

Abstract
Hint1 has recently emerged to be an important target of interest due to its involvement in the regulation of a broad range of CNS functions including opioid signaling, tolerance, neuropathic pain, and nicotine dependence. A series of inhibitors were rationally designed, synthesized, and tested for their inhibitory activity against hHint1 using isothermal titration calorimetry (ITC). The studies resulted in the development of the first small-molecule inhibitors of hHint1 with submicromolar binding affinities. A combination of thermodynamic and high-resolution X-ray crystallographic studies provides an insight into the biomolecular recognition of ligands by hHint1. These novel inhibitors have potential utility as molecular probes to better understand the role and function of hHint1 in the CNS.
Keywords: hHint1, human histidine triad nucleotide binding protein 1, ITC, isothermal titration calorimetry, CNS, central nervous system
Human histidine triad nucleotide binding protein 1 (hHint1) has emerged a protein of interest due to its widespread expression in the CNS and recently discovered potential as a new target for the treatment of pain.1,2 Human Hint1 belongs to the histidine triad (HIT) superfamily, which are characterized by their conserved sequence motif, His-X-His-X-His-XX, where X is a hydrophobic residue. Human Hint1 exists as a homodimer and possesses nucleoside phosphoramidase and acyl-AMP hydrolase activity, with a substrate preference for purine over pyrimidine nucleosides.3 Structural and kinetic studies have shown that hHint1 possess two identical and independent nucleotide-binding subunits.4−6 Each monomer consists of five antiparallel beta strands and two alpha helices. A conserved string of hydrophobic residues in or adjacent to the β-sheets creates a binding pocket (S1) for the nucleobase, while the aspartate (43) residue anchors the ribose sugar. The α-monophosphate group interacts with a conserved string of polar residues including the partially positive His114 and the nucleophilic His112 in the active site. The side chains of the nucleoside phosphoramidates or acyl-AMP can occupy a relative shallow and solvent accessible pocket containing the only tryptophan residue in hHint1. A nucleophilic histidine (His112) residue forming part of the active site triad of hHint1 is responsible for the catalysis. A detailed investigation of the kinetic mechanism of hHint1 has revealed that the mechanism proceeds by rapid formation of the nucleotidylated-His intermediate, followed by partially rate limiting water mediated hydrolysis and subsequent release of the nucleoside monophosphate from the active site.5 The nucleoside phosphoramidase activity of hHint1 has been shown to be necessary for the activation of several preclinical and clinically approved antiviral and anticancer phosphoramidate pronucleotides.7−10 In addition, Chou and Wagner et al. have demonstrated that lysyl t-RNA synthetase-generated lysyl-AMP is also a substrate for hHint1 in vitro.11
Hint proteins are highly conserved across all the kingdoms of life, suggesting that they have an important biological function. Hint1 has been implicated in the regulation of MITF/USF2 transcriptional activation complex in mast cells,12 t-RNA synthetase amino acid adenylation,11 apoptosis,13 and tumorigenicity.14 Hint1 is widely expressed in the region of brain primarily responsible for the modulation of pain [periaquaductal gray area (PAG)], addiction properties (nucleus accumbens), and the motor and sensory functions (cerebral cortex).1 In agreement with these results, alterations in gene function or aberrant expression of hHint1 have been found in the brain tissues of clinical patients suffering from schizophrenia and bipolar disorders.15 Moreover, Hint1–/– mice have been shown to exhibit hypersensitivity to amphetamine and decreased dependence on nicotine in self-administration studies.16,17 In addition, the NMDAR mediated feedback inhibition of the analgesic response has been demonstrated to be critically dependent on the coassociation of Hint1 with MOR and NMDAR.18 Hence, Hint1–/– mice have been shown to display an enhanced analgesic response to morphine.19 Nevertheless, the roles of the Hint1 active site and the potential endogenous substrate regulating its response in these signaling pathways have remained enigmatic.
