

Abstract
ATG4B or autophagin-1 is a cysteine protease that cleaves ATG8 family proteins. ATG4B plays essential roles in the autophagosome formation and the autophagy pathway. Herein we disclose the design and structural modifications of a series of fluoromethylketone (FMK)-based peptidomimetics as highly potent ATG4B inhibitors. Their structure–activity relationship (SAR) and protease selectivity are also discussed.
Keywords: ATG4B, autophagy, covalent inhibitor, fluoromethylketone, peptidomimetics
Autophagy is an evolutionarily conserved process essential for cell homeostasis and housekeeping by catabolizing aggregated proteins and damaged cellular components.1 The hallmark of autophagy is the formation of autophagosomes and subsequent fusion with lysosomes to achieve degradation of their contents. Dysregulation of autophagy has been recently described in the pathogenesis of a variety of diseases such as cancer, neurodegenerative and metabolic disorders, and viral infections.2,3 Modulation of autophagy has become a very active area of preclinical and clinical research, and particularly, there is high interest to identify potent and specific autophagy inhibitors.4
ATG4 or autophagins are a class of cytosolic cysteine proteases that cleave ATG8 family proteins, such as light chain 3 (LC3). ATG4 plays essential roles in the formation and maturation of autophagosomes.5,6 Among all four human ATG4 orthologues, ATG4B is functionally dominant, and it is the sole enzyme reported to efficiently cleave LC3 precursors and to deconjugate lipid from membrane bound LC3-phosphatidylethanolamine (LC3-PE).7 ATG4B has been considered as a potential therapeutic target in the development of chemosensitizer for the treatment of certain cancer types. Recently, a number of small molecule ATG4B inhibitors such as Z-l-Phe-chloromethylketone and NSC185058 were reported to have modulatory effects on the autophagy process.8−10 However, because of its high chemical reactivity, the chloromethylketone moiety is usually associated with significant cytotoxicity. NSC185058, however, is only a weak ATG4B inhibitor with an IC50 of 51 μM in an assay detecting the cleavage of LC3-GST.10
Previously, we reported the identification of Z-FA-FMK (1, Figure 1) as a covalent active-site directed ATG4B inhibitor from a TR-FRET based focused library screening.11 The hit expansion of 1 led to the discovery of Z-FG-FMK (2), which was 10-times more potent than 1 with an IC50 of 1.2 μM in the biochemical assay.11 Herein we disclose the structure-guided optimization of 2 toward highly potent fluoromethylketone (FMK)-based ATG4B inhibitors.
Figure 1.
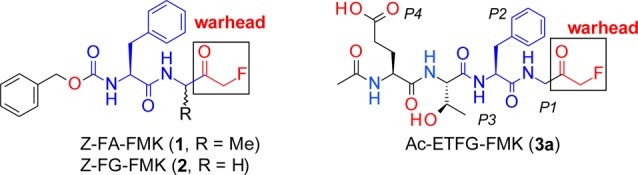
Structures of peptidomimetic ATG4B inhibitors.
As an early step in the autophagosome formation, ATG4B cleaves proLC3 to expose the C-terminal Gly120 for subsequent PE-conjugation and autophagosome membrane insertion.5 The cocrystal structures of catalytically inert C74S or H280A mutant Atg4B in complex with the LC3(1–124) peptide revealed that the substrate induces a conformational change of both the regulatory loop (258–262) and Trp142 resulting in an open protein conformation in which the C-terminal ETFG residues at the processing site of LC3 make several intermolecular contacts with the catalytic pocket of ATG4B.12 Molecular modeling13 of compounds 1 and 2 into the open conformation of ATG4B, using the TFG residues of LC3 as template, suggests a potential steric clash of the additional methyl group in 1 with Trp142, which likely explains the inferior anti-ATG4B activity of this molecule (Figure 2).
Figure 2.
