

Abstract

Optimization of pyridine-based noncatalytic site integrase inhibitors (NCINIs) based on compound 2 has led to the discovery of molecules capable of inhibiting virus harboring N124 variants of HIV integrase (IN) while maintaining minimal contribution of enterohepatic recirculation to clearance in rat. Structure–activity relationships at the C6 position established chemical space where the extent of enterohepatic recirculation in the rat is minimized. Desymmetrization of the C4 substituent allowed for potency optimization against virus having the N124 variant of integrase. Combination of these lessons led to the discovery of compound 20, having balanced serum-shifted antiviral potency and minimized excretion in to the biliary tract in rat, potentially representing a clinically viable starting point for a new treatment option for individuals infected with HIV.
Keywords: HIV integrase, NCINI, enterohepatic recirculation
Since the advent of combination antiretroviral therapy (cART) for the treatment of HIV infected individuals, there has been a sustained effort to discover new mechanistic treatment options for patients failing therapy due to emerging resistance or poor tolerability to first line regimens. This wave of innovation has seen the mechanistic options available to patients expand beyond inhibitors of HIV protease or HIV reverse transcriptase (NRTIs and NNRTIs) to include inhibitors of viral entry (CCR5 antagonists, fusion inhibitors)1 and HIV integrase (strand transfer inhibitors).2 We have previously reported on our progress toward a new HIV treatment option resulting in the discovery of a new mechanistic class that we have termed noncatalytic site integrase inhibitors (NCINIs).3−5 The most advanced molecule from this mechanistic class is BI 224436 (1), a potent inhibitor of HIV replication that was advanced into a phase Ia clinical trial. This molecule exhibited low apparent clearance in rat, in part due to enterohepatic recirculation of both the parent molecule and its corresponding acyl glucuronide (Table 1). Potential species differences in the extent of the contribution of enterohepatic recirculation to clearance introduced a large degree of uncertainty in predicting human pharmacokinetic parameters. Therefore, our medicinal chemistry strategy to provide a second compound from the NCINI class of inhibitors in order to mitigate the risk of attrition of BI 224436 focused on minimizing the contribution of enterohepatic recirculation to in vivo clearance. This resulted in the pyridine series of NCINIs exemplified by compound 2, a prototype molecule exhibiting minimal excretion into the biliary tract but suffering a lack of potency against the clinically relevant N124 variants of HIV integrase (ca. 17% of patient population).5,6 Herein, we describe our effort to optimize potency against the N124 variants of integrase while maintaining minimal contribution of enterohepatic recirculation to in vivo clearance.
Table 1. Profiles of Compounds 1 and 2.

| 1 | 2 | ||
|---|---|---|---|
| EC50a (nM) | T124 | 19 | 7.7 |
| EC50b (nM) | N124 | 30 | 810 |
| % excreted into bile @ T3h | 60 | 22 | |
| rat in vivo CL (%QH) | 0.7 | 0.6 | |
| rat in vivot1/2 (h) | 8.8 | 8.9 | |
| bile duct cannulated rat in vivo t1/2 (h) | 1.6 | 5.4 | |
NL4.3 virus (T124/T125).
NL4.3 virus (N124/T125).
