

Abstract
We report the design, synthesis, and evaluation of potent and selective inhibitors of aldo-keto reductase 1C3 (AKR1C3), an important enzyme in the regulatory pathway controlling proliferation, differentiation, and apoptosis in myeloid cells. Combination treatment with the nontoxic AKR1C3 inhibitors and etoposide or daunorubicin in acute myeloid leukemia cell lines, elicits a potent adjuvant effect, potentiating the cytotoxicity of etoposide by up to 6.25-fold and the cytotoxicity of daunorubicin by >10-fold. The results validate AKR1C3 inhibition as a common adjuvant target across multiple AML subtypes. These compounds in coadministration with chemotherapeutics in clinical use enhance therapeutic index and may avail chemotherapy as a treatment option to the pediatric and geriatric population currently unable to tolerate the side effects of cancer drug regimens.
Keywords: AKR1C3 Inhibitor, acute myeloid leukemia, etoposide, daunorubicin, synergism, adjuvant
The aldo-keto reductase family 1 member C (AKR1C) enzymes are oxidoreductases, which catalyze the NADPH-dependent reduction of aldehyde and ketone functionalities on a range of steroids, carbohydrates, and prostaglandins.1 The AKR1C3 enzyme, also known as prostaglandin (PG) F2α synthase,2 converts PGD2 to 11β-PGF2α, which acts to prevent myeloid differentiation and facilitates proliferation of tumor cells by preventing peroxisome proliferator-activated receptor (PPAR) γ activation.3,4 In the absence of AKR1C3, PGD2 can be converted to the PGJ2 series of prostanoids that activate PPARγ leading to differentiation and apoptosis.5
Acute myeloid leukemia (AML) is a hematologic neoplasm characterized by proliferation of poorly differentiated myeloid progenitor cells.6 The current treatment of AML involves chemotherapy with cytarabine combined with etoposide and/or anthracycline chemotherapeutics.7 Young age (<1)8 and older age (>60)7 are widely recognized risk factors for a major cause of therapeutic failure in AML, the inability to tolerate high toxicity associated with the most intensive chemotherapeutic treatment regimes, resulting in an increased rate of early death. Adjuvant therapies that increase response to the chemotherapeutic agent, yet have no innate toxicity, represent a promising therapeutic strategy allowing for lower dosing, and hence, less toxicity to nonmalignant cells while preserving the desired cytotoxic effect in cancerous cells.9 All-trans retinoic acid (ATRA) is an effective differentiation agent in acute promyelocytic leukemia (APL), but its effects are limited to this one subtype of AML.10 The identification of a therapeutic agent with activity across all AML subtypes would have substantial clinical implications.
Primary AML cells and the AML cell lines HL-60 (APL, M3 subtype) and KG1a (AML, M0 subtype) predominantly express AKR1C3 with median levels two orders of magnitude greater than the AKR1C1 isoform and more than three orders of magnitude greater than the AKR1C2 isoform.11−13 Strong expression of AKR1C3 is also detected in nonmalignant proliferating CD34+ve cells isolated from peripheral blood.13 Overall, this data identifies AKR1C3 as the primary AKR1C isoform encountered in myeloid progenitors and indicates a critical role in the regulation of myelopoiesis. Pharmacological inhibition of AKR1C3 holds the promise of an adjuvant effect, sensitizing leukemic cells to the cytotoxic action of chemotherapeutics delivered synergistically.
Combination of the weak and nonselective pan-AKR1C inhibitor medroxyprogesterone acetate (AKR1C3 pIC50 = 5.6) and bezafibrate demonstrated an approximate 2-fold potentiation of cytotoxic activity.14 A recent study reported that the specific AKR1C3 inhibitor 4-MDDT (pIC50 = 6.3) does not give the adjuvant effect at concentrations up to 50 μM that a pan-AKR1C inhibitor does, despite the low expression of other isoform in AML cells, casting doubt on the validity of AKR1C3 as a therapeutic target in AML.15
The structurally distinct natural product baccharin (1, Figure 1a) demonstrates highly potent inhibitory activity for AKR1C3 (pIC50 = 7.0). Critically, baccharin exhibits exquisite selectivity with absolutely no inhibition of the AKR1C1 or AKR1C2 isoforms.16 Hydrolysis of the ester moiety of 1 provides the known phenol drupanin (1a, Figure S7), which possesses attenuated AKR1C3 inhibitory activity (pIC50 = 4.8) with only 7-fold selectivity for AKR1C3 over the 1C2 isoform. Molecular modeling of baccharin in the active site of AKR1C3 predicts formation of a hydrogen bond between the ester carbonyl and the active site Tyr55 residue.16 Ester bond hydrolysis is a primary feature of metabolism and the magnitude in the reduction of potency for drupanin highlights the unsuitable pharmacokinetics of baccharin as a drug candidate or chemical probe.17
Figure 1.

