and increases in cholesterol catabolism by cholesterol 24‐hydroxylase leading to higher plasma levels of 24‐hydroxycholesterol in aged individuals (Lutjohann et al. 1996; Lund et al. 1999; Thelen et al. 2006). Although hippocampi from aged human and rodents exhibit these lipid alterations, it may be brain region specific due to alterations in lipoprotein transporters and receptors (Bu et al. 1994; Runquist et al. 1995). Along with changes in cholesterol, alterations in sphingomyelin (Trovo et al. 2011) and glycosylated sphingolipids known as gangliosides have also been reported in the aged brain (Posse de Chaves & Sipione, 2010). These lipid changes are most likely to be due to increased ceramide production, a signalling lipid that is a product of sphingomyelin and glycosphingolipid catabolism by sphingomyelinases (Valaperta et al. 2006; Sacket et al. 2009). Moreover, it has been shown that increased oxidative stress in the hippocampus and striatum upregulates sphingomyelinases thus increasing ceramide production (Denisova et al. 2001). Additional evidence demonstrates that ganglioside localization to MLRs is altered with age due to changes in the ceramide moiety within gangliosides (Wolf et al. 1998), suggesting that MLR lipid composition and MLR function are altered with age (Prinetti et al. 2001; Jiang et al. 2010; Trovo et al. 2011).
Figure 1. Aged‐related changes in cholesterol and gangliosides in the neuronal plasma membrane .
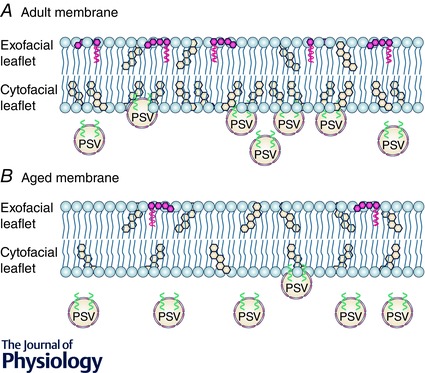
A, in young adult human and rodent neurons, approximately 85% of plasmalemmal cholesterol is found within the cytofacial leaflet while the majority of glycosphingolipids (e.g. GM1 gangliosides) are found in the exofacial leaflet. Both gangliosides and cholesterol enhance the negative curvature property of the plasma membrane, thus enhancing the fusogenicity with presynaptic vesicles (PSV). B, however, with age there is either a reduction in membrane gangliosides and cholesterol or a redistribution of cholesterol from the cyto‐ to the exofacial leaflet. These age‐related changes in cholesterol membrane content and distribution drastically impede PSV docking and fusion with the cytofacial leaflet, neurotransmitter release and postsynaptic signalling (e.g. NMDAR and AMPAR), synaptic plasticity (i.e. LTP) and strength, and behaviour.
Although an abundant amount of evidence supports age‐related changes in plasmalemmal lipid distribution (i.e. redistribution of cholesterol and sphingolipids), there still remains much controversy regarding this ‘flip–flop’ transbilayer diffusion rate specifically related to cholesterol (Steck & Lange, 2012). An elegant study conducted by Garg et al. using time‐resolved small‐angle neutron scattering to measure passive transfer of cholesterol between the two facial leaflets of the PM demonstrated that cholesterol, in its natural form, exhibited a transverse diffusion (i.e. flip–flop) in the bilayer with a half‐time of 200 min at 50°C (Garg et al. 2011). This finding is different from others, which showed half‐time values of minutes to seconds to milliseconds. Steck and Lange argue the following: (1) cholesterol intramembrane transfer may be too fast for the limited sensitivity of detection by the above‐mentioned methodology, and (2) the slow diffusion observed by Garg et al. may in fact reflect the heterogeneity in the intermembrane transfer kinetics of cholesterol. It is well known that different technologies can yield conflicting results. Cells and the lipid bilayer that encases them are highly dynamic. Therefore more in‐depth studies are necessary to yield greater consistency regarding intra‐ and intermembrane cholesterol transfer (and other lipids for that matter) in order to give us an overall better understanding of the role of cholesterol during both neuronal development and the ageing process.
Although changes in membrane lipid composition (e.g. cholesterol, sphingomyelin, gangliosides) are significant with age, how would these biochemical changes ultimately affect synaptic plasticity? Synaptic plasticity represents the ability to change synaptic connections in response to environmental cues and is the hallmark of learning and memory. With age, there is a severe decline in synaptic plasticity in the brain and specifically in the hippocampus (Barnes, 1979; Rosenzweig & Barnes, 2003). Two key components of memory formation are LTP (i.e. strengthening of synapses) and long‐term depression (LTD; i.e. the opposing process that involves weakening of synaptic connections). During age, LTP decays faster and there is a shift from LTP to LTD that ultimately leads to deficits in learning and memory (Barnes & McNaughton, 1980; Larson et al. 1986; Roman et al. 1987; Kelly et al. 2000).
Proper synaptic transmission necessary for LTP and LTD induction involves the fusion of synaptic vesicles (SVs) with the presynaptic membrane in order to facilitate neurotransmitter release and subsequent postsynaptic receptor activation and signalling, a trans‐synaptic event that declines with age (Ledesma et al. 2012). Membrane curvature is a key physical property for facilitating SV fusion (i.e. the more curved a membrane is, the greater its fusogenicity; Lentz et al. 1987). Specific lipids like cholesterol and sphingolipids are necessary for membrane curvature in addition to determining membrane fluidity. Modulation of membrane curvature by lipids is essential for SV fusion with the membrane and receptor diffusion within the membrane, both of which contribute to synaptic plasticity. Cholesterol is a major lipid of synaptic vesicle membranes (∼30%) and facilitates fusion by forming a high curvature stalk‐pore (Deutsch & Kelly, 1981; Churchward et al. 2005). Cholesterol promotes curvature of synaptic vesicles through its interaction with the integral membrane protein synaptophysin (Thiele et al. 2000). Moreover, experiments that involve cholesterol depletion resulted in impaired exocytosis of synaptic vesicles, decreased neurotransmitter release, and blunted synaptic plasticity (Chamberlain et al. 2001; Kudinov et al. 2006; Linetti et al. 2010). Therefore, age‐related decreases in membrane cholesterol or its redistribution from the cyto‐ to the exofacial leaflet of the plasma membrane ultimately contributes to decreased neurotransmitter release and reduced synaptic plasticity. In addition to its direct influence on membrane curvature, cholesterol also serves to concentrate SNARE proteins. Therefore, reduced cholesterol not only affects curvature but also may lead to instability of SNARE complexes (Chamberlain et al. 2001; Lang et al. 2001; Chamberlain & Gould, 2002).