Chemical genetics is a powerful approach to elucidate biological functions of genes or proteins of interest using screens of diverse and targeted small molecules. Screening small molecule libraries for a compound that induces a phenotype of interest represents forward chemical genetics, whereas the reverse approach uses small molecules targeted to a protein or gene of interest, to probe their biological function.20 Both approaches offer particular advantages of reversibility and the potential to modulate a single function. Recently, our laboratory has designed and synthesized a nonhydrolyzable analogue for hHint1 (compound 3, see Figure 1a), by replacing the acyl-adenylate or phosphoramidate backbone in the substrate (compounds 1 and 2, see Figure 1a) with a carbamate linker. Compound 3 inhibits hHint1 with a low micromolar binding affinity (Kd = 3.65 ± 1.0 μM).2 Using reverse chemical genetics, we demonstrated that compound 3 not only enhances morphine analgesia but also rescues and prevents the development of NMDAR mediated morphine tolerance in mice.2 Importantly, the increase in the analgesic response and reduction in morphine tolerance were only observed in the wild type mice and not in Hint1–/– animals.2 Thus, compound 3 acts as a selective modulator of Hint1 in vivo. Moreover, a single dose of 3 was able to reduce mechanical allodynia in animals for several days. Nevertheless, compound 3 suffers from poor solubility and low micromolar binding affinity for hHint1. Hence, the development of water-soluble analogues with a higher binding affinity would enhance the capability of the probe to serve as a pharmacological tool for elucidating the biological role of the Hint1 active site on the regulation of NMDAR.
Figure 1.

(a) Chemical structures of the hHint1 substrates (compounds 1 and 2) and a previously reported hHint1 inhibitor (compound 3). (b) Design of the new generation hHint1 inhibitors with an acyl-sulfamate or sulfamide backbone to improve solubility and potency over compound 3.
Replacement of the acyl-phosphate linker of enzyme substrates with a bioisosteric acyl-sulfamate or acyl-sulfamide backbone has been shown to be a successful strategy for the generation of water-soluble and potent inhibitors. For example, 5′-amino-5′-N-(biotinyl)sulfamoyl-5′-deoxyadenosine (Bio-AMS) is a bisubstrate subnanomolar inhibitor of biotin protein ligase and contains a acylsulfamide backbone. Bio-AMS displays potent antitubercular activity against multidrug-resistant Mycobacterium tuberculosis strains.21 Consequently, we report here our preliminary evaluation of nonhydrolyzable nucleotide analogues for hHint1 that contain an acyl-sulfamate or acyl-sulfamide linker that mimics the corresponding phosphoramidate and acyl-phosphate moieties.
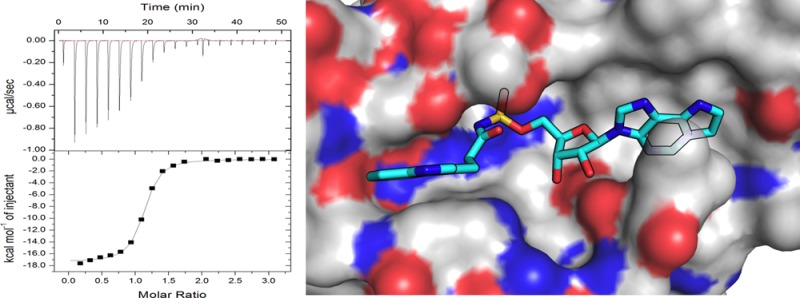
We began by comparing the effect of a nonhydrolyzable nucleotide analogue Bio-AMS with compound 3 on the activity of hHint1 using a fluorescence assay described previously.3 At a fixed saturating substrate concentration, both Bio-AMS and compound 3 exhibited a dose-dependent decrease in the activity of hHint1 with maximum half inhibitory concentration (IC50) values of 1.0 ± 0.3 and 25.5 ± 6.0 μM, respectively (Supplementary Figure 1). We next employed isothermal titration calorimetry (ITC) to investigate the nature of noncovalent interactions on the inhibitory activity of Bio-AMS on hHint1. The ITC studies provided an experimental dissociation constant (Kd) of 0.32 ± 0.1 μM with an n value of 1.0 ± 0.1 indicating one binding site per hHint1 monomer. Bio-AMS was found to bind approximately 11- and 209-fold more tightly than compound 3 and guanosine monophosphate (GMP), respectively, and to be dominated by enthalpy and not entropy (Supplementary Table 1).
