

ABSTRACT
Tick-borne transmission of bacterial pathogens in the order Rickettsiales is responsible for diverse infectious diseases, many of them severe, in humans and animals. Transmission dynamics differ among these pathogens and are reflected in the pathogen-vector interaction. Anaplasma marginale has been shown to establish and maintain infectivity within Dermacentor spp. for weeks to months while escaping the complex network of vacuolar peptidases that are responsible for digestion of the tick blood meal. How this prolonged maintenance of infectivity in a potentially hostile environment is achieved has been unknown. Using the natural vector Dermacentor andersoni, we demonstrated that A. marginale-infected tick vacuoles (AmVs) concurrently recruit markers of the early endosome (Rab5), recycling endosome (Rab4 and Rab11), and late endosome (Rab7), are maintained near neutral pH, do not fuse with lysosomes, exclude the protease cathepsin L, and engage the endoplasmic reticulum and Golgi apparatus for up to 21 days postinfection. Maintenance of this safe vacuolar niche requires active A. marginale protein synthesis; in its absence, the AmVs mature into acidic, protease-active phagolysosomes. Identification of this bacterially directed modeling of the tick midgut endosome provides a mechanistic basis for examination of the differences in transmission efficiency observed among A. marginale strains and among vector populations.
IMPORTANCE Ticks transmit a variety of intracellular bacterial pathogens that cause significant diseases in humans and animals. For successful transmission, these bacterial pathogens must first gain entry into the tick midgut digestive cells, avoid digestion, and establish a replicative niche without harming the tick vector. Little is known about how this replicative niche is established and maintained. Using the ruminant pathogen A. marginale and its natural tick vector, D. andersoni, this study characterized the features of the A. marginale niche in the tick midgut and demonstrates that A. marginale protein synthesis is required for the maintenance of this niche. This work opens a new line of inquiry about the pathogen effectors and their targets within the tick that mediate tick-pathogen interactions and ultimately serve as the determinants of pathogen success.
INTRODUCTION
Ticks are biological vectors of the obligate intracellular bacteria of the order Rickettsiales, including pathogens that cause severe diseases in humans and animals (1). Transmission dynamics determine the epidemiologic features of a given disease and vary among the pathogen-vector pairs. For example, Rickettsia rickettsii, the causative agent of Rocky Mountain spotted fever, is maintained in the tick vector Dermacentor andersoni through transovarial transmission, and thus each new generation of larval ticks is infected. Additionally, infection is maintained with each successive molt, circumventing the need for feeding on an infected mammalian host to maintain the pathogen through time. In contrast, Anaplasma marginale, responsible for anaplasmosis in ruminants, must be acquired and transstadially transmitted by each new generation of adult D. andersoni ticks (2). It is not transovarially transmitted, and the larvae and nymphs of D. andersoni are not exposed to A. marginale because they feed preferentially on small mammals. Adult male ticks feed preferentially on cattle, move between hosts while seeking mates, and thus are the epidemiologically important transmission vector. Consequently, A. marginale must persist in the tick for the period of time necessary for an adult tick to ingest two separate blood meals, which depending on conditions, may occur in separate seasons. Experimentally infected adult male D. andersoni ticks are able to transmit A. marginale for up to 26 days after acquisition feeding (2–4). The mechanisms by which pathogens maintain long-term infections within ticks remain poorly understood.
The tick midgut is the first barrier to ongoing transmission and presents a hostile environment for bacterial pathogens (5). Unlike other hematophagous arthropods, digestion of the blood meal in ticks takes place intracellularly (6–8). Many gaps exist in terms of basic characterization and understanding of the molecular machinery that regulates this process; however, ingested blood and other materials are sorted into vacuoles and digested by a complex network of peptidases. This network is induced upon blood feeding and degrades endocytosed material such as hemoglobin at acidic pH (7, 9), making the midgut epithelium a potentially harsh environment for intravacuolar bacteria. A. marginale successfully exploits this environment by establishing a replicative niche within intracytoplasmic vacuoles in tick midgut cells (10). How A. marginale or other tick-borne obligate intracellular bacterial pathogens survive in ticks and acquire the nutrients required for replication while escaping vacuolar degradation is unknown and represents a major knowledge gap in vector-pathogen biology.
In mammalian cells, vesicular trafficking is, in large part, mediated by Rab GTPases (11, 12), which conduct the vesicles and their cargo through the different compartments of the endosome and, for the nonrecycled cargo, ultimately to the phagolysosome (11–15). Obligate intracellular pathogens modulate the endosomal and secretory pathways in their mammalian hosts, commonly by altering the recruitment of endogenous Rab GTPases to the pathogen-containing vacuole. While the exact mechanisms employed and the resulting niche are varied among pathogens, intracellular pathogens share features of retaining and excluding different groups of Rab GTPase molecules to create a niche that avoids degradation and allows acquisition of nutrients (16).
Here, we characterize the essential features of A. marginale-containing vacuoles in the natural tick vector D. andersoni and test the hypothesis that active A. marginale protein synthesis is required for modulation of the maturation of the vacuole. The findings are discussed in the context of the tick-pathogen interface and the requirements for successful ongoing transmission.
MATERIALS AND METHODS
Cell lines, bacterial strains, and culture conditions.