Ideally, evolution of the pyridine-based NCINIs would result in a molecule with favorable alignment between serum-shifted potency against the N124 variants of IN, metabolic stability, and minimal contribution of enterohepatic recirculation to in vivo clearance. Preliminary data using compound 2 as a prototype molecule indicated that pyridine-based NCINIs initially suffered from a two log shift in potency against N124 variants compared to A124 or T124 variants of IN. Furthermore, compound 2 exhibited high serum shift, which when combined with antiviral potency and human PK prediction led to an unacceptable dose projection.7 Our optimization strategy began with an evaluation of the impact of a range of C6-substitutions on these parameters.8 At the outset of this effort, most potent analogues, such as compounds 2 and 3 use the phenyl group at C6 to project a substituent (an isoxazole in these cases) in a conformationally restricted way. We hypothesized that varying conformational flexibility of this linker may have an impact on the potency against virus harboring the N124 variant of IN. Introduction of five-membered heterocycles to give analogues such as compound 4 or an amide linker to give compound 5 gave inhibitors with similar potency (cf. compounds 3–5, Table 2), although serum shifts remained unacceptably high. An early investigation of the amide N-substituent revealed that aliphatic groups were very well tolerated and often led to improvements in potency, such as was the case for trans-4-methylcyclohex-1-yl analogue 6. When the symmetrical chlorophenyl C4-substituent replaced the chromane, potency and serum shift were maintained at similar levels (cf. compounds 6 and 7), but metabolic stability improved. Based on these findings, the structure–activity relationships (SARs) were expanded to include a large diversity of amide analogues. Through this effort, numerous examples of aliphatic amides that retain the general potency profile of compound 7 were identified, but independent SARs around serum shifted potency also emerged. In many cases, serum shift was reduced to a very low level, as exemplified by compound 8. This effort also revealed that polarity within the aliphatic substituent was not tolerated, as demonstrated by the alcohol and amine analogues 9 and 10, respectively. Anilide based inhibitors were also explored, and many examples with very similar potency profiles to the aliphatic amides (cf. compounds 7, 11, and 12) were found. Interestingly, the reversed amide 13 was well tolerated, exhibiting a profile very similar to compound 12. Cyclization of the amide found in compound 11 gave benzimidazole 14, which showed excellent potency against virus having the T124 variant of IN, although serum shift for this compound was found to be somewhat high. Substitution of the N1 atom of the benzimidazole had a marginal impact on antiviral potency, but generally provided analogues with lower serum shift values. Taken together, these results indicated that variation of the C6 position could not improve potency against the N124 IN containing virus to an appreciable extent, but serum shift could be improved in the context of both the C6-amide and C6-benzimidazole subseries.
Table 2. Preliminary SAR at the C6-Position.
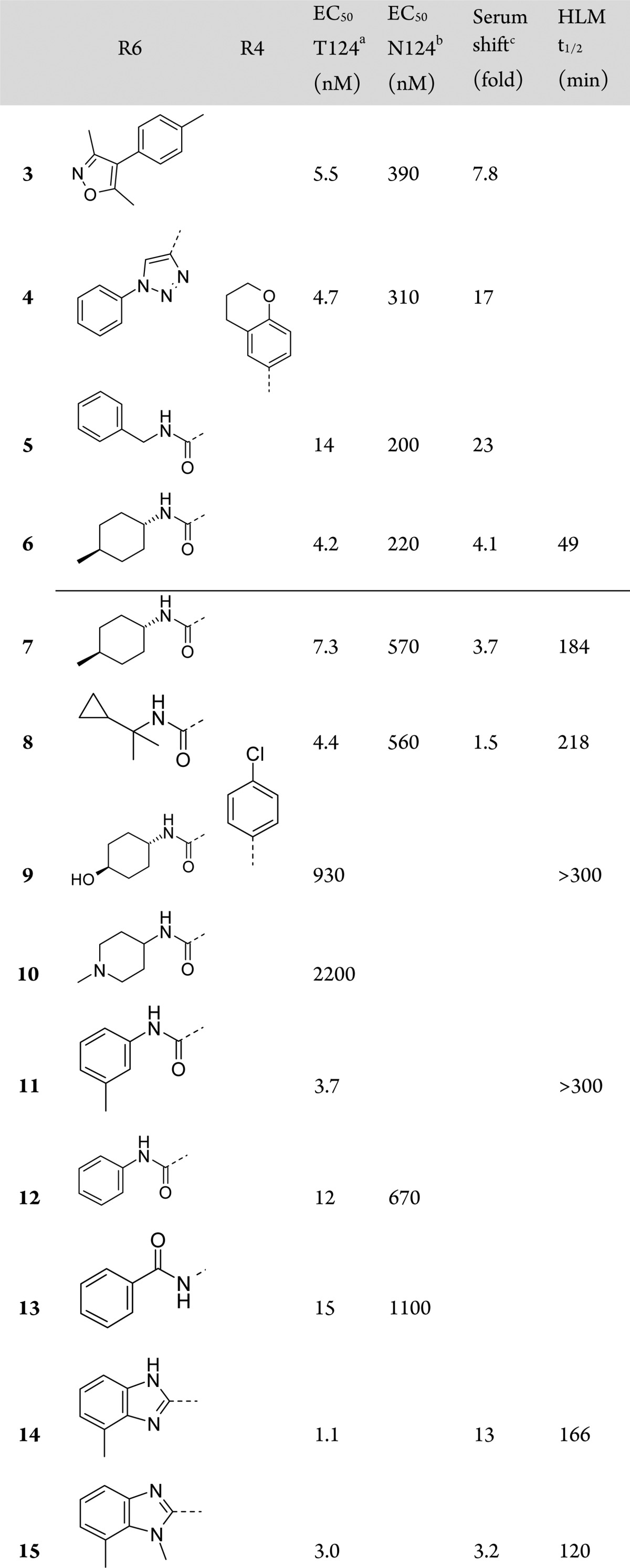
NL4.3 virus (T124/T125).
