

Abstract
Lung cancer cells harboring activating EGFR mutations acquire resistance to EGFR tyrosine kinase inhibitors (TKIs) by activating several bypass mechanisms, including MET amplification and overexpression. We show that a significant proportion of activated MET protein in EGFR TKI-resistant HCC827 lung cancer cells resides within the mitochondria. Targeting the total complement of MET in the plasma membrane and mitochondria should render these cells more susceptible to cell death and hence provide a means of circumventing drug resistance. Herein, the mitochondrial targeting triphenylphosphonium (TPP) moiety was introduced to the selective MET kinase inhibitor PHA665752. The resulting TPP analogue rapidly localized to the mitochondria of MET-overexpressing erlotinib-resistant HCC827 cells, partially suppressed the phosphorylation (Y1234/Y1235) of MET in the mitochondrial inner membrane and was as cytotoxic and apoptogenic as the parent compound. These findings provide support for the targeting of mitochondrial MET with a TPP-TKI conjugate as a means of restoring responsiveness to chemotherapy.
Keywords: Mitochondrial targeting, triphenylphosphonium conjugate, MET kinase, PHA665752, drug resistance, nonsmall cell lung cancer
Drug resistance to the EGFR tyrosine kinase inhibitors (TKIs) gefitinib and erlotinib seriously impedes the therapeutic usefulness of these drugs on nonsmall cell lung cancers (NSCLC) that harbor activating mutations at the catalytic domain of EGFR, the most common of which are found at exon 21 (L858R) and exon 19 (delE746-A750).1 Acquired resistance due to a single point mutation at the gatekeeper residue (T790M) on the kinase domain2 enhances affinity for ATP and displaces gefinitib and erlotinib, which are ATP-competitive inhibitors from the kinase binding pocket.3 Aside from acquired secondary EGFR mutations, resistance to TKIs may arise from constitutive activation of alternative signaling pathways that bypass or lie downstream of EGFR.4 The hepatocyte growth factor (HGF)-MET signaling pathway is frequently cited as a bypass mechanism co-opted by cancer cells with acquired or innate resistance to TKIs.5−7 MET is amplified in approximately 20% of tumors from NSCLC patients with acquired resistance8−10 and its presence correlates with poor prognosis in surgically resected patients.10 Engelman and co-workers described the focal amplification of the MET proto-oncogene in an NSCLC cell line that was resistant to gefitinib and the modest restoration of sensitivity when MET signaling was inhibited.11 Viewed in this context, inhibition of the HGF-MET axis may potentially reverse resistance to TKIs, hence restoring the curative efficacies of these drugs. Other strategies to address resistance to TKIs have been recently reviewed.12
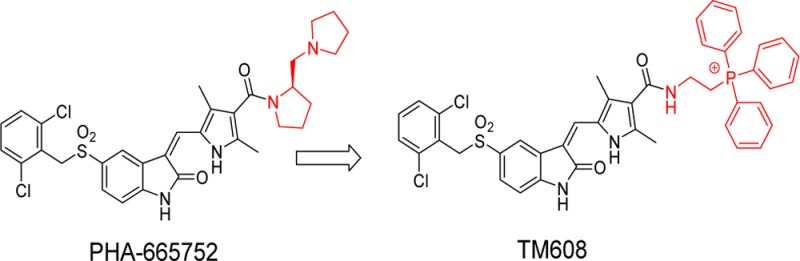
We have previously shown that MET overexpression in cancer cells is associated with MET protein localization to the mitochondria while also remaining on the plasma membrane.13 The presence of MET in the mitochondria shifted cellular metabolism to a state of heightened glycolysis, increased tricarboxylic acid (TCA) cycle activity, and oxidative phosphorylation.13 Such metabolic remodeling would conceivably enhance the fitness of cancer cells for survival by increasing ATP production and providing abundant TCA cycle intermediates for biomass production. Thus, we postulate that in these and other ways yet to be uncovered, mitochondrial MET increases the fitness of cancer cells for survival and may counteract anticancer therapies designed to achieve cell killing. The corollary is that inhibition of mitochondrial MET would serve to restore responsiveness to chemotherapy. However, current MET kinase inhibitors are designed to inhibit MET in plasma membranes, not MET sequestered within the mitochondria. Thus, if the total complement of MET that resides in plasma and mitochondrial membranes is to be inhibited, inhibitors should have a mitochondrial-targeting motif that would direct delivery to the organelle while not compromising the inherent ability of the entity to inhibit MET. To test this hypothesis, we prepared a probe molecule (TM608) in which a triphenylphosphonium (TPP) moiety is conjugated to a selective MET inhibitor, PHA665752 (Scheme 1A). TPP is one of the more widely employed mitochondrial directing motifs.14 TPP conjugates of several bioactive molecules (antioxidants, anticancer agents) have been reported to specifically accumulate in the mitochondria, with enhancement of the desired activity.15−17 The lipophilicity of the TPP cation coupled with extensive delocalization of the positive charge favors accumulation in the organelle due to the substantial potential difference that exists across the mitochondrial membranes.18
Scheme 1. (A) Modification of PHA665752 To Give TM608; (B) Synthesis of TM608.