Along with cholesterol, sphingolipids also affect membrane curvature. Sphingolipids interact with cholesterol (via the α smooth face of cholesterol (Garmy et al. 2005) while the β rough face of cholesterol intercalates with the helices of transmembrane proteins (Paila et al. 2009)); both are enriched in MLRs and in synaptic membranes (Fig. 2). Sphingolipid (originally coined sphingosine by J. L. W. Thudichum in 1884 based upon the enigmatic Greek mythological sphinx) is a lipid composed of a long base chain, a fatty acyl chain, and the sugar hexose (Thudichum, 1884). The more appropriate term sphingolipid was coined by Carter and colleagues to describe any lipids derived from sphingosine (Carter et al. 1947; Futerman & Hannun, 2004). SV fusion and synaptic transmission are highly influenced by the sphingolipid–cholesterol interaction, and age‐related changes in the sphingolipid to cholesterol ratio in both rodent and human hippocampal neurons reduces fusion efficiency through alterations in membrane curvature (Haque et al. 2001; Martin et al. 2008; Yamamoto et al. 2008; Trovo et al. 2011). Ganglioside, a type of glycosphingolipid found within MLRs and located at nerve endings, is normally enriched in the exofacial leaflet of the plasma membrane and contributes to the positive curvature of synaptic membranes (Leskawa et al. 1979; Salaun et al. 2004). Gangliosides enhance neurotransmitter release, and evidence shows that with age there is a decrease in gangliosides in the exofacial leaflet of neuronal membranes (Svennerholm et al. 1994; Posse de Chaves & Sipione, 2010).
Figure 2.
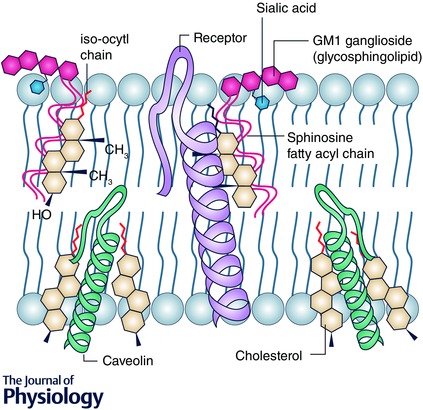
The physical interaction between cholesterol, sphingolipids and transmembrane proteins
Cholesterol physically interacts with sphingolipids and the transmembrane component of proteins. Cholesterol possesses an α smooth surface, which permits it to interact with sphingolipids, while the β rough surface (due to methyl groups on carbon 10 and 13 and the iso‐octyl chain link to carbon 17) facilitates insertion into the helices of transmembrane proteins. Due to its dissymmetrical physical properties, one molecule of cholesterol can interact with two distinct membrane molecules, such as sphingolipids and transmembrane proteins, within raft microdomains. This serves to create a more liquid ordered membrane parameter (i.e. decreased fluidity) characteristic of MLR.
Another critical component of synaptic plasticity and synaptic strength is neurotransmitter receptor diffusion. Both NMDAR and AMPAR signalling contribute to memory induction and consolidation within the hippocampus. The lateral diffusion of these receptors is critical to synaptic tuning and plasticity and is heavily dependent upon membrane lipid composition. MLRs regulate neurotransmitter receptor clustering and signal transduction (Head et al. 2008, 2011). Others have shown that cholesterol depletion from hippocampal synaptic membranes directly affects AMPAR trafficking and mobility through receptor destabilization (Hering et al. 2003; Renner et al. 2009). Furthermore, NMDAR‐dependent calcium influx and NMDAR‐mediated LTP in the hippocampus are greatly impaired upon cholesterol depletion (Frank et al. 2004, 2008). In fact, LTP itself increases cholesterol synthesis suggesting that LTP‐induced synaptic plasticity requires cholesterol; therefore age‐related reduction in cholesterol or asymmetric cholesterol redistribution could negatively impact synaptic plasticity (Kudinov et al. 2006). Gangliosides also contribute to AMPAR and NMDAR function by facilitating membrane fluidity and clustering necessary for synaptic signal transduction (Cole et al. 2010). Age‐related reduction in these lipids also negatively influences neurotransmitter receptor‐mediated synaptic plasticity.
Sphingolipids, cholesterol and MLRs regulate neurotransmitter receptor conformation, function and trafficking by directly binding to the transmembrane helices and extracellular loops of the receptors (Fantini & Barrantes, 2009). These lipids alter receptor conformation within the membrane (e.g. acetylcholine and serotonin receptors), and directly modulate neurotransmitter binding, signal transduction and even receptor trafficking. Interestingly, gangliosides demonstrate direct affinity for neurotransmitters and facilitate their attachment to the postsynaptic membranes, thus promoting synaptic transmission. Specifically, serotonin binds directly to gangliosides in a ‘catch neurotransmitter’ manner onto the postsynaptic membrane (i.e. ganglioside assisted neurotransmitter delivery), which serves to concentrate the neurotransmitter while at the same time preventing neurotransmitter aggregation (Fantini & Barrantes, 2009). With respect to receptors, gangliosides also regulate the conformational change of the receptor upon ligand binding in order to couple to heterotrimeric G proteins, as is the case with GPCRs such as the opioid receptor (Wu et al. 1997). Intriguingly, the addition of gangliosides to neurons enhances neurite outgrowth, axonal sprouting, and neuronal differentiation in vitro and in vivo (Gorio, 1986), suggesting that interventions that enhance membrane gangliosides may facilitate dendritic and axonal growth, enhance synaptic plasticity, and potentially regenerate neuronal connections, which ultimately improves functional neuronal networks.
Can genetic interventions that enhance MLRs specifically in neurons evoke structural and functional neuronal plasticity?
Ageing is a physiological process that is accompanied by a significant reduction in neuronal plasticity (Calabrese et al. 2013). Activating molecular mechanisms that evoke structural and functional plasticity have the capacity to improve function in the aged or neurodegenerative brain (Mesulam, 1999; Morrison & Baxter, 2014; Villeda et al. 2014). A key molecule that enhances neuronal growth is cAMP (Atkins et al. 2007, 2013; Murray et al. 2009; Titus et al. 2013 b); cAMP binds to protein kinase A (PKA), which then activates extracellular response kinase (ERK1/2) and phosphorylates cAMP response element binding protein (CREB). In addition, cAMP leads to exchange protein activated by cAMP (Epac) activation within growth cones (Ming et al. 1997; Murray et al. 2009). After injury or with age, there is a significant reduction in CREB phosphorylation and ERK1/2 activation, presumably due to a reduction in cAMP production (Atkins et al. 2007, 2009; Titus et al. 2013 a). Increasing cAMP levels by phosphodiesterase inhibition induces neuronal sprouting, reorganization of the neurons in the cortex and recovery of motor function after injury (Atkins et al. 2007; MacDonald et al. 2007). Therefore, genetic and pharmacological interventions that increase cAMP may improve motor and cognitive function in the aged brain (Atkins et al. 2007, 2013; Titus et al. 2013 a).