NL4.3 virus (N124/T125).
Determined by measurement of EC50 values ±50% human serum.
Based on the existing X-ray crystallographic information available at the time,3 the substituent at the C6-position was assumed to be in close proximity to residue 124.9 Due to the significant differences between the threonine and asparagine side chains, we hypothesized that the shift in potency between viruses with the N124 or T124 variants of IN maybe due to inferior affinity of the inhibitors for the N124 variant.10 In an attempt to increase binding affinity, the C6-substituents with the most favorable properties were selected and held constant for an attempt to optimize the C4-substituent. A similar strategy with quinoline based inhibitors had been successful in improving potency across multiple IN variants, and the key tactic employed in that effort was desymmetrization of the C4-arene. Utilizing the C4-substituent of compound 1, which was highly optimized for potency across all major variants of integrase, and a promising C6-amide such as that found in compound 6 gave the stable atropisomer 16,11 which exhibited good potency against the T124 variant but was inactive up to a concentration of 2.5 μM against the N124 variant. However, morpholine analogue 17, which exhibits free rotation about the C4-axis, regained potency against the N124 variant containing virus and exhibited good serum shift and excellent metabolic stability. Relocking the conformation about the C4-axis by addition of a methyl group at the ortho position of the C4-arene gave compound 18, which showed a slight improvement in serum-shifted antiviral potency accompanied by some loss in metabolic stability. Methylation of the morpholine nitrogen of compounds 17 and 18 produced compounds 19 and 20, respectively, which regained excellent metabolic stability and maintained good serum-shifted antiviral potency. Similar SAR was observed in the reversed amide subseries (e.g. compounds 21 and 22). With benzimidazoles installed at C6 (compounds 23–25), these SAR trends were also observed with antiviral potency against N124 containing virus breaching the 100 nM level, although metabolic stability was moderately compromised.
Having established that antiviral potency against N124 IN containing virus could be optimized, a range of structurally diverse analogues were evaluated in bile-duct cannulated rat pharmacokinetic experiments. For these studies, compounds with aligned potency and stability in human and rat liver microsome assays were selected. We evaluated the plasma exposure and the percent of parent excreted into the biliary tract as the acyl glucuronide at 3 h post i.v. dose.12 For promising compounds, full pharmacokinetic profiles were determined. For these compounds, the results comparing the extent to which the acyl glucuronide of parent was excreted into the biliary tract to the ratio of terminal half-life in rat to bile duct-cannulated rat are presented in Figure 1. In general, a trend toward a ratio of 1 for half-life in rat to half-life in bile duct cannulated rat as the extent of acyl glucuronide excreted into the biliary tract decreases is observed. For compounds with a ratio near unity, a clearance in vitro–in vivo correlation was observed, confirming the generality of this observation for the pyridine series.