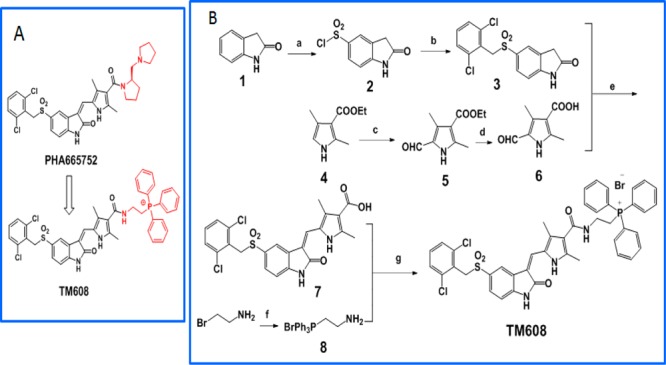
Reagents and conditions: (a) chlorosulfonic acid; (b) 2,6-dichlorobenzyl bromide, NaH2PO4, Na2SO3, 30–60 °C, acetone; (c) POCl3, DMF; (d) NaOH reflux; (e) piperidine, ethanol, 60 °C; (f) PPh3, 80 °C; (g) EDCI, DMAP, DMF, rt.
To rationally design TM608 so that kinase inhibitory activity is retained, we examined the orientation of PHA665752 in the ATP-binding pocket of MET (SI, Figure S1).19 Noting that PHA665752 binds to the pocket with its basic side chain directed toward the solvent space and away from the critical hinge region (Met1160, Pro1158) that is required for catalytic activity, we inserted the TPP moiety in place of the basic side chain and attached it to the core scaffold via an ethylaminocarbonyl linker. Redocking of TM608 showed that it was retained within the hinge region with the TPP side chain directed toward the solvent space (SI, Figure S1). Scheme 1B outlines the synthesis of TM608. Briefly, the pyrrole carboxylate 4 was formylated in a Vilsmeier reaction, hydrolyzed to yield the free acid 6 and condensed with indolinone 3 to give intermediate 7. EDC coupling between 7 and (2-aminoethyl) triphenyl phosphonium bromide 8 gave TM608 in satisfactory yields. Details are provided in SI.