In order to avoid potential off-target effects on enzymes utilizing adenosine and adenosine nucleotide based substrates, we sought to develop analogues of compound 3 containing an acyl-sulfamate or acyl-sulfamide backbone (see Figure 1b). The first inhibitor examined was based on the replacement of the carbamate backbone in 3 with a bioisosteric acyl-sulfamate backbone. The synthesis of compound 4 (Scheme 1) began with 5′-OH sulfamoylation of 2′,3′-O-isopropylidene guanosine (8) to provide intermediate 9. Coupling of 9 with the N-hydroxysuccinic acid ester of 3-indole propionic acid in the presence of DBU (1,8-diazabicyclo[5.4.0] undec-7-ene) afforded 10. Deprotection of the acetonide in compound 10 with aqueous TFA yielded the final compound 4. In similar fashion compound 5 with a butyric acid side chain was synthesized.
Scheme 1.

Reagents and conditions: (i) NH2SO2Cl, DMA, 85%; (ii) 22, DBU, DMF 55%; (iii) 80% aq. TFA quant.
Structural studies performed using X-ray crystallography or NMR provide a very important insight into the molecular recognition driving the interaction between a protein and a ligand. Using 2D-NMR studies, Shapiro and co-workers investigated and identified key interactions between nucleoside monophosphates and mouse Hint1.22 Their 1H–15N HSQC investigations revealed large chemical shift perturbations (Δδ > 0.2 ppm) for the residues surrounding the canopy holding the nucleobase and sugar upon addition of the nucleoside monophosphate. Isoleucine 44 in the S1 hydrophobic pocket exhibited the largest chemical shift difference of Δδ = 1.11 ppm indicating that nucleobase recognition maybe a key event in driving the molecular recognition of nucleotide based ligands by Hint1.
We chose to explore the impact of a hydrophobic nucleoside inhibitor with an acyl-sulfamate backbone by replacing the guanosine base in 4 with a tricyclic ethenoadenosine base. Compound 7 provides an additional advantage of stability toward cyclonucleoside formation when compared to an adenosine nucleobase due to the extensive delocalization of the N-3 nitrogen electrons into the tricyclic ring system. The synthesis of compound 7 (Scheme 2) began with the cyclization of exocyclic amine in 11 with chloroacetaldehyde in mildly acidic sodium acetate buffer at 40 °C to yield the fluorescent compound 12. The formation of 12 in the reaction mixture can be easily monitored on thin layer chromatography due to its fluorescent properties. Compound 12 was then treated with sulfamoyl chloride in the presence of triethylamine to yield compound 13. Coupling of 13 with the N-hydroxysuccinic acid ester of 3-indole propionic acid in the presence of DBU followed by deprotection of the acetonide with aqueous TFA yielded compound 7. To avoid potential decomposition due to the intrinsically acidic free acyl-sulfamate group, all the acyl-sulfamate compounds (4, 5, and 7) were prepared and purified as a triethylammonium salt using reverse phase chromatography. Stability studies performed on all the compounds using high performance liquid chromatography (HPLC) revealed no cyclonucleoside formation at 37 °C over 48 h in phosphate buffered saline (Supplemental Figure 3). This result is in striking contrast to the previously reported rapid decomposition of the acyl-sulfamate analogue of Bio-AMS via cyclonucleoside formation.21
Scheme 2.

Reagents and conditions: (i) chloroacetaldehyde, NaOAc 0.1 M, pH 6.5, 40 °C, 30%; (ii) NH2SO2Cl, DMA, 85%; (iii) 22, DBU, overnight, 55%; and 80% aq. TFA, 1 h quant.
