

Abstract
Metabotropic glutamate receptors (mGluRs) are important drug targets because of their involvement in several neurological diseases. Among mGluRs, mGlu5 is a particularly high-profile target because its positive or negative allosteric modulation can potentially treat schizophrenia or anxiety and chronic pain, respectively. Here, we computationally and experimentally probe the functional binding of a novel photoswitchable mGlu5 NAM, termed alloswitch-1, which loses its NAM functionality under violet light. We show alloswitch-1 binds deep in the allosteric pocket in a similar fashion to mavoglurant, the co-crystallized NAM in the mGlu5 transmembrane domain crystal structure. Alloswitch-1, like NAM 2-Methyl-6-(phenylethynyl)pyridine (MPEP), is significantly affected by P655M mutation deep in the allosteric pocket, eradicating its functionality. In MD simulations, we show alloswitch-1 and MPEP stabilize the co-crystallized water molecule located at the bottom of the allosteric site that is seemingly characteristic of the inactive receptor state. Furthermore, both NAMs form H-bonds with S809 on helix 7, which may constitute an important stabilizing interaction for NAM-induced mGlu5 inactivation. Alloswitch-1, through isomerization of its amide group from trans to cis is able to form an additional interaction with N747 on helix 5. This may be an important interaction for amide-containing mGlu5 NAMs, helping to stabilize their binding in a potentially unusual cis-amide state. Simulated conformational switching of alloswitch-1 in silico suggests photoisomerization of its azo group from trans to cis may be possible within the allosteric pocket. However, photoexcited alloswitch-1 binds in an unstable fashion, breaking H-bonds with the protein and destabilizing the co-crystallized water molecule. This suggests photoswitching may have destabilizing effects on mGlu5 binding and functionality.
Keywords: Allosteric modulation, docking, metabotropic glutamate receptor, molecular dynamics, mutation, protein structure, transmembrane domain
Introduction
As the most significant excitatory neurotransmitter in the CNS, glutamate is involved in a whole host of diverse neurological disorders such as chronic pain, epilepsy and Alzheimer’s [1-3]. It operates through two distinct kinds of transmembrane receptors: ionotropic cation channels for fast neuronal ionic transmission [4] and metabotropic glutamate (mGlu) G protein-coupled receptors (GPCRs) for sustained but slower modulation of neuronal synaptic activity [5-7]. The mGlu receptor (mGluR) family contains eight member subtypes each consisting of disulphide-linked dimers [7, 8]. Each protomer contains an extracellular Venus flytrap (VFT) domain where the orthosteric (glutamate) binding-site is located and a heptahelical transmembrane (TM) domain where allosteric binding-sites are found. Of all mGluRs, mGlu5 is a particularly attractive target for pharmacological modulation because its stimulation can potentially be a therapy for schizophrenia [9] whilst its inhibition can potentially treat anxiety [10], depression [11] and pain [12-14]. Despite different mGluR VFT domains having been crystallized since 2000 [15-17], the targeting of the orthosteric site of mGluRs with agonists or antagonists that compete with glutamate has yielded mixed results. This is partly because mGluRs contain very similar VFT domains making subtype selectivity a problem [18]. This has spurned the development of mGluR positive and negative allosteric modulators (PAMs and NAMs, respectively), which potentially offer better selectivity by binding in the TM domain where structural differences are more apparent amongst family members [19, 20]. As a further benefit, there is a reduced risk of mGluR over-sensitization as strict allosteric modulators (those with no intrinsic agonist activity) only modulate natural responses to glutamate and do not activate the receptor themselves [18]. This means spatial and temporal effects of glutamate can be maintained, which is a therapeutically desirable feature. However, until very recently, no mGluR TM domain (or even close homologue) had been crystallized, making high-level exploration of the allosteric site of mGlu5 difficult despite various homology models built on distantly related GPCRs [21-23]. However, with the release of the crystal structure of the TM domain of mGlu5 in an inactive state with bound NAM mavoglurant in 2014 [24], as well as the TM domain of mGlu1 [25], much more accurate studies of NAM and PAM binding to mGlu5 have become possible.
Here we take advantage of the mGlu5 TM domain crystal structure (henceforth referred to as mGlu5) to investigate, in silico and in vitro, mGlu5 NAM binding, functionality and potential mechanisms of receptor recognition with regards to negative allosteric action. For this we compare two mGlu5 NAMs with respect to co-crystallized mavoglurant [24]: the well-characterized 2-Methyl-6-(phenylethynyl)pyridine (MPEP) [26-28] and a novel photoswitchable compound called alloswitch-1, which has recently been developed [29] (see Fig. 1). Alloswitch-1 is similar to other mGlu5 NAMs in that it contains three aromatic rings connected by two spacer groups [21, 23, 30]. The first of these spacers is a rigid trans-azo group, which is similar in length to a rigid ethynyl group, previously identified as an important structural feature of several mGlu5 NAMs [31, 30] and also present in mavoglurant [24]. However, the azo group has the added capability of photoswitching under violet light into a cis-azo configuration and back to a normal trans-azo state under green light or thermally in the dark [29] (Fig.1). Experiments have shown that alloswitch-1 acts as a potent NAM in dark or under green light but loses its NAM potency under violet light when photoisomerized [29]. It is currently unknown whether this photo-induced loss of NAM functionality is due to a loss of ligand binding to mGlu5 or a more subtle effect such as a change in binding mode or a ligand-induced effect on receptor conformation. Indeed, it is also unknown whether photoswitching of alloswitch-1 can occur inside the allosteric site of mGlu5 whilst the ligand is bound or whether it only occurs in the solvent in an unbound state.
Fig. (1).
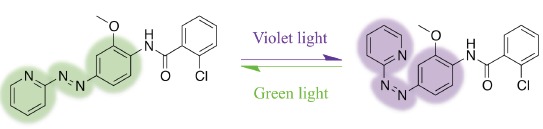
The chemical structure of alloswitch-1 and conversion to cis-azo during photoswitching with violet light (380-390 nm) and back to ground trans-azo state by either relaxation in dark or photoinduced with green light (490-500 nm).
Here, we attempt to answer some of these questions from a theoretical point of view by employing computational docking and molecular dynamics (MD) simulations. Furthermore, by combining experimental and computational approaches, we seek to establish the functional NAM binding-mode of the trans-azo conformation of alloswitch-1, as well as that of MPEP, and therefore by extension other similar mGlu5 NAMs. In particular, the second spacer group of alloswitch-1 consists of an amide, which is another common constituent of many mGlu5 NAMs [30] but its functional significance is not quite understood. We explore some theoretical possibilities why the conformational state of this amide group may be significant for the NAM functionality of alloswitch-1 and potentially other mGlu5 NAMs.
Results and Discussion
In order to investigate NAM functionality in mGlu5 in silico, the missing second intracellular and extracellular loops (ICL2 and ECL2) of the mGlu5 crystal structure (PDB id 4OO9) were constructed with MODELLER [32] and unnatural amino-acids in the TM domain converted back to wild-type with CHIMERA [33] (see Methods). As a control for validating a reliable NAM docking protocol for wt mGlu5, we redocked the co-crystallized NAM mavoglurant back into the empty allosteric binding pocket of the TM domain using Autodock-4.2 [34], seeking to reproduce the crystal binding pose. Fig. 2 shows the top-ranked docking solution of mavoglurant in wt mGlu5, with an RMSD of 0.2 Å compared to the crystal state. This best-ranked solution re-establishes all protein-ligand H-bonds and important interactions observed in the original crystal structure. In particular, the carbon-carbon triple bond of mavoglurant traverses a narrow region in the pocket between P655 on TM3 and S809 on TM7, enabling the methylated aromatic ring of mavoglurant to occupy a larger space at the bottom of the allosteric pocket, located between G624 on TM2, Y659 on TM3, A810 on TM7, and a co-crystallized water molecule (which forms a three-way H-bonding network with Y659, T781 and S809). We took this result as a validation of our docking protocol, which we then extended to other NAMs.
Fig. (2).
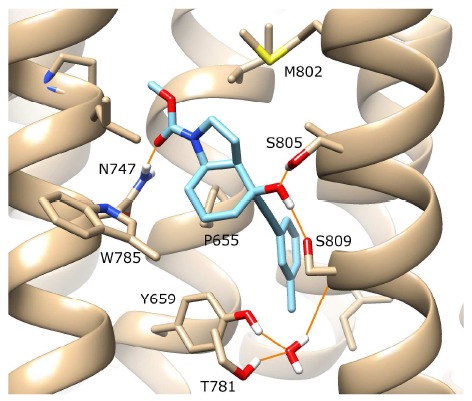
The best-ranked docking solution of mavoglurant (in blue) in the complete wt structure of mGlu5 (in beige). All protein-ligand H-bonds originally identified in the crystal structure (PDB id: 4OO9) were reformed during docking (orange lines, involving residues N747, S805, S809). For visualisation purposes, the backbone of TM6 is not shown, however the sidechains of residues W785 and T781 on TM6 are included (in the foreground), with the latter making an H-bond with a co-crystallized water molecule. Regarding helix position, TM5 is foreground left, TM7 is foreground right. In the background, TM4 is left, TM3 is central and TM2 is right.