With the probe molecule TM608 on hand, we proceeded to interrogate its ability to inhibit MET kinase in NSCLC cells that are resistant to TKIs. An isogenic pair of NSCLC cells (HCC827A and HCC827B) was used for this purpose. The parental HCC827A cells harbor an activating mutation in exon 19 of EGFR and is erlotinib-sensitive. Its derivative HCC827B is erlotinib-resistant and MET-enriched,20 but the location of MET, at its usual site at the cell membrane or additionally partitioned to other organelles, is not known. We were curious if the “extra” MET is localized to the mitochondria as this has been observed in other MET-amplified malignant cells.13 Hence, we carried out an immunoblotting analysis of activated MET (MET phosphorylated at the catalytic tyrosine residues Y1234/1235) in whole cell lysates comprising equal numbers (105) of HCC827A or HCC827B cells and the subcellular fractions derived from this number of cells as they were fractionated to obtain purified mitochondria. The signal from each lane in Figure 1 reflects phosphorylated MET (p-MET) present in the same number of cells (105 cells) but not derived from identical total protein loading because the amounts of protein would progressively decline as purification progressed. The subcellular fractions (heavy membranes were obtained by centrifuging whole cell lysates at 500g for 5 min and were enriched in plasma membranes; the flow-through fraction did not bind to the anti-TOM22 beads used to capture mitochondria) were derived at each sequential fractionation step leading to pure mitochondria. Quantification by densitometry confirmed that whole cell lysates of HCC827B cells had nearly six times more p-MET (Y1234/1235) than HCC827A cells (B/A ratio 5.71). Moreover, the increase in p-MET (Y1234/Y1235) was more pronounced in the mitochondria (B/A ratio 6.07) than in the other fractions.
Figure 1.

Expression of activated MET protein (phospho-Y1234/1235) in whole cells, heavy membranes, flow-through fraction and pure mitochondria derived from HCC827A and HCC827B cells. B/A ratios report the extent to which HCC827B cells or fractions were enriched in p-MET (Y1234/Y1235) compared to the corresponding HCC827A cells/fractions.
The susceptibility of the mitochondrial MET in HCC827B to pharmacological inhibition would depend on whether MET is located externally at the outer membrane or internally, within the inner mitochondrial membrane. To determine the site(s) at which MET was localized, we carried out controlled trypsinization of pure mitochondria derived from HCC827B cells and probed for the presence of α- and β-subunits of MET, TOM20 (an outer mitochondrial membrane protein), and SDHA (an inner mitochondrial membrane protein). As seen from Figure 2, trypsinization degraded TOM20 but had little effect on MET and SDHA. Thus, we concluded that MET was located at a similar intramitochondrial compartment as SDHA, namely, the inner mitochondrial membrane. Taken together, these experiments affirmed the presence of stable and high levels of activated MET sequestered within the inner membranes of the mitochondria of HCC827B cells. Hence HCC827B is a clinically relevant resistant phenotype of NSCLC on which investigations on TM608 could be pursued.
Figure 2.

Detection of MET and mitochondrial membrane proteins (TOM20, SDHA) in intact whole cells WC (Lane 1), intact pure mitochondria PM (Lane 2), and time-controlled trypsinized mitochondrial fractions from HCC827B cells (Lanes 3–5).
We then proceeded to determine the growth inhibitory activities (IC50) of PHA665752 and TM608 on HCC827A and HCC827B cells by the colorimetric tetrazolium assay. The sensitivity of the HCC827A cells to erlotinib was confirmed. In our hands, % viability at 1 μM erlotinib was 22.4% ± 2.8 (n = 3) versus 72.4% ± 4.4 (n = 3) on HCC827B cells. PHA-665752 and TM608 were equipotent on the erlotinib sensitive HCC827A cells with IC50 values (72 h) of 15.7 ± 0.4 μM (PHA665752) and 16.7 ± 1.0 μM (TM608) (p = 0.2156, unpaired Student’s t test). On the erlotinib-resistant high MET expressing HCC827B cells, TM608 was found to be modestly more potent, with IC50 of 5.3 ± 0.4 μM compared to 8.9 ± 0.6 μM for PHA665752 (p = 0.0013, unpaired Student’s t test). Interestingly, TM608 was less cytotoxic than PHA665752 when evaluated on TAMH (TGFα murine hepatocytes), a stable, nontumorigenic, and metabolically competent cell line that is widely used for toxicological evaluation.21 The 24 h IC50 of TM608 was 15.1 μM (±1.6) as compared to 8.9 μM (±1.2) for PHA665752.
