

Abstract
Many cortical areas play crucial roles in higher order brain functions such as pain and emotion-processing, decision-making, and cognition. Among them, anterior cingulate cortex (ACC) and insular cortex (IC) are two key areas. Glutamate mediates major excitatory transmission during long-term plasticity in both physiological and pathological conditions. Specifically related to nociceptive or pain behaviors, metabotropic glutamate subtype receptors (mGluRs) have been involved in different types of synaptic modulation and plasticity from periphery to the spinal cord. However, less is known about their functional roles in plasticity related to pain and its related behaviors within cortical regions. In this review, we first summarized previous studies of synaptic plasticity in both the ACC and IC, and discussed how mGluRs may be involved in both cortical long-term potentiation (LTP) and long-term depression (LTD)-especially in LTD. The activation of mGluRs contributes to the induction of LTD in both ACC and IC areas. The loss of LTD caused by peripheral amputation or nerve injury can be rescued by priming ACC or IC with activations of mGluR1 receptors. We also discussed the potential functional roles of mGluRs for pain-related behaviors. We propose that targeting mGluRs in the cortical areas including the ACC and IC may provide a new therapeutic strategy for the treatment of chronic pain, phantom pain or anxiety.
Keywords: Anterior cingulate cortex, insular cortex, long term depression, long term potentiation, metabotropic glutamate receptor, pain
Introduction
Pain is a key higher order brain function which controls emotion, affects decision making, and forms vital memory for survival. Human imaging and animal studies show that several cortical regions are related to pain and pain-triggered unpleasantness [1, 2]. Two major areas for pain perception are the anterior cingulate cortex (ACC) and insular cortex (IC) [3-5]. For example, in vivo electrophysiological recordings from adult rats and mice show that ACC neurons were activated by peripheral physiological stimulation or a peripheral injury model, such as digit amputation [6-8].
Glutamate contributes as a major excitatory neurotransmitter in the central nervous system (CNS) [9]. Glutamatergic receptors are essential in basal synaptic transmission and long-term plasticity in the CNS. There are two types of glutamatergic receptors, which are ionotropic and metabotropic glutamatergic receptors (mGluRs). α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPARs), N-methyl-D-aspartate receptors (NMDARs) and kainate receptors (KARs) belong in ionotropic glutamatergic receptors. AMPARs and KARs mediate fast excitatory transmission in the CNS, while NMDARs mediate a slow component of excitatory synaptic transmission. Among these ionotropic receptors, AMPARs and NMDARs have been most reviewed thus far. mGluRs, on the other hand, are members of G-protein coupled receptors (GPCRs), which can be classified into three groups: group I (mGluR1 and 5), group II (mGluR2 and 3) and group III (mGluR4, mGluR6, mGluR7 and mGluR8) [10, 11]. Different mGluRs are distributed in both the peripheral nervous system and the CNS. Most subtypes except of mGluR6 are widely expressed in pain-related areas of spinal cord and the brain [10, 11]. Group I mGluRs are mainly expressed in the postsynaptic membrane; group II mGluRs are located in both postsynaptic and presynaptic elements. Group III mGluRs are located to presynaptic terminals closed to or in the active zone of the synapse [10, 11].
Two major forms of long-term plasticity, long-term potentiation (LTP) and long-term depression (LTD) are synaptic models of learning, memory and pain [3, 12-15]. Its mechanisms have been primarily studied in subcortical areas, especially the hippocampus [13, 16, 17]. Both LTP and LTD have also been observed in cortical areas including the ACC and IC of adult animals [3, 18-20]. Interestingly, mice undergoing chronic pain had altered forms of LTP and LTD in the ACC [3, 21-24] and the IC [19, 25]. Although the functional roles of AMPARs, NMDARs and KARs on LTP and LTD have been well reviewed [3, 13, 16, 17], less is understood about the roles of mGluRs on cortical long-term plasticity and their function in pain-related behaviors. Here, we will explore the mechanisms of cortical long-term plasticity, its contribution to pain behaviors, and the possible role of mGluRs in both.
Cortical areas in pain perception
Five major cortical areas: the ACC, the IC, prefrontal cortex (PFC), primary somatosensory cortex and secondary somatosensory cortex consistently respond to pain stimuli [1, 2]. The importance of its activity during pain is easily demonstrated through brain imaging in humans [3, 4]. Bushnell et al. (2013) reported that a warm-cold grid, producing painful sensation, can selectively activate the ACC region, while warm or cold stimuli alone did not produce any pain or cause ACC activation [2]. Thermal noxious heat, cold and chemical stimuli all result in ACC neuronal activity [1]. Similar to the ACC, the IC is triggered by different painful or noxious stimuli [1, 2, 19, 26]. Thus, neural activities in the ACC and IC are thought to be critical for pain perception and unpleasantness [2]. Furthermore, both areas are involved in psychological pain, social exclusion and pain empathy, providing further evidence [27].
Long-term potentiation (LTP) in cortex as a cellular model for chronic pain
We have systematically characterized the mechanisms of LTP and their functional roles induced by peripheral injury, thereby triggering plastic changes in the ACC synapses [3, 15]. In vivo extracellular recordings reveal that digit amputation potentiate field excitatory postsynaptic potentials in adult rat ACC neurons [7]. In in vivo intracellular recordings, digit amputation also causes membrane depolarization of pyramidal neurons in the adult rat ACC [8]. Such potentiation or excitation lasts for a long time period, and consequently might generate altered neuronal spike activity in the brain without obvious peripheral sensory stimulation [28].