MPEP is often considered a standard mGlu5 NAM with which other molecules are compared as it has been well characterized experimentally with important receptor residues identified for binding e.g. W785, F788, S809 and A810 amongst others [21, 28, 35, 36]. Interestingly, like mavoglurant, MPEP contains a methylated aromatic ring. We therefore docked MPEP into mGlu5 with Autodock-4.2 to compare its binding mode with that of mavoglurant. As anticipated, MPEP docks in a very similar binding mode to mavoglurant (in all reported top 10 docking solutions) with its methylated pyridine ring occupying the same position as methylated benzene of mavoglurant (see Fig. 3A for the top-ranked solution). This is in agreement with another recent study where MPEP was docked into the crystal structure of mGlu5 [37]. Furthermore, like mavoglurant, S809 on TM7 is observed to make an H-bond with MPEP. However, unlike mavoglurant, S809 acts as a proton donor to the nucleophilic nitrogen on the pyrindine ring of MPEP. This H-bonding does not affect the rotameric position of the sidechain, which remains unchanged with respect to the crystal structure, but involves a rotation of its hydroxyl group. Other differences compared to mavoglurant include a lack of protein-ligand H-bonds with respect to N747 on TM5 and S805 on TM7. These “missing” interactions are a result of MPEP being shorter in length and not containing some of the chemical groups possessed by mavoglurant. The rest of the receptor, including the co-crystallized water molecule between TM3, TM6 and TM7 (identified by Marshall et al. [24]) remains unchanged with respect to the original crystal structure (except mutated residues reverted back to wt).
Fig. (3).
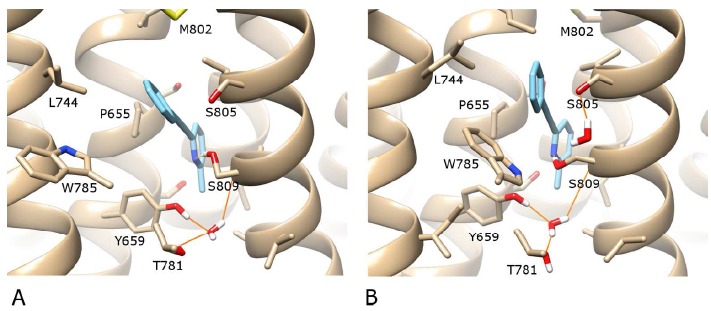
A) The best-ranked docking solution of MPEP (in blue) in the complete wt structure of mGlu5 (in beige). B) After 130 ns of MD simulation. H-bonds are shown as orange lines. A protein-ligand H-bond between MPEP and S809 is formed. For visualisation purposes, the backbone of TM6 is not shown, however the sidechains of residues W785 and T781 on TM6 are included (in the foreground). Regarding helix position, TM5 is foreground left, TM7 is foreground right. In the background, TM3 is centre-left and TM2 is centre-right.
In order to validate the predicted binding-mode of MPEP, we pursued two different exploratory lines: (i) experimentally by making mutations at the top and bottom of the allosteric pocket (see SI Fig. 1) and studying their effect on MPEP functionality, and (ii) computationally with an MD simulation, incorporating wt mGlu5 with bound MPEP in an explicit water/lipid-membrane environment for studying receptor-ligand behaviour and comparison with previously reported mutational data [28, 38]. As previously mentioned, the carbon-carbon triple bond of MPEP is predicted to traverse a narrow region at the bottom of the allosteric pocket between P655 and S809, like mavoglurant. As such, mutating P655 into a bulkier residue was anticipated to adversely affect MPEP binding and disrupt NAM functionality in mGlu5. This residue has previously been mutated to serine, reducing mGlu5 affinty for MPEP but not abolishing its binding or functionality [38]. It was therefore decided to mutate this residue to methionine as this is the equivalent residue in mGlu4, which would likely not compromise mGlu5 activity but be bulky enough to potentially abolish MPEP functionality (assuming the prediction of MPEP binding pose is correct). This hypothesis was tested by making the P655M mutation and determining antagonist activity of MPEP on the effect of orthosteric agonist quisqualate (see Methods). As predicted, the P655M mutation significantly affects MPEP functionality, effectively abolishing its NAM capability whilst also preserving normal receptor activity in response to agonist (see Fig. 4, SI Table 1). Presumably this mutation prevents MPEP from binding in its preferred orientation. In agreement with this hypothesis, re-docking MPEP and mavoglurant into a model of the P655M mutant is unable to replicate either of the predicted binding modes observed in the wt as methionine fills the narrow space where they previously bound (data not shown). These results support the notion that MPEP, like mavoglurant, binds in the lower cavity of the allosteric pocket. An additional Q647A mutation was made at the top of the allosteric site in mGlu5. The rationale for making this mutation is that the equivalent residue in the mGlu1 crystal structure is in close proximity to NAM FITM, which has a higher laying binding pose at the top of the allosteric pocket [25]. However, Q647 lies above mavoglurant in the mGlu5 crystal structure and does not interact with the ligand (SI (5.1MB, pdf) Fig. 1). As MPEP is predicted to follow the same binding pattern as mavoglurant, mutation of Q647 was not anticipated to affect MPEP activity. As expected, this mutation does not affect MPEP and does not affect normal agonist-induced activation of the receptor (Fig. 4, SI Table 1).
Fig. (4).
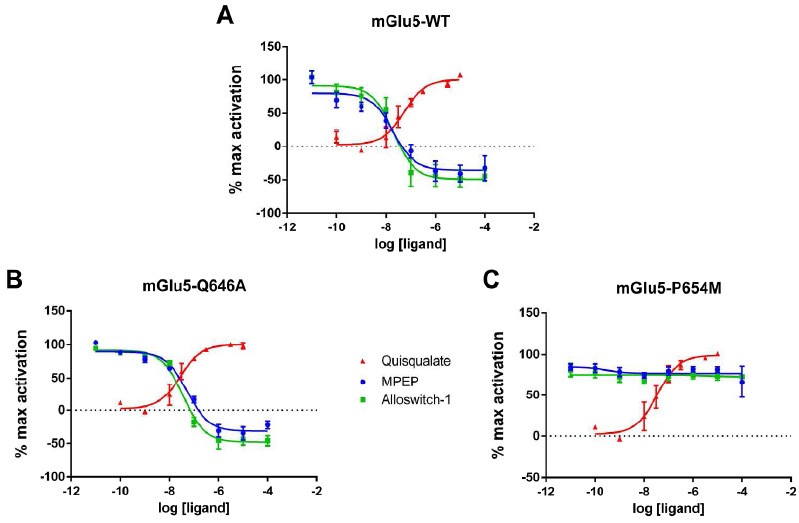
Pharmacological validation of MPEP and alloswitch-1 binding mode. Concentration-response curves of quisqualate (Red), MPEP (Blue) and alloswitch-1 (Green) in (A) wild-type rat mGlu5 receptors, (B) Q646A (equivalent to Q647A in human mGlu5) or (C) P654M (equivalent to P655M in human mGlu5) mutated rat mGlu5 receptors. MPEP and alloswitch-1 activities were evaluated in the presence of 30 nM of quisqualate. HEK293 cells were transiently transfected with the mGlu5 receptor constructs. Receptor activation following stimulation by different concentrations of ligands was determined by IP1 production (see methods). Each data point corresponds to means ± SEM of at least 3 experiments performed in triplicate.
In an MD simulation of docked MPEP in wt mGlu5, the ligand remains bound to the receptor in a stable orientation with little deviation over the course of 130 ns (see Fig. 3B and SI Fig. 2) with the final RMSD of MPEP pyridine ring, 0.5 Å, and the receptor, 2.0 Å. Furthermore, although the predicted H-bond between S809 and MPEP is temporarily broken mid-simulation as the rotameric state of S809 flips (forming an H-bond with the mainchain oxygen atom of S805), it is later recovered and maintained (SI Fig. 3). This suggests an S809-MPEP H-bond contributes to NAM functionality (as supported by previous S809 mutations, which reduce MPEP potency [28]) and also perhaps indicates that S809 can operate as an allosteric toggle switch. In addition, other features of interest in the mGlu5-MPEP simulation include (i) stabilization of the co-crystallized water molecule at the bottom of the allosteric pocket [24], which remains unchanged with respect to the receptor, (ii) introduction of a second water molecule into the allosteric pocket, which H-bonds between S805 and S809, in a similar manner to the hydroxyl group of mavoglurant (Fig. 2 and 3B), (iii) a partial inward movement of W785 sidechain in the allosteric pocket with respect to the outward orientation seen in the mavoglurant-bound crystal structure [24] (Fig. 3B), and (iv) an adjustment in the plane of MPEP that optimises hydrophobic interactions with L744 (TM5), W785, F788 (TM6) and M802 (TM7, see SI Fig. 4). Two of these residues in particular, W785 and F788, have been found to reduce the potency of MPEP when mutated to alanine [28]. Collectively, these features (observations from simulation and reported mutational data) point to a validation of the MPEP docking pose at the bottom of the allosteric pocket.