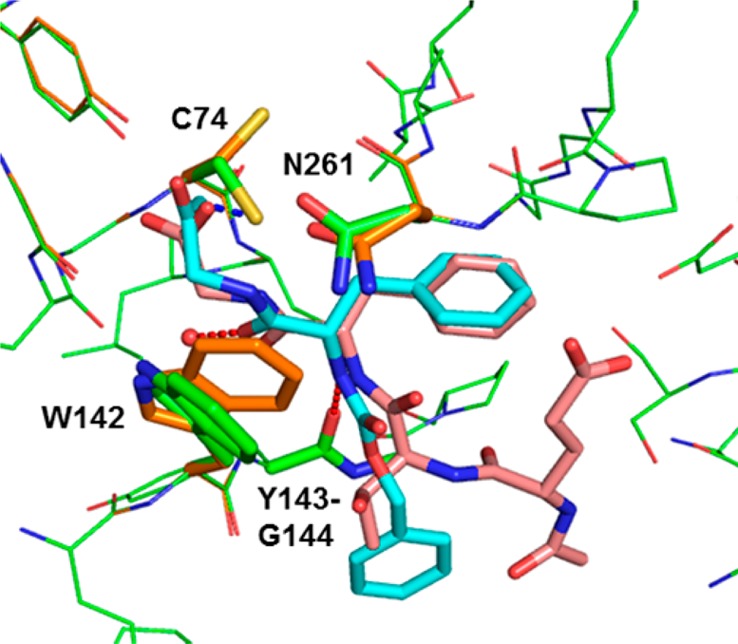
Overlay of the X-ray crystal structure (PDB code: 2Z0D)12 of the C-terminal LC3 peptide (Glu117-Gly120, salmon) in human ATG4B (orange) with a model of Z-FG-FMK 2 (cyan). Intermolecular hydrogen bonds for 2 are shown as red lines and the covalent interaction with Cys74 is shown in blue. Models were built using the modeling software MOLOC.13 See also Figure S1 in the Supporting Information for a surface representation of the binding site.
In the predicted binding mode, the FMK warhead makes a covalent bond with the S atom of Cys74 and the planar P1 glycine moiety stacks onto the π-system of the indole ring of Trp142. The benzyl substituent nicely overlaps with the P2 phenylalanine of LC3 and is engaged in dispersion interactions with lipophilic side chains in the S2 pocket as well as stacking interactions with the π-surface of a β-turn. The amide linker connecting P1 and P2 fragments uses its carbonyl oxygen atom to form a hydrogen bond to a buried water molecule. In contrast, in the amide linker between P2 and P3 the NH functionality makes a hydrogen bond with the carbonyl oxygen atom of Tyr143, and the carbonyl group is solvent-exposed. Additionally, the Cbz phenyl ring stacks onto the backbone amide of Tyr143–Gly144.
To investigate the SAR of the C-terminal LC3 peptides, we synthesized and tested a number of FMK-based peptidomimetics including Ac-ETFG-FMK (3a, Figure 1) for their ATG4B inhibitory activities. We find that 3a is about 15 times less active than 2 in the TR-FRET assay and inactive in the cellular based actin-LC3-dNGluc cleavage assay (or luciferase release assay, LRA),11 probably due to limited cell permeability (Table 1). Moreover, it should be noted that the FMK warhead at the P1 position is important for ATG4B inhibition as peptides like Ac-ETFG (3b) and Ac-TFG are inactive in both TR-FRET and LRA assays.
Table 1. Inhibition of ATG4B by 1–3 and 4a–n.
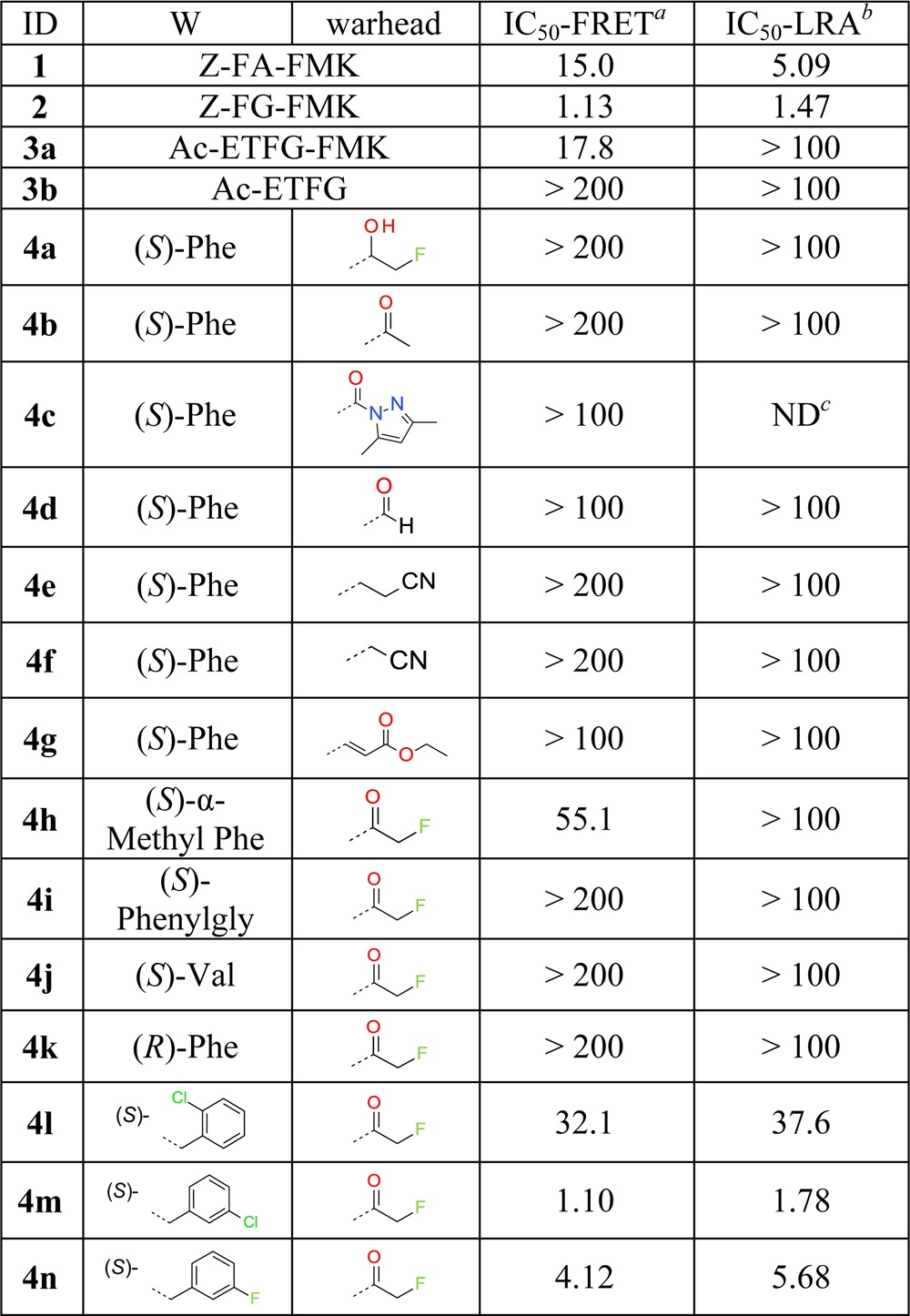
IC50s [μM] are mean values of TR-FRET experiments in duplicate runs, with variation ±10%.
IC50s [μM] are mean values determined by luciferase-release assay (LRA) with Actin-LC3B-dNGLuc construct used in HEK-293T cells. Experiments were run in duplicate with variation ±15%. Compounds were not cytotoxic to HEK-293T cells (CC50 > 100 μM) in 24 h treatment.