Cortical LTP of Excitatory Transmission
Various genetic, electrophysiological and pharma- cological approaches have been performed in order to investigate mechanisms of basic LTP in ACC synapses. Three different stimulation protocols can be used to induce LTP in ACC pyramidal neurons of adult mice brain slice preparation: the pairing training protocol (synaptic activity paired with postsynaptic depolarization), theta burst stimulation (TBS) and the spike-excitatory postsynaptic potential (EPSP) pairing protocol [18, 29]. Using whole-cell patch-clamp recording, the pairing protocol induced postsynaptic LTP mainly requires the activation of NMDARs [18]. In fEPSP recording, the LTP induced by TBS mediates NMDARs and L-type voltage-gated calcium channels (L-VGCCs) [30, 31]. NMDA receptors containing GluN2A or GluN2B subunits specifically in ACC synapses contribute to the NMDA receptor currents [18, 32]. Bath applications of GluN2A and GluN2B antagonist significantly blocked both NMDA receptor-mediated excitatory postsynaptic currents (EPSCs) and LTP; the application of either antagonist alone only partially reduced LTP [18]. Activations of NMDA receptors increase in postsynaptic calcium (Ca2+) in dendritic spines. Ca2+ is heavily involved in intracellular signaling, triggering a series of biochemical events that Ca2+ binds to calmodulin (CaM) activates Ca2+-stimulated signaling pathways [18]. A postsynaptic injection of a Ca2+ chelator, BAPTA blocked the induction of LTP, suggesting that elevated postsynaptic Ca2+ concentrations are critical for the LTP [18]. Ca2+-stimulated adenylyl cyclase subtype 1 (AC1), which is in neuron-specific is also implicated in LTP, an enzyme highly expressed in the ACC [33, 34]. In AC1 knockout (KO) mice, TBS or pairing stimulation induced LTPs are abolished [31]. Likewise, the administration of a selective AC1 inhibitor, NB001 significantly inhibited LTP [33].
Using genetic and pharmacological approaches, the functional roles of AMPA GluA1 and GluA2/3 have been investigated. GluA1-KO mice reduced LTP in the ACC [29]. LTP induction in the ACC requires the interactions between the C terminus of GluA1 and PDZ domain proteins since LTP was blocked by the GluA1 subunit C-terminal peptide analog Pep1-TGL [35, 36]. Therefore, the induction of LTP in the ACC requires the interactions between the C terminus of GluA1 and PDZ domain proteins. Synaptic delivery of the GluA1 subunit from extrasynaptic sites and GluA1-PDZ interactions is a key mechanism for the synaptic plasticity [37]. Application of a glutamate receptor antagonist, philanthotoxin 433 five minute after the induction of LTP inhibited synaptic potentiation without affecting basal AMPA receptor-mediated currents. These results suggest that Ca2+-permeable GluA2-lacking receptors are critical for the maintenance of LTP in the ACC [38]. By contrast, the GluA2/3-PDZ interaction did not affect LTP in the ACC [38].
Similar to the ACC, neurons in the IC also show LTP [19, 39]. Using whole-cell patch-clamp recordings, the pairing protocol produces LTP in pyramidal neurons in layer II/III of the IC [19]. As shown through pharmacological experiments, insular LTP requires NMDARs subunits GluN2A and GluN2B. The fEPSPs recordings showed that activations of both GluN2A and GluN2B subunits of the NMDARs, L-VGCC and mGluR1 are required for insular LTP produced by TBS [39]. The expression of insular LTP is also dependent on the recruitment of postsynaptic calcium-permeable AMPARs, which involves transient receptor potential vanilloid type 1-independent mechanisms [40].
Cortical LTP and Chronic Pain
In both the ACC and IC, our previous studies report that chronic pain in mice results in reduced LTP [19, 21]. Because the LTP related receptors and molecular are already activated, we can observe occlusion of cortical LTP.
As we reviewed above, the cingulate LTP requires activation of NMDARs. Chronic inflammation model mice upregulate the expression of NMDARs GluN2 and enhance the function of GluN2 receptors mediated currents [41]. Furthermore, bilateral microinjections of an NMDAR antagonist into the ACC decreased chronic inflammatory pain behavior [41]. In the IC, nerve injury model increase amount of synaptic NMDARs [19]. Local injections of an NMDAR antagonist or an GluN2B antagonist statistically reduced mechanical allodynia [19]. AMPARs in addition to NMDARs are also associated with long-term plasticity and pain behaviors in these regions [3]. In the ACC, nerve injury enhanced phosphorylation and redistribution of GluA1 [42]. Bilateral microinjections of AMPA/KA antagonist into the same area alleviated mechanical allodynia [42]. Moreover, layer V neurons projecting from the ACC to spinal cord are potentiated by nerve injury through GluA1 accumulation, which was demonstrated by an electron microscopic study [43, 44]. Finally, we microinjected a Ca2+-permeable AMPA receptors antagonist, NASPM into the ACC, which elevated mechanical threshold in the nerve injury model [43, 44]. Similar to the ACC, GluA1 phosphorylation in the IC contributes to nerve injury model [26]. Microinjections of an AMPA/KA antagonist or GluA1-containing AMPAR antagonist into the IC also increase mechanical withdrawal threshold induced by nerve injury model, suggesting that GluA1-containing AMPARs in the ACC and the IC are critical for nociceptive behaviors.