Both TM608 and PHA665752 induced apoptosis in HCC827B cells as seen from the elevated levels of apoptotic marker proteins (cleaved PARP, cleaved caspases 3 and 7) in cells treated with sublethal concentrations (4 μM) of either compound (Figure 3). The onset of apoptosis was delayed in TM608-treated cells but, once triggered, persisted up to the 60 h time point.
Figure 3.

Immunoblots of apoptotic markers (cleaved PARP, cleaved caspases 3 and 7) in whole cell lysates of HCC827B cells treated with 4 μM TM608 or 4 μM PHA665752 at 12, 24, 36, 48, and 60 h time points. Controls (C) were untreated cells at 0 h under similar conditions. GADPH was the loading control.
To determine if the presence of the TPP moiety would preferentially direct TM608 to the mitochondria, we treated HeLa cells to sublethal conditions of 4 μM TM608 or PHA665752 for 3 h, followed by treatment with the mitochondrion-specific dye MitoTracker Red (MTR) and visualization by confocal microscopy. Both TM608 and PHA665752 emitted green fluorescence but only TM608 colocalized with MTR in the mitochondria (Figure 4).
Figure 4.
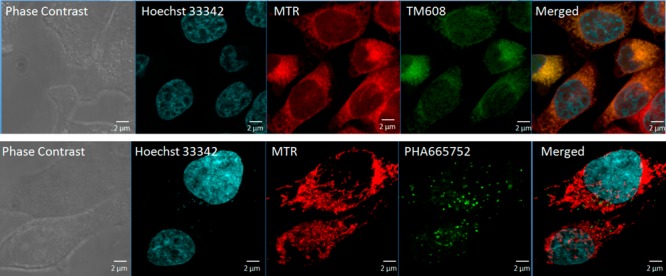
Confocal microscopy images of HeLa cells treated with 4 μM TM608 (1st row) and 4 μM PHA665752 (2nd row) for 3 h and stained with MitoTracker Red and Hoechst 33342 for visualization of mitochondria and nuclei, respectively. PHA665752 and TM608 are fluorescent (Ex 460 nm, Em 520 nm). Overlap of the fluorescence of TM608 (green) and MTR (red) is evident from the Merged Panel.
The localization of TM608 in the mitochondria was further investigated by a flow cytometric analysis of purified mitochondria isolated from HCC827B cells that were treated with PHA665752 or TM608 (4 μM, 1 h) in the absence or presence of MTR for mitochondrial staining (Figure 5).
Figure 5.
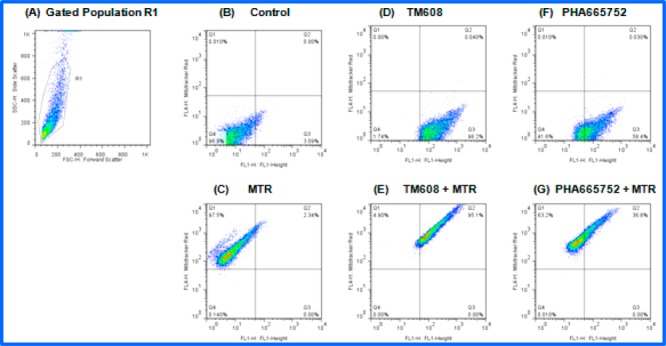
Representative flow cytometry dot plots from double staining experiments using MTR and test compounds (TM608/PHA665752) in purified mitochondria of HCC827B cells. (A) Selection of mitochondria based on forward scatter (x-axis) and side scatter (y-axis) plot. Events within the gate R1 were selected for analysis. (B) Unstained control sample (DMSO). (C–G) Fluorescence of samples stained with MTR, TM608, PHA665752, MTR + TM608, and MTR + PHA665752 for events within R1.