DAE100T cells isolated from D. andersoni embryonated eggs (a generous gift from U. G. Munderloh, Department of Entomology, University of Minnesota) were used in all in vitro experiments and were maintained in flasks at 34°C (Sigma-Aldrich) in L-15B complete medium as previously described (17, 18). For in vitro experiments, DAE100T cells were grown to confluence on chambered glass slides (Nunc) or coverslips and were synchronously infected with the St. Maries strain of A. marginale (19), except those used for the pH determination studies. Because the pH determination studies required live imaging, DAE100T cells were infected with red fluorescent protein (RFP)-tagged A. marginale (Virginia strain) isolates (a generous gift from A. Barbet, University of Florida) (20).
Evaluation of A. marginale sensitivity to gentamicin.
In order to verify the efficacy of gentamicin at 50 μg/ml in inhibiting the infectivity of extracellular A. marginale and the lack of killing of intracellular A. marginale, three technical replicates of cell-free A. marginale isolates were incubated in media with and without 50 μg/ml of gentamicin (Gibco, Gaithersburg, MD) for 15 min. The gentamicin-exposed and gentamicin-unexposed A. marginale isolates were then inoculated into monolayers of DAE100T cells and were allowed to infect them for 1 h. After infection, the gentamicin-treated and gentamicin-untreated A. marginale isolates were maintained with and without 50 μg/ml gentamicin. At days 3, 5, and 7 postinoculation, the resulting mean percentages of A. marginale-infected DAE100T in each group were determined by counting the number of infected cells per 100 cells on methanol-fixed, Giemsa-stained cytospins (Shandon Southern Instruments, Sewickley, PA). No A. marginale colonies were detected at any time point in the D. andersoni cells that were inoculated with gentamicin-treated, cell-free A. marginale isolates, indicating complete inhibition of infectivity. In comparison, over 70% of the cells that were inoculated with untreated cell-free A. marginale were infected. The mean percentages of infected cells maintained with and without gentamicin did not significantly change over time in the gentamicin-treated (mean, 72.7%; standard deviation [SD], 1.5) and untreated (mean, 78.4%; SD, 1.6) cells (factor B [see “Statistical analyses” below]; P = 0.190). Thus, exposure to 50 μg/ml gentamicin resulted in a complete loss of infectivity in cell-free A. marginale but had no significant impact on the growth of intracellular A. marginale. The absence of antibiotic-induced cytotoxicity was verified by comparing viable cell counts between culture cells maintained in the gentamicin-free medium and the medium that contained gentamicin using trypan blue.
Synchronized infections of culture cells.
To achieve synchronized infections, a tick-cell-free A. marginale inoculum was prepared by passing heavily infected DAE100T cells through a 27-gauge needle to rupture the cells and release the bacteria into the L-15B complete medium (21). The medium was then filtered through a 5.0-μm-pore-size syringe filter (Whatman) to remove large cellular debris. Complete rupture of the cells was verified by staining the filtrate with trypan blue and evaluating it by light microscopy. To calculate a multiplicity of infection (MOI), the number of A. marginale cells in the resulting filtrate was enumerated using a Petroff-Hausser counting chamber. A single cell-free A. marginale inoculum was divided and used for all experiments. An approximate MOI of 10 was used in each experiment. After 1 h to allow for infection, cells were washed and maintained in a medium containing 50 μg/ml of gentamicin to remove extracellular bacteria and prevent reinfection of cells for the length of the experiment. In experiments performed to determine the role of bacterial protein synthesis, 20 μg/ml tetracycline or a similar volume of 70% ethanol as a vehicle control was added to the medium for 1 h prior to imaging to inhibit bacterial protein synthesis. Cells for all of the experiments, except the pH determination, were fixed in 4% paraformaldehyde (Sigma-Aldrich) after 1 h, 5 h, 24 h, 5 days, 10 days, and 21 days of infection and then processed for confocal microscopy. All experiments were done and assessed in triplicate.
Antibodies.
Primary antibodies to detect A. marginale included mouse monoclonal antibodies directed against major surface protein 5 (Msp5) (AnaF16C1) at 1 μg/ml and major surface protein 2 (Msp2) (AnaR4901) at 2 μg/ml in Western blotting and immunofluorescence assays, respectively (22, 23). Horseradish peroxidase (HRP)-conjugated anti-mouse IgG(H+L) (Kirkegaard and Perry Labs) at 0.1 μg/ml served as the secondary antibody in Western blot assays. Goat anti-mouse IgG(H+L) Alexa Fluor 488 (Invitrogen) at 5 μg/ml served as the secondary antibody in immunofluorescence assays.
All of the antibodies that were used to detect Rab-GTPases and organelle markers were obtained from Abcam as affinity-purified rabbit polyclonal antibodies and were used at 1 μg/ml in Western blot assays and 2 μg/ml in immunofluorescent assays. Anti-Rab5 (ab31261) and anti-Rab7 (ab187868) antibodies were used to detect the early endosome and late endosome, respectively. Anti-Rab4 (ab13252) and anti-Rab11 (ab65200) antibodies were used to detect the recycling endosome. Anti-lysosome-associated endosomal protein 1 (LAMP1) (ab30687) and anti-cathepsin L and V antibodies (ab6314) were used to detect lysosomes. Anti-calnexin (ab22595) and anti-calreticulin (ab2907) antibodies were used to detect the endoplasmic reticulum (ER), and anti-58K Golgi protein (formimidoyltransferase cyclodeaminase) and anti-GM130 (ab30637) antibodies were used to detect the Golgi apparatus. The anti-Rab5, Rab11, Rab7, LAMP1, calnexin, and 58K Golgi protein antibodies were used on tick sections. A rabbit IgG polyclonal isotype control (ab171870) and a mouse IgG isotype control (ab37355) at 2 μg/ml were used as isotype controls to test the primary antibody specificity. In immunofluorescence assays, an anti-rabbit IgG(H+L) Alexa Fluor 594 conjugate (Invitrogen) at 5 μg/ml was used as the secondary antibody.