Targeting AC1 which is associated with cortical LTP is a potential candidate for pain therapy for three major reasons [24, 33, 34]. Firstly, AC1 is stimulated in a Ca2+-CaM-dependent manner, downstream from NMDARs. Secondly, AC1 is involved in chronic pain-related neuronal plasticity in the ACC and spinal cord. Finally, AC1-KO mice reduce or block behavioral sensitization to non-noxious mechanical stimuli in animal models of chronic pain including neuropathic and inflammatory pain [34]. NB001 has been identified as a selective AC1 inhibitor [33]. NB001 dose-dependently suppresses cAMP production triggered in ACC slices, suggesting its effectiveness in inhibiting activity-dependent cAMP under physiological or pathological conditions in adult rodents. In adult mice ACC slices, postsynaptic application of the inhibitor significantly reduced the induction of LTP in ACC pyramidal neurons [33]. NB001 also inhibited a pairing protocol induced LTP in the spinal cord dorsal horn. These results demonstrate the critical role of AC1 in both spinal and ACC LTP. This cellular mechanism involving AC1 likely contributes to neuropathic pain behavior. We have previously shown that AC1-KO mice had significantly reduced behavioral allodynia in neuropathic pain and inflammatory pain of animal models [34]. In support of these genetic studies, administration of NB001 (intraperitoneal) significantly caused analgesic effects in CFA induced chronic inflammatory pain model [33].
A downstream signaling protein from AC1, protein kinase Mζ (PKMζ), is essential for maintaining pain-related LTP [24]. This atypical isoform of PKC, has been identified in many brain regions, such as the frontal cortex [45]. Specifically in the ACC of nerve injured mice, the activity of PKMζ was upregulated, enhancing either protein phosphorylation or synthesis [24]. The activity of AC1 is critical in upregulation of PKM since AC1-KO mice did not display significant changes in neither the levels nor the phosphorylation of PKMζ (p-PKMζ). A possible function of PKMζ is to maintain ACC late phase LTP (L-LTP), which was erased by a selective PKMζ inhibitor, ζ inhibitory peptide (ZIP) [24]. Since PKMζ plays a critical role in cognitive learning and memory, it is unlikely a drug target for controlling chronic pain.
Cortical LTD and chronic pain
LTD mechanisms, like LTP, have been well-characterized in subcortical areas, especially in the hippocampus [17, 46]. At least two major forms of LTD in the hippocampus have been investigated by using various recordings and protocols [17]: NMDA-dependent LTD and mGluR-dependent LTD.
Cortical LTD
Similar to the hippocampus, two LTD types - NMDAR- and mGluR-dependent LTD - have been reported in cortical areas including the ACC and IC [20, 22, 23, 47]. Using whole-cell patch-clamp recording, NMDAR-dependent LTD can be observed. The presynaptic stimulation paired with postsynaptic depolarization (1 Hz, 300 pulses, holding membrane potential at -45 mV) produces NMDA receptor-dependent LTD in layer II/III pyramidal neurons [20, 38]. The activations of GluN2A and GluN2B subunits is required for this NMDA-dependent LTD, followed by postsynaptic Ca2+ influx [38]. Furthermore, expression of cingulate LTD is crucial to AMPA GluA2 subunit not GluA3 as well as the interaction between the GluA2/3 C-terminal and its PDZ binding partners [38]. For instance, specific GluA2/3-PICKI and GluA2/3-GRIP/ABP interactions require the LTD in the ACC [20, 38].
Cortical LTD and Chronic Pain
LTD is associated with learning and memory, and pain [3, 17]. Interestingly, rodents undergoing chronic pain models such as peripheral injury have altered or reduced LTD in both the ACC and IC. In the digit amputation model, both NMDAR- and mGluR-dependent LTD forms were either modified or blocked in the ACC and the IC (Fig. 1) [22, 23, 47]. Specifically in the ACC of bone cancer pain model mice, NMDA receptor-dependent LTD was blocked [48]. Digit amputation in mice also diminished mGluR-dependent LTD after two weeks (Fig. 1) [22, 23]. Similar to the ACC, this amputation model reduced LTD in the IC induced by low frequency stimulation [25]. Chronic pain model mice enhance the expression and functions of NMDARs in the cortex [19, 41]. Such postsynaptic excitability may cause loss function of LTD. All these findings provide a possible dis-inhibitory mechanism for chronic pain condition in cortical area [3]. In other words, loss function of undergoing LTD might allow to maintain excitation for an extended period of time in cortical synapses.
Fig. (1).
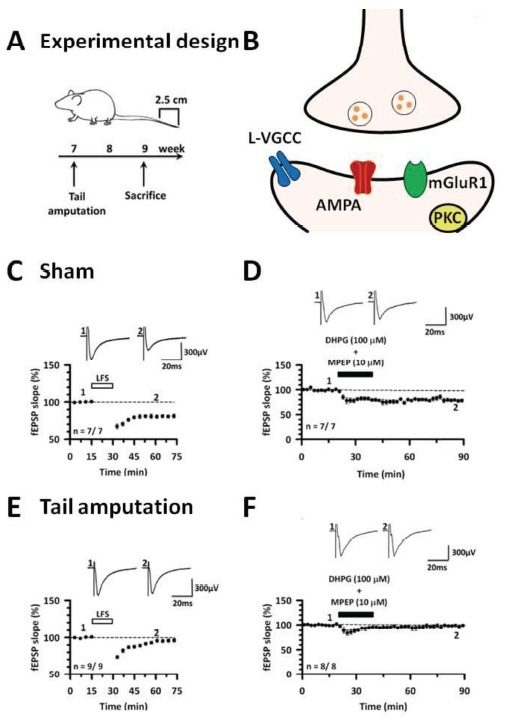
The cingulate LTD and occlusion by tail amputation. (A) Experimental design of cingulate LTD and tail amputation. (B) The mechanisms of cingulate LTD using fEPSP recording. (C) Low-frequency stimulation (1 Hz for 15 min) produces LTD in the ACC. (D) The activation of mGluR1 by a group I mGluR agonist DHPG (100 μM) together with a mGluR5 antagonist MPEP (10 μM) show cingulate LTD in sham mice. (E) The 2 weeks of tail amputation model mice show impaired cingulate LTD. (F) Twenty minute after DHPG with MPEP show no chemical induced LTD in the ACC of the amputation model mice. Modified from [22].