Figure 1.

Ratio of i.v. t1/2 (rat) and i.v. t1/2 (bile duct cannulated rat) as a function of %parent excreted as the acyl glucuronide into the biliary tract.
Based on their preliminary antiviral profiles, compounds 20 and 23 were characterized in an expanded battery of in vitro and in vivo experiments. Both compounds exhibit excellent antiviral potency against virus with T124 variants of IN and reasonable serum shift values (Table 3). Further profiling revealed these two compounds showed favorable permeability in the Caco-2 assay and excellent aqueous solubility (Table 4). Metabolic stability was assessed in human and rat hepatocyte incubations, and generally low levels of hepatic clearance were observed. Both compounds showed similarly low clearance in rat pharmacokinetic experiments with volumes of distribution in the expected range for a carboxylic acid. However, while compound 20 showed very low total excretion into the biliary tract, compound 23 was highly excreted indicating the low clearance of this compound likely has a significant contribution from enterohepatic recirculation. This was a general observation for the C6-benzimidazole derivatives, and so, inhibitors related to compound 23 were deprioritized.
Table 3. C4- and C6-Position Combinations.
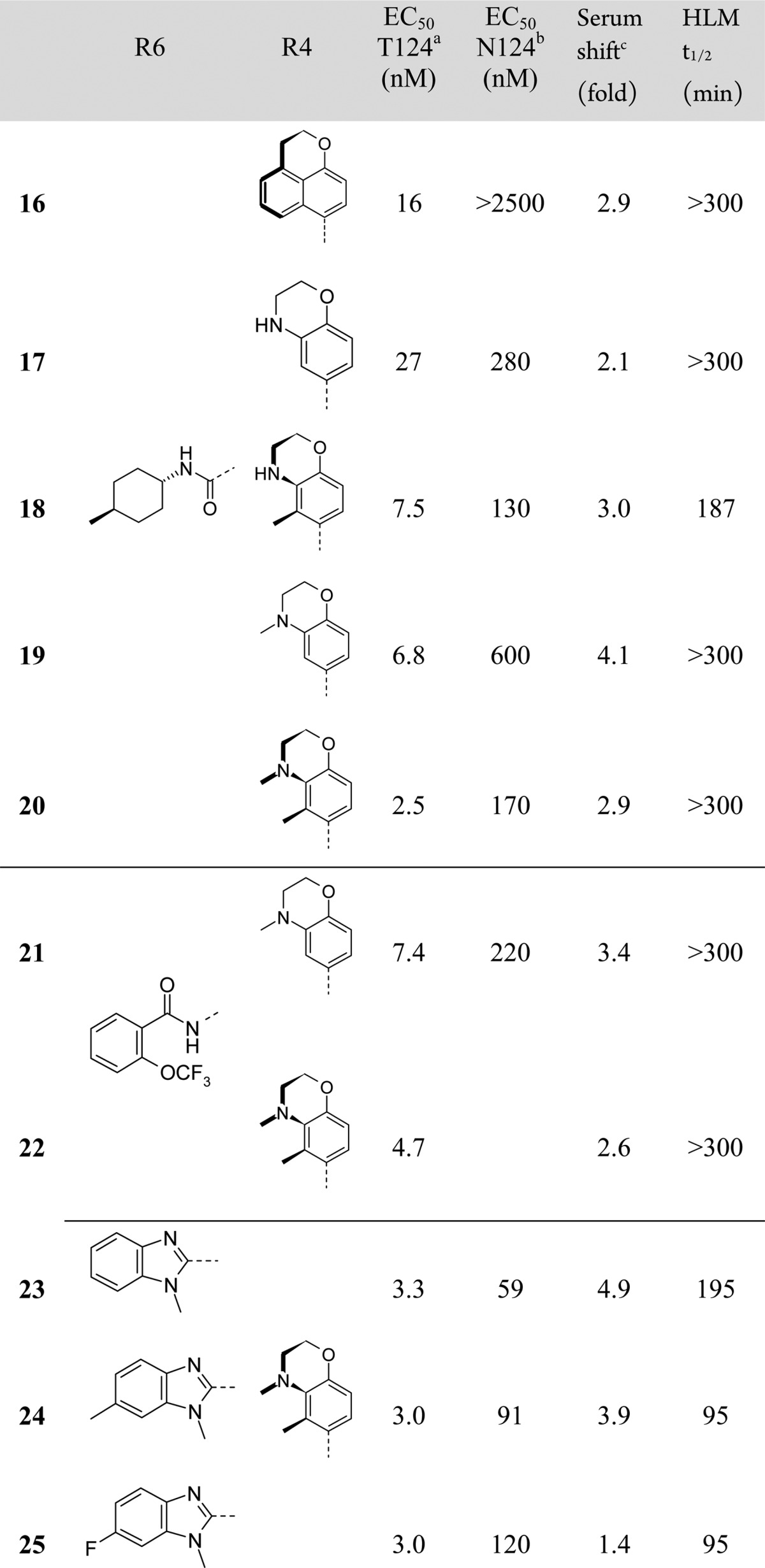
NL4.3 virus (T124/T125).
NL4.3 virus (N124/T125).
Determined by measurement of EC50 values ±50% human serum.
Table 4. Selected in Vitro ADME and Rat Pharmacokinetic Data for Compounds 20 and 23.