Western blotting and LC-MS/MS.
Western blotting and liquid chromatography-tandem mass spectrometry (LC-MS/MS) were performed to ensure that the anti-Rab-GTPase antibodies and organelle markers did not cross-react with A. marginale proteins and that they recognized the appropriate D. andersoni proteins. For Western blot assays, 50 μg of total proteins from lysates of DAE100T cells and 50 μg of uninfected and 50 μg of A. marginale-infected bovine erythrocytes, which were washed to remove leukocytes, were analyzed by immunoblotting as previously described (24), with the exception that the separated proteins were transferred to a polyvinylidene fluoride membrane (Millipore) rather than to nitrocellulose. No proteins were detected in either A. marginale-infected or uninfected erythrocytes, and the antibodies did not cross-react with each other or bind nonspecifically to the culture cells and A. marginale (Fig. 1A and B). All antibodies recognized bands of the appropriate size in D. andersoni cells. Bands were analyzed by LC-MS/MS as previously described (25–27) at the University of Idaho Mass Spectrometry Core. Analysis of peptides was done using reverse-phase liquid chromatography on a Waters nanoAcquity ultra performance liquid chromatograph. Peptide tandem mass spectrometry (MS/MS) spectral data were generated using a Waters Micromass Q-Tof Premier quadrupole time of flight mass spectrometer using a nanospray electrospray ionization inlet controlled with MassLynx v4.1 software. Data acquired using a label-free expression profiling method were compared to the NCBI eukaryotic protein database using ProteinLynx Global Server (PLGS) software v2.3 (Waters Corp.). Protein identifications were based on one or more tryptic peptides that yielded a PLGS score greater than 18 corresponding to a probability of 95% or greater that the peptide match was not a random event. All peptides had significant PLGS scores (P < 0.05) for the expected protein using LC-MS/MS (Table 1).
FIG 1.

Western blot detection of endosomal and secretory pathway proteins from D. andersoni and specificity of immunofluorescence assays. (A) Equivalent amounts of protein (50 μg) from DAE100T cells (lane 1), uninfected erythrocytes (lane 2), and A. marginale-infected erythrocytes (lane 3) are in each lane. In panel i, the anti-Msp5 antibody detects a protein of the expected size (20 kDa), confirming the presence of A. marginale in the infected erythrocytes. In panels ii to x, the anti-Rab and anti-organelle antibodies detect bands of the expected size in DAE100T cells (lane 1) as follows: Rab5 (26 kDa), Rab4 (23 kDa), Rab11 (24 kDa), Rab7 (23 kDa), LAMP1 (120 kDa), cathepsin L (37 kDa), calnexin (67 kDa), calreticulin (50 kDa), and 58K Golgi protein, as indicated by the asterisks in panels ii, iii, iv, and v. None of the antibodies recognize A. marginale (lane 3). (B) To demonstrate the specificity of the primary and secondary antibodies, A. marginale-infected DAE100T cells were incubated with only secondary antibodies (no primary) or primary antibodies targeting cellular markers (anti-Rab5 antibody shown here), A. marginale Msp2, or murine or rabbit isotype controls (IgG-IC). All assays were performed using anti-rabbit IgG (red) and anti-murine IgG (green) secondary antibodies.
TABLE 1.
Identification of targeted tick proteins by Western blotting and LC-MS/MS
| Antibody target | Molecular mass (kDa)a | Accession no.b | BLAST homolog descriptionb | PLGS scorec | No. of peptide matchesd | Coverage (%)e |
|---|---|---|---|---|---|---|
| Rab5 | 26 | JAB84144.1 | Putative Ras-related protein Rab5b (Ixodes ricinus) | 265 | 5 | 33 |
| Rab4 | 23 | AAA80151.1 | Rab4 (Dictyostelium discoideum) | 208 | 2 | 12 |
| Rab11 | 24 | JAA58910.1 | Putative Ras-related protein Rab11a (Rhipicephalus pulchellus) | 192 | 5 | 23 |
| Rab7 | 23 | JAB83106.1 | Putative Rab7 member Ras oncoprotein family (Ixodes ricinus) | 707 | 8 | 49 |
| LAMP1 | 120 | XP_007466423.1 | Predicted lysosome-associated membrane glycoprotein 1 (Lipotes vexillifer) | 89 | 4 | 17 |
| Cathepsin L | 37 | JAC35239.1 | Putative cathepsin l-like cysteine proteinase b (Amblyomma triste) | 179 | 4 | 19 |
| Calnexin | 67 | EEH07274.1 | Calnexin (Histoplasma capsulatum) | 123 | 5 | 13 |
| Calreticulin | 50 | AAR29942.1 | Calreticulin (Dermacentor andersoni) | 512 | 6 | 18 |
| 58K Golgi protein | 58 | NP_001192328.1 | Formimidoyltransferase cyclodeaminase/58K Golgi protein (Bos taurus) | 111 | 4 | 6 |
Molecular mass of band on SDS-PAGE.
Homolog with the highest Protein Lynx Global Server (PLGS) score as identified by the Basic Local Alignment Search Tool (BLAST).
PLGS ion scores of ≥18 correspond to a probability ≥95% that the peptide match is not random and indicate identity or extensive homology.
Number of peptides identified from each protein.
Percentage of coverage of target protein by the identified peptide.
Tick infection and processing.