Mitochondria were distinguished from the background by their light scattering properties deduced from side scatter versus forward scatter plots. The selected gated population (R1, Figure 5A) was analyzed for MTR fluorescence, test compound (TM608 or PHA665752) fluorescence, and combined MTR-test compound fluorescence. More than 98% of the events within R1 were MTR positive (Figure 5C), confirming that mitochondria were being examined. Almost the same proportion of R1 events (98%) were TM608 positive (Figure 5D), an indication that the mitochondria were stained with TM608. The proportion of R1 events that were “double-stained” with MTR and TM608 was exceedingly high (Q2 95%, Figure 5E), confirming the colocalization of MTR and TM608 in the mitochondria. Interestingly, a small proportion (Q2 35%) of the R1 events depicted double staining of MTR and PHA665752 (Figure 5G), which implied that some PHA665752 had gained access into the mitochondria, albeit at levels that were greatly exceeded (2.5×) by TM608. Thus, evidence from two different approaches point to the mitochondrial localization of TM608.
We then proceeded to determine if the localization of TM608 in the mitochondria would suppress the activation of mitochondrial MET in HCC827B cells. Whole cells and purified mitochondria were separately treated with TM608 or PHA665752 at 4 μM for 1 and 1.5 h. Immunoblotting of whole cell lysates and mitochondrial fractions showed that MET protein expression was not altered by TM608 or PHA-665752 under these conditions (Figure 6A). However, PHA665752 and TM608 suppressed MET phosphorylation at Y1234/1235 in whole cell lysates. The suppression was predictably complete in lysates treated with PHA665752 but partial in the case of TM608. It would seem that the presence of TPP had compromised the fit of TM608 at the kinase site, thus partially attenuating its MET inhibitory activity.
Figure 6.

Immunoblots of MET and phospho-MET (Y1234/Y1235) in whole cell lysates and purified mitochondrial proteins of MET-overexpressing HCC827B cells. (A) Cells and pure mitochondria isolated from cells were separately treated with PHA665752 or TM608 (4 μM) for 1 and 1.5 h. (B) Cells were treated with PHA665752 or TM608 (4 μM) for 24 h, after which whole cell lysates and mitochondria were isolated from treated cells and probed for phospho-MET. In both (A) and (B), 10 μg of protein was loaded in each lane. SDHA and actin were loading controls for mitochondria and whole cells, respectively.
When we probed mitochondrial fractions that were initially isolated from HCC827B cells and then treated with either TM608 or PHA665752 over the same time period, no evidence of phospho-MET inhibition was detected for either compound (Figure 6A). Such an outcome was anticipated for PHA665752 in view of its limited presence in the mitochondria but was unexpected for TM608. We reasoned that TM608 would require a longer treatment time to manifest its suppressive effects on MET because it is a weaker MET inhibitor than PHA665752. Hence, HCC827B cells were exposed to TM608 or PHA665752 (4 μM) for 24 h, after which mitochondria were isolated from the treated cells and probed for p-MET Y1234/1235 (Figure 6B). The longer treatment time necessitated a change in the experimental sequence (treatment followed by isolation of mitochondria) as the structural integrity of isolated mitochondria would be lost if the previous method (isolation followed by treatment) was employed. Whole cells treated with either compound for the extended time were also probed for p-MET Y1234/1235. The results on the whole cell lysates were essentially similar to those obtained at the shorter time intervals, thus confirming that TM608 partially inhibited the activation of MET in HCC827B cells.
When we examined the treated mitochondrial fractions, TM608 was now found to significantly suppress MET phosphorylation, but to our surprise, PHA665752 also diminished the phosphorylation of mitochondrial MET. To explain these unusual findings, we recalled that the preceding mitochondrial colocalization investigations by flow cytometry detected some PHA665752 in the mitochondria (Figure 5G), which would imply that even without a mitochondrial targeting entity, PHA665752 gains access to the mitochondria, possibly due to its lipophilicity and charged state. Prolonging the exposure time would have allowed more of it to accumulate within the mitochondria and potently inhibit MET in that organelle, as observed here. In the case of TM608, the mitochondrial targeting motif accelerated its entry into the mitochondria but hampered inhibition. Consequently, a longer time was necessary to augment its MET inhibitory activity, which was likewise reflected in other downstream events, as evidenced from the slower onset of TM608-induced apoptosis that nonetheless went to completion (Figure 3). Rapid localization to the mitochondria would advantageously blunt extra-mitochondrial off-target effects, and this could have contributed to the cytotoxicity advantage displayed by TM608 on TAMH cells.