Replacement of the 5′-oxygen atom in the acyl-sulfamate with a nitrogen affords an acyl-sulfamide backbone, which increases the pKa of the backbone NH by 2–3 units. The increased pKa has been shown to increase the stability and the binding affinity of Bio-AMS toward biotin protein ligase.20 Hence, we designed compound 6 to investigate the impact of the enhanced negative charge of the backbone on the binding affinity of hHint1. The synthesis of compound 6 (Scheme 3) began with protection of the exocyclic nitrogen on compound 8 with N,N-dimethylformamide dimethylacetal to yield N,N-dimethyl aminomethylene-2′-3′-O-isopropylidene guanosine (14). Iodination of the 5′-hydroxy group in 14 with methyltriphenoxyphosphonium iodide (MTPI) in a Moffat reaction afforded compound 14a (see Supporting Information). Displacement of the iodide in 14a with sodium azide followed by reduction under the Staudinger reaction conditions yielded compound 16 with a 5′-amine on the ribose sugar. Next, the corresponding 5′-amino nucleoside was converted to the 5′-sulfamide by refluxing compound 16 with sulfamide (NH2SO2NH2) in 1,4-dioxane for 2 h.23 Surprisingly, this step also resulted in the removal of the N,N-dimethyl aminomethylene of the exocyclic amine along with the formation of the desired product. The crude 5′-sulfamide nucleoside was then stirred in sodium hydroxide/methanol solution to completely deprotect the N,N-dimethyl aminomethylene group to afford 17 in an overall yield of 34% over two steps. Coupling of 17 to the N-hydroxysuccinic acid ester of 3-indole propionic acid in the presence of DBU followed by the deprotection of the acetonide with aqueous TFA (trifluoroacetic acid) yielded the final compound 6. All the compounds prepared above displayed superior aqueous solubility compared to compound 3. Stock solutions up to 5 mM for the guanosine analogues (compounds 4, 5, and 6) and 20 mM for 7 were easily prepared in aqueous buffers.
Scheme 3.

Reagents and conditions: (i) MTPI, THF, −70 °C for 30 min and then RT for 4 h 92%; (ii) NaN3, DMF, RT overnight, 55%; (iii) triphenyl phosphine/aq dioxane, triethylamine, 50 °C, 3 h, 54%; (iv) NH2SO2NH2, 1,4-dioxane reflux for 2 h; and 4 N NaOH/MeOH for 2 h, 33%; (v) 22, DBU in DMF, overnight, 55%; and 80% aq. TFA, 1 h quant.
Our next step was to evaluate the in vitro binding affinity of the new series of analogues for hHint1 using isothermal calorimetry (ITC). In comparison to 3, compound 4 displayed a 4.5-fold increase in binding affinity with a measured dissociation constant of 0.81 ± 0.11 μM for hHint1 (Table 1). The increased binding affinity of 4 is likely due to electrostatic and/or hydrogen-bonding interactions of the acyl-sulfamate backbone, with polar side chains in the active site, as indicated by the increased gain in the enthalpic component over 3. Increasing the pKa of the backbone with a sulfamide in compound 6 did not alter the binding affinity of compound 1 as indicated by their similar dissociation constants. Attachment of an indole side chain intramolecularly to a nucleoside has been shown to dynamically quench the fluorescence of the indole side chain due to stacking interaction of the indole ring on the nucleobase.24 Therefore, one might predict that compounds 4 and 5 are likely to encounter a higher entropic penalty upon binding to hHint1. Consequently, one might propose that removal of the indole group in compound 5 would likely increase the binding affinity by decreasing the entropic cost of binding to hHint1. Surprisingly, the dissociation constant for compound 5 for hHint1 was found to be in a 3–4-fold greater in comparison to 4 (Table 1). Comparing the thermodynamic parameters of 4 and 5 revealed no significant differences in the entropy of binding. However, compound 5 displayed a nearly 2 kcal mol–1 decrease in the enthalpy of binding. These results indicate that increasing interactions associated with the active site can improve the ligand binding affinity. Consistent with this observation, compound 7 in which a tricyclic nucleobase has been incorporated, resulted in an increased binding affinity with a measured dissociation constant of 0.23 ± 0.01 μM. Compound 7 displayed an increase in binding affinity of 16- and 291-fold over compound 3 and GMP, respectively. In comparison with 4, a nearly 1 kcal mol–1 increase in the enthalpy of binding was observed for 7, with no observable difference in the entropic component. These results indicate that, while ligand and active site desolvation is important, the interactions of the ligand with the active site appear to dominate biomolecular recognition of the ligands by hHint1.