We next proceeded to dock the active isomer of alloswitch-1 into mGlu5 in its original crystal structure conformation. Alloswitch-1 is an unusual molecule because it contains two spacer groups between three aromatic rings that can theoretically display either cis or trans dispositions (Fig. 1). One of these is a rigid azo group which normally resides in a lower-energy trans-position but can isomerize into a higher-energy cis-state when violet light is applied (forming a non-active isomer) [29]. The other is a partially flexible amide group, which can be promoted from low-energy trans-states to higher-energy cis-states when conditions are favourable, such as those seen in protein folding (predominantly but not exclusively involving prolines) [39] or organic molecules with stabilizing intra-molecular interactions e.g. pentafluorophenyl-containing ligands [40]. Accordingly, but somewhat unexpectedly, when alloswitch-1 in the trans-azo configuration, corresponding to the active form in the dark, is docked into the original crystal structure of mGlu5 with Autodock-4.2 (see Methods), the ligand adopts a cis-amide state in 9 out of 10 docking solutions. As might be expected, in 7 of the best docking solutions (SI Fig. 5), including the top-ranked solution (Fig. 5A), the orientation of the pyridine ring of alloswitch-1 adopts the same orientation as the pyridine ring of MPEP, binding deep in the allosteric site. Furthermore, the azo-group adopts the same orientation as the carbon triple bond in MPEP (and mavoglurant), traversing the narrow gap between P655 on TM3 and S809 on TM7. This binding mode appears to be confirmed by experiment which shows that alloswitch-1, like MPEP, is signficantly affected by the P655M mutation (Fig. 4, SI Table 1). More unusually however, in the higher region of the allosteric pocket, the amide bond prefers a cis conformation facilitating a ligand “L” shape and maximizing favourable interactions with the receptor (Fig. 5A). In this way, interactions with N747 on TM5 can be formed (N747 has been shown to be important for the affinity of amide-containing NAMs, of which alloswitch-1 is an example, but not other non-amide NAMs [41]) as well as avoiding unfavourable interactions with F788 on TM6 (SI (5.1MB, pdf) Fig. 6). As this result is replicated in the majority of docking solutions, it is unlikely to be an artefact. Although it is possible to find a much lower-ranked docking solution where alloswitch-1 adopts a more conventional trans-amide conformation, this has the consequence of forcing the ligand much deeper into the allosteric pocket of mGlu5 than that explored by mavoglurant or MPEP, creating unfavourable interactions with Y659 and the co-crystallized water molecule (SI Fig. 5). This would seem inconsistent with the crystal structure binding-mode of mGlu5 NAMs as well as from a purely energetic point of view. These results suggest a ligand cis-amide is the more likely conformation when the receptor is in its crystal state.
Fig. (5).
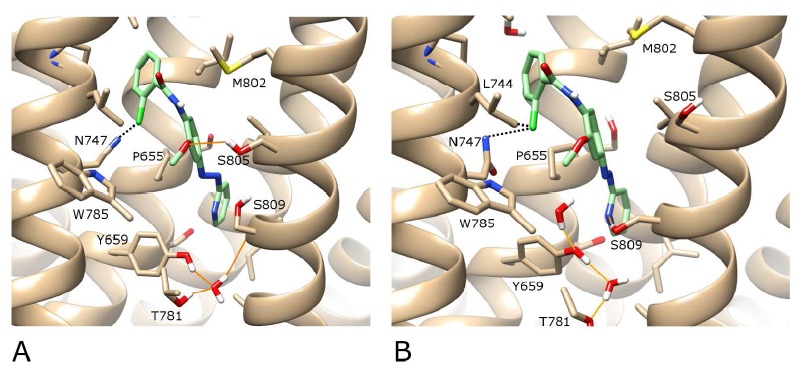
A) The best-ranked docking solution of the active isomer of alloswitch-1 (in green) in the complete wt structure of mGlu5 (in beige) where the ligand adopts a cis-amide conformation. B) After 100 ns of MD simulation “in the dark”. H-bonds are shown as orange lines and halogen interactions as dotted black lines. A protein-ligand H-bond between alloswitch-1 and S809 is formed during MD. For visualisation purposes, the backbone of TM6 is not shown, however the sidechains of residues W785 and T781 on TM6 are included (in the foreground). Regarding helix position, TM5 is foreground left, TM7 is foreground right. In the background, TM3 is centre-left and TM2 is centre-right.
Fig. (6).
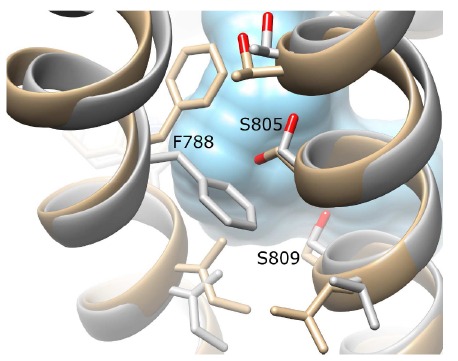
Two alternative conformations of mGlu5: the original crystal structure (PDB id: 4OO9) (beige) and an alternative state with a wider allosteric pocket identified by ROSETTA relaxation (grey). The volume of the allosteric pocket in the original crystal structure is in blue. Different sidechain positions between TM6 (left) and TM7 (right) on the extracellular side enable an enlargement of the allosteric pocket, primarily through concomitant movement of F788 on TM6 and S805 on TM7.
In a similar manner to that performed with MPEP, the top-ranked docking solution of alloswitch-1 (with cis-amide) was subjected to an MD simulation over 100 ns. During the simulation, the ligand remains stably bound to mGlu5 in its cis-amide conformation (final RMSD of ligand pyridine ring compared to docking: 1.0 Å, and receptor RMSD: 2.1 Å, SI Fig. 7). In addition, the methoxy group of alloswitch-1 binds between L744 on TM5 and W785, F788 on TM6. This maintains W785 in an outward orientation as seen in the mavoglurant-bound crystal structure and also means the ligand methoxy group rests partially out of plane with respect to the rest of the molecule (SI Fig. 6). Furthermore, the co-crystallized water molecule at the bottom of the allosteric pocket [24] remains stable over 100 ns (as in the MPEP-containing MD simulation), however a second water molecule enters the allosteric pocket, binding between alloswitch-1, Y659 (TM3) and W785 (TM6). Perhaps most interestingly of all, and despite not being directly predicted by docking, S809 forms a stable H-bond with the pyridine ring of alloswitch-1. This occurs early in the MD simulation as the ligand pyridine ring quickly but subtlely shifts its position in the allosteric pocket (Fig. 5B, SI Fig. 8). This binding-mode closely mimics the predicted binding-mode of MPEP, where an interaction between S809 and ligand pyridine ring has previously been suggested as functionally relevant [28]. An optimized conformation of the active (trans-azo) isomer of alloswitch-1 with cis-amide was also separately generated by density functional theory (DFT) using Gaussian [42] (independently of the receptor, see Methods and SI Fig. 9). This was compared with the top-ranked docking solution of alloswitch-1 in mGlu5 before and after 100 ns of MD. The top docking solution of alloswitch-1 contains an amide dihedral angle of -8.7° and after MD, this shifts to 4.2°. This is in close agreement with the lowest-energy conformation generated by DFT, which contains a dihedral angle of 6.8° (SI (5.1MB, pdf) Fig. 9). This suggests alloswitch-1 is able to adopt a near-optimal cis conformation within the allosteric pocket.
Fig. (7).
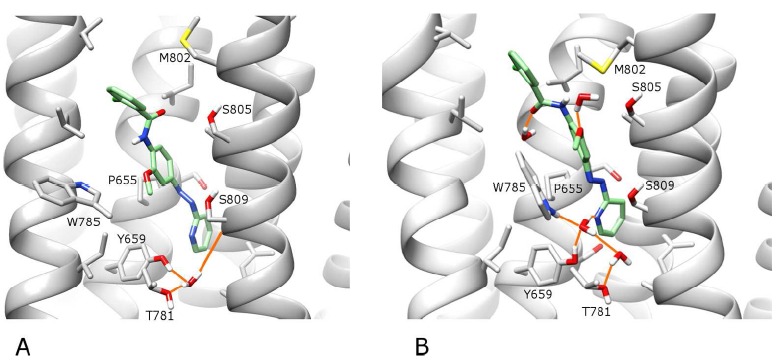
A) The best-ranked docking solution of the active isomer of alloswitch-1 (in green) in an alternative ROSETTA conformation of wt mGlu5 (in grey). B) After 100 ns of MD simulation “in the dark”. H-bonds are shown as orange lines. For visualisation purposes, the backbone of TM6 is not shown, however the sidechains of residues W785 and T781 on TM6 are included (in the foreground). Regarding helix position, TM5 is foreground left, TM7 is foreground right. In the background, TM3 is centre-left and TM2 is centre-right.
Fig. (8).
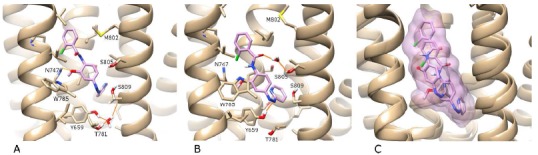
A) The best-ranked docking solution of photoexcited alloswitch-1 (in violet) in mGlu5 (in beige) “in violet light”. B) After 100 ns of MD simulation “in violet light”. C) Pronounced lateral oscillation and rocking of photoexcited alloswitch-1 in the first 10 ns of MD simulation (two individual conformations displayed within a combined volume). H-bonds are shown as orange lines. For visualisation purposes, the backbone of TM6 is not shown, however the sidechains of residues W785 and T781 on TM6 are included (in the foreground). Regarding helix position, TM5 is foreground left, TM7 is foreground right. In the background, TM3 is centre-left and TM2 is centre-right.
Fig. (9).