MET activates multiple signaling pathways and is intrinsically networked for cross-talk with other receptor tyrosine kinases (RTKs).22 We have observed a hitherto unrecognized facet of MET kinase activity, namely, its localization to the mitochondria of cancer cells, which are high MET expressors13 and the ensuing alteration of cellular functions that promote survival and hence resistance to TKIs. Our finding resonates with that of Ding et al., who showed that ERBB2 translocated to mitochondria of cancer cells and that localization was associated with greater resistance to cell killing by trastuzumab, a monoclonal antibody that blocks plasma membrane ERBB2 kinase activation.23 The similarity of observations from these independent studies on two different RTKs (MET and ERBB2) hint at an expanded view of how these kinases affect cellular functions beyond the canonical model of signaling from the plasma membrane and the potential of targeting mitochondrial RTKs as a means of exploiting a distinct vulnerability of cancer cells.
In conclusion, our findings provide proof of concept that TM608, a first example of a TPP-TKI conjugate, rapidly accumulates in mitochondria and suppresses the activation of MET in high MET expressing, erlotinib-resistant HCC827B cells (total cell lysates and mitochondrial fractions). That inhibition of MET activation by TM608 was time dependent but incomplete, points to the direction future synthetic efforts should take, namely, to reconcile targeted delivery with robust potency in final analogues. It is however telling that, in spite of its weaker inhibitory activity, TM608 was as potent and apoptogenic as PHA665752 in abrogating NSCLC cell viability. Directed accessibility to the mitochondria as afforded by a targeting moiety like TPP could conceivably compensate for deficient activity if uptake is rapid, cumulative, and selective.
Acknowledgments
This work was supported by the National Cancer Centre to O.L.K. and Biomedical Research Council (R148000706733) to M.L.G. The authors gratefully acknowledge Professor Tetsuya Mitsudomi and Professor Kenichi Suda (Kinki University, Japan) for their gift of the isogenic pair of nonsmall cell lung cancer HCC827 cells and the Drug Development Unit, National University of Singapore for carrying out the cytotoxicity determinations.
Glossary
ABBREVIATIONS
- EGFR
epidermal growth factor receptor
- NSCLC
nonsmall cell lung cancer
- HGF
hepatocyte growth factor
- TKI
tyrosine kinase inhibitors
- TCA
tricarboxylic acid
- TPP
triphenylphosphonium
- EDC
1-ethyl-3-(3-(dimethylamino)propyl)-carbodiimide
- TOM
translocase of outer membrane
- SDHA
succinate dehydrogenase complex subunit A
- PARP
poly ADP ribose polymerase
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.6b00223.
Docking of PHA665752 and TM608 in the Met kinase binding pocket (2WKM); synthetic protocols, spectroscopic data, and purity determination of TM608; protocols for cell culture, evaluation of cell viability, cytotoxicity determinations, protein immunoblotting, live cell imaging, mitochondrial isolation, flow cytometry, determination of activated MET, and localization of mitochondrial MET (PDF)
Author Contributions
The manuscript was written through contributions of all authors.
The authors declare no competing financial interest.