MGLURS mediated LTP in the ACC and IC
Major forms of LTP investigated in the ACC and IC are NMDA receptor dependent [3, 18, 19]. Zhao et al. has previously reported that cortical LTP induced by three major protocols (pairing protocol, spike timing and TBS) is blocked by AP-5, an NMDA receptor antagonist in the ACC [18]. Whole-cell patch-clamp recordings reveal that in layer II/III pyramidal neurons of the IC, pairing protocol induced LTP is also blocked by AP-5 [19]. Recently, we reported a form of cortical presynaptic LTP (pre-LTP) in the ACC [49, 50]. Using whole-cell patch-clamp recordings, this cortical pre-LTP does not require mGluRs because a broad mGluRs antagonist (MCPG, 500 μM) did not show any effect on the cortical pre-LTP [49]. Using field EPSP analysis, pre-LTP was elicited through combining 2 Hz low frequency stimulation (for 2 min) and a GluK1 agonist (ATPA, 1 μM) [50]. This pre-LTP requires mGluRs because a broad mGluRs antagonist reduced the pre-LTP in the ACC [50]. Future studies are clearly necessary to fully investigate this mGluR-dependent LTP.
MGLURS LTD in the ACC and IC
In addition to NMDA-dependent cortical LTD, the mGluR-dependent LTD is observed in both the ACC and IC [22, 23, 25, 47]. In the ACC, a low frequency stimulation (1 Hz for 15 min) produced LTD by field EPSP recordings [22, 23]. The stimulation induced LTD was subsequently inhibited by a non-selective mGluRs antagonist MCPG, but not by a NMDA antagonist AP-5 [22, 23]. Since mGluR1 antagonist LY367385, not a mGluR5 antagonist, reduces the cortical LTD, mGluR1 is specific to the LTD mechanisms that L-VGCC is involved in mGluR-related LTD (Fig. 1) [22, 23]. Additionally, the mGluR1 dependent LTD requires PKC, not the PKA or CaMKII signaling pathways [22]. Recently, we employed multi-electrode array in order to examine the mapping of neural circuits of LTD in the ACC [22]. The mGluR-dependent LTD decreases the numbers of LTD showing channels. Similar to the cingulate LTD, low frequency stimulation also produces LTD during multi-electrode array recordings from adult mice the IC [25, 47]. The insular LTD requires both NMDARs and mGluRs which is a different subtype of group I mGluRs may be involved in insular LTD (Fig. 1) [25, 47].
MGLURS mediated metaplasticity
Metaplasticity is a term referred to the plasticity of plasticity itself [51]. A unique feature of group I mGluRs is their roles in metaplasticity, where their activity can effect subsequent synaptic plasticity [52]. In the hippocampus, it is known that activations of group I mGluRs by DHPG have priming effects on synaptic plasticity, especially LTP. Our group tested to see whether priming can influence the ability to induce LTD in the ACC and IC [22, 39]. A mouse model of chronic pain induces metaplasticity that mGluR-dependent LTD is altered by digit amputation rodents in the ACC as well as the IC [22, 23, 25]. In the ACC from the digit amputation model mice, impairment of ACC LTD could be rescued by priming with the selective activation of mGluR1. Similar to ACC synapses, prior activation of group I mGluRs can produce a form of metaplasticity that restores the low frequency stimulation-evoked LTD in the IC from tail-amputated mice (Fig. 2) [25].
Fig. (2).
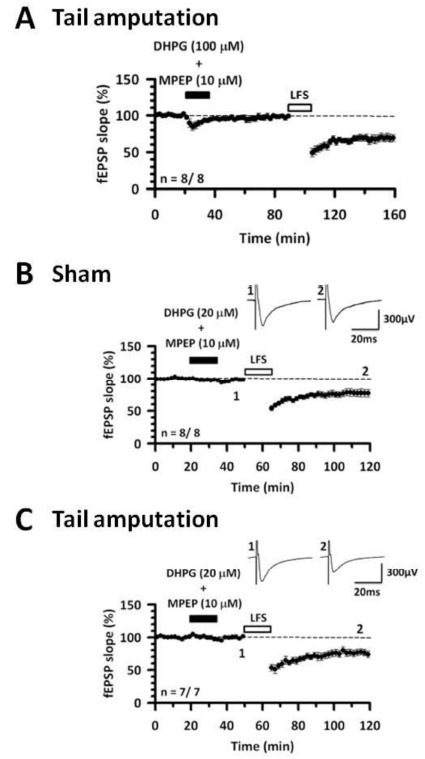
Recovery of the cingulate LTD in tail amputation model mice by priming. (A) High concentration of DHPG (100 μM) together with MPEP (10 μM) were applied for 20 min in the tail amputation model mice. After washout for 50 min, low-frequency stimulation (1 Hz for 15 min) shows cingulate LTD. (B) DHPG (20 μM) together with MPEP (10 μM) were applied for 15 min in sham group. After washout for 15 min, the low-frequency stimulation produces LTD. (C) In tail amputation model mice, DHPG with MPEP were given for 15 min. After washout for 15 min, the stimulation induces cingulate LTD. Modified from [29].