All protocols involving the use of animals were approved by the University of Idaho Institutional Animal Care and Use Committees (ASAF number 2013-66). Adult male D. andersoni ticks (Reynolds Creek colony, USDA-ARS-Animal Disease Research Unit [ADRU]) were fed for 7 days during increasing A. marginale St. Maries strain bacteremia or on an uninfected calf. Ticks were then held at 26°C for 7 days to allow for clearance of the blood meal from the midgut lumen. The infected and control D. andersoni ticks were fixed whole in 4% paraformaldehyde, embedded in paraffin, and thin sectioned onto glass slides. Following deparaffinization and rehydration, the sections were treated with heated sodium citrate buffer (pH 6.0) to recover antigens and were then processed in triplicate for confocal microscopy.
Confocal microscopy.
For endocytic pathway and organelle marker localization studies, fixed infected and uninfected control DAE100T cells and thin paraffin sections of infected and uninfected ticks were labeled for indirect immunofluorescence microscopy. Briefly, the cells and sections were blocked and permeabilized with 1% bovine serum albumin (BSA) and 0.05% Triton X-100 in phosphate-buffered saline (PBS) for 30 min and then double labeled with either the anti-A. marginale and anti-Rab-GTPases or the anti-organelle antibodies listed above. Additionally, infected cells were incubated with no primary antibody, with only one primary antibody at a time, or with an isotype control listed above followed by the secondary antibodies to control for nonspecific antibody binding and antibody cross-reaction. Coverslips were mounted with ProLong Gold antifade (Invitrogen).
For determination of the pH of the A. marginale-infected vacuoles (AmVs), live DAE100T cells that were infected with the RFP-tagged A. marginale (Virginia strain) isolates and grown for 7 days were incubated in L-15B complete medium only or a medium with 20 μg/ml of tetracycline or a similar volume of 70% ethanol as the vehicle control for 1 h. LysoSensor Green DND-153 and LysoSensor Green DND-189 (Invitrogen) at a concentration of 10 μM were then added to the media for 2 h at 34°C. Live cells were evaluated by confocal microscopy.
Imaging included evaluation of marker localization on individual 1- to 2-μm-thick planes in a Z series and was performed using a Leica TCS SP8 X confocal laser-scanning microscope under an HC PL APO CS2 63×/1.20 WATER objective running Leica application suite X. Three-dimensional renderings of Z-planes were evaluated when additional spatial details were required to verify a positive or negative call. Detection of green fluorescence from antibodies bound to Msp2 signaled the location of the AmVs. Red fluorescence was used for cellular markers. The numbers of AmVs labeled with each marker in 100 infected cells from randomly selected fields were counted. Statistical analyses were conducted on the mean of three technical replicates.
Electron microscopy.
Infected DAE100T and uninfected control cells were fixed in 2% paraformaldehyde and 2% glutaraldehyde in 0.1 M cacodylate buffer at 4°C. Tissues were rinsed in 0.1 M cacodylate buffer, postfixed in 1% OsO4 for 2 h at room temperature, and then rinsed in cacodylate buffer. Following dehydration in an ethanol gradient, samples were infiltrated with acetone and embedded in Spurr's resin. Thin sections (90 nm) were placed on nickel grids and stained in 4% uranyl acetate for 10 min and in Reynolds lead for 3 min. Sections were examined on an FEI Tecnai G2 20 Twin transmission electron microscope fitted with a 4K Eagle camera.
Statistical analyses.
Statistical analyses were performed using SigmaPlot v11.0 (Systat Software, Inc.). The average of three technical replicates was analyzed for each experiment. The data for gentamicin sensitivity assays were analyzed by two-way repeated measures analysis of variance (ANOVA) followed by pairwise multiple comparison procedures (Holm-Sidak method). Incubations of cell-free A. marginale in the media with and without gentamicin were considered the two levels of factor A, and the cultures of the infected D. andersoni cells in media with and without gentamicin were considered the two levels of factor B. Measurements were repeated on 3 days, 5 days, and 7 days postinoculation.
For the immunofluorescent assays, the means of the percentages of AmVs labeled with each marker at each time point were compared using a two-way ANOVA with interaction followed by multiple pairwise comparison testing. The organelle marker was considered factor A, and the time postinoculation was considered factor B. The Holm-Sidak method was used to correct the family-wise error rate for post hoc multiple comparisons. Differences were considered statistically significant at a P value of ≤0.05. A two-way profile plot of the least mean squares, including the pairwise comparison significances from the two-way ANOVA, was used to assess the relative impact of each factor level. Statistically significant differences between tetracycline-treated cells and untreated cells were determined using a one-way ANOVA with the same rejection criteria. Student's t tests with statistical significance set at a P value of ≤0.05 were used to evaluate data from the experiments determining AmV pH.
RESULTS
AmVs label concurrently with early, recycling, and late endosome markers, retain early and recycling endosome markers for long periods, and do not fuse with lysosomes.
We determined whether the endosomal markers representing the early, late, and recycling endosome and lysosome (red fluorescence) were recruited to the AmV in D. andersoni cells at 1 h, 5 h, 1 day, 5 days, 10 days, and 21 days postinoculation. Images from 1 h, 5 h, and 1 day are similar, as are images from 10 days and 21 days. Representative images are presented (Fig. 2 to 7). We defined each individual AmV as positively labeled with its respective fluorescent antibody when there was either segmental or circumferential punctate intensity around the A. marginale morulae in any plane of a Z series. This is shown in the first panel of Fig. 1B, 2 to 7, and 12 to 15 in grayscale and in the merged panels of those figures in red/yellow color. None of the anti-Rab or antiorganelle antibodies bound to A. marginale. A representative example of this is shown for anti-Rab5 antibody in Fig. 1B. Similarly, the anti-Msp2 antibody did not bind tick cell proteins. No signal was present in the absence of primary antibodies (Fig. 1B, left). Additionally, no secondary antibody fluorescent signal was detected in cells incubated with isotype controls (Fig. 1B, right).