Supplementary Material
References
- Yamamoto H.; Toyoka S.; Mitsudomi T. Impact of EGFR mutation analysis in non-small cell lung cancer. Lung Cancer 2009, 63, 315–321. 10.1016/j.lungcan.2008.06.021. [DOI] [PubMed] [Google Scholar]
- Gazdar A. F. Activating and resistance mutations of EGFR in non-small cell lung cancer: role in clinical response to EGFR tyrosine kinase inhibitors. Oncogene 2009, 28, S24–S31. 10.1038/onc.2009.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun C. H.; Mengwasser K. E.; Toms A. V.; Woo M. S.; Greulich H.; Wong K. K.; Meyerson M.; Eck M. J. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 2070–2075. 10.1073/pnas.0709662105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelman J. A.; Settleman J. Acquired resistance to tyrosine kinase inhibitors during cancer therapy. Curr. Opin. Genet. Dev. 2008, 18, 73–79. 10.1016/j.gde.2008.01.004. [DOI] [PubMed] [Google Scholar]
- Wilson T. R.; Fridlyand J.; Yan Y.; Penuel E.; Burton L.; Chan E.; Peng J.; Lin E.; Wang Y.; Sosman J.; Ribas A.; Li J.; Moffat J.; Sutherlin D. P.; Koeppen H.; Merchant M.; Neve R.; Settleman J. Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors. Nature 2012, 487, 505–509. 10.1038/nature11249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C. T.; Kim H.; Liska D.; Gao S.; Christensen J. G.; Weiser M. R. MET activation mediates resistance to lapatinib inhibition of HER2-amplified gastric cancer cells. Mol. Cancer Ther. 2012, 11, 660–669. 10.1158/1535-7163.MCT-11-0754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corso S.; Giordano S. Cell-autonomous and non-cell-autonomous mechanisms of HGF/MET-driven resistance to targeted therapies: from basic research to a clinical perspective. Cancer Discovery 2013, 3, 978–992. 10.1158/2159-8290.CD-13-0040. [DOI] [PubMed] [Google Scholar]
- Pao W.; Girard N. New driver mutations in non-small cell lung cancer. Lancet Oncol. 2011, 12, 175–180. 10.1016/S1470-2045(10)70087-5. [DOI] [PubMed] [Google Scholar]
- Beau-Faller M.; Ruppert A. M.; Voegeli A. C.; Neuville A.; Meyer N.; Guerin E.; Legrain M.; Mennecier B.; Wihlm J. M.; Massard G.; Quoix E.; Oudet P.; Gaub M. P. MET gene copy number in non-small cell lung cancer: molecular analysis in a targeted non-small cell lung cancer inhibitor naïve cohort. J. Thorac. Oncol. 2008, 3, 331–339. 10.1097/JTO.0b013e318168d9d4. [DOI] [PubMed] [Google Scholar]
- Cappuzzo F.; Marchetti A.; Skokan M.; Rossi E.; Gajapathy S.; Felicioni L.; Del Grammastro M.; Sciarrotta M. G.; Buttitta F.; Incarbone M.; Toschi L.; Finocchiaro G.; Destro A.; Terracciano L.; Roncalli M.; Alloisio M.; Santoro A.; Varella-Garcia M. Increased MET gene copy number negatively affects survival of surgically resected non-small cell lung cancer patients. J. Clin. Oncol. 2009, 27, 1667–1674. 10.1200/JCO.2008.19.1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelman J. A.; Zejnullahu K.; Mitsudomi T.; Song Y.; Hyland C.; Park J. O.; Lindeman N.; Gale C.-M.; Zhao X.; Christensen J.; Kosaka T.; Holmes A. J.; Rogers A. M.; Cappuzzo F.; Mok T.; Lee C.; Johnson B. E.; Cantley L. C.; Janne P. A. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007, 316, 1039–1043. 10.1126/science.1141478. [DOI] [PubMed] [Google Scholar]
- Maina F. Strategies to overcome drug resistance of Receptor Tyrosine Kinase-addicted cancer cells. Curr. Med. Chem. 2014, 21, 1607–1617. 10.2174/09298673113209990222. [DOI] [PubMed] [Google Scholar]