ND, not determined.
In the first optimization phase we focused on modifications of the warhead and the Phe residue at the P2 position of 2. Thus, analogues 4a–n were synthesized (see the Supporting Information) and tested in both TR-FRET and LRA assays. The resulting SAR reveals that formation of a covalent bond to Cys74 is critical for anti-ATG4B activity as direct structural changes of the FMK moiety completely abolish anti-ATG4B activities (examples 4a–b in Table 1), in agreement with the results of 3a and 3b. In addition, we find that many covalent warheads such as pyrazole amide (example 4c), aldehyde (4d), nitrile (4e–f), and Michael acceptor-like 4g are not tolerated either. We hypothesize that this is due to an insufficient fit to the binding site around Cys74 and/or too low reactivity for the formation of a covalent bond.
The chemistry space at the P2 position was explored with a variety of amino acids. For example, (S)-α-methyl substitution and amino acids such as (S)-phenylglycine, (S)-valine, and (R)-Phe were incorporated but resulted in a complete loss of anti-ATG4B activity (examples 4h–k). We find that halogen substituents can be tolerated at the meta-position of the phenyl group of 2. For example, meta-Cl analogue 4m shows an IC50 of 1.1 μM in the TR-FRET assay. In contrast, substituents at ortho- and para-positions are clearly detrimental to anti-ATG4B activity (examples including 4l).
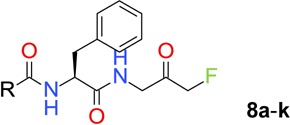
Next, structural explorations were carried out to optimize the terminal N-Cbz group. By using the method shown in Scheme 1, (S)-N-Boc phenylalanine was coupled with 1-amino-3-fluoro-propan-2-ol hydrochloride (5) in the presence of HATU and DIPEA to give amide product. The N-Boc protective group was removed by treating the intermediate with a solution of HCl in EtOAc, and the resulting amine hydrochloride salt 6 was used for further functional transformations. For instance, amine 6 was coupled with selected carboxylic acids to give amides 7. The FMK group was generated by treating 7 with Dess–Martin periodinane (DMP), and inhibitors 8a–k were evaluated for their anti-ATG4B activities.