At the leading edge of neurite extension, within growth cones, there exists several pro‐growth pathways that converge upon cAMP: NMDAR, AMPAR, TrkB and GPCRs such as dopamine 1 receptor (D1R) and serotonin receptors (5‐HT6 and 5‐HT7) (Gao et al. 2003; Schmitt et al. 2004; Wayman et al. 2004, 2006; Kong et al. 2007; Chytrova et al. 2008; Fantini & Barrantes, 2009). These receptors localize to MLR within the growth cone (Fantini & Barrantes, 2009; Head et al. 2014). We have previously shown that neuron‐targeted Cav‐1 over‐expression enhanced MLR formation (as indicated by increased cholera toxin‐B (CT‐B) detection in buoyant membrane fractions) and increased protein expression of neuronal receptors in CT‐B‐enriched membrane fractions (Head et al. 2011). Furthermore, neuron‐targeted Cav‐1 enhanced NMDAR and TrkB‐mediated signal transduction and increased cAMP production following receptor agonism (D1R, 5‐HT6 and NMDAR) or direct activation of adenylyl cyclase (with forskolin) (Head et al. 2011). The substantial increase in cAMP may underlie the pro‐growth properties of increasing Cav‐1 and MLRs specifically in neurons. More recent data from our group show that delivery of neuron‐targeted Cav‐1 using an adeno‐associated virus serotype 9 (AAV9‐SynCav1) into the hippocampus enhances structural and functional plasticity in granule cell neurons and in CA1 pyramidal cell neurons and increases hippocampus‐dependent fear learning and memory in adult (6 months) and aged mice (20 months) (Egawa et al. 2014). Post mortem biochemical analysis of these mice revealed enhanced CT‐B in MLR fractions and increased TrkB protein expression in these CT‐B positive MLR fractions (similar to what we have previously demonstrated in primary neurons in vitro; Head et al. 2011), suggesting that neuron‐targeted overexpression of Cav‐1 increases MLR formation and pro‐growth receptor localization to MLRs. Thus interventions that enhance MLR formation specifically in neuronal plasma membranes may significantly increase structural and functional plasticity and vastly accelerate motor and cognitive recovery in the aged or neurodegenerative brain (Fig. 3).
Figure 3. Age‐related loss in synaptic signalling may be restored by increasing MLR formation .
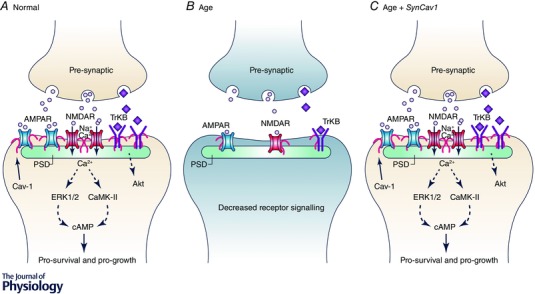
A, in the healthy brain, synaptic transmission is dependent upon the appropriate neuronal receptors (e.g. AMPAR, NMDAR, and TrkB), their localization to the postsynaptic density, and release of neurotransmitter from the presynaptic cleft. MLRs, which are critical membrane components of both the pre‐ and postsynaptic membrane, scaffold these receptors in part via the cholesterol binding protein Cav‐1. B, however, with age there is a drastic decrease in neuronal MLRs, Cav‐1, and MLR‐associated neuronal receptors, which results in decreased synaptic transmission (reduced cAMP formation), progressive neurodegeneration, and increased behavioural dysfunction. C, most highly targeted therapies are often ineffective in eliciting the desired response, likely to be due to decreased expression of key receptors (e.g. AMPARs, NMDARs, TrkB, GPCRs) and inadequate production of second messenger (cAMP). Genetic interventions that enhance and/or restore MLR formation (i.e. neuron‐targeted Cav‐1 or SynCav1) can potentially re‐establish an active signalling platform that regulates cytoskeletal dynamics, enhances structural and functional neuroplasticity, and improves cognition in the ageing and neurodegenerative brain.
Conclusion
Restoration of synaptic plasticity and preservation of cognitive function in the aged brain remains a major medical challenge for the ageing population. Understanding how age‐related changes affect the synaptic membrane protein clustering within neurons may yield therapeutic targets for the purpose of restoring functional MLRs, neuronal membrane expansion, and synapse formation that are necessary to improve function in the ageing brain. Genetic interventions that re‐establish the proper subcellular membrane regions necessary to restore pro‐growth and pro‐survival signalling have the potential to not only reduce neuronal loss and enhance endogenous brain repair, but also to increase the efficacy of pharmacological agents designed to improve functional outcome. Enhancement of MLRs within neuronal membranes in combination with pharmacological agents that enhance cAMP (i.e. PDE inhibitors, selective serotinin re‐uptake inhibitors (SSRIs) or serotonin dopamine re‐uptake inhibitors (SDRIs)), TrkB agonism, or exercise therapy which enhances BDNF may prove efficacious in promoting functional and structural plasticity, pruning and refining neuritic growth, and improving cognitive function in the aged brain.
Additional information
Competing interests
The authors declare no conflicting financial interest.
Funding
Work in the authors’ laboratories is supported by Veteran Affairs Merit Award from the Department of Veterans Affairs BX001225 (to B.P.H.) and National Institutes of Health, Bethesda, MD, USA, NS073653 (to B.P.H.).
Biographies
Junji Egawa carries out research focused on caveolin‐1 (Cav‐1) and membrane lipid rafts (MLRs) in the ageing brain and traumatic brain injury model in vivo. He received the John D. Michenfelder New Investigator Award for his research at the 2014 SNACC meeting and also received the best abstracts award for Cav‐1 effects on the TBI model at the 2014 ASA meeting.

Matthew L. Pearn carries out research focused on the detrimental effects of anaesthetic exposure in vulnerable states, such as extreme ages (i.e. neonates and elderly), after injury (i.e. traumatic brain injury and stroke), and with preexisting cognitive deficits (i.e. Alzheimer's disease and Down Syndrome).
Brian P. Lemkuil is an Associate Director of Neurocritical Care, and Clinical Director of Neuroanesthesia and the Neuroanesthesia Fellowship.
Piyush M. Patel carries out research focused on the molecular mechanisms that underlie anaesthetic‐mediated cytoskeletal disruption, neurite loss, dendritic spine loss and synapse loss in postnatal developing neurons.
Brian P. Head carries out research focused on the cellular mechanisms by which MLRs and Cav‐1 promote neuritogenesis, axonal growth and dendritic arborization in neuronal models (primary neurons, hippocampal and cortical neurons in vivo, and human iPSC‐derived neurons). In 2012 he was bestowed the Presidential Early Career Awards for Scientists and Engineers (PECASE), the highest honor given by the United States Government to science and engineering professionals.
This review was presented at the symposium “Membrane/lipid rafts: Age‐associated changes in membrane biology and function”, which took place at Ageing and Degeneration: A Physiological Perspective in Edinburgh, UK, 10–11 April 2015.