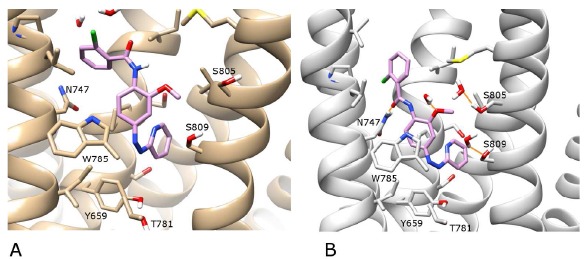
A) The photoexcited conformation of alloswitch-1 (in violet) in mGlu5 (in beige) after 200 ns of MD (0-100 ns in “dark”, 100-200 ns photoexcited in “light”). B) The photoexcited conformation of alloswitch-1 (in violet) in an alternative receptor conformation (in grey) after 200 ns of MD (0-100 ns in “dark”, 100-200 ns photoexcited in “light”). H-bonds are shown as orange lines. For visualisation purposes, the backbone of TM6 is not shown, however the sidechains of residues W785 and T781 on TM6 are included (in the foreground). Regarding helix position, TM5 is foreground left, TM7 is foreground right. In the background, TM3 is centre-left and TM2 is centre-right.
As the proposed binding-mode of alloswitch-1 with a cis-amide is an unusual feature and as GPCRs are dynamic molecules with potentially different signalling conformational states [43], an alternative conformation of mGlu5 was derived from the original crystal structure in order to explore other feasible binding modes of alloswitch-1. This alternative receptor conformation was found with ROSETTA conformational sampling (minus mavoglurant, see Methods) and deemed to be still in an inactive receptor state. Although similar to the crystal structure, it has a wider allosteric pocket (SI Fig. 10) created by different side-chain packing between TM6 and TM7 (Fig. 6). In particular, this involves an outward flipping of F788 sidechain on TM6 and an inward motion of adjacent S805 sidechain on TM7. However, this alternative conformation still preserves the co-crystallized water molecule between Y659, T781 and S809 in the allosteric pocket and other intracellular interactions observed in the original mGlu5 crystal structure [24]. Alloswitch-1 was re-docked into this alternative mGlu5 conformation with all top 10 docking solutions identifying a ligand conformation containing a trans-azo and trans-amide with a high degree of structural overlap (max RMSD range of 2.3 Å; SI Fig. 11). In the top-ranked solution (Fig. 7A), the ligand pyridine ring occupies the bottom of the allosteric pocket as seen previously in the cis-amide conformation, although no specific protein-ligand H-bonds are predicted, nor interaction with N747 on TM5 (predicted docking score remains less than the predicted cis-amide conformation; SI Table 2). In an MD simulation of 100 ns, the trans-amide conformation of alloswitch-1 binds in a stable manner with the bound co-crystal water molecule [24] stabilized as in previous simulations (final RMSD of ligand pyridine ring compared to docking: 1.3 Å, and receptor RMSD: 2.2 Å; SI Fig. 12). However, three additional water molecules enter the allosteric pocket as its volume is higher in this conformation than in the original crystal structure (250 Å3 compared to 213 Å3 as calculated with POVME [44]). In addition, the directionality of the water-mediated H-bonding between TM3 and TM6 is altered (Fig. 7B) as Y659 (TM3) and T781 (TM6) become connected by two water molecules rather than one. Also deserving mention, with alloswitch-1 in a trans-amide conformation, S809 does not make a stable H-bond with the ligand pyridine ring (Figs. 7B and SI 13). This is because the plane of alloswitch-1 is changed and no longer adopts an optimal orientation for H-bond formation. This appears to be because the planar (trans-amide) conformation of alloswitch-1 places different constraints on the pyridine ring at the bottom of the allosteric pocket. Ultimately whether this planarity and lack of protein-ligand H-bonding would affect alloswitch-1 functionality is unknown and open to speculation. However, this conformation seemingly has less in common with the predicted binding-mode of MPEP (and mavoglurant) than that of bound alloswitch-1 with a cis-amide. Therefore it is possible that the trans-amide conformation of alloswitch-1 may not be as functional in terms of negative allosterism, although theoretically free to bind mGlu5 assuming the receptor adopts a suitable conformation.
Fig. (10).
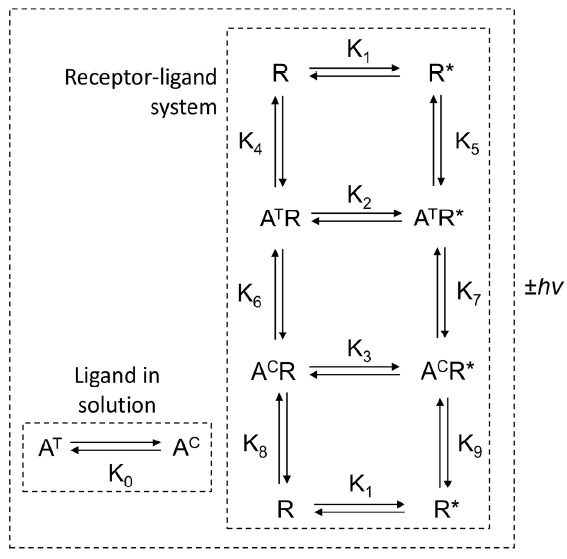
Pathways of alloswitch-1 (A) binding/unbinding to mGlu5 (R). The receptor displays two states: R (inactive) and R* (active). The ligand adopts two photoisomeric states: AT (trans-azo: bioactive state) and AC (cis-azo: inactive photoisomerized state) with addition of light energy (+hv). The mathematical model includes the possibility of photosimorerization from trans- to cis-azo both in solution and in the receptor allosteric pocket. Selection of trans- and cis-azo alloswitch-1 isomers by the inactive and active receptor states are also included in the mathematical formalism (see [47] for discussion on ligand induction and conformational selection functional mechanisms). Obviously, in dark or green light conditions the proportion of cis-azo isomer is negligible both in solution and in the receptor.
In order to further probe the possibility of a cis-amide conformation of alloswitch-1 being the preferred bioactive state, a preliminary analysis of force-field potential energies was made in the respective MD simulations of the cis-amide and trans-amide ligand-mGlu5 complexes (SI Table 3). These analyses show the bound cis-amide state of alloswitch-1 is +3.5 kcal/mol higher than the bound trans-amide state on average (this is in relatively good agreement with high-level DFT calculations outside of the receptor, which show the cis-amide state is +4.4 kcal/mol higher than the trans-amide state; SI Fig. 9). Conversely, protein-ligand interactions with the cis-amide state are -3.6 kcal/mol lower than with the trans-amide state on average (however it should be noted that these are not strict free-energy of binding calculations and may over-emphasise protein-ligand interaction energies). Taken together, these calculations suggest that although the cis-amide state of alloswitch-1 is energetically less favourable, more favourable interactions with the protein may effectively cancel out this penalty. Thus, both amide conformations of alloswitch-1, cis and trans, may be equally feasible in energetic terms inside the allosteric pocket of mGlu5.
As an additional validation along more general lines, the prevalence of cis-amides in crystallized organic compounds was assessed by screening two structural databases: Cambridge Structural Database [45] (CSD; >500,000 small-molecule compounds in isolation) and Protein Data Bank [46] (PDB; 19,209 non-redundant protein-ligand entries). In the PDB, 22 non-cyclic ligands (dihedral angle =0° +/-20°) out of a total of 4,178 amide-containing ligands were identified as having cis-amides (three examples shown in SI Fig. 14). In the CSD, 87 compounds were identified as containing non-cyclic cis-amides amongst a total of 32,806 amide-containing compounds (non-powders, non-solvents, see SI Fig. 15 for examples). This shows that under normal crystallograhic conditions, cis-amides are relatively rare but still possible if selective conditions exist. Furthermore, ligands with cis-amides may be more common when complexed with proteins, which theoretically could help stabilize their relatively higher-energy conformation (~0.5% occurrence in PDB compared to ~0.3% occurrence in CSD). This adds indirect evidence that mGlu5 could theoretically stabilize a cis-amide conformation of alloswitch-1 in the allosteric pocket by providing suitable mitigating interactions, such as an interaction with N747 on TM5, a residue known to influence the functionality of amide-containing NAMs [41].
So far we have only considered the behaviour of the active isomer of alloswitch-1 but this compound also possesses a photoisomerizable azo group (Fig. 1). Under normal physiological conditions this group resides in trans but can switch to cis under violet light. Photoswitching appears to alter the biological activity of alloswitch-1 as its NAM activity is lost under violet light but regained when the light stimulus is removed or green light is applied [29]. However, the structural basis for this physiological effect is not clear and many questions remain unanswered. For example, can the photoisomerized conformation of alloswitch-1 bind mGlu5? Can photoswitching of alloswitch-1 occur inside the allosteric pocket if the ligand is already bound in its NAM state? If so, what happens to the ligand-receptor complex? We set out to answer some of these possibilities by simulating mGlu5 with a bound photoswitched (cis-azo) conformation of alloswitch-1 using two different computational strategies. In the first approach, we determined a likely conformation of photoexcited alloswitch-1 independently of the receptor (see Methods) and then attempted to dock this conformation into mGlu5. We followed this with an MD simulation to assess protein-ligand stability in a continued photoexcited state. This effectively simulates a photoswitching event occurring outside of the pocket followed by binding to the receptor in a photoisomerized state. In a second approach, we set out to simulate potential photoswitching inside the allosteric pocket of mGlu5 with alloswitch-1 already bound in its normal NAM functional state i.e. photoswitching during an ongoing MD simulation with “application of violet light”. For this approach we extended two previous MD simulations of alloswitch-1 bound to mGlu5 in cis-amide and trans-amide conformations and implemented photoswitching of the azo group from trans to cis, followed by 100 ns simulation in the photoexcited state (see Methods for details).