Scheme 1. Synthesis of FMK-Based ATG4B Inhibitors 8a–k and 9a–h.

Reagents and conditions: (a) HATU, DIPEA, DMF, rt, 97%; (b) HCl, EtOAc, 30 °C, 30 min, quant.; (c) carboxylic acids, HATU, DMF, rt; (d) DMP, DCM, rt.
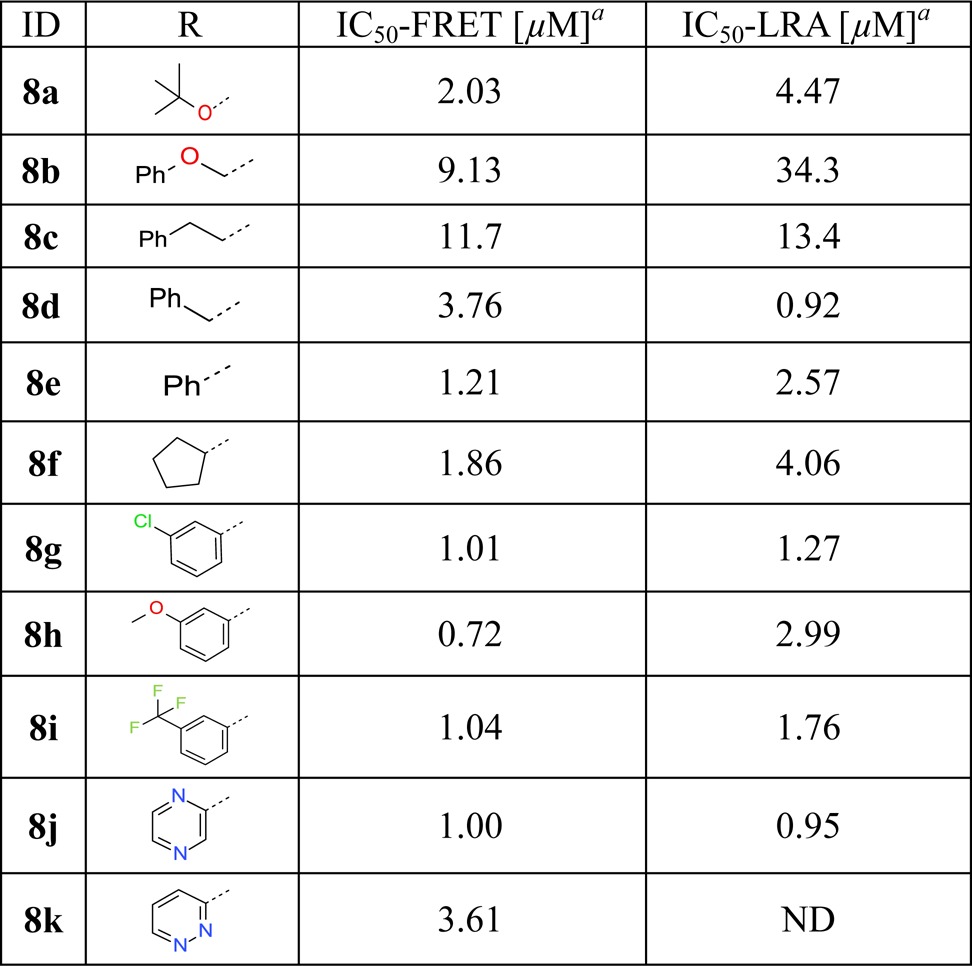
We find that the N-Boc analogue 8a shows comparable activity to 2 with an IC50 of 2.0 μM in TR-FRET assay (Table 2). Importantly, the carbamate moiety can be replaced by more rigid and smaller amide moieties, exemplified by benzamide analogue 8e and cyclopentanecarboxamide 8f, which demonstrate good anti-ATG4B activities with IC50 values of 1.21 and 1.86 μM, respectively. Pyrazine-2-carboxamide analogue 8j shows promising potency and physicochemical properties, such as a high water solubility of 129 μg/mL in a lyophilized solubility assay (LYSA). Further structural explorations suggest that aryl substituents, especially those at the meta-substitution, are well tolerated revealing a rather flat SAR (examples 8g–i).
Table 2. Inhibition of ATG4B by 8a–ka.