References
- Allen JA, Halverson‐Tamboli RA & Rasenick MM (2007). Lipid raft microdomains and neurotransmitter signalling. Nat Rev Neurosci 8, 128–140. [DOI] [PubMed] [Google Scholar]
- Atkins CM, Cepero ML, Kang Y, Liebl DJ & Dietrich WD (2013). Effects of early rolipram treatment on histopathological outcome after controlled cortical impact injury in mice. Neurosci Lett 532, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkins CM, Falo MC, Alonso OF, Bramlett HM & Dietrich WD (2009). Deficits in ERK and CREB activation in the hippocampus after traumatic brain injury. Neurosci Lett 459, 52–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkins CM, Oliva AA Jr, Alonso OF, Pearse DD, Bramlett HM & Dietrich WD (2007). Modulation of the cAMP signaling pathway after traumatic brain injury. Exp Neurol 208, 145–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aziz R & Steffens DC (2013). What are the causes of late‐life depression? Psychiatr Clin North Am 36, 497–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes CA (1979). Memory deficits associated with senescence: a neurophysiological and behavioral study in the rat. J Comp Physiol Psychol 93, 74–104. [DOI] [PubMed] [Google Scholar]
- Barnes CA & McNaughton BL (1980). Physiological compensation for loss of afferent synapses in rat hippocampal granule cells during senescence. J Physiol 309, 473–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassani S & Passafaro M (2012). TSPAN7: A new player in excitatory synapse maturation and function. Bioarchitecture 2, 95–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begley DJ & Brightman MW (2003). Structural and functional aspects of the blood‐brain barrier. Prog Drug Res 61, 39–78. [DOI] [PubMed] [Google Scholar]
- Bjorkhem I & Diczfalusy U (2004). 24(S), 25‐epoxycholesterol–a potential friend. Arterioscler Thromb Vasc Biol 24, 2209–2210. [DOI] [PubMed] [Google Scholar]
- Bjorkhem I, Leoni V & Meaney S (2010). Genetic connections between neurological disorders and cholesterol metabolism. J Lipid Res 51, 2489–2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjorkhem I, Lutjohann D, Diczfalusy U, Stahle L, Ahlborg G & Wahren J (1998). Cholesterol homeostasis in human brain: turnover of 24S‐hydroxycholesterol and evidence for a cerebral origin of most of this oxysterol in the circulation. J Lipid Res 39, 1594–1600. [PubMed] [Google Scholar]
- Breceda EY & Dromerick AW (2013). Motor rehabilitation in stroke and traumatic brain injury: stimulating and intense. Curr Opin Neurol 26, 595–601. [DOI] [PubMed] [Google Scholar]
- Brown MS & Goldstein JL (1997). The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane‐bound transcription factor. Cell 89, 331–340. [DOI] [PubMed] [Google Scholar]
- Bu G, Maksymovitch EA, Nerbonne JM & Schwartz AL (1994). Expression and function of the low density lipoprotein receptor‐related protein (LRP) in mammalian central neurons. J Biol Chem 269, 18521–18528. [PubMed] [Google Scholar]
- Bulloj A, Leal MC, Surace EI, Zhang X, Xu H, Ledesma MD, Castano EM & Morelli L (2008). Detergent resistant membrane‐associated IDE in brain tissue and cultured cells: Relevance to Abeta and insulin degradation. Mol Neurodegener 3, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burger M & Seidel K (1958). [Chemical biomorphosis of the human brain and sciatic nerve: a survey]. Z Alternsforsch 12, 52–79. [PubMed] [Google Scholar]
- Calabrese F, Guidotti G, Racagni G & Riva MA (2013). Reduced neuroplasticity in aged rats: a role for the neurotrophin brain‐derived neurotrophic factor. Neurobiol Aging 34, 2768–2776. [DOI] [PubMed] [Google Scholar]
- Carter HE, Haines WJ et al (1947). Biochemistry of the sphingolipides; preparation of sphingolipides from beef brain and spinal cord. J Biol Chem 169, 77–82. [PubMed] [Google Scholar]
- Cecchi C, Rosati F, Pensalfini A, Formigli L, Nosi D, Liguri G, Dichiara F, Morello M, Danza G, Pieraccini G, Peri A, Serio M & Stefani M (2008). Seladin‐1/DHCR24 protects neuroblastoma cells against Abeta toxicity by increasing membrane cholesterol content. J Cell Mol Med 12, 1990–2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamberlain LH, Burgoyne RD & Gould GW (2001). SNARE proteins are highly enriched in lipid rafts in PC12 cells: implications for the spatial control of exocytosis. Proc Natl Acad Sci USA 98, 5619–5624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamberlain LH & Gould GW (2002). The vesicle‐ and target‐SNARE proteins that mediate Glut4 vesicle fusion are localized in detergent‐insoluble lipid rafts present on distinct intracellular membranes. J Biol Chem 277, 49750–49754. [DOI] [PubMed] [Google Scholar]
- Churchward MA, Rogasevskaia T, Hofgen J, Bau J & Coorssen JR (2005). Cholesterol facilitates the native mechanism of Ca2+‐triggered membrane fusion. J Cell Sci 118, 4833–4848. [DOI] [PubMed] [Google Scholar]
- Chytrova G, Ying Z & Gomez‐Pinilla F (2008). Exercise normalizes levels of MAG and Nogo‐A growth inhibitors after brain trauma. Eur J Neurosci 27, 1–11. [DOI] [PubMed] [Google Scholar]
- Cole AA, Dosemeci A & Reese TS (2010). Co‐segregation of AMPA receptors with GM1 ganglioside in synaptosomal membrane subfractions. Biochem J 427, 535–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conte V, Raghupathi R, Watson DJ, Fujimoto S, Royo NC, Marklund N, Stocchetti N & McIntosh TK (2008). TrkB gene transfer does not alter hippocampal neuronal loss and cognitive deficits following traumatic brain injury in mice. Restor Neurol Neurosci 26, 45–56. [PMC free article] [PubMed] [Google Scholar]
- Crutcher KA (2002). Aging and neuronal plasticity: lessons from a model. Auton Neurosci 96, 25–32. [DOI] [PubMed] [Google Scholar]
- Denisova NA, Cantuti‐Castelvetri I, Hassan WN, Paulson KE & Joseph JA (2001). Role of membrane lipids in regulation of vulnerability to oxidative stress in PC12 cells: implication for aging. Free Radic Biol Med 30, 671–678. [DOI] [PubMed] [Google Scholar]
- Dent EW & Gertler FB (2003). Cytoskeletal dynamics and transport in growth cone motility and axon guidance. Neuron 40, 209–227. [DOI] [PubMed] [Google Scholar]