Synergistic activity of AKR1C3 inhibitors with etoposide in HL-60 cells following 72 h coincubation. Values are the mean ± SD (n = 6). The two-tailed t test analysis was used to compare the statistical difference between control and treatments; ns, not significant; *p < 0.01, **p < 0.05, ***p < 0.001, ****p < 0.0001.
We sought to evaluate the suitability of baccharin and rationally designed hydrolytically more stable derivatives as chemical probes to evaluate adjuvant effects in AML cell lines. To this end, we replaced the ester bond of 1 with the hydrolytically more stable amide bioisostere (2, Scheme 1). Commercially available 4-iodoaniline (5) was brominated and the desired meta-bromide (6) was obtained as the major product by column chromatography. Selective Heck reaction with tert-butyl acrylate was achieved by exploiting the greater reactivity of the iodo substituent over the bromide,18 providing cinnamic ester (7). Dimethylaminopyridine catalyzed addition of a suitably functionalized acid chloride installed the amide bond (8); subsequent Suzuki–Miyaura reaction19 with prenyl pinacolborane provided ester (9), which underwent hydrolysis by exposure to silica gel,20 to yield the amide derivative of baccharin (2). Ring analogues of the hit compound were synthesized based on the premise that reducing the distance of the hydrogen bond between the AKR1C3 active site Tyr55 residue and the side chain ester carbonyl would result in enhanced potency coupled with enhanced steric congestion around the ester providing greater hydrolytic stability. Commercially available 3-hydroxycinnamic acid (10, Scheme 2) was protected as the methyl ester (11). Subsequent bromination with molecular bromine provided 4-bromo-3-hydroxycinamic ester (12), which was readily separable from regioisomers by column chromatography. Subsequent Suzuki–Miyaura coupling with prenyl pinacolborane and hydrolysis provided cinnamic acid (14), which underwent addition of a suitably functionalized acid chloride to yield meta-ester derivative (3). In a similar fashion, the meta-amide derivative (4, Scheme 3) was accessed utilizing the same synthetic sequence but beginning with commercial 2-bromo-5-iodoaniline (15).
Scheme 1. Synthesis of para-Amide Derivative 2.

Reagents and conditions: (i) Br2, AcOH, 44%; (ii) tert-butyl acrylate, NEt3, P(Ph)3, Pd(OAc)2, PhMe, reflux, 64%; (iii) 3-phenylpropanoyl chloride, NEt3, DMAP, DCM, 70 °C, 10%; (iv) prenylboronic acid pinacol ester, Pd(dppf)Cl2, Cs2CO3, DMF, 90 °C, 44%; (v) SiO2, PhMe, reflux, 50%.
Scheme 2. Synthesis of meta-Ester Derivative 3.

Reagents and conditions: (i) MeOH, H2SO4, reflux, 93%; ii Br2, AcOH, RT, 35%; (iii) prenylboronic acid pinacol ester, Pd(dppf)Cl2, Cs2CO3, DMF, 90 °C, 27%; (iv) NaOH, H2O, reflux, 99%; (v) 3-phenylpropanoyl chloride, DMAP, NEt3, DCM, RT, 82%.
Scheme 3. Synthesis of meta-Amide Derivative 4.

Reagents and conditions: (i) tert-butyl acrylate, Pd(OAc)2, P(Ph)3, NEt3, PhMe, 110 °C, 64%; (ii) 3-phenylpropanoyl chloride, DMAP, NEt3, DCM, 70 °C, 88%; (iii) prenylboronlc acid pinacol ester, Pd(dppf)Cl2, Cs2CO3, DMF, 90 °C, 42%; (iv) SiO2, PhMe, reflux, 27%.