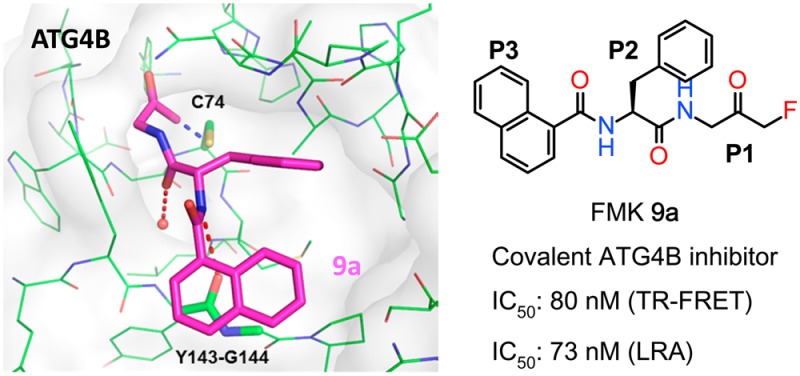
Subsequently, bicyclic aromatic groups were used at the P3 position, and the naphthalene-1-carboxamide analogue 9a showed strong inhibition of ATG4B, with IC50 values of 80 and 73 nM in the TR-FRET and cellular-based LRA assays, respectively (Table 3). In comparison, β-substituted analogue 9b is more than 10 times less active than 9a probably due to less optimal π-stacking of the β-naphthyl group with the backbone amide. The physicochemical properties of 9a show moderate solubility (LYSA: 41 μg/mL) and high human and mouse liver microsome clearances of 13.9 and 70 mL min–1 kg–1, respectively. Finally, halogen-substitutions and a nitrogen atom walk were carried out on the α-naphthyl group. We find that 5-F substituted analogue 9d has the best anti-ATG4B activity in our series with an IC50 of 15 nM in the cellular LRA assay. We also note that aza-decorations are well tolerated at the 3- and 4-positions but not at the 8-position of the naphthyl group (examples 9e–g). Both quinoline-4-carboxamide analogue 9e and cinnoline-4-carboxamide analogue 9g show strong anti-ATG4B activities and improved solubility compared to 9a with LYSA values of 188 and 230 μg/mL, respectively. In addition, they show increased intrinsic metabolic stability compared to 9a. For example, the human and mouse liver microsome clearances of 9g are about 9.9 and 39 mL min–1 kg–1, respectively.
Table 3. Inhibition of ATG4B by 9a–ja.
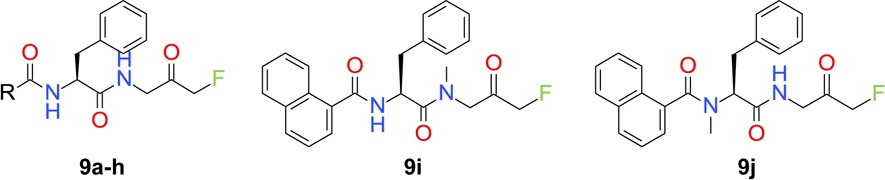
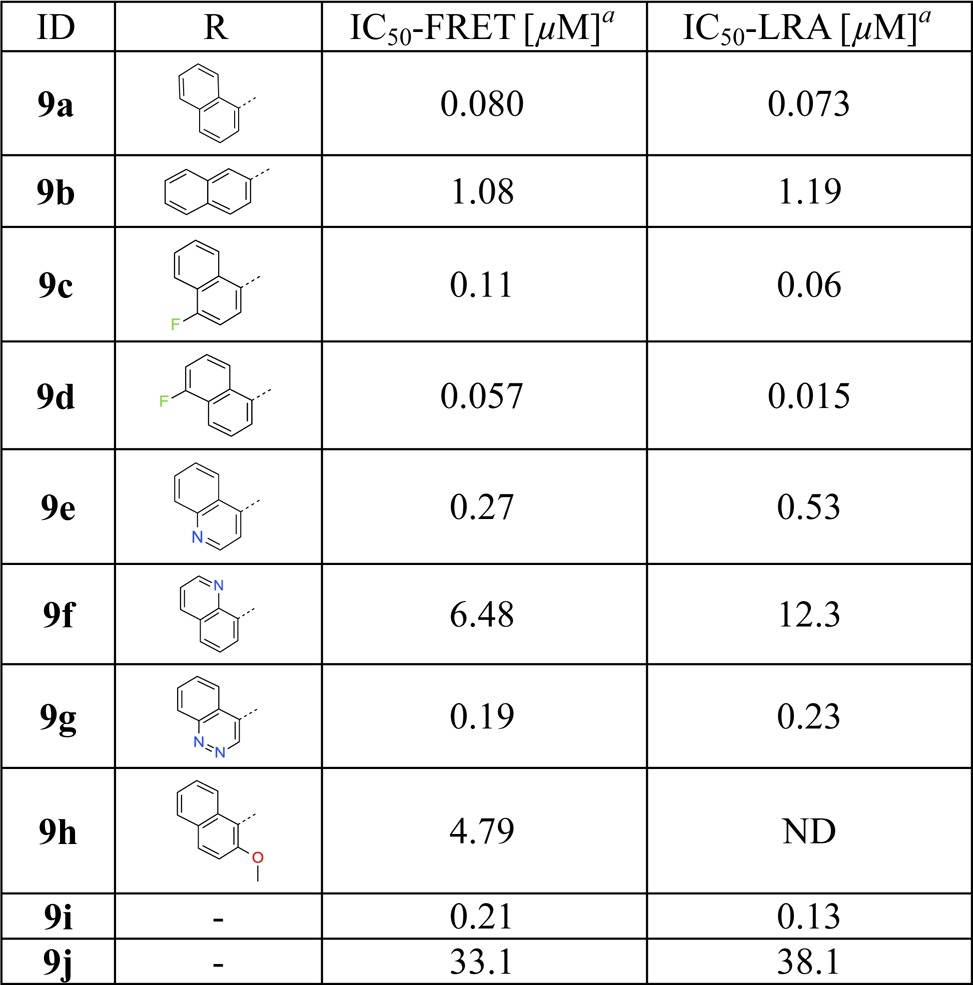
ATG4B has several solvent-exposed cysteine residues. Nevertheless, ATG4B formed a 1:1 association with FMK analogues after 4 h of coincubation.11 A subsequent LC–MS/MS study indicated that 9a formed an irreversible covalent bond with the more reactive thiol group of Cys74 located at the catalytic site of ATG4B. This finding and additional SAR supports our hypothetical binding mode (Figure 2). For example, in our model the P1 amide NH group is not involved in direct ATG4B interactions, while the P2 NH moiety and the backbone carbonyl oxygen of Tyr143 form a hydrogen bond. In line with this binding mode, blocking of both ligand NH groups through N-methylation (9i and 9j) is well tolerated at the P1 position (9i) but not on the phenylalanine P2 part (9j, Table 3). Based on a binding model of 9a in ATG4B (Figure S2) we postulate