- Dent EW, Gupton SL & Gertler FB (2011). The growth cone cytoskeleton in axon outgrowth and guidance. Cold Spring Harb Perspect Biol 3, a001800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutsch JW & Kelly RB (1981). Lipids of synaptic vesicles: relevance to the mechanism of membrane fusion. Biochemistry 20, 378–385. [DOI] [PubMed] [Google Scholar]
- Egawa J, Schilling JM, Mandyam CD, Patel PM & Head BP (2014). Neuron‐targeted Caveolin‐1 remodels hippocampal neurons and enhances hippocampal plasticity and cognition. In Abstracts From the 42nd Annual Meeting of the Society for Neuroscience in Anesthesiology and Critical Care, New Orleans, LA, October 10, 2014. J Neurosurg Anesthesiol 26, 478. [Google Scholar]
- Fantini J & Barrantes FJ (2009). Sphingolipid/cholesterol regulation of neurotransmitter receptor conformation and function. Biochim Biophys Acta 1788, 2345–2361. [DOI] [PubMed] [Google Scholar]
- Fjell AM, McEvoy L, Holland D, Dale AM, Walhovd KB & Alzheimer Disease Neuroimaging Initiative (2014). What is normal in normal aging? Effects of aging, amyloid and Alzheimer's disease on the cerebral cortex and the hippocampus. Prog Neurobiol 117C, 20–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fjell AM, Walhovd KB, Fennema‐Notestine C, McEvoy LK, Hagler DJ, Holland D, Brewer JB & Dale AM (2009). One‐year brain atrophy evident in healthy aging. J Neurosci 29, 15223–15231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fjell AM, Westlye LT, Grydeland H, Amlien I, Espeseth T, Reinvang I, Raz N, Holland D, Dale AM, Walhovd KB & Alzheimer Disease Neuroimaging Initiative (2013). Critical ages in the life course of the adult brain: nonlinear subcortical aging. Neurobiol Aging 34, 2239–2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank C, Giammarioli AM, Pepponi R, Fiorentini C & Rufini S (2004). Cholesterol perturbing agents inhibit NMDA‐dependent calcium influx in rat hippocampal primary culture. FEBS Lett 566, 25–29. [DOI] [PubMed] [Google Scholar]
- Frank C, Rufini S, Tancredi V, Forcina R, Grossi D & D'Arcangelo G (2008). Cholesterol depletion inhibits synaptic transmission and synaptic plasticity in rat hippocampus. Exp Neurol 212, 407–414. [DOI] [PubMed] [Google Scholar]
- Futerman AH & Hannun YA (2004). The complex life of simple sphingolipids. EMBO Rep 5, 777–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Nikulina E, Mellado W & Filbin MT (2003). Neurotrophins elevate cAMP to reach a threshold required to overcome inhibition by MAG through extracellular signal‐regulated kinase‐dependent inhibition of phosphodiesterase. J Neurosci 23, 11770–11777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg S, Porcar L, Woodka AC, Butler PD & Perez‐Salas U (2011). Noninvasive neutron scattering measurements reveal slower cholesterol transport in model lipid membranes. Biophys J 101, 370–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garmy N, Taieb N, Yahi N & Fantini J (2005). Interaction of cholesterol with sphingosine: physicochemical characterization and impact on intestinal absorption. J Lipid Res 46, 36–45. [DOI] [PubMed] [Google Scholar]
- Gil C, Cubi R, Blasi J & Aguilera J (2006). Synaptic proteins associate with a sub‐set of lipid rafts when isolated from nerve endings at physiological temperature. Biochem Biophys Res Commun 348, 1334–1342. [DOI] [PubMed] [Google Scholar]
- Gorio A (1986). Ganglioside enhancement of neuronal differentiation, plasticity, and repair. CRC Crit Rev Clin Neurobiol 2, 241–296. [PubMed] [Google Scholar]
- Gresack JE, Risbrough VB, Scott CN, Coste S, Stenzel‐Poore M, Geyer MA & Powell SB (2010). Isolation rearing‐induced deficits in contextual fear learning do not require CRF(2) receptors. Behav Brain Res 209, 80–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall A & Lalli G (2010). Rho and Ras GTPases in axon growth, guidance, and branching. Cold Spring Harb Perspect Biol 2, a001818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haque ME, McIntosh TJ & Lentz BR (2001). Influence of lipid composition on physical properties and peg‐mediated fusion of curved and uncurved model membrane vesicles: “nature's own” fusogenic lipid bilayer. Biochemistry 40, 4340–4348. [DOI] [PubMed] [Google Scholar]
- Head BP, Hu Y, Finley JC, Saldana MD, Bonds JA, Miyanohara A, Niesman IR, Ali SS, Murray F, Insel PA, Roth DM, Patel HH & Patel PM (2011). Neuron‐targeted caveolin‐1 protein enhances signaling and promotes arborization of primary neurons. J Biol Chem 286, 33310–33321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Head BP, Patel HH & Insel PA (2014). Interaction of membrane/lipid rafts with the cytoskeleton: Impact on signaling and function: Membrane/lipid rafts, mediators of cytoskeletal arrangement and cell signaling. Biochim Biophys Acta 1838, 532–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Head BP, Patel HH, Tsutsumi YM, Hu Y, Mejia T, Mora RC, Insel PA, Roth DM, Drummond JC & Patel PM (2008). Caveolin‐1 expression is essential for N‐methyl‐D‐aspartate receptor‐mediated Src and extracellular signal‐regulated kinase 1/2 activation and protection of primary neurons from ischemic cell death. FASEB J 22, 828–840. [DOI] [PubMed] [Google Scholar]
- Head BP, Peart JN, Panneerselvam M, Yokoyama T, Pearn ML, Niesman IR, Bonds JA, Schilling JM, Miyanohara A, Headrick J, Ali SS, Roth DM, Patel PM & Patel HH (2010). Loss of caveolin‐1 accelerates neurodegeneration and aging. PLoS One 5, e15697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henley JM & Wilkinson KA (2013). AMPA receptor trafficking and the mechanisms underlying synaptic plasticity and cognitive aging. Dialogues Clin Neurosci 15, 11–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hering H, Lin CC & Sheng M (2003). Lipid rafts in the maintenance of synapses, dendritic spines, and surface AMPA receptor stability. J Neurosci 23, 3262–3271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heverin M, Maioli S, Pham T, Mateos L, Camporesi E, Ali Z, Winblad B, Cedazo‐Minguez A & Bjorkhem I (2015). 27‐Hydroxycholesterol mediates negative effects of dietary cholesterol on cognition in mice. Behav Brain Res 278, 356–359. [DOI] [PubMed] [Google Scholar]
- Heverin M, Meaney S, Lutjohann D, Diczfalusy U, Wahren J & Bjorkhem I (2005). Crossing the barrier: net flux of 27‐hydroxycholesterol into the human brain. J Lipid Res 46, 1047–1052. [DOI] [PubMed] [Google Scholar]