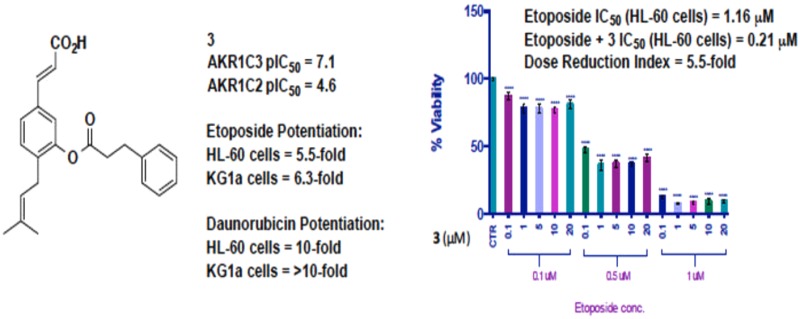
The inhibitory activities of the synthesized baccharin derivatives against AKR1C3 and the highly homologous isoform AKR1C2 were determined (Table 1).21 Synthetic baccharin (1) exhibited potency for AKR1C3 inhibition (pIC50 = 7.0) and excellent selectivity (510-fold) as expected. The para-amide derivative (2) retained some potency and selectivity (pIC50 = 6.4 with 89-fold selectivity) for AKR1C3. The meta-ester derivative (3) represents the best combination of potent inhibitor (pIC50 = 7.1) and selectivity (261-fold for AKR1C3). The meta-amide derivative (4) is the most potent AKR1C3 inhibitor of this scaffold class (pIC50 = 7.2) with 109-fold increase in affinity for AKR1C3.
Table 1. AKR1C3 Inhibition Activity and Selectivity of Synthesized Compounds.
| AKR1C3 | AKR1C2 | fold selectivity | |
|---|---|---|---|
| compd | pIC50 | pIC50 | AKR1C3 |
| 1 | 7.0 | 4.3 | 510 |
| 1a | 4.8 | 3.9 | 7 |
| 2 | 6.4 | 4.4 | 89 |
| 3 | 7.1 | 4.6 | 261 |
| 4 | 7.2 | 5.1 | 109 |
| MPAa,32 | 5.632 | 5.4 | 0.66 |
MPA: medroxyprogesterone acetate.
This selection of compounds enable direct comparison of adjuvant activity for the four AKR1C3 inhibitors (1–4), which exhibit potent inhibitory activity and selectivity over AKR1C2 while incorporating hydrolytically labile and stable linkers to a pharmacophoric moiety.
To investigate the general cytotoxicity of baccharin (1) against a range of cancer cell lines, the compound was submitted to the National Cancer Institute 60 cancer cell line assay (Figure S1).22 The results, in agreement with others,15 show little cytotoxic activity associated with the inhibition of AKR1C3 at a concentration of 10 μM. Baccharin (1) and the derivative AKR1C3 inhibitors (2 and 3) demonstrate no toxicity to the HL-60 human APL cell line at concentrations up to 100 μM (Figure S2). The highly potent meta-amide derivative 4 was significantly (p < 0.0001) cytotoxic at 50 μM. At the lower concentrations employed in this study (0.1–1 μM) 4 showed only a 8% reduction of cell viability. The toxicity of AKR1C3 inhibitor 4 may be attributed to its high potency for AKR1C3 enzyme inhibition that leads to cell death, as has been reported for similarly potent AKR1C3 inhibitors in prostate cancer cells.23 When the AKR1C3 inhibitors were exposed to the KG1a AML cell line (AML M0 subtype), which has much greater expression of AKR1C3 (Figure S3), cytotoxicity was observed at concentrations above 25 μM for inhibitors 1–3 indicating direct toxicity to AML cells upon inhibition of AKR1C3 (Figure S4). Potent inhibitor 4 showed 20% reduction of cell viability at just 1 μM in the KG1a cell line.
The clinically approved drug etoposide, employed as a second line chemotherapeutic to manage AML24,25 was chosen as the cytotoxic agent. The use of etoposide ensures that any potentiation of cytotoxicity would be attributable to AKR1C3 inhibition rather than prevention of AKR1C3-mediated metabolism of the chemotherapeutic agent. A dose–response curve of etoposide was obtained in HL-60 and KG1a cells (Figures S5 and S6). In accordance with the literature, 0.1 μM of etoposide provides no cytotoxic effect in HL-60 cells.26 The IC50 value of etoposide was calculated as 1.16 and 6.70 μM for the HL-60 and KG1a cell lines, respectively (Table 2).