that the improved activities of naphthyl analogues of this topology are due to more efficient stacking of the extended bicyclic π-system with the backbone amide group of Tyr143–Gly144.14 This should be sensitive to the relative orientation of the naphthyl ring to the plane of the adjacent amide linker. This is indeed observed as the preferred orthogonal orientation in the case of α-substitution (9a) leads to significantly better ATG4B activity than for β-substitution (9b) in which the preferred orientation is close to planar.
FMK analogues 1, 8e, and 9a were selected for target selectivity profiling against a panel of proteases (Table 4). These ATG4B inhibitors have no activity against the aspartic protease, serine proteases, metalloproteases, and the 20S proteasome from our panel. In addition, they show very low inhibition of caspases, which belong to the cysteine protease family. However, the FMK analogues hit a few cysteine proteases such as cathepsin B and calpain with IC50(app) values <1 μM. While the sequence identity is very low between ATG4B and these cysteine proteases, the result can likely be explained by the significant reactivity of the FMK group and the relatively open binding pockets of these proteases. Compound 9a is slightly more active on ATG4B compared to cathepsin B and calpain. In this study, we did not attempt to further increase the selectivity window through target-based molecular design.
Table 4. Selectivity of FMK-Based ATG4B Inhibitorsa.
| protease family | protease | 1 | 8e | 9a | |
|---|---|---|---|---|---|
| Asp. protease | cathepsin D | >100 | >100 | >100 | |
| Ser. protease | cathepsin G | >100 | >100 | >100 | |
| tryptase, βII | >100 | >100 | >100 | ||
| neutrophil elastase | >100 | >100 | >100 | ||
| Compl. protease | 20S proteasome | >100 | >100 | >100 | |
| Cys. proteases | caspase | caspase 2 | >100 | >100 | >100 |
| caspase 3 | 43.5 | >100 | 75.1 | ||
| caspase 7, 8, 9 | >100 | >100 | >100 | ||
| cathepsin | cathepsin B | 0.3 | 0.7 | 0.2 | |
| others | calpain | 0.6 | 0.6 | 0.096 | |
| metalloprotease | ACE, ECE-1 | >100 | >100 | >100 | |
| MMP1, MMP3, MMP9 | >100 | >100 | >100 | ||
IC50 apparent [μM] are mean values of protease inhibition from duplicate runs, with variation ±15%. Assays were performed by Genscript according to their standard protocols, see the Supporting Information (www.genscript.com).