- Guo T.; Zhu Y.; Gan C. S.; Lee S. S.; Zhu J.; Wang H.; Li X.; Christensen J.; Huang S.; Kon O. L.; Sze S. K. Quantitative proteomics discloses MET expression in mitochondria as a direct target of MET kinase inhibitor in cancer cells. Mol. Cell. Proteomics 2010, 9, 2629–2641. 10.1074/mcp.M110.001776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madak J.; Neamati N. Membrane permeable lipophilic cations as mitochondrial directing groups. Curr. Top. Med. Chem. 2015, 15, 745–766. 10.2174/1568026615666150302105622. [DOI] [PubMed] [Google Scholar]
- Millard M.; Gallagher J. D.; Olenyuk B. Z.; Neamati N. A selective mitochondrial-targeted chlorambucil with remarkable cytotoxicity in breast and pancreatic cancers. J. Med. Chem. 2013, 56, 9170–9179. 10.1021/jm4012438. [DOI] [PubMed] [Google Scholar]
- Lee C. W.; Park H. K.; Jeong H.; Lim J.; Lee A.; Cheon K. Y.; Kim C.; Thomas A. P.; Bae B.; Kim N. D.; Kim S. H.; Suh P.; Ryu J.; Kang B. H. Development of a mitochondria-targeted Hsp90 inhibitor based on the crystal structures of human TRAP1. J. Am. Chem. Soc. 2015, 137, 4358–4367. 10.1021/ja511893n. [DOI] [PubMed] [Google Scholar]
- Liang B.; Shao W.; Zhu C.; Wen G.; Yue X.; Wang R.; Quan J.; Du J.; Bu X. Mitochondria-targeted approach: remarkably enhanced cellular bioactivities of TPP2a as selective inhibitor and probe toward TrxR. ACS Chem. Biol. 2016, 11, 425–434. 10.1021/acschembio.5b00708. [DOI] [PubMed] [Google Scholar]
- Ross M. F.; Prime T. A.; Abakumova I.; James A. M.; Porteous C. M.; Smith R. A. J.; Murphy M. P. Rapid and extensive uptake and activation of hydrophobic triphenylphosphonium cations within cells. Biochem. J. 2008, 411, 633–645. 10.1042/BJ20080063. [DOI] [PubMed] [Google Scholar]
- Cui J. J.; Tran-Dube M.; Shen H.; Nambu M.; Kung P. P.; Pairish M.; Jia L.; Meng J.; Funk L.; Botrous I.; McTigue M.; Grodsky N.; Ryan K.; Padrique E.; Alton G.; Timofeevski S.; Yamazaki S.; Li Q.; Zou H.; Christensen J.; Mroczkowski B.; Bender S.; Kania R. S.; Edwards M. P. Structure based drug design of crizotinib (PF-02341066), a potent and selective inhibitor of mesenchymal-epithelial transition factor (c-MET) kinase and anaplastic lymphoma kinase (ALK). J. Med. Chem. 2011, 54, 6342–6363. 10.1021/jm2007613. [DOI] [PubMed] [Google Scholar]
- Suda K.; Murakami I.; Katayama T.; Tomizawa K.; Osada H.; Sekido Y.; Maehara Y.; Yatabe Y.; Mitsudomi T. Reciprocal and complementary role of MET amplification and EGFR T790M mutation in acquired resistance to kinase inhibitors in lung cancer. Clin. Cancer Res. 2010, 16, 5489–5498. 10.1158/1078-0432.CCR-10-1371. [DOI] [PubMed] [Google Scholar]
- Pierce R.; Franklin C. C.; Campbell J. S.; Tonge R. P.; Chen W.; Fausto N.; Nelson S. D.; Bruschi S. A. Cell culture model for acetaminophen-induced hepatocyte death in vivo. Biochem. Pharmacol. 2002, 64, 413–424. 10.1016/S0006-2952(02)01180-2. [DOI] [PubMed] [Google Scholar]
- Peters S.; Adjei A. A. MET: A promising anticancer therapeutic target. Nat. Rev. Clin. Oncol. 2012, 9, 314–326. 10.1038/nrclinonc.2012.71. [DOI] [PubMed] [Google Scholar]
- Ding Y.; Liu Z.; Desai S.; Zhao Y.; Liu H.; Pannell L. K.; Yi H.; Wright E. R.; Owen L. B.; Dean-Colomb W.; Fodstad O.; Lu J.; LeDoux S. P.; Wilson G. L.; Tan M. Receptor tyrosine kinase ErbB2 translocates into mitochondria and regulates cellular metabolism. Nat. Commun. 2012, 3, 1271–1280. 10.1038/ncomms2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.