Functional roles of MGLURS in nociceptive behaviors and pain-related synaptic changes
Different mGluRs are expressed in the periphery and the CNS. Most mGluR subtypes are widely distributed in pain-related brain areas, and are related to the induction, expression and maintenance of nociception and chronic pain [10, 11, 53-55]. Group I, II and III are classified into Gq, Gi/o and Gi/o proteins, respectively. Here, we summarize the functional roles of mGluRs in pain-related areas including cortex, subcortex, brainstem, spinal cord and the periphery for various behavioral tests in acute nociceptive and chronic pain model animals. In most pharmacological studies about mGluRs on nociceptive behaviors in acute or chronic pain model, selective or non-selective mGluR agonists and antagonists were mainly applied. Local injections of a mGluR agonist or antagonist show the effects in an area-specific manner. In general, activations of group II and III mGluRs or blocking of group I mGluRs produces analgesic and/or anti-allodynic effects.
Cortical mGluR
mGluRs are expressed in the whole brain, playing important roles in synaptic transmission. A study conducted by Shigemoto and Mizuno reported the distributions of mGluRs in neocortical area [11]. In the neocortex, mGluR1, 2, 3, 5 and 7a are found in layer I and II. Layer III and IV have mGluR2, 3 and 5. The mGluR2, 3, 5 and 7a are distributed in layer V, while layer VI has mGluR2 and 3 subtypes. In particular, the ACC mainly has mGluR2, 3, 5, 7a isoforms [10, 11]. In the IC, however, the distributions and types of mGluRs are unclear.
Only a few studies have been conducted which focus on the roles of mGluRs in nociception and chronic pain within the cortex. It is still unclear as to how mGluRs are involved in basal transmission and long-term synaptic plasticity. As far as we know, only one report looks at their pain-related functions in the cortex. Calejesan et al. reported that a microinjection of a non-selective mGluR agonist into the ACC enhanced the tail flick reflex due to heat stimuli [56]. This study suggests that mGluRs in the cortex contributes to nociceptive behaviors. Further studies are needed to understand the functional roles of mGluRs in the ACC and IC.
mGluR Function in Pain-related Synaptic Changes
In contrast to the association of long-term plasticity in pain, there are few studies about cortical mGluR functions in pain-related changes. Although the possible synaptic mechanisms of mGluRs on basal synaptic transmission are still unclear in pain-related changes, an in vivo electro- physiological study suggests the functional roles of mGluRs in pain-related changes. In vivo unit recording shows that arthritis pain model deactivates the neural activity in mPFC [57]. The pain-related inhibition in mPFC is mediated by the activation of γ-aminobutyric acid (GABA) ergic neurons because pharmacological inhibition of GABAA receptors increases the suppressed neural activity. In normal condition, mGluRs are not involved in the neural activity of mPFC neurons. However, blocking mGluR1 in the mPFC increases the suppressed responses in arthritis pain model. Therefore, it is likely that mGluR1 activates GABAergic neurons in mPFC of arthritis pain model.
ACKNOWLEDGEMENTS
This work was supported by grants from the EJLB-CIHR Michael Smith Chair in Neurosciences and Mental Health, Canada Research Chair, Canadian Institute for Health Research operating Grants (MOP-124807), NSERC Discovery Grant (RGPIN 402555) and the Azrieli Neurodevelopmental Research Program and Brain Canada. K.K. was supported by the postdoctoral fellowship from Fragile X research foundation of Canada.
LIST OF ABBREVIATIONS
- AC1
adenylyl cyclase 1
- ACC
anterior cingulate cortex
- AMPARs
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors
- cAMP
Adenosine-3′,5′-cyclic monophosphate
- Ca2+
calcium
- CaM
calmodulin
- CNS
central nervous system
- EPSCs
excitatory postsynaptic currents
- EPSP
excitatory postsynaptic potential
- FMRP
fragile X mental retardation protein
- GPCRs
G-protein coupled receptors
- GABA
γ-aminobutyric acid
- IC
insular cortex
- KARs
kainate receptors
- KO
knockout
- LTD
long term depression
- LTP
long term potentiation
- L-VGCC
L-type voltage gated calcium channel
- mGluRs
metabotropic glutamate receptors
- NMDA
N-methyl-D-aspartate receptors
- mPFC
medial prefrontal cortex
- PKA
protein kinase A
- PKC
protein kinase C
- PKMζ
protein kinase Mζ
- PNS
peripheral nerves system
- Pre-LTP
presynaptic LTP
- Post-LTP
postsynaptic LTP
- TBS
theta burst stimulation
CONFLICT OF INTEREST
The authors declare that they have no competing interests.