Regarding the first photoswitching approach, an optimized conformation of the photoexcited state of alloswitch-1 was generated by density functional theory (DFT) using Gaussian [42] (independently of the receptor, see Methods and SI Fig. 16). These calculations place the cis-azo dihedral angle at 12.0°, i.e. partially planar. This conformation was docked into the original crystal structure of mGlu5 with Autodock-4.2 (see Methods). Although the docking score of the top-ranked solution is not as favourable as previously docked active (trans-azo) isomers (~7-fold less; SI Table 2), the non-active (cis-azo) photoisomer still docks respectably in the allosteric site, suggesting binding to the receptor is possible. However, all 10 docking solutions are unable to position the ligand pyridine ring at the bottom of the cavity formed between G624 on TM2, Y659 on TM3 and A810 on TM7 (top-ranked solution shown in Fig. 8A). This is in contrast to the pyridine rings of the trans-azo state of alloswitch-1 and MPEP, respectively, which both fill this lower cavity. In addition, the ligand methoxy group of the photoexcited state is swivelled towards TM5 and its amide group is in trans. During a 100 ns MD simulation with the azo group maintained in cis (to simulate a continued photoexcited state), the ligand experiences considerable positional fluctuation, moving vertically and rocking in the allosteric pocket, particularly during the first 10 ns of the simulation (Figs. 8C and SI 17). At the same time, the allosteric pocket experiences structural disruption. In particular, the co-crystallized water molecule [24] at the bottom of the allosteric pocket is lost during the simulation and not rebound. As such, the water-mediated H-bonding network connecting TM3 (Y659), TM6 (T781) and TM7 (S809) is broken and not re-formed. In addition, W785 on TM6 is distorted and pushed outwards into an exaggerated conformation, which is not observed in any other previous simulation or crystal structure, suggesting this is an unfavourable state (Fig. 8B). In an energetic analysis of the simulation, the cis-azo photoisomer of alloswitch-1 is +6.8 kcal/mol higher than the trans-azo isomer (SI Table 3) with protein-ligand interactions +2.0 kcal/mol higher than the active NAM isomer-mGlu5 complex. Taken together, these results indicate the photoexcited protein-ligand complex is possible but less energetically stable with concomitant destabilizing effects on receptor conformation.
In a second approach we set out to investigate alloswitch-1 photoswitching mid-simulation inside of the allosteric pocket, with particular focus on receptor destabilization. In two MD simulations, with alloswitch-1 in cis-amide and trans-amide conformations respectively, changing the azo from trans to cis induces a sudden shift in ligand orientation and position, as photoexcited alloswitch-1 moves higher in the allosteric pocket (closer to exit), experiencing increased rocking and lateral fluctuation (SI Figs. 7 and SI 12). In both cases, the middle aromatic ring of alloswitch-1 swivels to accommodate the cis-azo conformation within the pocket, with the methoxy group changing its position by pointing to TM7 rather than TM6 and the cis-azo dihedral angle shifting to ~0° (from a theoretical optimal angle of 12°) due to tightness in the allosteric pocket. Interestingly, in both cases, the same destabilizing effect on the receptor is observed as before, in that the water-mediated H-bonding network connecting TM3 (Y659), TM6 (T781) and TM7 (S809) is broken and not re-formed (Fig. 9) as the co-crystallized water [24] is lost from the bottom of the allosteric pocket. A very outward orientation of W785 is again observed (Fig. 9), particularly in the trans-amide conformation.
According to these simulations, it appears theoretically possible for a photoswitched state of alloswitch-1 to bind mGlu5 although in a different orientation to the unswitched state (conventional NAM mode) as it does not reach as far into the allosteric pocket. Furthermore, it appears theoretically possible from our simulations, which should be taken with caution as preliminary calculations, that photoswitching might occur in the allosteric pocket of mGlu5. However, tightness of the allosteric pocket means the photoexcited conformation may be more strained than would be ideal, which perhaps could contribute further to protein-ligand instability in the photoswitched state. What is not yet clear is whether different transition states in between the trans- and cis-azo conformations of alloswitch-1 could also bind mGlu5. As the transition pathway of photoswitching is potentially complex, theoretically combining elements of incremental bond rotation and inversion, it is currently not possible to answer this question confidently as molecular mechanics is unable to simulate different light energy conditions or excited electronic states. More experiments and higher-level simulations are necessary to address this possibility in terms of transitional kinetics. However, in terms of a theoretically “instantaneous” photoswitch without transition states (see Methods), both trans- and cis-azo conformations appear feasible in terms of receptor binding. Regarding the receptor, the presence of a co-crystallized water molecule has been suggested to be important for the NAM activity of mavoglurant by stabilizing the inactive state of mGlu5 [24]. It is therefore interesting to observe that simulations of mGlu5 containing two conventional but different NAMs: MPEP and unswitched alloswitch-1 both bind in a stable fashion and stabilize this water molecule in the allosteric pocket whilst also forming an H-bond with S809 (like mavoglurant). Conversely, the photoswitched isomer of alloswitch-1, which has been experimentally proven to not behave as a NAM [29], destablizes this co-crystallized water molecule and does not form an H-bond with S809. Does this mean that the inactive state of mGlu5 is therefore also destabilized? Possibly yes, but this is speculative. It is currently difficult to say what the exact functional behaviour of the photoswitched isomer of alloswitch-1 is. Certainly the crystallized receptor state is destabilized but there may be other functional “inactive” states that do not require a bound water molecule between TM3/6/7 in the allosteric pocket. It may also be possible that photoexcited alloswitch-1 acts as a SAM with no obvious functional effects other than preventing the binding of other NAMs.
It should be noted that although it appears theoretically possible from a computational point of view that photoisomerized alloswitch-1 binds the allosteric pocket of mGlu5, in reality there are several different pathways of binding/unbinding available to the ligand in either of its photoisomeric states with the receptor, itself in different states (see Fig. 10). This means that the dynamics of alloswitch-1 may be complicated indeed, with many factors influencing its behaviour. For example, it is possible that duration of binding of the photoexcited state (assuming it occurs) is insignificant compared to other processes, such as conventional ligand unbinding of the bioactive NAM state and photoisomerization outside of the pocket. It is also possible that re-binding of the photoexcited state might be prevented by dynamic interactions with receptor extracellular loops that partially obstruct the allosteric pocket [24]. Further experimental information combined with additional computational simulations are needed to test these possibilities and explore the reaction kinetics in more detail.
Conclusions
Alloswitch-1 is an interesting and novel mGlu5 NAM, similar to other mGlu5 NAMs in that it possesses three aromatic rings connected by two spacer groups. However, unlike other mGlu5 NAMs, one of these spacers is a photo-sensitive azo group that photoisomerizes from trans to cis and back again under violet and green light, respectively. Recent experimental evidence has shown that alloswitch-1 is a potent NAM under dark/green light conditions but loses its NAM functionality under violet light [29]. Here, we propose from a structural point of view that the photoswitch of alloswitch-1 alters the ligand’s binding mode, reduces its binding affinity and stability, and disrupts receptor conformation, particularly with regard to the co-bound water molecule in the allosteric pocket that has been suggested as important for stabilizing the inactive NAM-bound state [24]. Molecular simulations suggest binding of the photoswitched isomer of alloswitch-1 is possible, as well as theoretical “instantaneous” photoswitching in the allosteric pocket of mGlu5 while the ligand is bound, although further experimental evidence is required to support this possibility. Furthermore, by probing MPEP binding, it appears that mavoglurant, MPEP and alloswitch-1 all share a common binding-mode by stabilizing a receptor conformation that contains a bound water molecule at the bottom of the allosteric pocket, connecting residues on TM3, TM6, and TM7, and characteristic of the NAM-bound inactive state [24]. By simulating photoswitching of alloswitch-1, this water-mediated connection is lost in the receptor, which may destabilize its inactive state. Finally, alloswitch-1 has the theoretical capability of isomerizing its amide group between cis and trans conformations. This is interesting because an amide group is a characteristic feature of several mGlu5 NAMs and its presence may provide a necessary means of flexibilty for these particular ligands to adopt their functional binding states. Simulations suggest a cis-amide conformation is energetically equivalent to the trans-amide state because more favourable protein-ligand interactions are formed with residues known to be functionality important for NAM functionality e.g. N747 on TM5 and S809 on TM7 [28]. These favourable protein-ligand interactions may compensate the higher energy state of the cis-amide and at the same time fulfill functionally important mechanisms of receptor inactivation. On a more general note, cis configurations are an interesting possibility in amide-containing ligands (not normally considered by default in docking programs) as crystallographic structures suggest they can sometimes be responsible for important protein or self interactions.