- Igbavboa U, Avdulov NA, Schroeder F & Wood WG (1996). Increasing age alters transbilayer fluidity and cholesterol asymmetry in synaptic plasma membranes of mice. J Neurochem 66, 1717–1725. [DOI] [PubMed] [Google Scholar]
- Jiang L, Fang J, Moore DS, Gogichaeva NV, Galeva NA, Michaelis ML & Zaidi A (2010). Age‐associated changes in synaptic lipid raft proteins revealed by two‐dimensional fluorescence difference gel electrophoresis. Neurobiol Aging 31, 2146–2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurevics H & Morell P (1995). Cholesterol for synthesis of myelin is made locally, not imported into brain. J Neurochem 64, 895–901. [DOI] [PubMed] [Google Scholar]
- Jurevics HA, Kidwai FZ & Morell P (1997). Sources of cholesterol during development of the rat fetus and fetal organs. J Lipid Res 38, 723–733. [PubMed] [Google Scholar]
- Kalaitzakis ME, Graeber MB, Gentleman SM & Pearce RK (2008). Striatal beta‐amyloid deposition in Parkinson disease with dementia. J Neuropathol Exp Neurol 67, 155–161. [DOI] [PubMed] [Google Scholar]
- Kamiguchi H (2006). The region‐specific activities of lipid rafts during axon growth and guidance. J Neurochem 98, 330–335. [DOI] [PubMed] [Google Scholar]
- Kao PF, Davis DA, Banigan MG, Vanderburg CR, Seshadri S & Delalle I (2010). Modulators of cytoskeletal reorganization in CA1 hippocampal neurons show increased expression in patients at mid‐stage Alzheimer's disease. PLoS One 5, e13337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly A, Maguire C & Lynch MA (2000). Deficits in nerve growth factor release and tyrosine receptor kinase phosphorylation are associated with age‐related impairment in long‐term potentiation in the dentate gyrus. Neuroscience 95, 359–365. [DOI] [PubMed] [Google Scholar]
- Kong MM, Hasbi A, Mattocks M, Fan T, O'Dowd BF & George SR (2007). Regulation of D1 dopamine receptor trafficking and signaling by caveolin‐1. Mol Pharmacol 72, 1157–1170. [DOI] [PubMed] [Google Scholar]
- Kudinov AR, Kudinova NV & Berezov TT (2006). [Cholesterol is an important molecule in the processes of the synaptic plasticity and degeneration of neurons]. Vestn Ross Akad Med Nauk 2006, 61–66. [PubMed] [Google Scholar]
- Lang T, Bruns D, Wenzel D, Riedel D, Holroyd P, Thiele C & Jahn R (2001). SNAREs are concentrated in cholesterol‐dependent clusters that define docking and fusion sites for exocytosis. EMBO J 20, 2202–2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson J, Wong D & Lynch G (1986). Patterned stimulation at the theta frequency is optimal for the induction of hippocampal long‐term potentiation. Brain Res 368, 347–350. [DOI] [PubMed] [Google Scholar]
- Leal SL & Yassa MA (2013). Perturbations of neural circuitry in aging, mild cognitive impairment, and Alzheimer's disease. Ageing Res Rev 12, 823–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledesma MD, Martin MG & Dotti CG (2012). Lipid changes in the aged brain: effect on synaptic function and neuronal survival. Prog Lipid Res 51, 23–35. [DOI] [PubMed] [Google Scholar]
- Lentz BR, Carpenter TJ & Alford DR (1987). Spontaneous fusion of phosphatidylcholine small unilamellar vesicles in the fluid phase. Biochemistry 26, 5389–5397. [DOI] [PubMed] [Google Scholar]
- Leskawa KC, Yohe HC, Matsumoto M & Rosenberg A (1979). Large‐scale preparation of synaptosomes from bovine brain using a zonal rotor technique. Neurochem Res 4, 483–504. [DOI] [PubMed] [Google Scholar]
- Linetti A, Fratangeli A, Taverna E, Valnegri P, Francolini M, Cappello V, Matteoli M, Passafaro M & Rosa P (2010). Cholesterol reduction impairs exocytosis of synaptic vesicles. J Cell Sci 123, 595–605. [DOI] [PubMed] [Google Scholar]
- Liscum L, Cummings RD, Anderson RG, DeMartino GN, Goldstein JL & Brown MS (1983). 3‐Hydroxy‐3‐methylglutaryl‐CoA reductase: a transmembrane glycoprotein of the endoplasmic reticulum with N‐linked “high‐mannose” oligosaccharides. Proc Natl Acad Sci USA 80, 7165–7169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowery LA & Van Vactor D (2009). The trip of the tip: understanding the growth cone machinery. Nat Rev Mol Cell Biol 10, 332–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund EG, Guileyardo JM & Russell DW (1999). cDNA cloning of cholesterol 24‐hydroxylase, a mediator of cholesterol homeostasis in the brain. Proc Natl Acad Sci USA 96, 7238–7243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund EG, Xie C, Kotti T, Turley SD, Dietschy JM & Russell DW (2003). Knockout of the cholesterol 24‐hydroxylase gene in mice reveals a brain‐specific mechanism of cholesterol turnover. J Biol Chem 278, 22980–22988. [DOI] [PubMed] [Google Scholar]
- Lutjohann D, Breuer O, Ahlborg G, Nennesmo I, Siden A, Diczfalusy U & Bjorkhem I (1996). Cholesterol homeostasis in human brain: evidence for an age‐dependent flux of 24S‐hydroxycholesterol from the brain into the circulation. Proc Natl Acad Sci USA 93, 9799–9804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald E, Van der Lee H, Pocock D, Cole C, Thomas N, VandenBerg PM, Bourtchouladze R & Kleim JA (2007). A novel phosphodiesterase type 4 inhibitor, HT‐0712, enhances rehabilitation‐dependent motor recovery and cortical reorganization after focal cortical ischemia. Neurorehabil Neural Repair 21, 486–496. [DOI] [PubMed] [Google Scholar]
- Mailman T, Hariharan M & Karten B (2011). Inhibition of neuronal cholesterol biosynthesis with lovastatin leads to impaired synaptic vesicle release even in the presence of lipoproteins or geranylgeraniol. J Neurochem 119, 1002–1015. [DOI] [PubMed] [Google Scholar]
- Maioli S, Bavner A, Ali Z, Heverin M, Ismail MA, Puerta E, Olin M, Saeed A, Shafaati M, Parini P, Cedazo‐Minguez A & Bjorkhem I (2013). Is it possible to improve memory function by upregulation of the cholesterol 24S‐hydroxylase (CYP46A1) in the brain? PLoS One 8, e68534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal PK, Pettegrew JW, Masliah E, Hamilton RL & Mandal R (2006). Interaction between Abeta peptide and alpha synuclein: molecular mechanisms in overlapping pathology of Alzheimer's and Parkinson's in dementia with Lewy body disease. Neurochem Res 31, 1153–1162. [DOI] [PubMed] [Google Scholar]