FIG 2.

AmVs accumulate and retain the early endosomal compartment marker Rab5. Indirect immunofluorescence localization of Rab5 using anti-Rab5 antibodies (red) in D. andersoni cells that were synchronously infected with A. marginale labeled with anti-Msp2 antibodies (green) or mock infected with medium (uninfected). Recruitment of Rab5 is not apparent at 1 h postinoculation. Rab5 accumulates and is detected as circumferential staining (arrowhead) on the majority of AmV membranes 5 days postinoculation and is retained at 10 days postinoculation (arrowhead). Inhibition of bacterial protein synthesis (Tetracycline Rx) results in the loss of Rab5 labeling of the AmVs. The uninfected control shows the pattern of staining of Rab5 in D. andersoni cells. The box in the Merge+4′,6-diamidino-2-phenylindole (DAPI) image outlines the area of magnification in the Enlarged column. Scale bars represent 10 μm on a 63×/1.20 objective. The white arrowhead in the first column indicates the location of the AmV and the area enlarged in the inset. All images are at the same magnification, with the exception of the insets and last column, which are enlarged 3-fold.
FIG 7.

AmVs do not accumulate the lysosome protease cathepsin L throughout infection. Indirect immunofluorescence localization of cathepsin L using anti-cathepsin L antibodies (red) in D. andersoni cells that were synchronously infected with A. marginale labeled with anti-Msp2 antibodies (green) or mock infected with medium (uninfected). Labeling with cathepsin L in AmVs is not apparent at 1 h, 5 days, and 10 days postinoculation. Inhibition of bacterial protein synthesis (Tetracycline Rx) resulted in cathepsin L labeling within the majority of AmVs. The uninfected control shows the pattern of staining of cathepsin L in D. andersoni cells. The box in the Merge+DAPI image outlines the area of magnification in the Enlarged column. Scale bars represent 10 μm on a 63×/1.20 objective. All images are at the same magnification, with the exception of the insets and last column, which are enlarged 3-fold. The white arrowhead in the first column indicates the location of the AmV and the area enlarged in the inset.
FIG 12.

AmVs accumulate and retain the endoplasmic reticulum marker calnexin. Indirect immunofluorescence localization of calnexin using an anti-calnexin antibody (red) in D. andersoni cells that were synchronously infected with A. marginale labeled with anti-Msp2 antibodies (green) or mock infected with medium (uninfected). Calnexin accumulates around AmVs 5 days postinoculation and is retained at 10 days postinoculation. The box in the Merge+DAPI image outlines the area of magnification in the Enlarged column. The uninfected control shows the pattern of calnexin staining in D. andersoni cells. Scale bars represent 10 μm on a 63×/1.20 objective. The white arrowhead in the first column indicates the location of the AmV and the area enlarged in the inset. All images are at the same magnification, with the exception of the insets and last column, which are enlarged 3-fold.
FIG 15.
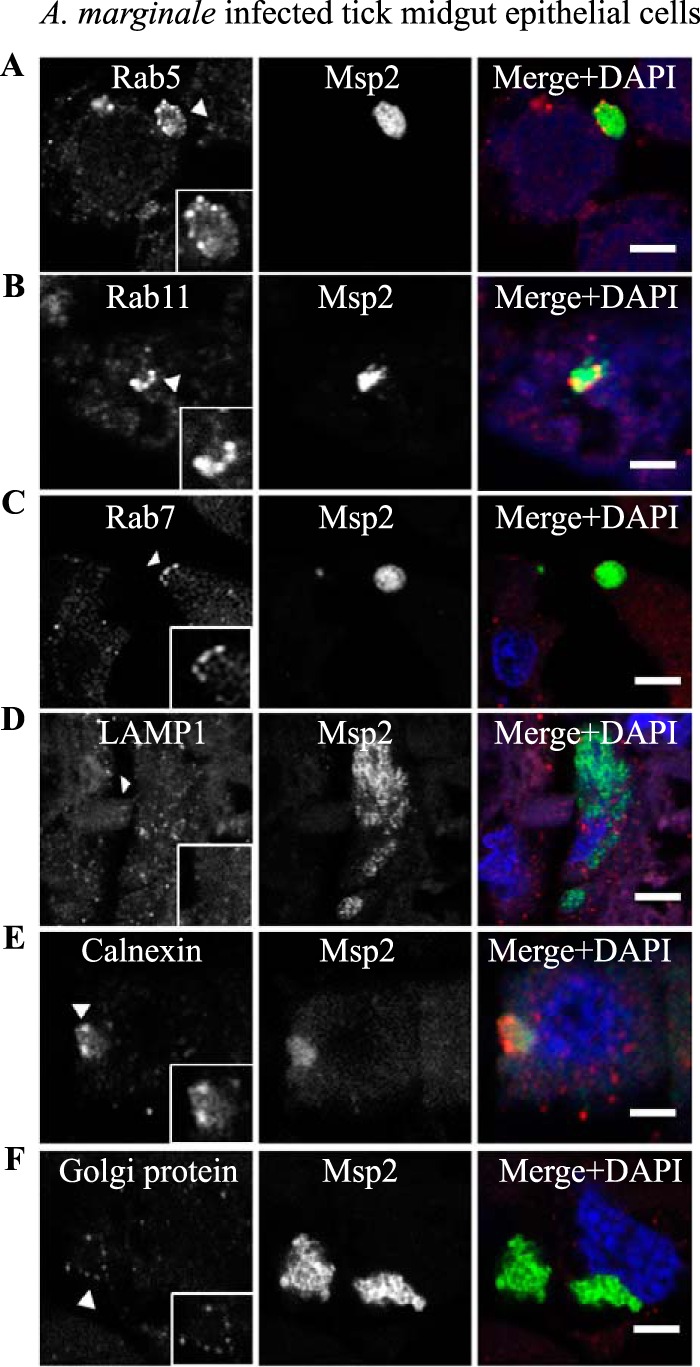
Aberrant labeling of AmVs with early, recycling, and late endosome markers and association with secretory markers in D. andersoni in vivo. The characteristics of the AmVs were evaluated in tissue sections from A. marginale (green)-infected D. andersoni midguts. AmVs in the tick midgut recruited the early endosome marker Rab5 (A), the recycling endosome marker Rab11 (B), and the late endosome marker Rab7 (C). The AmVs did not associate with the lysosome marker LAMP1 (D). The endoplasmic reticulum marker calnexin (E) and the Golgi marker 58K Golgi protein (F) associated with the AmVs. All cellular markers are in red. Scale bars represent 10 μm on a 63×/1.20 objective.