Methods
Experimental
HEK293 cells were transiently transfected with mGlu5 receptors by electroporation. Receptors were cotransfected with EAAC1, a glutamate transporter, to avoid the influence of extracellular glutamate. The receptor activity was monitored through measurements of inositol monophosphate (IP1) production. IP1 production was determined using the IP-One HTRF kit (CisBio Bioassays) according to the manufacturer’s recommendations [48]. All points were performed in triplicate. Mutant receptors were obtained using the Quick-Change strategy (Stratagene, La Jolla, CA, USA) and all mutations were verified by sequencing. Experimental data were fitted using the Hill equation with Prism software (GraphPad, La Jolla, CA, USA). Quisqualate and MPEP were purchased from Tocris Bioscience (Bristol, UK). Alloswitch-1 was synthesized in the laboratory of A. Llebaria following described literature procedures [29]. All solutions were prepared just before experiments.
Computational
Completion of wt TM mGlu5 Structure
The TM mGlu5-lysozyme fusion crystal structure (PDB id 4OO9) was completed by first cleaving lysozyme and removing solvent and fatty acid molecules from the structure, then modelling missing second intracellular and extracellular loops (ICL2 and ECL2) with MODELLER [32] and converting unnatural amino-acids and thermostabilising mutations in the TM domain back to wild-type with CHIMERA [33] (C634, C691, E579, N667, I669, G675, T742, S753). This was done by selecting the most appropriate best-fit wt rotamer with highest possible statistical probability. Co-crystallized water molecules were maintained in the structure and mavoglurant ligand was removed.
Docking of NAMs
Coordinates of mGlu5 were extracted from OPM database [49], PDB id: 4OO9 [24], reorientated in an implicit membrane. Cis and trans optimized conformations of the azo and amide groups for alloswitch-1 were respectively obtained by Density Functional Theory (DFT) at the B3LYP/6-31+G(d) level using Gaussian [42]. Coordinates for MPEP and mavoglurant were generated with Maestro [50]. Autodock 4.2 was used with “-B amide” flag to give flexibility to ligand amide groups and flag “-U nphs_lps_nonstdres” to maintain co-crystallized waters in the receptor structure. Grid points were extended to cover total allosteric pocket volume. For docking cis-azo alloswitch-1 (photoswitched state), full ligand flexibility was ensured except the azo dihedral angle, which was maintained at +12° as indicated by DFT calculations. The final docking conformation of each molecule represents the top-ranked hit identified by Autodock based on best docking score (expressed as predicted Ki) in the largest docking cluster (out of a total of 10 alternative docking solutions). Energy minimization (1000 steps of steepest descent and 100 steps of conjugate gradient) of docked structures was performed with CHIMERA [33] and the AMBER-12SB force-field [51] to relax final docked structures.
Conformational Sampling of mGlu5
Through structural inspection of the mGlu5 crystal structure (PDB id: 4OO9), residue F788 on TM6 was identified as influential in determining the conformational shape and volume of the allosteric pocket of mGlu5. Therefore an alternative conformation of mGlu5 for docking alloswitch-1 was identified by adjusting the rotameric position of F788 from inward to outward pointing in the mGlu5 crystal structure using CHIMERA [33] (selection of second most probable rotamer). This structure was then relaxed with ROSETTA [52] (minus ligand) in an implicit membrane, generating up to 1000 possible conformations, clustered at 2 Å, and the highest scoring conformation in the largest cluster selected as the final structure. The co-crystallized water molecule at the bottom of the allosteric pocket, as well as F788 sidechain, were positionally constrained during this process.
Molecular Dynamics (MD)
All MD simulations were performed with NAMD [53] v2.9 on the CESCA Supercomputer using the CHARMM-36 force-field [54]. Membrane (POPC)/solvent (TIP3P) simulation boxes were built with VMD Membrane Builder [55], with protein insertion in the membrane performed with an inhouse VMD-script. All ligands were parameterized with CHARMM General Force Field (CGenFF) [56] using the ParamChem webserver (https://cgenff.paramchem.org/). Ions were added to neutralize the system and salt was added at a concentration of 0.2 M. The system was initially minimized and then equilibrated with an 8.0 ns MD run with harmonic restraints on the protein progressively relaxed every 2.0 ns from 10 kcal mol–1 Å–2 to 0 kcal mol–1 Å–2. MD simulations were then run for 100-200 ns (production run) for each mGlu5-NAM system. In total, four different simulation systems were constructed: TM domain of mGlu5 with (i) docked MPEP, (ii) docked alloswitch-1 with cis-amide and trans-azo (i.e. “dark” state), (iii) docked alloswitch-1 with trans-amide and cis-azo (i.e. photoswitched state) and (iv) an alternative conformation of the TM domain of mGlu5 with docked alloswitch-1 with trans-amide and trans-azo (i.e. alternative “dark” state). In two MD simulations (ii) and (iv), after 100 ns, the azo dihedral angle of alloswitch-1 was changed from trans to cis (azo dihedral angle of 12° was presumed for cis state as calculated with DFT) to simulate ligand photoswitching. Dihedral angle change was implemented in one complete rotational movement within a single MD timestep i.e. 2 fs. We describe this as an “instantaneous” photoswitch as no transition state between trans- and cis-azo is explored. Both simulations (ii) and (iv) were continued for another 100 ns with the ligand in a cis-azo state, to simulate as closely as possible sustained photoswitching conditions. During this time, the azo dihedral angle is free to rotate within its cis angle range (-20° to +20°) and within the natural confines of the protein. No confines were placed on the amide group of alloswitch-1 in either simulation, which is free to rotate within the natural confines of the protein.
MD Analysis
For each MD simulation, VMD plugin [55] “NAMDenergy” was used to calculate average bonded and non-bonded energies of the ligand and non-bonded energies of protein-ligand interactions. The VMD plugin “RMSD Trajectory Tool” was used to calculate RMSD values for ligand pyridine ring atoms and protein backbone atoms in each simulation. VMD plugin “Hydrogen Bonds” was used to analyze the formation/breaking of the protein-ligand H-bond between S809 sidechain and ligand pyridine ring in each simulation.
Database Searching
Crystallized non-cyclic cis-amide containing-ligands (amide dihedral angle: 0°±20°) were identified in the CSD [45] by using the Mogul tool [57] v1.7 and non-cyclic cis-amide ligands were identified in the PDB [46] by geometric matching (with an inhouse script) against representative non-redundant ligands in PDBeChem [58] using an idealized cis-amide search fragment (i.e. with 0° amide dihedral angle).
Structure Visualisation
Chimera [33] was used to visualise structures, calculate H-bonds and generate images. POVME [44] was used to calculate the volume of the allosteric pocket in mGlu5 by using default parameters where possible (see http://nbcr.ucsd.edu/data/sw/hosted/POVME/) with grid points adjusted to fully map the allosteric site of mGlu5.
SUPPORTING INFORMATION
The supplementary section contains RMSD and H-bond plots with energetic analyses from MD simulations; docking scores; examples of cis-amide containing compounds identified in the CSD [45] and PDB; Density Functional Theory (DFT) calculations, and experimental EC50 values.
ACKNOWLEDGEMENTS
This study was supported in part by Ministerio de Economía y Competitividad (SAF2010-19257, ERA-NET NEURON PCIN-2013-018-C03-02) and Fundació La Marató de TV3 (Refs. 110230, 110231 and 110232). We acknowledge the support of Generalitat de Catalunya (2009SGR-1072) and CSIC. Computations were performed at the Center for Scientific and Academic Services of Catalonia (CESCA). The authors participate in the European COST Action CM1207 (GLISTEN: GPCR-Ligand Interactions, Structures, and Transmembrane Signalling: a European Research Network).
LIST OF ABBREVIATIONS
- GPCR
G protein-coupled receptor
- TM
transmembrane
- VFT
Venus flytrap
- mGlu5
metabotropic glutamate receptor 5
- NAM
negative allosteric modulator
- ECL2
extracellular loop 2
- ICL2
intracellular loop 2
- RMSD
root mean square deviation
- ND
not determined.
SUPPLEMENTARY MATERIAL
Supplementary material is available on the publisher’s web site along with the published article.