- Martin M, Dotti CG & Ledesma MD (2010). Brain cholesterol in normal and pathological aging. Biochim Biophys Acta 1801, 934–944. [DOI] [PubMed] [Google Scholar]
- Martin MG, Perga S, Trovo L, Rasola A, Holm P, Rantamaki T, Harkany T, Castren E, Chiara F & Dotti CG (2008). Cholesterol loss enhances TrkB signaling in hippocampal neurons aging in vitro. Mol Biol Cell 19, 2101–2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin MG, Pfrieger F & Dotti CG (2014). Cholesterol in brain disease: sometimes determinant and frequently implicated. EMBO Rep 15, 1036–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesulam MM (1999). Neuroplasticity failure in Alzheimer's disease: bridging the gap between plaques and tangles. Neuron 24, 521–529. [DOI] [PubMed] [Google Scholar]
- Ming GL, Song HJ, Berninger B, Holt CE, Tessier‐Lavigne M & Poo MM (1997). cAMP‐dependent growth cone guidance by netrin‐1. Neuron 19, 1225–1235. [DOI] [PubMed] [Google Scholar]
- Mishra M, Akatsu H & Heese K (2011). The novel protein MANI modulates neurogenesis and neurite‐cone growth. J Cell Mol Med 15, 1713–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison JH & Baxter MG (2014). Synaptic health. JAMA Psychiatry 71, 835–837. [DOI] [PubMed] [Google Scholar]
- Murray AJ, Tucker SJ & Shewan DA (2009). cAMP‐dependent axon guidance is distinctly regulated by Epac and protein kinase A. J Neurosci 29, 15434–15444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann AK, Itano MS & Jacobson K (2010). Understanding lipid rafts and other related membrane domains. F1000 Biol Rep 2, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niethammer P, Delling M, Sytnyk V, Dityatev A, Fukami K & Schachner M (2002). Cosignaling of NCAM via lipid rafts and the FGF receptor is required for neuritogenesis. J Cell Biol 157, 521–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohyama Y, Meaney S, Heverin M, Ekstrom L, Brafman A, Shafir M, Andersson U, Olin M, Eggertsen G, Diczfalusy U, Feinstein E & Bjorkhem I (2006). Studies on the transcriptional regulation of cholesterol 24‐hydroxylase (CYP46A1): marked insensitivity toward different regulatory axes. J Biol Chem 281, 3810–3820. [DOI] [PubMed] [Google Scholar]
- Osono Y, Woollett LA, Herz J & Dietschy JM (1995). Role of the low density lipoprotein receptor in the flux of cholesterol through the plasma and across the tissues of the mouse. J Clin Invest 95, 1124–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paila YD, Tiwari S & Chattopadhyay A (2009). Are specific nonannular cholesterol binding sites present in G‐protein coupled receptors? Biochim Biophys Acta 1788, 295–302. [DOI] [PubMed] [Google Scholar]
- Paul G, Zachrisson O, Varrone A, Almqvist P, Jerling M, Lind G, Rehncrona S, Linderoth B, Bjartmarz H, Shafer LL, Coffey R, Svensson M, Mercer KJ, Forsberg A, Halldin C, Svenningsson P, Widner H, Frisen J, Palhagen S & Haegerstrand A (2015). Safety and tolerability of intracerebroventricular PDGF‐BB in Parkinson's disease patients. J Clin Invest 125, 1339–1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peri A & Serio M (2008). Neuroprotective effects of the Alzheimer's disease‐related gene seladin‐1. J Mol Endocrinol 41, 251–261. [DOI] [PubMed] [Google Scholar]
- Phillips C, Baktir MA, Srivatsan M & Salehi A (2014). Neuroprotective effects of physical activity on the brain: a closer look at trophic factor signaling. Front Cell Neurosci 8, 170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posse de Chaves E & Sipione S (2010). Sphingolipids and gangliosides of the nervous system in membrane function and dysfunction. FEBS Lett 584, 1748–1759. [DOI] [PubMed] [Google Scholar]
- Prinetti A, Chigorno V, Prioni S, Loberto N, Marano N, Tettamanti G & Sonnino S (2001). Changes in the lipid turnover, composition, and organization, as sphingolipid‐enriched membrane domains, in rat cerebellar granule cells developing in vitro. J Biol Chem 276, 21136–21145. [DOI] [PubMed] [Google Scholar]
- Remy F, Mirrashed F, Campbell B & Richter W (2004). Mental calculation impairment in Alzheimer's disease: a functional magnetic resonance imaging study. Neurosci Lett 358, 25–28. [DOI] [PubMed] [Google Scholar]
- Renner M, Choquet D & Triller A (2009). Control of the postsynaptic membrane viscosity. J Neurosci 29, 2926–2937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roman F, Staubli U & Lynch G (1987). Evidence for synaptic potentiation in a cortical network during learning. Brain Res 418, 221–226. [DOI] [PubMed] [Google Scholar]
- Rosenzweig ES & Barnes CA (2003). Impact of aging on hippocampal function: plasticity, network dynamics, and cognition. Prog Neurobiol 69, 143–179. [DOI] [PubMed] [Google Scholar]
- Rouser G & Yamamoto A (1968). Curvilinear regression course of human brain lipid composition changes with age. Lipids 3, 284–287. [DOI] [PubMed] [Google Scholar]
- Runquist M, Parmryd I, Thelin A, Chojnacki T & Dallner G (1995). Distribution of branch point prenyltransferases in regions of bovine brain. J Neurochem 65, 2299–2306. [DOI] [PubMed] [Google Scholar]
- Russell DW, Halford RW, Ramirez DM, Shah R & Kotti T (2009). Cholesterol 24‐hydroxylase: an enzyme of cholesterol turnover in the brain. Annu Rev Biochem 78, 1017–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacket SJ, Chung HY, Okajima F & Im DS (2009). Increase in sphingolipid catabolic enzyme activity during aging. Acta Pharmacol Sin 30, 1454–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salaun C, James DJ & Chamberlain LH (2004). Lipid rafts and the regulation of exocytosis. Traffic 5, 255–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schilling JM, Cui W, Godoy JC, Risbrough VB, Niesman IR, Roth DM, Patel PM, Drummond JC, Patel HH, Zemljic‐Harpf AE & Head BP (2014). Long‐term atorvastatin treatment leads to alterations in behavior, cognition, and hippocampal biochemistry. Behav Brain Res 267, 6–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt JM, Wayman GA, Nozaki N & Soderling TR (2004). Calcium activation of ERK mediated by calmodulin kinase I. J Biol Chem 279, 24064–24072. [DOI] [PubMed] [Google Scholar]