In summary, we discovered a series of highly potent FMK-based covalent ATG4B inhibitors by structural modifications of a focused-library screening hit. Naphthalene-1-carboxamide analogue 9a and its derivatives inhibit ATG4B with IC50 values <100 nM in both a TR-FRET assay and a cell-based luciferase-release assay. The FMK class of ATG4B inhibitors are inactive in a panel of different proteases involving cysteine proteases like caspase 2, 3, 7, 8, and 9. However, they inhibit cathepsin B and calpain with submicromolar IC50 values. The SAR of the current FMK series and biostructure differences between ATG4B and other cysteine-proteases support further structural modifications to increase target selectivity. An LC-MS/MS study demonstrated that analogue 9a forms an irreversible and selective covalent bond with Cys74 and thus inactivates ATG4B proteolytic activity. A binding mode of our series in ATG4B was constructed by molecular modeling revealing key protein–ligand interactions. Efforts including modulation of warhead reactivity and bioisostere replacements of the P1 amide group as well as autophagy modulation effects by analogues of 9a will be the subject of future publications.
Experimental Methods
Molecular Docking of FMK Analogues
Model building was performed with the cocrystal structure of human ATG4B with LC3 (1–120) as template (PDB code: 2Z0D)12 using the following procedure: (1) in silico mutation of the TFG residues of LC3 into Z-FA(G)-FMK; (2) rebuilding of A280H using the structure 2Z0E(12) to reproduce the human sequence; (3) removal of the fluorine atom of Z-FA(G)-FMK and formation of a covalent bond between the sulfur atom of Cys74 and the reactive methylene unit of FMK. Here, the second, alternate conformation of the side chain of Cys74 in the crystal structure 2Z0D was used as this is closer to the FMK group. (4) Minimization of the ligand within the mostly rigid protein allowing for relaxation of only few selected residues (e.g., Tyr54, Cys71, Trp142, Asn261) in close contact to the small molecule.
TR-FRET Assay
Five microliters of serial-diluted test compounds (1% DMSO) in assay buffer was added into 5 μL of purified ATG4B (final 0.1 nM) in 384-well plate, and the solution was incubated at rt for 30 min. Then, 5 μL of His-GATE-16-GST (final 20 nM) was added into the wells, and the solution was incubated at rt for another 30 min. Five microliters of detection solution (final 2 nM of Eu-anti-His and 20 nM of Ulight-anti-GST) was added, and the resulting mixture was incubated at rt for 40 min. The fluorescence signal was measured with Envision4 (Ex, 340 nm; Em, 665/615 nm) and data was analyzed by Genedata Screener (Genedata AG).
Luciferase Release Assay (LRA)
A similar procedure has been reported before.15 Basically, HEK-293T cells (human embryonic kidney cells, ATCC, CRL-11268) were seeded in 6-well plate with a density of 70% confluence. One microgram of enzyme expression plasmid and 2 μg of substrate expression plasmid were cotransfected using 9 μL of X-tremeGENE HP DNA Transfection Reagent (Roche diagnostics cat#06366236001). Three combinations (pCNDA3.1 vector and dNGluc, HTRA4 and Actin-dNGluc, and ATG4B and Actin-LC3-dNGluc) were used in the transfections. After 24 h, the transfected cells were trypsinized, suspended, and transferred to 96-well plates (Corning, can#3599), serial-diluted compounds in assay buffer were added into the platesm and the mixture was incubated for 24 h. Fifty microliters of cell supernatant was transferred to a 96-well white plate (Corning, Cat#3917), and luciferase signal was detected by using BioLux Gaussia luciferase assay kit (NEW ENGLAND BioLabs, Cat#E3300L) and measured with Envision4.
Acknowledgments
We are grateful to Qinshan Gao, Yujuan Gao, and Jin Ge for purification of final compounds.
Glossary
ABBREVIATIONS
- ATG4B
autophagy-related gene 4B
- Cbz
carbobenzyloxy or benzyloxy carbamate
- DMP
Dess–Martin periodinane
- LC3
light chain 3
- LRA
luciferase release assay
- LYSA
lyophilized solubility assay
- PE
phosphatidylethanolamine
- TR-FRET
time-resolved fluorescence energy transfer
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.6b00208.