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
REFERENCES
- 1.Niswender C.M., Conn P.J. Metabotropic glutamate receptors: physiology, pharmacology, and disease. Annu. Rev. Pharmacol.Toxicol. 2010;50:295–322. doi: 10.1146/annurev.pharmtox.011008.145533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Featherstone D.E. Intercellular glutamate signaling in the nervous system and beyond. ACS Chem. Neurosci. 2010;1(1):4–12. doi: 10.1021/cn900006n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nicoletti F., Bockaert J., Collingridge G.L., Conn P.J., Ferraguti F., Schoepp D.D., Wroblewski J.T., Pin J.P. Metabotropic glutamate receptors: from the workbench to the bedside. Neuropharmacology. 2011;60(7-8):1017–1041. doi: 10.1016/j.neuropharm.2010.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dingledine R., Borges K., Bowie D., Traynelis S.F. The glutamate receptor ion channels. Pharmacol. Rev. 1999;51(1):7–61. [PubMed] [Google Scholar]
- 5.Sugiyama H., Ito I., Hirono C. A new type of glutamate receptor linked to inositol phospholipid metabolism. Nature. 1987;325(6104):531–533. doi: 10.1038/325531a0. [DOI] [PubMed] [Google Scholar]
- 6.Schoepp D.D., Jane D.E., Monn J.A. Pharmacological agents acting at subtypes of metabotropic glutamate receptors. Neuropharmacology. 1999;38(10):1431–1476. doi: 10.1016/S0028-3908(99)00092-1. [DOI] [PubMed] [Google Scholar]
- 7.Rondard P., Pin J.P. Dynamics and modulation of metabotropic glutamate receptors. Curr. Opin. Pharmacol. 2015;20:95–101. doi: 10.1016/j.coph.2014.12.001. [DOI] [PubMed] [Google Scholar]
- 8.Kniazeff J., Prezeau L., Rondard P., Pin J.P., Goudet C. Dimers and beyond: The functional puzzles of class C GPCRs. Pharmacol. Ther. 2011;130(1):9–25. doi: 10.1016/j.pharmthera.2011.01.006. [DOI] [PubMed] [Google Scholar]
- 9.Conn P.J., Lindsley C.W., Jones C.K. Activation of metabotropic glutamate receptors as a novel approach for the treatment of schizophrenia. Trends Pharmacol. Sci. 2009;30(1):25–31. doi: 10.1016/j.tips.2008.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Swanson C.J., Bures M., Johnson M.P., Linden A.M., Monn J.A., Schoepp D.D. Metabotropic glutamate receptors as novel targets for anxiety and stress disorders. Nat. Rev. Drug Discov. 2005;4(2):131–144. doi: 10.1038/nrd1630. [DOI] [PubMed] [Google Scholar]
- 11.Palucha A., Pilc A. Metabotropic glutamate receptor ligands as possible anxiolytic and antidepressant drugs. Pharmacol. Ther. 2007;115(1):116–147. doi: 10.1016/j.pharmthera.2007.04.007. [DOI] [PubMed] [Google Scholar]
- 12.Walker K., Bowes M., Panesar M., Davis A., Gentry C., Kesingland A., Gasparini F., Spooren W., Stoehr N., Pagano A., Flor P.J., Vranesic I., Lingenhoehl K., Johnson E.C., Varney M., Urban L., Kuhn R. Metabotropic glutamate receptor subtype 5 (mGlu5) and nociceptive function. I. Selective blockade of mGlu5 receptors in models of acute, persistent and chronic pain. Neuropharmacology. 2001;40(1):1–9. doi: 10.1016/S0028-3908(00)00113-1. [DOI] [PubMed] [Google Scholar]
- 13.Chiechio S., Nicoletti F. Metabotropic glutamate receptors and the control of chronic pain. Curr. Opin. Pharmacol. 2012;12(1):28–34. doi: 10.1016/j.coph.2011.10.010. [DOI] [PubMed] [Google Scholar]
- 14.Palazzo E., Marabese I., de Novellis V., Rossi F., Maione S. Supraspinal metabotropic glutamate receptors: a target for pain relief and beyond. Eur. J. Neurosci. 2014;39(3):444–454. doi: 10.1111/ejn.12398. [DOI] [PubMed] [Google Scholar]
- 15.Kunishima N., Shimada Y., Tsuji Y., Sato T., Yamamoto M., Kumasaka T., Nakanishi S., Jingami H., Morikawa K. Structural basis of glutamate recognition by a dimeric metabotropic glutamate receptor. Nature. 2000;407(6807):971–977. doi: 10.1038/35039564. [DOI] [PubMed] [Google Scholar]
- 16.Tsuchiya D., Kunishima N., Kamiya N., Jingami H., Morikawa K. Structural views of the ligand-binding cores of a metabotropic glutamate receptor complexed with an antagonist and both glutamate and Gd3+. Proc. Natl. Acad. Sci. USA. 2002;99(5):2660–2665. doi: 10.1073/pnas.052708599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Muto T., Tsuchiya D., Morikawa K., Jingami H. Structures of the extracellular regions of the group II/III metabotropic glutamate receptors. Proc. Natl. Acad. Sci. USA. 2007;104(10):3759–3764. doi: 10.1073/pnas.0611577104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Flor P.J., Acher F.C. Orthosteric versus allosteric GPCR activation: the great challenge of group-III mGluRs. Biochem.Pharmacol. 2012;84(4):414–424. doi: 10.1016/j.bcp.2012.04.013. [DOI] [PubMed] [Google Scholar]
- 19.Conn P.J., Christopoulos A., Lindsley C.W. Allosteric modulators of GPCRs: a novel approach for the treatment of CNS disorders. Nat. Rev. Drug Discov. 2009;8(1):41–54. doi: 10.1038/nrd2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Burford N.T., Watson J., Bertekap R., Alt A. Strategies for the identification of allosteric modulators of G-protein-coupled receptors. Biochem. Pharmacol. 2011;81(6):691–702. doi: 10.1016/j.bcp.2010.12.012. [DOI] [PubMed] [Google Scholar]
- 21.Kaae B.H., Harpsoe K., Kvist T., Mathiesen J.M., Molck C., Gloriam D., Jimenez H.N., Uberti M.A., Nielsen S.M., Nielsen B., Brauner-Osborne H., Sauerberg P., Clausen R.P., Madsen U. Structure-activity relationships for negative allosteric mGluR5 modulators. ChemMedChem. 2012;7(3):440–451. doi: 10.1002/cmdc.201100578. [DOI] [PubMed] [Google Scholar]
- 22.Gregory K.J., Nguyen E.D., Reiff S.D., Squire E.F., Stauffer S.R., Lindsley C.W., Meiler J., Conn P.J. Probing the metabotropic glutamate receptor 5 (mGlu(5)) positive allosteric modulator (PAM) binding pocket: discovery of point mutations that engender a "molecular switch" in PAM pharmacology. Mol.Pharmacol. 2013;83(5):991–1006. doi: 10.1124/mol.112.083949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dalton J.A., Gomez-Santacana X., Llebaria A., Giraldo J. Computational analysis of negative and positive allosteric modulator binding and function in metabotropic glutamate receptor 5 (in)activation. J. Chem. Inf. Model. 2014;54(5):1476–1487. doi: 10.1021/ci500127c. [DOI] [PubMed] [Google Scholar]
- 24.Dore A.S., Okrasa K., Patel J.C., Serrano-Vega M., Bennett K., Cooke R.M., Errey J.C., Jazayeri A., Khan S., Tehan B., Weir M., Wiggin G.R., Marshall F.H. Structure of class C GPCR metabotropic glutamate receptor 5 transmembrane domain. Nature. 2014;511(7511):557–562. doi: 10.1038/nature13396. [DOI] [PubMed] [Google Scholar]
- 25.Wu H., Wang C., Gregory K.J., Han G.W., Cho H.P., Xia Y., Niswender C.M., Katritch V., Meiler J., Cherezov V., Conn P.J., Stevens R.C. Structure of a class C GPCR metabotropic glutamate receptor 1 bound to an allosteric modulator. Science. 2014;344(6179):58–64. doi: 10.1126/science.1249489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Micheli F. Methylphenylethynylpyridine (MPEP) Novartis. Curr. Opin. Investig. Drugs. 2000;1(3):355–359. [PubMed] [Google Scholar]
- 27.O'Leary D.M., Movsesyan V., Vicini S., Faden A.I. Selective mGluR5 antagonists MPEP and SIB-1893 decrease NMDA or glutamate-mediated neuronal toxicity through actions that reflect NMDA receptor antagonism. Br. J. Pharmacol. 2000;131(7):1429–1437. doi: 10.1038/sj.bjp.0703715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Molck C., Harpsoe K., Gloriam D.E., Clausen R.P., Madsen U., Pedersen L.O., Jimenez H.N., Nielsen S.M., Mathiesen J.M., Brauner-Osborne H. Pharmacological characterization and modeling of the binding sites of novel 1, 3-bis(pyridinylethynyl) benzenes as metabotropic glutamate receptor 5-selective negative allosteric modulators. Mol. Pharmacol. 2012;82(5):929–937. doi: 10.1124/mol.112.078808. [DOI] [PubMed] [Google Scholar]
- 29.Pittolo S., Gomez-Santacana X., Eckelt K., Rovira X., Dalton J., Goudet C., Pin J.P., Llobet A., Giraldo J., Llebaria A., Gorostiza P. An allosteric modulator to control endogenous G protein-coupled receptors with light. Nat. Chem. Biol. 2014;10(10):813–815. doi: 10.1038/nchembio.1612. [DOI] [PubMed] [Google Scholar]
- 30.Emmitte K.A. mGlu5 negative allosteric modulators: a patent review (2010-2012). Expert Opin. Ther. Pat. 2013;23(4):393–408. doi: 10.1517/13543776.2013.760544. [DOI] [PubMed] [Google Scholar]
- 31.Gomez-Santacana X., Rovira X., Dalton J.A., Goudet C., Pin J.P., Gorostiza P., Giraldo J., Llebaria A. A double effect molecular switch leads to a novel potent negative allosteric modulator of metabotropic glutamate receptor 5. MedChemComm. 2014;5(10):1548–1554. doi: 10.1039/C4MD00208C. [DOI] [Google Scholar]