Table 2. Adjuvant Effect of Compounds 1–4 To Potentiate the Effect of Etoposide in Human AML Cell Lines upon Cotreatment at 72 h.
| AKR1C3 | HL-60 |
KG1a |
|||||
|---|---|---|---|---|---|---|---|
| compd | pIC50 | CIa | DRIb | IC50c (μM) | CIa | DRIb | IC50c (μM) |
| 1 | 7.0 | 0.44 | 2.25 | 0.51 | N/A | N/A | N/A |
| 2 | 6.4 | 0.38 | 2.56 | 0.45 | 0.42 | 2.34 | 2.88 |
| 3 | 7.1 | 0.28 | 3.46 | 0.33 | 0.16 | 6.25 | 1.08 |
| 4 | 7.2 | 0.45 | 2.17 | 0.53 | 0.37 | 2.73 | 2.47 |
| etoposide | N/A | N/A | N/A | 1.16 | N/A | N/A | 6.70 |
Combination index.
Dose reduction index.
Calculated for etoposide + AKR1C3 inhibitor (μM). N/A: not applicable.
Combination treatment of HL-60 cells with a range of concentrations of etoposide and AKR1C3 inhibitor was performed to determine the effect on cell viability (Figure 1). Etoposide treatment of HL-60 cells alone at 1 μM reduces cell viability by 40%. The natural product hit compound baccharin (1) at a concentration of 0.1 μM when delivered synergistically with etoposide (1 μM) provided a significant (p < 0.0001) potentiation of etoposide cytotoxicity providing an overall reduction of cell viability by 70% (Figure 1a). When amide analogue (2), the lowest potency AKR1C3 inhibitor of the four lead compounds (pIC50 = 6.4), was delivered synergistically at 0.1 μM concentration with 1 μM etoposide, a potentiation effect was observed that provided an overall 57% reduction of cell viability (Figure 1b). The ring analogue (3) with greater AKR1C3 inhibition potency (pIC50 7.1) at a concentration of 0.1 μM provided an 80% reduction in cell viability in combination with 1 μM etoposide (Figure 1c). The most potent AKR1C3 inhibitor, amide analogue (4) (pIC50 = 7.2), provided a 52% reduction in cell viability at 0.1 μM along with 1 μM etoposide. A significant (p < 0.0001) adjuvant effect was observed when 4 was incubated with just 0.5 μM of etoposide. This concentration of etoposide provides a 20% reduction of cell viability alone, which was potentiated by the action of 4 at 0.1 μM to provide a 45% reduction of cell viability (Figure 1d). When a comparison is made at this concentration of etoposide with the less active inhibitor 2, a reduction of cell viability of only 25% is observed. These data illustrate that all four AKR1C3 inhibitors (1–4) show a potent adjuvant effect.
The adjuvant effect was enhanced in KG1a cells when compounds 2–4 were incubated with etoposide (Figure 2a–c). The cotreatment experiments demonstrated potentiation of etoposide toxicity at 1 μM dose from 18% cell viability reduction (etoposide alone) to 42% when dosed with 0.1 μM of inhibitor 2 (Figure 2a). Ring analogues 3 and 4 at 0.1 μM reduced KG1a cell viability by 54% and 40%, respectively, with 1 μM etoposide (Figure 2b,c).
Figure 2.

Synergistic activity of AKR1C3 inhibitors with etoposide in KG1a cells. Seventy-two hours of coincubation with etoposide. Values are the mean ± SD (n = 6). The two-tailed t test analysis was used to compare the statistical difference between control and treatments; ns, not significant; *p < 0.01, **p < 0.05, ***p < 0.001, ****p < 0.0001.
To assess if this adjuvant effect was indeed attributable to AKR1C3 inhibition we employed the hydrolysis product of 1, the low potency (pIC50 = 4.8) and nonselective (7-fold selectivity for AKR1C3) inhibitor drupanin (1a) as a control compound. The observed adjuvant effect diminished substantially, in parallel with AKR1C3 inhibition activity, when 1a was coincubated with etoposide in HL-60 cells. Compound 1a demonstrated simple additive effects, providing no statistically significant adjuvant effect (p = 0.8512) (Figure S7).
To further discern if inhibition of AKR1C3 is indeed responsible for sensitizing AML cells to the cytotoxic action of etoposide, AKR1C3 inhibitors were next incubated in HL-60 cells for 24 h prior to the addition of etoposide. After a further 72 h, cell viability was measured (Figure 3a–d). All four AKR1C3 inhibitors demonstrated a greater potentiation effect after 24 h pretreatment in contrast to the cotreatments, showing a near complete abrogation of cell viability (80–95% cell viability reduction) when combined with 1 μM etoposide. This observation can be attributed to the greater inhibition of AKR1C3 before addition of etoposide.