Markers of the early (Rab5; Fig. 2), recycling (Rab4 and Rab11; Fig. 3 and 4, respectively), and late endosome (Rab7; Fig. 5) were recruited to the AmVs. Qualitatively, the intensity of the staining generally increased through time. For an individual AmV, the intensity of the staining at 1 h was inconsistent and subtle, and on day 5, it was consistent and subtle with short segmental to punctate regions of colocalization for Rab5, Rab4, and Rab7. Nearly circumferential staining of the AmV by Rab11 was apparent by day 5 and became more punctate through time. The lysosomal LAMP1 (Fig. 6) and cathepsin L (Fig. 7) did not colocalize with the AmV at any time point. Quantitatively, increasing numbers of AmVs colocalized with Rab5, Rab4, Rab11, and Rab7 from 5 h to 10 days postinoculation, with little increase between days 10 and 21.
FIG 3.

AmVs accumulate and retain the recycling endosomal compartment marker Rab4. Indirect immunofluorescence localization of Rab4 using anti-Rab4 antibodies (red) in D. andersoni cells that were synchronously infected with A. marginale labeled with anti-Msp2 antibodies (green) or mock infected with medium (uninfected). Recruitment of Rab4 is not apparent 1 h postinoculation. Rab4 accumulates on the majority of AmVs 5 days postinoculation and is retained at 10 days postinoculation. Inhibition of bacterial protein synthesis (Tetracycline Rx) results in loss of Rab4 labeling of the AmVs. The uninfected control shows the pattern of staining of Rab4 in D. andersoni cells. The box in the Merge+DAPI image outlines the area of magnification in the Enlarged column. Scale bars represent 10 μm on a 63×/1.20 objective. The white arrowhead in the first column indicates the location of the AmV and the area enlarged in the inset. All images are at the same magnification, with the exception of the insets and last column, which are enlarged 3-fold.
FIG 4.

AmVs accumulate and retain the recycling endosomal compartment marker Rab11. Indirect immunofluorescence localization of Rab11 using anti-Rab11 antibodies (red) in D. andersoni cells that were synchronously infected with A. marginale labeled with anti-Msp2 antibodies (green) or mock infected with medium (uninfected). Recruitment of Rab11 is not apparent 1 h postinoculation. Rab11 accumulates on the majority of AmVs 5 days postinoculation and is retained at 10 days postinoculation. Inhibition of bacterial protein synthesis (Tetracycline Rx) results in loss of Rab11 labeling of the AmVs. The uninfected control shows the pattern of staining of Rab11 in D. andersoni cells. The box in the Merge+DAPI image outlines the area of magnification in the Enlarged column. Scale bars represent 10 μm on a 63×/1.20 objective. The white arrowhead in the first column indicates the location of the AmV and the area enlarged in the inset. All images are at the same magnification, with the exception of the insets and last column, which are enlarged 3-fold.
FIG 5.

AmVs accumulate and retain the late endosomal compartment marker Rab7. Indirect immunofluorescence localization of Rab7 using anti-Rab7 antibodies (red) in D. andersoni cells that were synchronously infected with A. marginale labeled with anti-Msp2 antibodies (green) or mock infected with medium (uninfected). Labeling with Rab7 is not apparent 1 h postinoculation. Rab7 accumulates on the majority of AmVs 5 days postinoculation and is retained at 10 days postinoculation. Inhibition of bacterial protein synthesis (Tetracycline Rx) did not impact Rab7 labeling of the AmVs. The uninfected control shows the pattern of staining of Rab7 in D. andersoni cells. The box in the Merge+DAPI image outlines the area of magnification in the Enlarged column. Scale bars represent 10 μm on a 63×/1.20 objective. The white arrowhead in the first column indicates the location of the AmV and the area enlarged in the inset. All images are at the same magnification, with the exception of the insets and last column, which are enlarged 3-fold.
FIG 6.