- 32.Sali A., Blundell T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993;234(3):779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 33.Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E. UCSF chimera - A visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25(13):1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 34.Morris G.M., Huey R., Lindstrom W., Sanner M.F., Belew R.K., Goodsell D.S., Olson A.J. AutoDock4 and AutoDock Tools4: Automated docking with selective receptor flexibility. J.Comput. Chem. 2009;30(16):2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pagano A., Ruegg D., Litschig S., Stoehr N., Stierlin C., Heinrich M., Floersheim P., Prezeau L., Carroll F., Pin J.P., Cambria A., Vranesic I., Flor P.J., Gasparini F., Kuhn R. The non-competitive antagonists 2-methyl-6-(phenylethynyl)pyridine and 7-hydroxyiminocyclopropan[b]chromen-1a-carboxylic acid ethyl ester interact with overlapping binding pockets in the transmembrane region of group I metabotropic glutamate receptors. J. Biol. Chem. 2000;275(43):33750–33758. doi: 10.1074/jbc.M006230200. [DOI] [PubMed] [Google Scholar]
- 36.Wood M.R., Hopkins C.R., Brogan J.T., Conn P.J., Lindsley C.W. “Molecular switches” on mGluR allosteric ligands that modulate modes of pharmacology. Biochemistry. 2011;50(13):2403–2410. doi: 10.1021/bi200129s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bennett K.A., Dore A.S., Christopher J.A., Weiss D.R., Marshall F.H. Structures of mGluRs shed light on the challenges of drug development of allosteric modulators. Curr. Opin. Pharmacol. 2014;20C:1–7. doi: 10.1016/j.coph.2014.09.022. [DOI] [PubMed] [Google Scholar]
- 38.Malherbe P., Kratochwil N., Zenner M.T., Piussi J., Diener C., Kratzeisen C., Fischer C., Porter R.H. Mutational analysis and molecular modeling of the binding pocket of the metabotropic glutamate 5 receptor negative modulator 2-methyl-6-(phenylethynyl)-pyridine. Mol. Pharmacol. 2003;64(4):823–832. doi: 10.1124/mol.64.4.823. [DOI] [PubMed] [Google Scholar]
- 39.Craveur P., Joseph A.P., Poulain P., de Brevern A.G., Rebehmed J. Cis-trans isomerization of omega dihedrals in proteins. Amino Acids. 2013;45(2):279–289. doi: 10.1007/s00726-013-1511-3. [DOI] [PubMed] [Google Scholar]
- 40.Forbes C.C., Beatty A.M., Smith B.D. Using pentafluorophenyl as a Lewis acid to stabilize a cis secondary amide conformation. Org. Lett. 2001;3(22):3595–3598. doi: 10.1021/ol016733l. [DOI] [PubMed] [Google Scholar]
- 41.Gregory K.J., Nguyen E.D., Malosh C., Mendenhall J.L., Zic J.Z., Bates B.S., Noetzel M.J., Squire E.F., Turner E.M., Rook J.M., Emmitte K.A., Stauffer S.R., Lindsley C.W., Meiler J., Conn P.J. Identification of specific ligand-receptor interactions that govern binding and cooperativity of diverse modulators to a common metabotropic glutamate receptor 5 allosteric site. ACS Chem. Neurosci. 2014;5(4):282–295. doi: 10.1021/cn400225x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Frisch M.J., Trucks G.W., Schlegel H.B., Scuseria G.E., Robb M.A., Cheeseman J.R., Scalmani G., Barone V., Mennucci B., Petersson G.A., Nakatsuji H., Caricato M., Li X., Hratchian H.P., Izmaylov A.F., Bloino J., Zheng G., Sonnenberg J.L., Hada M., Ehara M., Toyota K., Fukuda R., Hasegawa J., Ishida M., Nakajima T., Honda Y., Kitao O., Nakai H., Vreven T., Montgomery J.A., Jr, Peralta J.E., Ogliaro F., Bearpark M.J., Heyd J., Brothers E.N., Kudin K.N., Staroverov V.N., Kobayashi R., Normand J., Raghavachari K., Rendell A.P., Burant J.C., Iyengar S.S., Tomasi J., Cossi M., Rega N., Millam N.J., Klene M., Knox J.E., Cross J.B., Bakken V., Adamo C., Jaramillo J., Gomperts R., Stratmann R.E., Yazyev O., Austin A.J., Cammi R., Pomelli C., Ochterski J.W., Martin R.L., Morokuma K., Zakrzewski V.G., Voth G.A., Salvador P., Dannenberg J.J., Dapprich S., Daniels A.D., Farkas Ö., Foresman J.B., Ortiz J.V., Cioslowski J., Fox D.J. Gaussian 09. Wallingford, CT, USA: Gaussian, Inc.; 2009. [Google Scholar]
- 43.Katritch V., Cherezov V., Stevens R.C. Structure-function of the G protein-coupled receptor superfamily. Annu. Rev. Pharmacol. Toxicol. 2013;53:531–556. doi: 10.1146/annurev-pharmtox-032112-135923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Durrant J.D., de Oliveira C.A.F., McCammon J.A. An algorithm for measuring binding-pocket volumes. J. Mol. Graph.Model. 2011;29(5):773–776. doi: 10.1016/j.jmgm.2010.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Allen F.H. The Cambridge Structural Database: a quarter of a million crystal structures and rising. Acta Crystallographica.Section B. Struct. Sci. 2002;58(3-1):380–388. doi: 10.1107/s0108768102003890. [DOI] [PubMed] [Google Scholar]
- 46.Bernstein F.C., Koetzle T.F., Williams G.J., Meyer E.F., Jr, Brice M.D., Rodgers J.R., Kennard O., Shimanouchi T., Tasumi M. The Protein Data Bank: a computer-based archival file for macromolecular structures. J. Mol. Biol. 1977;112(3):535–542. doi: 10.1016/S0022-2836(77)80200-3. [DOI] [PubMed] [Google Scholar]
- 47.Giraldo J. Agonist induction, conformational selection, and mutant receptors. FEBS Lett. 2004;556(1-3):13–18. doi: 10.1016/S0014-5793(03)01404-2. [DOI] [PubMed] [Google Scholar]
- 48.Trinquet E., Fink M., Bazin H., Grillet F., Maurin F., Bourrier E., Ansanay H., Leroy C., Michaud A., Durroux T., Maurel D., Malhaire F., Goudet C., Pin J.P., Naval M., Hernout O., Chretien F., Chapleur Y., Mathis G. D-myo-inositol 1-phosphate as a surrogate of D-myo-inositol 1, 4, 5-tris phosphate to monitor G protein-coupled receptor activation. Anal. Biochem. 2006;358(1):126–135. doi: 10.1016/j.ab.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 49.Lomize M.A., Lomize A.L., Pogozheva I.D., Mosberg H.I. OPM: orientations of proteins in membranes database. Bioinformatics. 2006;22(5):623–625. doi: 10.1093/bioinformatics/btk023. [DOI] [PubMed] [Google Scholar]
- 50.Release S. 2014-2: Maestro, version 9.8. New York, NY: Schrödinger, LLC; 2014. [Google Scholar]
- 51.Case D.A., Berryman J.T., Betz R.M. , Cai Q., Cerutti D.S., Cheatham,III T.E., Darden T.A., Duke R.E., Gohlke H., Goetz A.W., Gusarov S., Homeyer N., Janowski P., Kaus J., Kolossváry I., Kovalenko A., Lee T.S., LeGrand S., Luchko T., Luo R., Madej B., Merz K.M., Paesani F., Roe D.R., Roitberg A., Sagui C., Salomon-Ferrer R., Seabra G., Simmerling C.L., Smith W., Swails J., Walker R.C., Wang J., Wolf R.M., Wu X., Kollman P.A. AMBER 14. San Francisco: University of California; 2014. [Google Scholar]
- 52.Bonneau R., Baker D. Ab initio protein structure prediction: progress and prospects. Annu. Rev. Biophys. Biomol. Struct. 2001;30:173–189. doi: 10.1146/annurev.biophys.30.1.173. [DOI] [PubMed] [Google Scholar]
- 53.Phillips J.C., Braun R., Wang W., Gumbart J., Tajkhorshid E., Villa E., Chipot C., Skeel R.D., Kale L., Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005;26(16):1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Best R. B., Zhu X., Shim J., Lopes P. E., Mittal J., Feig M., Mackerell A.D., Jr Optimization of the additive CHARMM allatom protein force field targeting improved sampling of the backbone phi, psi and side-chain chi(1) and chi(2) dihedral angles. J. Chem. heory Comput. 2012;8(9):3257–3273. doi: 10.1021/ct300400x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. 1996. [DOI] [PubMed]
- 56.Vanommeslaeghe K., MacKerell A.D., Jr, Automation of the CHARMM General Force Field (CGenFF) I: bond perception and atom typing. J. Chem. Inf. Model. 2012;52(12):3144–3154. doi: 10.1021/ci300363c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bruno I.J., Cole J.C., Kessler M., Luo J., Motherwell W.D., Purkis L.H., Smith B.R., Taylor R., Cooper R.I., Harris S.E., Orpen A.G. Retrieval of crystallographically-derived molecular geometry information. J. Chem. Inf. Comput. Sci. 2004;44(6):2133–2144. doi: 10.1021/ci049780b. [DOI] [PubMed] [Google Scholar]
- 58.Golovin A., Oldfield T.J., Tate J.G., Velankar S., Barton G.J., Boutselakis H., Dimitropoulos D., Fillon J., Hussain A., Ionides J.M., John M., Keller P.A., Krissinel E., McNeil P., Naim A., Newman R., Pajon A., Pineda J., Rachedi A., Copeland J., Sitnov A., Sobhany S., Suarez-Uruena A., Swaminathan G.J., Tagari M., Tromm S., Vranken W., Henrick K. E-MSD. 2004.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material is available on the publisher’s web site along with the published article.