REFERENCES
- 1.Apkarian A.V., Bushnell M.C., Treede R.D., Zubieta J.K. Human brain mechanisms of pain perception and regulation in health and disease. Eur. J. Pain. 2005;9(4):463–484. doi: 10.1016/j.ejpain.2004.11.001. [DOI] [PubMed] [Google Scholar]
- 2.Bushnell M.C., Ceko M., Low L.A. Cognitive and emotional control of pain and its disruption in chronic pain. Nat. Rev. Neurosci. 2013;14(7):502–511. doi: 10.1038/nrn3516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhuo M. Cortical excitation and chronic pain. Trends Neurosci. 2008;31(4):199–207. doi: 10.1016/j.tins.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 4.Vogt B.A. Pain and emotion interactions in subregions of the cingulate gyrus. Nat. Rev. Neurosci. 2005;6:533–544. doi: 10.1038/nrn1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Craig A.D. How do you feel--now? The anterior insula and human awareness. Nat. Rev. Neurosci. 2009;10(1):59–70. doi: 10.1038/nrn2555. [DOI] [PubMed] [Google Scholar]
- 6.Koga K., Li X., Chen T., Steenland H., Descalzi G., Zhuo M. In vivo whole-cell patch-clamp recording of sensory synaptic responses of cingulate pyramidal neurons to noxious mechanical stimuli in adult mice. Mol. Pain. 2010;6(62):1–10. doi: 10.1186/1744-8069-6-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wei F., Zhuo M. Potentiation of sensory responses in the anterior cingulate cortex following digit amputation in the anaesthetised rat. J. Physiol. 2001;532(3):823–833. doi: 10.1111/j.1469-7793.2001.0823e.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu M.F., Pang Z.P., Zhuo M., Xu Z.C. Prolonged membrane potential depolarization in cingulate pyramidal cells after digit amputation in adult rats. Mol. Pain. 2005;1(23):1–5. doi: 10.1186/1744-8069-1-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Anggono V., Huganir R.L. Regulation of AMPA receptor trafficking and synaptic plasticity. Curr. Opin. Neurobiol. 2012;22(3):461–469. doi: 10.1016/j.conb.2011.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ferraguti F., Shigemoto R. Metabotropic glutamate receptors. Cell Tissue Res. 2006;326(2):483–504. doi: 10.1007/s00441-006-0266-5. [DOI] [PubMed] [Google Scholar]
- 11.Shigemoto R., Mizuno N. Chapter III Metabotropic glutamate receptors — immunocytochemical and in situ hybridization analyses; Handbook of Chemical Neuroanatomy. Elsevier B.V: Amsterdam. 2000;18:63–98. [Google Scholar]
- 12.Bliss T.V., Collingridge G.L. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361(6407):31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- 13.Nicoll R.A., Schmitz D. Synaptic plasticity at hippocampal mossy fibre synapses. Nat. Rev. Neurosci. 2005;6(11):863–876. doi: 10.1038/nrn1786. [DOI] [PubMed] [Google Scholar]
- 14.Kandel E.R., Kandel E.R. The molecular biology of memory: cAMP, PKA, CRE, CREB-1, CREB-2, and CPEB. Mol. Brain. 2012;5(14):1–12. doi: 10.1186/1756-6606-5-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhuo M. Long-term potentiation in the anterior cingulate cortex and chronic pain. Philos. Trans.R. Soc. Lond. B. Biol. Sci. 2013 doi: 10.1098/rstb.2013.0146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bliss T.V., Collingridge G.L. Expression of NMDA receptor-dependent LTP in the hippocampus: bridging the divide. Mol. Brain. 2013;6(5):1–14. doi: 10.1186/1756-6606-6-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Collingridge G.L., Peineau S., Howland J.G., Wang Y.T. Long-term depression in the CNS. Nat. Rev. Neurosci. 2010;11(7):459–473. doi: 10.1038/nrn2867. [DOI] [PubMed] [Google Scholar]
- 18.Zhao M.G., Toyoda H., Lee Y., Wu L., Ko S., Zhang X., Jia Y., Shum F., Xu H., Li B.M., Kaang B.K., Zhuo M. Roles of NMDA NR2B subtype receptor in prefrontal long-term potentiation and contextual fear memory. Neuron. 2005;47(6):859–872. doi: 10.1016/j.neuron.2005.08.014. [DOI] [PubMed] [Google Scholar]
- 19.Qiu S., Chen T., Koga K., Guo Y.Y., Xu H., Song Q., Wang J.J., Descalzi G., Kaang B.K., Luo J.H., Zhuo M., Zhao M.G. An increase in synaptic NMDA receptors in the insular cortex contributes to neuropathic pain. Sci. Signal. 2013;6(275):1–11. doi: 10.1126/scisignal.2003778. [DOI] [PubMed] [Google Scholar]
- 20.Toyoda H., Zhao M.G., Zhuo M. Roles of NMDA receptor NR2A and NR2B subtypes for long-term depression in the anterior cingulate cortex. Eur. J. Neurosci. 2005;22(2):485–494. doi: 10.1111/j.1460-9568.2005.04236.x. [DOI] [PubMed] [Google Scholar]