Table 1. Thermodynamic Parameters and Dissociation Constants of hHint1–Ligand Complexes Determined Using ITC.
| compd | Kd (μM) | ΔH (kcal mol–1) | –TΔS (kcal mol–1) | ΔG (kcal mol–1) |
|---|---|---|---|---|
| 3a | 3.65 ± 1.00 | –13.54 ± 1.00 | 9.54 ± 4.17 | –4.1 ± 2.0 |
| 4 | 0.81 ± 0.11 | –16.51 ± 0.17 | 8.05 ± 0.88 | –8.46 ± 0.4 |
| 5 | 2.90 ± 0.25 | –13.59 ± 1.12 | 7.71 ± 0.27 | –5.81 ± 1.0 |
| 6 | 0.92 ± 0.07 | –14.75 ± 0.12 | 6.57 ± 0.17 | –8.24 ± 0.12 |
| 7 | 0.23 ± 0.01 | –17.31 ± 0.05 | 8.19 ± 0.13 | –9.13 ± 0.11 |
| GMPb | 67 ± 7.9 |
To identify the molecular interaction most critical to the potency of compound 7 and Bio-AMS, we solved the cocrystal structures with hHint1 at 1.6 and 1.25 Å resolution, respectively (Figure 2 and Supplemental Table 2). Compared to the structure of AMP (pdb: 3TW2),25 an additional hydrogen bond between the carbonyl of the acyl-sulfamate of 7 and active site Ser107 was observed. Interestingly, this interaction was not found in the structure with Bio-AMS (Supplementary Table 2). When compared with the hHint1-AMP structure, a rotation around the 5′-O–S bond is observed for 7 compared to the 5′-O–P for AMP binding, thus positioning the Ser107 further away and 2.6 Å from the carbonyl. These results are consistent with the gain in the binding affinity and the observed increase in the enthalpic contribution to binding (Table 1). They also are consistent with the preference of hHint1 for acyl-nucleoside monophosphate (NMP) substrates, suggesting a role for Ser107 in stabilizing negative charge development on the substrate carbonyl during catalysis. Examination of the ribose ring revealed that, as observed for all Hint nucleotide structures, active site binding of the ribose sugar 2′,3′-hydroxyl are driven by hydrogen bond interactions with the side chain oxygen atoms of Asp43 (2.6 and 2.4-Å). With regard to the 5′ side-chain of compound 7, stabilizing van der Waals interactions were observed between the linking methylenes and the indole ring of Trp123 (Figure 2A). In addition, the planar tricyclic ring of the nucleobase is well accommodated by the hydrophobic S1 pocket (which largely comprises Ile18, Phe19, Ile22, Ile27, and Ile44). When compared to the AMP bound structure, minor changes were observed in the side chain of the isoleucines in the S1 pocket (Figure 2B), with no significant variation in the protein backbone structure. Moreover, no significant changes in the overall conformation of the protein were observed when compared to the apo or nucleotide bound structures.
Figure 2.
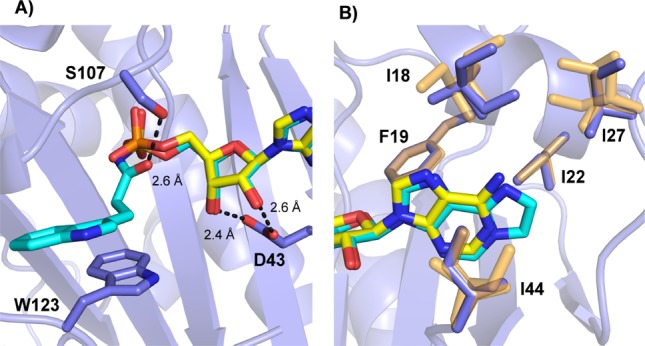
High-resolution X-ray crystal structure analysis of AMP (yellow; pdb: 3TW2) and overlaid with the compound 7 (cyan) in interaction with hHint1 (blue; pdb: 5I2E) complex. (A) H-bond interaction of the sugar and side chain are shown in dotted black lines. (B) Different orientations of isoleucine side chains observed in the hydrophobic nucleotide-binding pocket for AMP and compound 7 bound hHint1 structure is shown in yellow and blue, respectively.