Figure 3.

Synergistic activity of AKR1C3 inhibitors with etoposide in HL-60 cells (pretreatment). (a–d) Twenty-four hours of pretreatment of AKR1C3 inhibitor followed by 72 h exposure to etoposide. Values are the mean ± SD (n = 6). The two-tailed t test analysis was used to compare statistical difference between control and treatment; ns, not significant; **p < 0.05, ***p < 0.001, ****p < 0.0001. (e) Quantification of synergistic activity.
To quantify the degree of synergism, the results of the cotreatment and pretreatment experiments were analyzed by CompuSyn software (Paramus, NJ) based on the median effect principle or Chou–Talalay method.27,28 The combination index (CI) and dose reduction index (DRI) values were calculated at a constant ratio of etoposide to AKR1C3 inhibitor at 50% cytotoxic effect (Fa = 0.5). In all experiments, CI values were found to be <1, indicative of synergism. As outlined in Table 2, cotreatment of each AKR1C3 inhibitor with etoposide in AML cell lines demonstrated a synergistic drug action. In HL-60 cells, up to 3.5-fold reduction in dosing of etoposide was observed in cotreatment experiments with AKR1C3 inhibitors, and the dose reduction was increased up to 5.8-fold upon 24 h pretreatment with AKR1C3 inhibitor. CI values exhibited a narrow range from 0.16 (2) to 0.23 (1 and 4), indicative of strong synergism in the pretreatment experiments in HL-60 cells (Figure 3e). A stronger synergistic action was observed in KG1a cells, which have much greater AKR1C3 expression, wherein a dose reduction of up to 6.25-fold was observed upon cotreatment. CI and DRI indices ranged from 0.16 (3)–0.42 (2) and 2.34 (2)–6.25 (3), respectively (Table 2).
The data presented in Table 2 provides a clear quantification of synergistic activity of the AKR1C3 inhibitors by computer simulation. From the calculated DRI values, meta-ester 3 confers greatest adjuvant activity upon synergistic treatment with etoposide. The cytotoxicity of etoposide is enhanced by 3.5-fold in HL-60 cells and 6.25-fold in KG1a cells, resulting in new calculated IC50 values (3 and etoposide) of just 0.33 and 1.08 μM in HL-60 and KG1a cells, respectively. These data can perhaps be rationalized to the enhanced stability of meta-ester 3 to hydrolysis. The hit compound baccharin is metabolically labile; hydrolysis is known to provide an inactive compound (1a). The meta-ester 3 experiences greater steric congestion around the labile ester bond and, as such, would be expected to be more resistant to hydrolysis than 1. Inhibitor 2, by the nature of the amide bond, would be expected to be more stable to hydrolysis than esters 1 and 3 and provides the greatest potentiation effect in pretreatment experiments in HL-60 cells, yet is a less potent AKR1C3 inhibitor. Amide 4, while the most potent AKR1C3 inhibitor, possesses innate cytotoxicity that reduces the DRI index, accounting for the observed CI value of 0.45. Comparison of cell viability data (Figure 1) resulting from combination of AKR1C3 inhibitor (0.1 μM) and 1 μM etoposide in HL-60 cells with 72 h incubation show a linear relationship between AKR1C3 inhibition potency and reduction in cell viability. Compound 3 (pIC50 = 7.1) with etoposide provides 80% cell death, compound 1 (pIC50 = 7.0) with etoposide provides 70% cell death, and compound 4 (pIC50 = 6.4) with etoposide provides 57% cell death. Indeed, this is supported by data from cotreatment experiments in both HL-60 and KG1a cells; amide 2 provides a lower DRI (2.56 and 2.34, respectively) than the more potent meta-ester 3 (3.46 and 6.25, respectively) relating AKR1C3 inhibition potency to potentiation effect.
To establish the scope of synergistic activity across other chemotherapeutics, daunorubicin was employed. Pretreatment experiments were performed in HL-60 and KG1a cells with compound 3 followed by daunorubicin incubation for 72 h. A dose reduction of approximately 10-fold was observed, reducing IC50 from 42 to 4.2 nM and from 1.77 to 0.2 μM in the respective cell lines (Figure S8).