Synthetic procedures and analytical data for compounds 2–5, 8a–k, and 9a–j; predicted binding models of Z-FG-FMK (2) and 9a; general assay protocols for evaluation of protease selectivity (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Kobayashi S. Choose delicately and reuse adequately: the newly revealed process of autophagy. Biol. Pharm. Bull. 2015, 38, 1098–103. 10.1248/bpb.b15-00096. [DOI] [PubMed] [Google Scholar]
- Rubinsztein D. C.; Codogno P.; Levine B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat. Rev. Drug Discovery 2012, 11, 709–30. 10.1038/nrd3802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A.; Singh U. K.; Chaudhary A. Targeting autophagy to overcome drug resistance in cancer therapy. Future Med. Chem. 2015, 7, 1535–42. 10.4155/fmc.15.88. [DOI] [PubMed] [Google Scholar]
- Cheng Y.; Ren X.; Hait W. N.; Yang J. M. Therapeutic targeting of autophagy in disease: biology and pharmacology. Pharmacol. Rev. 2013, 65, 1162–97. 10.1124/pr.112.007120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L.; Li J.; Ouyang L.; Liu B.; Cheng Y. Unraveling the roles of Atg4 proteases from autophagy modulation to targeted cancer therapy. Cancer Lett. 2016, 373, 19–26. 10.1016/j.canlet.2016.01.022. [DOI] [PubMed] [Google Scholar]
- Yang Z.; Wilkie-Grantham R. P.; Yanagi T.; Shu C. W.; Matsuzawa S.; Reed J. C. ATG4B (Autophagin-1) phosphorylation modulates autophagy. J. Biol. Chem. 2015, 290, 26549–61. 10.1074/jbc.M115.658088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran E.; Chow A.; Goda T.; Wong A.; Blakely K.; Rocha M.; Taeb S.; Hoang V. C.; Liu S. K.; Emmenegger U. Context-dependent role of ATG4B as target for autophagy inhibition in prostate cancer therapy. Biochem. Biophys. Res. Commun. 2013, 441, 726–31. 10.1016/j.bbrc.2013.10.117. [DOI] [PubMed] [Google Scholar]
- Shu C. W.; Madiraju C.; Zhai D.; Welsh K.; Diaz P.; Sergienko E.; Sano R.; Reed J. C. High-throughput fluorescence assay for small-molecule inhibitors of autophagins/Atg4. J. Biomol. Screening 2011, 16, 174–82. 10.1177/1087057110392996. [DOI] [PubMed] [Google Scholar]
- Nguyen T. G.; Honson N. S.; Arns S.; Davis T. L.; Dhe-Paganon S.; Kovacic S.; Kumar N. S.; Pfeifer T. A.; Young R. N. Development of fluorescent substrates and assays for the key autophagy-related cysteine protease enzyme, ATG4B. Assay Drug Dev. Technol. 2014, 12, 176–89. 10.1089/adt.2013.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akin D.; Wang S. K.; Habibzadegah-Tari P.; Law B.; Ostrov D.; Li M.; Yin X. M.; Kim J. S.; Horenstein N.; Dunn W. A. Jr. A novel ATG4B antagonist inhibits autophagy and has a negative impact on osteosarcoma tumors. Autophagy 2014, 10, 2021–35. 10.4161/auto.32229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu D.; Xu Z.; Han L.; Liu C.; Zhou Z.; Qiu Z.; Lin X.; Tang G.; Shen H.; Aebi J.; Riemer C.; Kuhn B.; Stahl M.; Mark D.; Qin N.; Ding H. Identification of new ATG4B inhibitors based on a novel high-throughput screening platform. J. Biomol. Screening 2016, 10.1177/1087057116639202. [DOI] [PubMed] [Google Scholar]
- Satoo K.; Noda N. N.; Kumeta H.; Fujioka Y.; Mizushima N.; Ohsumi Y.; Inagaki F. The structure of Atg4B-LC3 complex reveals the mechanism of LC3 processing and delipidation during autophagy. EMBO J. 2009, 28, 1341–50. 10.1038/emboj.2009.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber P.; Müller K. MAB, a generally applicable molecular force field for structure modelling in medicinal chemistry. J. Comput.-Aided Mol. Des. 1995, 9, 251–268. 10.1007/BF00124456. [DOI] [PubMed] [Google Scholar]
- Harder M.; Kuhn B.; Diederich F. Efficient stacking on protein amide fragments. ChemMedChem 2013, 8, 397–404. 10.1002/cmdc.201200512. [DOI] [PubMed] [Google Scholar]
- Ketteler R.; Seed B. Quantitation of autophagy by luciferase release assay. Autophagy 2008, 4, 801–806. 10.4161/auto.6401. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.