In conclusion, we have designed, synthesized, and evaluated a series of nucleotidomimitic inhibitors of hHint1 with improved solubility and binding to hHint1. These studies, which focused on the optimization of the backbone linker, side chain, and nucleobase, resulted in the identification of the acyl-sulfamate 7, which exhibits approximately 16- and 300-fold higher binding affinity toward hHint1 than 3 and GMP, respectively. High-resolution X-ray crystal structure analysis of hHint1 in complex with 7 revealed an additional hydrogen bond and a gain in van der Waals interactions over the hHint1-AMP complex. Results from ITC and SAR studies indicate that a favorable enthalpic contribution and a fine balance between hydrophilicity/hydrophobicity of the ligand are likely necessary for maintaining the binding affinity of ligands toward hHint1. It is possible that a part of these enthalpic contributions originate from nonclassical hydrophobic effects as seen with the complexation of arenes and aromatic substrates with biological receptors in water.26−28 Future investigations on the free energy of solvation of the ligands, as well as water map calculations on the protein–ligand complex, may assist in defining the role of multiple hydrophobic effects on ligand molecular recognition by the hHint1 active site.26,27 Ongoing studies of the in vivo effects of these ligands on morphine analgesia and tolerance, as well as neuropathic pain, are currently under investigation and will be reported in due course.
Acknowledgments
We thank Dr. Courtney Aldrich lab at the University of Minnesota for providing the Bio-AMS for the initial SAR studies. The authors would also like to acknowledge IMCA-CAT beamline 17-ID at the Advanced Photon Source supported by the companies of the Industrial Macromolecular Crystallography Association through a contract with Hauptman-Woodward Medical Research Institute. Isothermal titration calorimetry was carried out using an ITC-200 microcalorimeter, funded by the NIH Shared Instrumentation Grant S10-OD017982.
Glossary
ABBREVIATIONS
- hHint1
human histidine triad nucleotide binding protein 1
- NMDAR
N-methyl d-aspartate receptor
- MOR
μ-opioid receptor
- CNS
central nervous system
- DBU
1,8-diazabicyclo[5.4.0] undec-7-ene
- TFA
trifluoroacetic acid
- MTPI
methyltriphenoxyphosphonium iodide
- DMA
dimethyl acetamide
- EDC
1-ethyl-3-(3-(dimethylamino)propyl)carbodiimide
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.6b00169.
Details of the synthetic, HPLC, and crystallographic procedures (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Liu Q.; Puche A. C.; Wang J. B. Distribution and expression of protein kinase C interactive protein (PKCI/HINT1) in mouse central nervous system (CNS). Neurochem. Res. 2008, 33, 1263–1276. 10.1007/s11064-007-9578-4. [DOI] [PubMed] [Google Scholar]
- Garzón J.; Herrero-Labrador R.; Rodríguez-Muñoz M.; Shah R.; Vicente-Sánchez A.; Wagner C. R.; Sánchez-Blázquez P. HINT1 protein: a new therapeutic target to enhance opioid antinociception and block mechanical allodynia. Neuropharmacology 2015, 89, 412–423. 10.1016/j.neuropharm.2014.10.022. [DOI] [PubMed] [Google Scholar]
- Chou T. F.; Baraniak J.; Kaczmarek R.; Zhou X.; Cheng J.; Ghosh B.; Wagner C. R. Phosphoramidate pronucleotides: a comparison of the phosphoramidase substrate specificity of human and Escherichia coli histidine triad nucleotide binding proteins. Mol. Pharmaceutics 2007, 4, 208–217. 10.1021/mp060070y. [DOI] [PubMed] [Google Scholar]