A substantial AKR1C3 induction in cells with low endogenous AKR1C3 levels is observed upon treatment with the anthracycline class of chemotherapeutics.29 This effect has been attributed to anthracycline resistance in various cancers.30 KG1a and HL-60 cells were incubated with AKR1C3 inhibitor 4 at 1, 5, and 10 μM concentration, and the expression levels of AKR1C3 were determined (Figure S3). Treatment with AKR1C3 inhibitor 4 resulted in increased AKR1C3 expression in both cell lines, which was more pronounced in HL-60 cells, after 48 h incubation. After 72 h incubation, reduced AKR1C3 expression was observed. The data are in agreement with prior reports that show a transient increase in AKR1C3 expression immediately preceding differentiation.31
AKR1C3 inhibitors derived from modification of a natural product have enhanced biological stability and exhibit extremely selective and potent activity. Proof-of-concept that AKR1C3 inhibitors derived from baccharin have a synergistic effect sensitizing AML cells to the chemotherapeutic effects of etoposide and daunorubicin is demonstrated. Treatment with the nontoxic AKR1C3 inhibitors in two in vitro models of AML with coadministration of the clinical chemotherapeutics etoposide or daunorubicin results in a synergistic drug action. The IC50 value of etoposide was reduced from 1.16 to 0.21 μM in HL-60 cells and from 6.70 to 1.08 μM in KG1a cells. The potency of daunorubicin is potentiated by up to 10-fold in both HL-60 and KG1a cells. This is contrary to recent reports detailing selective AKR1C3 inhibitors do not perform an adjuvant role compared to pan-AKR1C isoform inhibitors.15
Our results suggest that a strategy developing small molecule AKR1C3 inhibitors may yield powerful adjuvant agents for the synergistic treatment of leukemia, especially since favorable toxicity and pharmacokinetic properties can exist within the baccharin structure. The identified highly potent and selective derivatives represent valuable lead compounds to understand the structural features required for AKR1C3 inhibitory activity and selectivity, and how these properties interrelate to further our understanding of the role, which AKR1C3 plays to enable a potentiation of chemotherapeutic effect within AML.
Acknowledgments
We thank Dr. J. Lu and Dr. S. Srivastava for valuable advice.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.6b00163.
Full experimental details and characterization for all final compounds and descriptions of biological assays (PDF)
This work was supported by the NCI and NIEHS (R01-CA90744 and P30-ES013508 to T.M.P) from the National Institutes of Health and by Texas Tech University Health Sciences Center (to P.C.T).
The authors declare no competing financial interest.
Supplementary Material
References
- Rizner T. L.; Penning T. M. Role of aldo-keto reductase family 1 (AKR1) enzymes in human steroid metabolism. Steroids 2014, 79, 49–63. 10.1016/j.steroids.2013.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuura K.; Shiraishi H.; Hara A.; Sato K.; Deyashiki Y.; Ninomiya M.; Sakai S. Identification of a principal mRNA species for human 3 alpha-hydroxysteroid dehydrogenase isoform (AKR1C3) that exhibits high prostaglandin D-2 11-ketoreductase activity. J. Biochem. 1998, 124, 940–946. 10.1093/oxfordjournals.jbchem.a022211. [DOI] [PubMed] [Google Scholar]
- Reginato M. J.; Krakow S. L.; Bailey S. T.; Lazar M. K. Prostaglandins promote and block adipogenesis through opposing effects on peroxisome proliferator-activated receptor gamma. J. Biol. Chem. 1998, 273, 1855–1858. 10.1074/jbc.273.4.1855. [DOI] [PubMed] [Google Scholar]