- Shafaati M, Marutle A, Pettersson H, Lovgren‐Sandblom A, Olin M, Pikuleva I, Winblad B, Nordberg A & Bjorkhem I (2011). Marked accumulation of 27‐hydroxycholesterol in the brains of Alzheimer's patients with the Swedish APP 670/671 mutation. J Lipid Res 52, 1004–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soderberg M, Edlund C, Kristensson K & Dallner G (1990). Lipid compositions of different regions of the human brain during aging. J Neurochem 54, 415–423. [DOI] [PubMed] [Google Scholar]
- Sodero AO, Trovo L, Iannilli F, Van Veldhoven P, Dotti CG & Martin MG (2011). Regulation of tyrosine kinase B activity by the Cyp46/cholesterol loss pathway in mature hippocampal neurons: relevance for neuronal survival under stress and in aging. J Neurochem 116, 747–755. [DOI] [PubMed] [Google Scholar]
- Steck TL & Lange Y (2012). How slow is the transbilayer diffusion (flip‐flop) of cholesterol? Biophys J 102, 945–946; author reply 947–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiess M & Bradke F (2011). Neuronal polarization: the cytoskeleton leads the way. Dev Neurobiol 71, 430–444. [DOI] [PubMed] [Google Scholar]
- Suzuki T (2002). Lipid rafts at postsynaptic sites: distribution, function and linkage to postsynaptic density. Neurosci Res 44, 1–9. [DOI] [PubMed] [Google Scholar]
- Svennerholm L, Bostrom K, Jungbjer B & Olsson L (1994). Membrane lipids of adult human brain: lipid composition of frontal and temporal lobe in subjects of age 20 to 100 years. J Neurochem 63, 1802–1811. [DOI] [PubMed] [Google Scholar]
- Thelen KM, Falkai P, Bayer TA & Lutjohann D (2006). Cholesterol synthesis rate in human hippocampus declines with aging. Neurosci Lett 403, 15–19. [DOI] [PubMed] [Google Scholar]
- Thiele C, Hannah MJ, Fahrenholz F & Huttner WB (2000). Cholesterol binds to synaptophysin and is required for biogenesis of synaptic vesicles. Nat Cell Biol 2, 42–49. [DOI] [PubMed] [Google Scholar]
- Thudichum JLW (1884). A Treatise on the Chemical Constitution of the Brain. Bailliere, Tindall and Cox, London. [Google Scholar]
- Titus DJ, Furones C, Kang Y & Atkins CM (2013. a). Age‐dependent alterations in cAMP signaling contribute to synaptic plasticity deficits following traumatic brain injury. Neuroscience 231, 182–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Titus DJ, Sakurai A, Kang Y, Furones C, Jergova S, Santos R, Sick TJ & Atkins CM (2013. b). Phosphodiesterase inhibition rescues chronic cognitive deficits induced by traumatic brain injury. J Neurosci 33, 5216–5226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong J, Furukawa Y, Sherwin A, Hornykiewicz O & Kish SJ (2011). Heterogeneous intrastriatal pattern of proteins regulating axon growth in normal adult human brain. Neurobiol Dis 41, 458–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trovo L, Van Veldhoven PP, Martin MG & Dotti CG (2011). Sphingomyelin upregulation in mature neurons contributes to TrkB activity by Rac1 endocytosis. J Cell Sci 124, 1308–1315. [DOI] [PubMed] [Google Scholar]
- Turley SD, Burns DK & Dietschy JM (1998). Preferential utilization of newly synthesized cholesterol for brain growth in neonatal lambs. Am J Physiol Endocrinol Metab 274, E1099–1105. [DOI] [PubMed] [Google Scholar]
- Valaperta R, Chigorno V, Basso L, Prinetti A, Bresciani R, Preti A, Miyagi T & Sonnino S (2006). Plasma membrane production of ceramide from ganglioside GM3 in human fibroblasts. FASEB J 20, 1227–1229. [DOI] [PubMed] [Google Scholar]
- Vance JE & Hayashi H (2010). Formation and function of apolipoprotein E‐containing lipoproteins in the nervous system. Biochim Biophys Acta 1801, 806–818. [DOI] [PubMed] [Google Scholar]
- Vanmierlo T, Bloks VW, van Vark‐van der Zee LC, Rutten K, Kerksiek A, Friedrichs S, Sijbrands E, Steinbusch HW, Kuipers F, Lutjohann D & Mulder M (2009). Alterations in brain cholesterol metabolism in the APPSLxPS1mut mouse, a model for Alzheimer's disease. J Alzheimers Dis 19, 117–127.20061631 [Google Scholar]
- Villeda SA, Plambeck KE, Middeldorp J, Castellano JM, Mosher KI, Luo J, Smith LK, Bieri G, Lin K, Berdnik D, Wabl R, Udeochu J, Wheatley EG, Zou B, Simmons DA, Xie XS, Longo FM & Wyss‐Coray T (2014). Young blood reverses age‐related impairments in cognitive function and synaptic plasticity in mice. Nat Med 20, 659–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasser CR & Kavalali ET (2009). Leaky synapses: regulation of spontaneous neurotransmission in central synapses. Neuroscience 158, 177–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wayman GA, Impey S, Marks D, Saneyoshi T, Grant WF, Derkach V & Soderling TR (2006). Activity‐dependent dendritic arborization mediated by CaM‐kinase I activation and enhanced CREB‐dependent transcription of Wnt‐2. Neuron 50, 897–909. [DOI] [PubMed] [Google Scholar]
- Wayman GA, Kaech S, Grant WF, Davare M, Impey S, Tokumitsu H, Nozaki N, Banker G & Soderling TR (2004). Regulation of axonal extension and growth cone motility by calmodulin‐dependent protein kinase I. J Neurosci 24, 3786–3794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf AA, Jobling MG, Wimer‐Mackin S, Ferguson‐Maltzman M, Madara JL, Holmes RK & Lencer WI (1998). Ganglioside structure dictates signal transduction by cholera toxin and association with caveolae‐like membrane domains in polarized epithelia. J Cell Biol 141, 917–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood WG, Igbavboa U, Muller WE & Eckert GP (2011). Cholesterol asymmetry in synaptic plasma membranes. J Neurochem 116, 684–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G, Lu ZH & Ledeen RW (1997). Interaction of the delta‐opioid receptor with GM1 ganglioside: conversion from inhibitory to excitatory mode. Brain Res Mol Brain Res 44, 341–346. [DOI] [PubMed] [Google Scholar]
- Xie C, Lund EG, Turley SD, Russell DW & Dietschy JM (2003). Quantitation of two pathways for cholesterol excretion from the brain in normal mice and mice with neurodegeneration. J Lipid Res 44, 1780–1789. [DOI] [PubMed] [Google Scholar]
- Yamamoto N, Matsubara T, Sato T & Yanagisawa K (2008). Age‐dependent high‐density clustering of GM1 ganglioside at presynaptic neuritic terminals promotes amyloid beta‐protein fibrillogenesis. Biochim Biophys Acta 1778, 2717–2726. [DOI] [PubMed] [Google Scholar]