AmVs do not accumulate the lysosome marker LAMP1 throughout infection. Indirect immunofluorescence localization of LAMP1 using anti-LAMP1 antibodies (red) in D. andersoni cells that were synchronously infected with A. marginale labeled with anti-Msp2 antibodies (green) or mock infected with medium (uninfected). Labeling with LAMP1 is not apparent on AmVs at 1 h, 5 days, and 10 days postinoculation. Inhibition of bacterial protein synthesis (Tetracycline Rx) resulted in the labeling of the majority of AmVs. The uninfected control shows the pattern of staining of LAMP1 in D. andersoni cells. The box in the Merge+DAPI image outlines the area of magnification in the Enlarged column. Scale bars represent 10 μm on a 63×/1.20 objective. The white arrowhead in the first column indicates the location of the AmV and the area enlarged in the inset. All images are at the same magnification, with the exception of the insets and last column, which are enlarged 3-fold.
Quantitative data are presented in a bar graph (Fig. 8A). The effects of each marker and time and the relevant significant differences are presented in a profile analysis for ease of visual comparison across many groups (Fig. 8B). The mean numbers of AmVs that were labeled with Rab5, Rab4, Rab11, and Rab7 at a given time point between days 1 and 21 were similar to each other and statistically significantly different from LAMP1 and cathepsin L with a P value of <0.001 for each pairwise comparison (Fig. 8A and B). Between 1 h and 1 day, small numbers of vacuoles were labeled with each of the markers, with an average of 3% positive AmVs for any given marker at 1 h (Fig. 8A and B). By 5 days postinoculation, the majority of AmVs were labeled with all of the markers except LAMP1 and cathepsin L. On average, by day 5, 70% of AmVs accumulated Rab5, a GTPase whose conserved function is the formation of the early endosomal compartment (Fig. 2 and 5), and 59% and 61% of AmVs accumulated Rab4 and Rab11, respectively, whose conserved transport functions are endocytic recycling (Fig. 3, 4, and 8). Interestingly, on average, 58% of AmVs accumulated Rab7, a marker of the late endosome compartment (Fig. 5 and 8), whose conserved transport functions include moving vesicles from the early endosome compartment to the late endosome, lysosome biogenesis, and phagosome maturation (12). The percentage of AmVs labeled with the early, recycling, and late endosome markers increased until day 10, with minimal changes between day 10 and day 21 (Fig. 8A). The increase in positively labeled AmVs was statistically significant (P < 0.001) from days 1 to 10, with no significant differences from days 10 to 21 (Fig. 8B). Despite the early recruitment and retention of Rab7, the AmVs did not fuse with the lysosome as indicated by the failure of significant numbers of AmVs to be labeled with LAMP1 (Fig. 6 and 8) at any time point. Additionally, cathepsin L, one of the predominant intracellular lysosomal enzymes of ticks (7), did not localize significantly in the vacuoles that contained A. marginale (Fig. 7 and 8).
FIG 8.
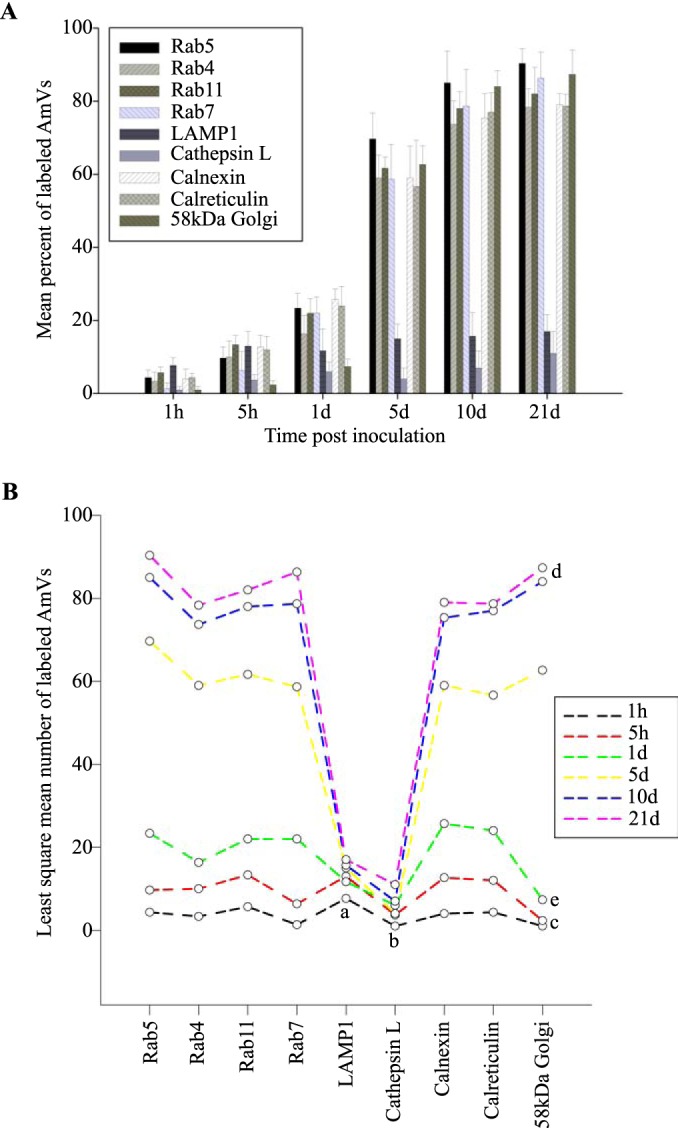
Statistical analysis of the mean number of AmVs labeled with a Rab or organelle marker through time. (A) Mean of the percentages (± standard deviation [SD]) of infected D. andersoni AmVs immunolabeled with a specified fluorescent marker. Cells were synchronously infected with A. marginale from 1 h to 21 days. Progressively increasing numbers of AmVs labeled concurrently with early, recycling, and late endosome and organelle markers but not with LAMP1 or cathepsin L. All recruited markers were retained up to 21 days of infection. (B) Profile plot of the least means squares of the data in panel A demonstrating the relative between-group and within-group effects of time postinoculation and recruitment of the markers. From a two-way ANOVA followed by pairwise comparisons, all LAMP1 (a) and all cathepsin L (b) means were not different from each other but were significantly different from all other markers measured after 1 day. For each of the markers other than LAMP1 and cathepsin L, the means at 1 h and 5 h (c) were similar to each other as were means at 10 days and 21days (e). The means at 1 h and 5 h as well as the means at 10 days and 21 days were statistically significantly different from the means at 1 day and 5 days, with the exception of Rab4 and Rab11 means at 5 h and 1 day (d). The means of the labeled AmVs for each of the markers at a given mean at each time point were not significantly different, except when compared to LAMP1 and cathepsin L. All statistically significant pairwise comparisons had a P value of <0.001.