- Adeniji A. O.; Chen M.; Penning T. M. AKR1C3 as a target in castrate resistant prostate cancer. J. Steroid Biochem. Mol. Biol. 2013, 137, 136–149. 10.1016/j.jsbmb.2013.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straus D. S.; Glass C. K. Cyclopentenone prostaglandins: New insights on biological activities and cellular targets. Med. Res. Rev. 2001, 21, 185–210. 10.1002/med.1006. [DOI] [PubMed] [Google Scholar]
- Shipley J. L.; Butera J. N. Acute myelogenous leukemia. Exp. Hematol. 2009, 37, 649–658. 10.1016/j.exphem.2009.04.002. [DOI] [PubMed] [Google Scholar]
- Kuendgen A.; Germing U. Emerging treatment strategies for acute myeloid leukemia (AML) in the elderly. Cancer Treat. Rev. 2009, 35, 97–120. 10.1016/j.ctrv.2008.09.001. [DOI] [PubMed] [Google Scholar]
- Tomizawa D.; Tawa A.; Watanabe T.; Saito A. M.; Kudo K.; Taga T.; Iwamoto S.; Shimada A.; Terui K.; Moritake H.; Kinoshita A.; Takahashi H.; Nakayama H.; Kiyokawa N.; Isoyama K.; Mizutani S.; Hara J.; Horibe K.; Nakahata T.; Adachi S. Appropriate dose reduction in induction therapy is essential for the treatment of infants with acute myeloid leukemia: a report from the Japanese Pediatric Leukemia/Lymphoma Study Group. Int. J. Hematol. 2013, 98, 578–88. 10.1007/s12185-013-1429-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown G.; Hughes P. Retinoid differentiation therapy for common types of acute myeloid leukemia. Leuk. Res. Treat. 2012, 2012, 939021. 10.1155/2012/939021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang M.; Ye Y.-c.; Chen S.; Chai J.-R.; Lu J.-X.; Zhoa L.; Gu L.-J.; Wang Z.-Y. Use of all-trans retinoic acid in the treatment of acute promyelocytic leukemia. Blood 1988, 72, 567–572. [PubMed] [Google Scholar]
- Mills K. I.; Gilkes A. F.; Sweeney M.; Choudhry M. A.; Woodgate L. J.; Bunce C. M.; Brown G.; Burnett A. K. Identification of a retinoic acid responsive aldoketoreductase expressed in HL60 leukaemic cells. FEBS Lett. 1998, 440, 158–162. 10.1016/S0014-5793(98)01435-5. [DOI] [PubMed] [Google Scholar]
- Nagase T.; Miyajima N.; Tanaka A.; Sazuka T.; Seki N.; Sato S.; Tabata S.; Ishikawa K.; Kawarabayasi Y.; Kotani H.; et al. Prediction of the coding sequences of unidentified human genes. III. The coding sequences of 40 new genes (KIAA0081-KIAA0120) deduced by analysis of cDNA clones from human cell line KG-1 (supplement). DNA Res. 1995, 2, 51–9. 10.1093/dnares/2.1.51. [DOI] [PubMed] [Google Scholar]
- Birtwistle L.; Hayden R. E.; Khanim F. L.; Green R. M.; Pearce C.; Davies N. J.; Wake N.; Schrewe H.; Ride J. P.; Chipman J. K.; Bunce C. M. The aldo-keto reductase AKR1C3 contributes to 7,12-dimethylbenz(a)anthracene-3,4-dihydrodiol mediated oxidative DNA damage in myeloid cells: Implications for leukemogenesis. Mutat. Res., Fundam. Mol. Mech. Mutagen. 2009, 662, 67–74. 10.1016/j.mrfmmm.2008.12.010. [DOI] [PubMed] [Google Scholar]
- Khanim F. L.; Hayden R. E.; Birtwistle J.; Lodi A.; Tiziani S.; Davies N. J.; Ride J. P.; Viant M. R.; Gunther U. L.; Mountford J. C.; Schrewe H.; Green R. M.; Murray J. A.; Drayson M. T.; Bunce C. M. Combined bezafibrate and medroxyprogesterone acetate: potential novel therapy for acute myeloid leukaemia. PLoS One 2009, 4, e8147. 10.1371/journal.pone.0008147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanim F.; Davies N.; Velica P.; Hayden R.; Ride J.; Pararasa C.; Chong M. G.; Gunther U.; Veerapen N.; Winn P.; Farmer R.; Trivier E.; Rigoreau L.; Drayson M.; Bunce C. Selective AKR1C3 inhibitors do not recapitulate the anti-leukaemic activities of the pan-AKR1C inhibitor medroxyprogesterone acetate. Br. J. Cancer 2014, 110, 1506–16. 10.1038/bjc.2014.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endo S.; Matsunaga T.; Kanamori A.; Otsuji Y.; Nagai H.; Sundaram K.; El-Kabbani O.; Toyooka N.; Ohta S.; Hara A. Selective inhibition of human type-5 17beta-hydroxysteroid dehydrogenase (AKR1C3) by baccharin, a component of Brazilian propolis. J. Nat. Prod. 2012, 75, 716–21. 10.1021/np201002x. [DOI] [PubMed] [Google Scholar]