| Caco-2 Papp × 10–6 cm/s | solubility pH = 6.8 mg/mL | HHEP (%QH) | RHEP (%QH) | rat i.v. Cl (%QH) | rat Vss (L/kg) | total excretion into bilea(%) | |
|---|---|---|---|---|---|---|---|
| 20 | 39.8 | 0.93 | 17 | 7 | 23 | 1.4 | 20 |
| 23 | 10.6 | >1 | 24 | 19 | 24 | 1.6 | 70 |
Sum of acylglucuronide and parent excreted into bile.
The profiling of compound 20 was expanded to include its effectiveness against infection of peripheral blood mononuclear cells with viruses harboring all of the major variants of IN. As illustrated in Table 5, compound 20 showed excellent potency against viruses with T124 and A124 variants of IN and EC50 values of 27 and 42 nM against the N124/T125 and N124/A125 strains, respectively.13 No detectable inhibition of the cytochrome P450 enzymes was observed, indicating a low risk for P450-mediated drug–drug interactions. The half-lives for compound 20 were similar for intact and bile-duct cannulated rats, consistent with results presented in Table 4, in view of the data summarized by Figure 1. Metabolic stability in incubations with dog and monkey hepatocytes indicated low to moderate clearance across species. As with rat, these observations translated to low to moderate clearance in vivo. Taken together, the data presented for compound 20 indicates that favorable alignment of serum shifted EC50s and a pharmacokinetic profile that includes minimized excretion into the biliary tract can be achieved for pyridine-based NCINIs.
Table 5. Profile of Compound 20.