- Wang J.; Fang P.; Schimmel P.; Guo M. Side chain independent recognition of aminoacyl adenylates by the Hint1 transcription suppressor. J. Phys. Chem. B 2012, 116, 6798–6805. 10.1021/jp212457w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X.; Chou T. F.; Aubol B. E.; Park C. J.; Wolfenden R.; Adams J.; Wagner C. R. Kinetic mechanism of human histidine triad nucleotide binding protein 1. Biochemistry 2013, 52, 3588–3600. 10.1021/bi301616c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lima C. D.; Klein M. G.; Hendrickson W. A. Structure-based analysis of catalysis and substrate definition in the HIT protein family. Science 1997, 278, 286–290. 10.1126/science.278.5336.286. [DOI] [PubMed] [Google Scholar]
- Murakami E.; Tolstykh T.; Bao H.; Niu C.; Steuer H. M.; Bao D.; Chang W.; Espiritu C.; Bansal S.; Lam A. M.; Otto M. J.; Sofia M. J.; Furman P. A. Mechanism of activation of PSI-7851 and its diastereoisomer PSI-7977. J. Biol. Chem. 2010, 285, 34337–34347. 10.1074/jbc.M110.161802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drontle D. P.; Wagner C. R. Designing a pronucleotide stratagem: lessons from amino acid phosphoramidates of anticancer and antiviral pyrimidines. Mini-Rev. Med. Chem. 2004, 4, 409–419. 10.2174/1389557043403945. [DOI] [PubMed] [Google Scholar]
- Li S.; Jia Y.; Jacobson B.; McCauley J.; Kratzke R.; Bitterman P. B.; Wagner C. R. Treatment of breast and lung cancer cells with a N-7 benzyl guanosine monophosphate tryptamine phosphoramidate pronucleotide (4Ei-1) results in chemosensitization to gemcitabine and induced eIF4E proteasomal degradation. Mol. Pharmaceutics 2013, 10, 523–531. 10.1021/mp300699d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abraham T. W.; Kalman T. I.; McIntee E. J.; Wagner C. R. Synthesis and biological activity of aromatic amino acid phosphoramidates of 5-fluoro-2′-deoxyuridine and 1-beta-arabinofuranosylcytosine: evidence of phosphoramidase activity. J. Med. Chem. 1996, 39, 4569–4575. 10.1021/jm9603680. [DOI] [PubMed] [Google Scholar]
- Chou T. F.; Wagner C. R. Lysyl-tRNA synthetase-generated lysyl-adenylate is a substrate for histidine triad nucleotide binding proteins. J. Biol. Chem. 2007, 282, 4719–4727. 10.1074/jbc.M610530200. [DOI] [PubMed] [Google Scholar]
- Lee Y. N.; Nechushtan H.; Figov N.; Razin E. The function of lysyl-tRNA synthetase and Ap4A as signaling regulators of MITF activity in FcepsilonRI-activated mast cells. Immunity 2004, 20, 145–151. 10.1016/S1074-7613(04)00020-2. [DOI] [PubMed] [Google Scholar]
- Li H.; Zhang Y.; Su T.; Santella R. M.; Weinstein I. B. Hint1 is a haplo-insufficient tumor suppressor in mice. Oncogene 2006, 25, 713–721. 10.1038/sj.onc.1209111. [DOI] [PubMed] [Google Scholar]
- Wang L.; Zhang Y.; Li H.; Xu Z.; Santella R. M.; Weinstein I. B. Hint1 inhibits growth and activator protein-1 activity in human colon cancer cells. Cancer Res. 2007, 67, 4700–4708. 10.1158/0008-5472.CAN-06-4645. [DOI] [PubMed] [Google Scholar]
- Varadarajulu J.; Schmitt A.; Falkai P.; Alsaif M.; Turck C. W.; Martins-de-Souza D. Differential expression of HINT1 in schizophrenia brain tissue. Eur. Arch Psychiatry Clin Neurosci. 2012, 262, 167–172. 10.1007/s00406-011-0216-4. [DOI] [PubMed] [Google Scholar]
- Barbier E.; Zapata A.; Oh E.; Liu Q.; Zhu F.; Undie A.; Shippenberg T.; Wang J. B. Supersensitivity to amphetamine in protein kinase-C interacting protein/HINT1 knockout mice. Neuropsychopharmacology 2007, 32, 1774–1782. 10.1038/sj.npp.1301301. [DOI] [PubMed] [Google Scholar]