- Lin J. H.; Lu A. Y. Role of pharmacokinetics and metabolism in drug discovery and development. Pharmacol. Rev. 1997, 49, 403–49. [PubMed] [Google Scholar]
- Beletskaya I. P.; Cheprakov A. V. The heck reaction as a sharpening stone of palladium catalysis. Chem. Rev. 2000, 100, 3009–66. 10.1021/cr9903048. [DOI] [PubMed] [Google Scholar]
- Miyaura N.; Suzuki A. Palladium-Catalyzed Cross-Coupling Reactions of Organoboron Compounds. Chem. Rev. 1995, 95, 2457–2483. 10.1021/cr00039a007. [DOI] [Google Scholar]
- Jackson R. W. A mild and selective method for the cleavage of tert-butyl esters. Tetrahedron Lett. 2001, 42, 5163–5165. 10.1016/S0040-4039(01)00980-7. [DOI] [Google Scholar]
- Zang T.; Verma K.; Chen M.; Jin Y.; Trippier P. C.; Penning T. M. Screening baccharin analogs as selective inhibitors against type 5 17beta-hydroxysteroid dehydrogenase (AKR1C3). Chem.-Biol. Interact. 2015, 234, 339–348. 10.1016/j.cbi.2014.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoemaker R. H. The NCI60 human tumour cell line anticancer drug screen. Nat. Rev. Cancer 2006, 6, 813–823. 10.1038/nrc1951. [DOI] [PubMed] [Google Scholar]
- Kikuchi A.; Furutani T.; Azami H.; Watanabe K.; Niimi T.; Kamiyama Y.; Kuromitsu S.; Baskin-Bey E.; Heeringa M.; Ouatas T. In vitro and in vivo characterisation of ASP9521: a novel, selective, orally bioavailable inhibitor of 17β-hydroxysteroid dehydrogenase type 5 (17βHSD5; AKR1C3). Invest. New Drugs 2014, 32, 860–870. 10.1007/s10637-014-0130-5. [DOI] [PubMed] [Google Scholar]
- Kohrt H. E.; Patel S.; Ho M.; Owen T.; Pollyea D. A.; Majeti R.; Gotlib J.; Coutre S.; Liedtke M.; Berube C. Second-line mitoxantrone, etoposide, and cytarabine for acute myeloid leukemia: A single-center experience. Am. J. Hematol. 2010, 85, 877–881. 10.1002/ajh.21857. [DOI] [PubMed] [Google Scholar]
- Archimbaud E.; Leblond V.; Michallet M.; Cordonnier C.; Fenaux P.; Travade P.; Dreyfus F.; Jaubert J.; Devaux Y.; Fiere D. Intensive sequential chemotherapy with mitoxantrone and continuous infusion etoposide and cytarabine for previously treated acute myelogenous leukemia. Blood 1991, 77, 1894–1900. [PubMed] [Google Scholar]
- Liu W. M.; Oakley P. R.; Joel S. P. Exposure to low concentrations of etoposide reduces the apoptotic capability of leukaemic cell lines. Leukemia 2002, 16, 1705–12. 10.1038/sj.leu.2402621. [DOI] [PubMed] [Google Scholar]
- Chou T. C. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol. Rev. 2006, 58, 621–81. 10.1124/pr.58.3.10. [DOI] [PubMed] [Google Scholar]
- Chou T. C. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010, 70, 440–6. 10.1158/0008-5472.CAN-09-1947. [DOI] [PubMed] [Google Scholar]
- Hofman J.; Malcekova B.; Skarka A.; Novotna E.; Wsol V. Anthracycline resistance mediated by reductive metabolism in cancer cells: the role of aldo-keto reductase 1C3. Toxicol. Appl. Pharmacol. 2014, 278, 238–48. 10.1016/j.taap.2014.04.027. [DOI] [PubMed] [Google Scholar]
- Heibein A. D.; Guo B.; Sprowl J. A.; Maclean D. A.; Parissenti A. M. Role of aldo-keto reductases and other doxorubicin pharmacokinetic genes in doxorubicin resistance, DNA binding, and subcellular localization. BMC Cancer 2012, 12, 381. 10.1186/1471-2407-12-381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsunaga T.; Hosogai M.; Arakaki M.; Endo S.; El-Kabbani O.; Hara A. 9,10-phenanthrenequinone induces monocytic differentiation of U937 cells through regulating expression of aldo-keto reductase 1C3. Biol. Pharm. Bull. 2012, 35, 1598–602. 10.1248/bpb.b12-00237. [DOI] [PubMed] [Google Scholar]
- Beranic N.; Gobec S.; Rizner T. L. Progestins as inhibitors of the human 20-ketosteroid reductases, AKR1C1 and AKR1C3. Chem.-Biol. Interact. 2011, 191, 227–33. 10.1016/j.cbi.2010.12.012. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.