| EC50 (nM) | IN Variant | |
| p24 ELISA | T124/T125 | 0.5 |
| T124/A125 | 0.4 | |
| A124/T125 | 0.6 | |
| A124/A125 | 0.9 | |
| N124/T125 | 27 | |
| N124/A125 | 42 | |
| CYP450 IC50 (μM) | 3A4 | >30 |
| 2D6 | >30 | |
| 1A2 | >30 | |
| 2C19 | >30 | |
| 2C9 | >30 | |
| rat t1/2/BDC rat t1/2 | 1.3 | |
| dog hepatocytes (%QH) | 31 | |
| monkey hepatocytes (%QH) | 31 | |
| dog i.v. Cl (%QH) | 10 | |
| monkey i.v. Cl (%QH) | 30 | |
In conclusion, the continued optimization of pyridine-based NCINIs has led to the discovery of molecules capable of inhibiting virus harboring N124 variants of IN while maintaining minimal contribution of enterohepatic recirculation to clearance in rat, which may have implications on the development of the NCINI class for therapeutic use in treating HIV infected individuals. Following the original disclosure of C6-phenyl and C6-benzimidazole modified NCINIs in 2010,8 the Kvaratskhelia group selected resistance mutants of HIV bearing the T124N amino acid substitution under selection pressure from a close analogue of compound 14.9 Given that variants of HIV having the N124 substitution are naturally occurring in the patient population, an appropriate level of potency against these viruses is critical to the use of pyridine-based NCINIs in any new treatment regimen. Compounds 20 and 23 demonstrate that it is possible to significantly inhibit these viruses with pyridine-based NCINIs, providing evidence that, with further optimization, this class of replication inhibitor may represent a clinically viable treatment option for individuals infected with HIV.
Acknowledgments
The authors thank Sonia Tremblay and Elizabeth Wardrop for EC50 determinations; Céline Plouffe for Kd-app determinations; Hugo Poirier for Caco-2 permeability data; Josie DeMarte for microsomal stability data; Federico Columbo for hepatocyte stability determinations; Laibin Luo, Danhui Sun, and Eduard Bugan for logD and solubility determinations; Edith Bellavance and Marieve Dupuis for pharmacokinetic experiments; and Michael Cordingley, Richard Bethell, and Paul Edwards for their leadership and support.
Glossary
ABBREVIATIONS
- HLM
human liver microsomes
- HHEP
human hepatocyte
- RHEP
rat hepotaocyte
- CYP450
cytochrome P450
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.6b00194.
Summary of synthetic approaches, characterization data for key compounds, correlation of apparent Kd values and EC50 values, comparison between the energy barriers for free rotation of the quinoline and pyridine C4-biaryl bonds, and summary of screening results in bile duct cannulated and intact rat PK experiments (PDF)
Author Present Address
† Boehringer Ingelheim Pharmaceuticals, Inc., 900 Ridgebury Road, Ridgefield, Connecticut 06877, United States.
The authors declare no competing financial interest.
Supplementary Material
References
- For a recent overview see:Kramer V. G; Wainberg M. A. Resistance against inhibitors of HIV-1 entry into target cells. Future Virol. 2015, 10, 97–112. 10.2217/fvl.14.104. [DOI] [Google Scholar]
- For a recent overview, see:Li Y.; Xuan S.; Feng Y.; Yan A. Targeting HIV-1 integrase with strand transfer inhibitors. Drug Discovery Today 2015, 20, 435–449. 10.1016/j.drudis.2014.12.001. [DOI] [PubMed] [Google Scholar]
- Fader L. D.; Malenfant E.; Parisien M.; Carson R.; Bilodeau F.; Landry S.; Pesant M.; Brochu C.; Morin S.; Chabot C.; Halmos T.; Bousquet Y.; Bailey M. D.; Kawai S. H.; Coulombe R.; LaPlante S.; Jakalian A.; Bhardwaj P. K.; Wernic D.; Schroeder P.; Amad M.; Edwards P.; Garneau M.; Duan J.; Cordingley M.; Bethell R.; Mason S. W.; Bös M.; Bonneau P.; Poupart M.-A.; Faucher A.-M.; Simoneau B.; Fenwick C.; Yoakim C.; Tsantrizos Y. Discovery of BI 224436, a Non-Catalytic Site Integrase Inhibitor (NCINI) of HIV-1. ACS Med. Chem. Lett. 2014, 5, 422–427. 10.1021/ml500002n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenwick C.; Amad M.; Bailey M. D.; Bethell R.; Bös M.; Bonneau P.; Cordingley M.; Coulombe R.; Duan J.; Edwards P.; Fader L. D.; Faucher A.-M.; Garneau M.; Jakalian A.; Kawai S.; Lamorte L.; LaPlante S.; Luo L.; Mason S.; Poupart M.-A.; Rioux N.; Schroeder P.; Simoneau B.; Tremblay S.; Tsantrizos Y.; Witvrouw M.; Yoakim C. Preclinical Profile of BI 224436, a Novel HIV-1 Non-Catalytic Site Integrase Inhibitor. Antimicrob. Agents Chemother. 2014, 58, 3233–3244. 10.1128/AAC.02719-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fader L. D.; Carson R. J.; Morin S.; Bilodeau F.; Chabot C.; Halmos T.; Bailey M. D.; Kawai S. H.; Coulombe R.; Laplante S.; Mekhssian K.; Jakalian A.; Garneau M.; Duan J.; Mason S. W.; Simoneau B.; Fenwick C.; Tsantrizos Y. S.; Yoakim C. Minimizing the contribution of enterohepatic recirculation to clearance in rat for the NCINI class of inhibitors of HIV. ACS Med. Chem. Lett. 2014, 5, 711–716. 10.1021/ml500110j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- All EC50 values were determined by luciferase reporter gene assay as previously described and are the average of at least two experiments. See refs (4) and (5).