- 21.Zhao M.G., Ko S.W., Wu L.J., Toyoda H., Xu H., Quan J., Li J., Jia Y., Ren M., Xu Z.C., Zhuo M. Enhanced presynaptic neurotransmitter release in the anterior cingulate cortex of mice with chronic pain. J. Neurosci. 2006;26:8923–8930. doi: 10.1523/JNEUROSCI.2103-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kang S.J., Liu M.G., Chen T., Ko H.G., Baek G.C., Lee H.R., Lee K., Collingridge G.L., Kaang B.K., Zhuo M. Plasticity of metabotropic glutamate receptor-dependent long-term depression in the anterior cingulate cortex after amputation. J. Neurosci. 2012;32(33):11318–11329. doi: 10.1523/JNEUROSCI.0146-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wei F., Li P., Zhuo M. Loss of synaptic depression in mammalian anterior cingulate cortex after amputation. J. Neurosci. 1999;19:9346–9354. doi: 10.1523/JNEUROSCI.19-21-09346.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li X.Y., Ko H.G., Chen T., Descalzi G., Koga K., Wang H., Kim S.S., Shang Y., Kwak C., Park S.W., Shim J., Lee K., Collingridge G.L., Kaang B.K., Zhuo M. Alleviating neuropathic pain hypersensitivity by inhibiting PKMzeta in the anterior cingulate cortex. Science. 2010;330(6009):1400–1404. doi: 10.1126/science.1191792. [DOI] [PubMed] [Google Scholar]
- 25.Liu M.G., Zhuo M. Loss of long-term depression in the insular cortex after tail amputation in adult mice. Mol. Pain. 2014;10(1):1–14. doi: 10.1186/1744-8069-10-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Qiu S., Zhang M., Liu Y., Guo Y., Zhao H., Song Q. ; Zhao M., Huganir R.L., Luo J., Xu H., Zhuo M. GluA1 Phosphorylation Contributes to Postsynaptic Amplification of Neuropathic Pain in the Insular Cortex. J. Neurosci. 2014;34(40):13505–13515. doi: 10.1523/JNEUROSCI.1431-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Singer T., Seymour B., O'Doherty J., Kaube H., Dolan R.J., Frith C.D. Empathy for pain involves the affective but not sensory components of pain. Science. 2004;303(5661):1157–1162. doi: 10.1126/science.1093535. [DOI] [PubMed] [Google Scholar]
- 28.Li X.Y., Wang N., Wang Y.J., Zuo Z.X., Koga K., Luo F., Zhuo M. Long-term temporal imprecision of information coding in the anterior cingulate cortex of mice with peripheral inflammation or nerve injury. J. Neurosci. 2014;34(32):10675–10687. doi: 10.1523/JNEUROSCI.5166-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Toyoda H., Zhao M.G., Ulzhöfer B., Wu L.J.H., X., Seeburg P.H., Sprengel R., Kuner R., Zhuo M. Roles of the AMPA receptor subunit GluA1 but not GluA2 in synaptic potentiation and activation of ERK in the anterior cingulate cortex. Mol. Pain. 2009;5(46):1–15. doi: 10.1186/1744-8069-5-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen T., Lu J.S., Song Q., Liu M.G., Koga K., Descalzi G., Li Y.Q., Zhuo M. Pharmacological rescue of cortical synaptic and network potentiation in a mouse model for Fragile X syndrome. Neuropsychopharmacology. 2014;39(8):1955–1967. doi: 10.1038/npp.2014.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liauw J., Wu L.J., Zhuo M. Calcium-stimulated adenylyl cyclases required for long-term potentiation in the anterior cingulate cortex. J. Neurophysiol. 2005;94(1):878–882. doi: 10.1152/jn.01205.2004. [DOI] [PubMed] [Google Scholar]
- 32.Wu L.J., Xu H., Ren M., Cao X., Zhuo M. Pharmacological isolation of postsynaptic currents mediated by NR2A- and NR2B-containing NMDA receptors in the anterior cingulate cortex. Mol. Pain. 2007;3(11):1–6. doi: 10.1186/1744-8069-3-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang H., Xu H., Wu L.J., Kim S.S., Chen T., Koga K., Descalzi G., Gong B., Vadakkan K.I., Zhang X., Kaang B.K., Zhuo M. Identification of an adenylyl cyclase inhibitor for treating neuropathic and inflammatory pain. Sci. Transl. Med. 2011;3(65):1–13. doi: 10.1126/scitranslmed.3001269. [DOI] [PubMed] [Google Scholar]
- 34.Wei F., Qiu C.S., Kim S.J., Muglia L., Maas J.W., Pineda V.V., Xu H.M., Chen Z.F., Storm D.R., Muglia L.J., Zhuo M. Genetic elimination of behavioral sensitization in mice lacking calmodulin-stimulated adenylyl cyclases. Neuron. 2002;36(4):713–726. doi: 10.1016/S0896-6273(02)01019-X. [DOI] [PubMed] [Google Scholar]
- 35.Hayashi Y., Shi S.H., Esteban J.A., Piccini A., Poncer J.C., Malinow R. Driving AMPA receptors into synapses by LTP and CaMKII: requirement for GluR1 and PDZ domain interaction. Science. 2000;287(5461):2262–2267. doi: 10.1126/science.287.5461.2262. [DOI] [PubMed] [Google Scholar]
- 36.Passafaro M., Piëch V., Sheng M. Subunit-specific temporal and spatial patterns of AMPA receptor exocytosis in hippocampal neurons. Nat. Neurosci. 2001;4(9):917–926. doi: 10.1038/nn0901-917. [DOI] [PubMed] [Google Scholar]
- 37.Toyoda H., Wu L.J., Zhao M.G., Xu H., Zhuo M. Time-dependent postsynaptic AMPA GluR1 receptor recruitment in the cingulate synaptic potentiation. Dev. Neurobiol. 2007;67(4):498–509. doi: 10.1002/dneu.20380. [DOI] [PubMed] [Google Scholar]
- 38.Toyoda H., Wu L.J., Zhao M.G., Xu H., Jia Z., Zhuo M. Long-term depression requires postsynaptic AMPA GluR2 receptor in adult mouse cingulate cortex. J. Cell. Physiol. 2007;211(2):336–343. doi: 10.1002/jcp.20940. [DOI] [PubMed] [Google Scholar]