- Jackson K. J.; Wang J. B.; Barbier E.; Damaj M. I.; Chen X. The histidine triad nucleotide binding 1 protein is involved in nicotine reward and physical nicotine withdrawal in mice. Neurosci. Lett. 2013, 550, 129–133. 10.1016/j.neulet.2013.06.027. [DOI] [PubMed] [Google Scholar]
- Rodríguez-Muñoz M.; Sánchez-Blázquez P.; Vicente-Sánchez A.; Bailón C.; Martín-Aznar B.; Garzón J. The histidine triad nucleotide-binding protein 1 supports mu-opioid receptor-glutamate NMDA receptor cross-regulation. Cell. Mol. Life Sci. 2011, 68, 2933–2949. 10.1007/s00018-010-0598-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guang W.; Wang H.; Su T.; Weinstein I. B.; Wang J. B. Role of mPKCI, a novel mu-opioid receptor interactive protein, in receptor desensitization, phosphorylation, and morphine-induced analgesia. Mol. Pharmacol. 2004, 66, 1285–1292. 10.1124/mol.66.5.. [DOI] [PubMed] [Google Scholar]
- Kawasumi M.; Nghiem P. Chemical genetics: elucidating biological systems with small-molecule compounds. J. Invest. Dermatol. 2007, 127, 1577–1584. 10.1038/sj.jid.5700853. [DOI] [PubMed] [Google Scholar]
- Duckworth B. P.; Geders T. W.; Tiwari D.; Boshoff H. I.; Sibbald P. A.; Barry C. E.; Schnappinger D.; Finzel B. C.; Aldrich C. C. Bisubstrate adenylation inhibitors of biotin protein ligase from Mycobacterium tuberculosis. Chem. Biol. 2011, 18, 1432–1441. 10.1016/j.chembiol.2011.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai G.; Feng B.; Wang J. B.; Pozharski E.; Shapiro M. Studies on ligand binding to histidine triad nucleotide binding protein 1. Bioorg. Med. Chem. 2010, 18, 6756–6762. 10.1016/j.bmc.2010.07.051. [DOI] [PubMed] [Google Scholar]
- Dawadi S.; Viswanathan K.; Boshoff H. I.; Barry C. E.; Aldrich C. C. Investigation and conformational analysis of fluorinated nucleoside antibiotics targeting siderophore biosynthesis. J. Org. Chem. 2015, 80, 4835–4850. 10.1021/acs.joc.5b00550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruber B. A.; Leonard N. J. Dynamic and static quenching of 1,N6-ethenoadenine fluorescence in nicotinamide 1,N6-ethenoadenine dinucleotide and in 1,N6-etheno-9-(3-(indol-3-yl) propyl) adenine. Proc. Natl. Acad. Sci. U. S. A. 1975, 72, 3966–3969. 10.1073/pnas.72.10.3966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolot R.; Ozga M.; Włodarczyk A.; Krakowiak A.; Nawrot B. A new crystal form of human histidine triad nucleotide-binding protein 1 (hHINT1) in complex with adenosine 5′-monophosphate at 1.38 Å resolution. Acta Crystallogr., Sect. F: Struct. Biol. Cryst. Commun. 2012, 68, 883–888. 10.1107/S1744309112029491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder P. W.; Mecinovic J.; Moustakas D. T.; Thomas S. W.; Harder M.; Mack E. T.; Lockett M. R.; Héroux A.; Sherman W.; Whitesides G. M. Mechanism of the hydrophobic effect in the biomolecular recognition of arylsulfonamides by carbonic anhydrase. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 17889–17894. 10.1073/pnas.1114107108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breiten B.; Lockett M. R.; Sherman W.; Fujita S.; Al-Sayah M.; Lange H.; Bowers C. M.; Heroux A.; Krilov G.; Whitesides G. M. Water networks contribute to enthalpy/entropy compensation in protein-ligand binding. J. Am. Chem. Soc. 2013, 135, 15579–15584. 10.1021/ja4075776. [DOI] [PubMed] [Google Scholar]
- Persch E.; Dumele O.; Diederich F. Molecular recognition in chemical and biological systems. Angew. Chem., Int. Ed. 2015, 54, 3290–3327. 10.1002/anie.201408487. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.