- For treatment of patients infected with virus harboring the T124 variant of IN with compound 2, the efficacious exposure at Cmin can be estimated to be 30 × serum-shifted EC50 = 2.2 μM. Human PK can be predicted in the following way: Cl = human hepatocyte stability; F(%) = 100 – Cl(%QH); Vd = cross-species average (rat, dog, cyno); assume ka = 1. Based on these assumptions, the dose required to cover Cmin = 2.2 μM for 24 h = 31 g.
- All compounds presented in this manuscript were prepared as previously described. See:Yoakim C.; Bailey M. D.; Bilodeau F.; Carson R. J.; Fader L.; Kawai S.; Laplante S.; Simoneau B.; Surprenant S.; Thibeault C.; Tsantrizos Y. S.. Preparation of 2-alkoxy-2-(pyridin-3-yl)acetic acid derivatives as inhibitors of human immunodeficiency virus (HIV) replication. PCT Int. Appl. WO/2010/130034.
- After the original disclosure of the pyridine series of integrase inhibitors (ref (8)), this assumption was confirmed by X-ray crystallography. See:Sharma A.; Slaughter A.; Jena N.; Feng L.; Kessl J. J.; Fadel H. J.; Malani N.; Male F.; Wu L.; Poeschla E.; Bushman F. D.; Fuchs J. R.; Kvaratskhelia M. A New Class of Multimerization Selective Inhibitors of HIV-1 Integrase. PLoS Pathog. 2014, 10, e1004171. 10.1371/journal.ppat.1004171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Throughout the course of this work, affinity was approximated using a displacement assay that utilized a T124 variant of IN. The derived apparent Kd values correlated with EC50 values. See Supporting Information.
- For a comparison between the energy barriers for free rotation of the quinoline and pyridine C4-biaryl bonds, see Supporting Information.
- See Supporting Information for a summary of screening results.
- For in depth antiviral potency evaluation, EC50 values were determined by ELISA formatted viral replication assay that quantifies the viral protein p24. For example, seeSturino C. F.; Bousquet Y.; James C. A.; DeRoy P.; Duplessis M.; Edwards P. J.; Halmos T.; Minville J.; Morency L.; Morin S.; Thavonekham B.; Tremblay M.; Duan J.; Ribadeneira M.; Garneau M.; Pelletier A.; Tremblay S.; Lamorte L.; Bethell R.; Cordingley M. G.; Rajotte D.; Simoneau B. Identification of potent and orally bioavailable nucleotide competing reverse transcriptase inhibitors: in vitro and in vivo optimization of a series of benzofurano[3,2-d]pyrimidin-2-one derived inhibitors. Bioorg. Med. Chem. Lett. 2013, 23, 3967–75. 10.1016/j.bmcl.2013.04.043. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.