- 39.Liu M.G., Kang S.J., Shi T.Y., Koga K., Zhang M.M., Collingridge G.L., Kaang B.K., Zhuo M. Long-term potentiation of synaptic transmission in the adult mouse insular cortex: multi-electrode array recordings. J. Neurophysiol. 2013;110(2):505–521. doi: 10.1152/jn.01104.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu M.G., Zhuo M. No requirement of TRPV1 in long-term potentiation or long-term depression in the anterior cingulate cortex. Mol. Brain. 2014;7(27):1–13. doi: 10.1186/1756-6606-7-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wu L.J., Toyoda H., Zhao M.G., Lee Y.S., Tang J., Ko S.W., Jia Y.H., Shum F.W., Zerbinatti C.V., Bu G., Wei F., Xu T.L., Muglia L.J., Chen Z.F., Auberson Y.P., Kaang B.K., Zhuo M. Upregulation of forebrain NMDA NR2B receptors contributes to behavioral sensitization after inflammation. J. Neurosci. 2005;25:11107–11116. doi: 10.1523/JNEUROSCI.1678-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xu H.M., Wu L.J., Wang H., Zhang X., Vadakkan K.I., Kim S.S., Steenland H.W., Zhuo M. Presynaptic and postsynaptic amplifications of neuropathic pain in the anterior cingulate cortex. J. Neurosci. 2008;28(29):7445–7453. doi: 10.1523/JNEUROSCI.1812-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen T., Koga K., Descalzi G., Qiu S., Wang J., Zhang L.S., Zhang Z.J., He X.B., Qin X., Xu F.Q., Hu J., Wei F., Huganir R.L., Li Y.Q., Zhuo M. Postsynaptic potentiation of corticospinal projecting neurons in the anterior cingulate cortex after nerve injury. Mol. Pain. 2014;10(33):1–16. doi: 10.1186/1744-8069-10-S1-P1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen T., Wang W., Dong Y.L., Zhang M.M., Wang J., Koga K., Liao Y.H., Li J.L., Budisantoso T., Shigemoto R., Itakura M., Huganir R.L., Li Y.Q., Zhuo M. Postsynaptic insertion of AMPA receptor onto cortical pyramidal neurons in the anterior cingulate cortex after peripheral nerve injury. Mol. Brain. 2014;7(76):1–12. doi: 10.1186/s13041-014-0076-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Naik M.U., Benedikz E., Hernandez I., Libien J., Hrabe J., Valsamis M., Dow-Edwards D., Osman M., Sacktor T.C. Distribution of protein kinase Mzeta and the complete protein kinase C isoform family in rat brain. 2000 doi: 10.1002/1096-9861(20001016)426:2<243::aid-cne6>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 46.Malenka R.C., Bear M.F. LTP and LTD: an embarrassment of riches. Neuron. 2004;44(1):5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- 47.Liu M.G., Koga K., Guo Y.Y., Kang S.J., Collingridge G.L., Kaang B.K., Zhao M.G., Zhuo M. Long-term depression of synaptic transmission in the adult mouse insular cortex in vitro. Eur. J. Neurosci. 2013;38(8):3128–3145. doi: 10.1111/ejn.12330. [DOI] [PubMed] [Google Scholar]
- 48.Chiou C.S., Huang C.C., Liang Y.C., Tsai Y.C., Hsu K.S. Impairment of long-term depression in the anterior cingulate cortex of mice with bone cancer pain. Pain. 2012;153(10):2097–2108. doi: 10.1016/j.pain.2012.06.031. [DOI] [PubMed] [Google Scholar]
- 49.Koga K., Descalzi G., Chen T., Ko H.G., Lu J., Li S., Son J., Kim T.H., Kwak C., Huganir R.L., Zhao M.G., Kaang B.K., Collingridge G.L., Zhuo M. Co-existence of two forms of LTP in ACC provides a synaptic mechanism for the interactions between anxiety and chronic pain. Neuron. 2015;85(2):377–389. doi: 10.1016/j.neuron.2014.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Koga K., Liu M.G., Qiu S., Song Q., O'Den G., Chen T., Zhuo M. Impaired presynaptic long-term potentiation in the anterior cingulate cortex of Fmr1 knockout mice. J. Neurosci. 2015;35(5):2033–2043. doi: 10.1523/JNEUROSCI.2644-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Abraham W.C., Bear M.F. Metaplasticity: the plasticity of synaptic plasticity. Trends Neurosci. 1996;19(4):126–130. doi: 10.1016/S0166-2236(96)80018-X. [DOI] [PubMed] [Google Scholar]
- 52.Abraham W.C. Metaplasticity: tuning synapses and networks for plasticity. Nat. Rev. Neurosci. 2008;9(5):1–13. doi: 10.1038/nrn2356. [DOI] [PubMed] [Google Scholar]
- 53.Chiechio S., Nicoletti F. Metabotropic glutamate receptors and the control of chronic pain. Curr. Opin. Pharmacol. 2012;12(1):28–34. doi: 10.1016/j.coph.2011.10.010. [DOI] [PubMed] [Google Scholar]
- 54.Palazzo E., Marabese I., de Novellis V., Rossi F., Maione S. Supraspinal metabotropic glutamate receptors: a target for pain relief and beyond. Eur. J. Neurosci. 2014;39(3):444–454. doi: 10.1111/ejn.12398. [DOI] [PubMed] [Google Scholar]
- 55.Li W., Neugebauer V. Differential changes of group II and group III mGluR function in central amygdala neurons in a model of arthritic pain. J. Neurophysiol. 2006;96(4):1803–1815. doi: 10.1152/jn.00495.2006. [DOI] [PubMed] [Google Scholar]
- 56.Calejesan A.A., Kim S.J., Zhuo M. Descending facilitatory modulation of a behavioral nociceptive response by stimulation in the adult rat anterior cingulate cortex. Eur. J. Pain. 2000;4(1):83–96. doi: 10.1053/eujp.1999.0158. [DOI] [PubMed] [Google Scholar]
- 57.Ji G., Neugebauer V. Pain-related deactivation of medial prefrontal cortical neurons involves mGluR1 and GABA(A) receptors. J. Neurophysiol. 2011;106(5):2642–2652. doi: 10.1152/jn.00461.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]