AmVs have a neutral pH.
The recruitment of markers to AmVs that define all three endosome compartments and the failure to fuse with the acidic lysosomes and label with acid proteases indicated the failure of normal endosome maturation. Given that acidification is an important component of normal endosomal maturation, we investigated whether this too was perturbed. We determined the pH of the AmVs in live D. andersoni cells infected with RFP-tagged A. marginale (Virginia strain) isolates using LysoSensor pH dyes, which provide semiquantitative readouts. LysoSensor Green DND-189 is nonfluorescent around a neutral pH (pH 7) and has a maximum fluorescent intensity inside acidic compartments around a pH of 2 to 5, whereas LysoSensor Green DND-153 is brightly fluorescent around a neutral pH (pH 7 to 8). These two dyes were used in parallel to bracket the AmV pH. LysoSensor DND-153 accumulated and had high fluorescent intensity in an average of 62% of AmVs (Fig. 9A, top). In the inverse experiment, the acidic organelle LysoSensorDND-189 had detectable fluorescent intensity in only 12% of untreated AmVs on average, a significantly lower percentage than the neutral pH indicator (P < 0.001) (Fig. 9B, top). Combined, these findings indicated that AmVs were near neutral pH and not acidic pH.
FIG 9.
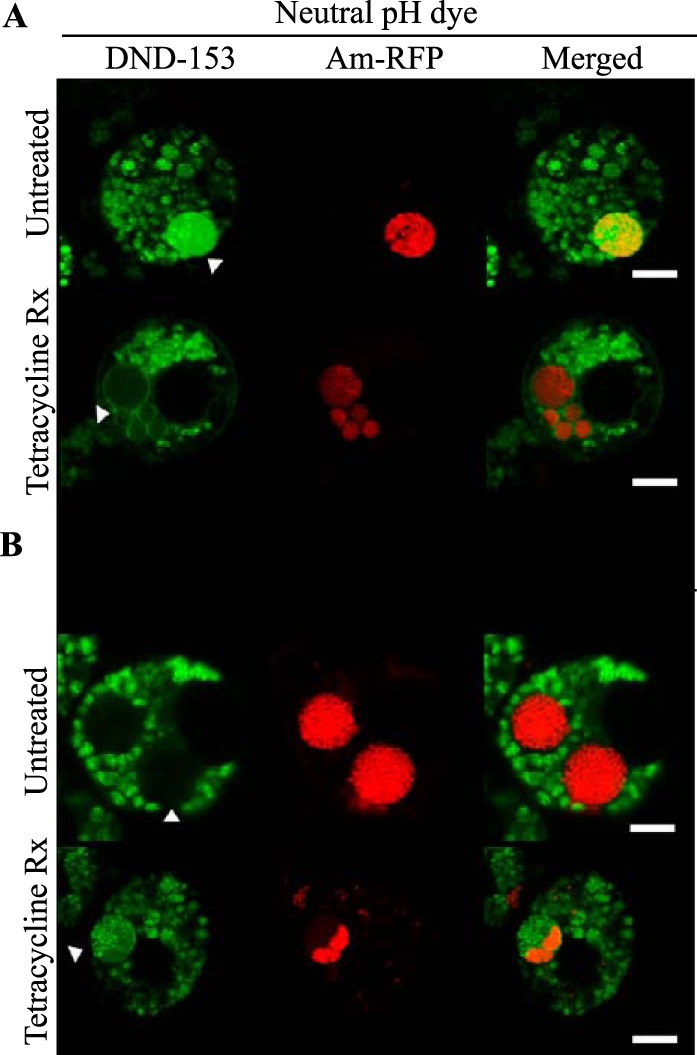
AmVs have a neutral pH and require bacterial protein synthesis to maintain neutral pH. D. andersoni cells that were synchronously infected with RFP-tagged A. marginale (Am-RFP) were treated with 20 μg/ml of tetracycline or a similar volume of 70% ethanol as a vehicle control for 1 h. (A) Untreated AmVs accumulated the fluorescent neutral pH dye LysoSensor Green DND-153. Bacterial protein synthesis inhibition (Tetracycline Rx) significantly reduced accumulation of the fluorescent neutral pH dye in the AmV, suggesting acidification of the vacuole. (B) In an inverse experiment, untreated AmVs failed to accumulate the fluorescent acidic pH dye LysoSensor Green DND-189, except when bacterial protein synthesis was inhibited (Tetracycline Rx).
Bacterial protein synthesis is required for long-term retention of early and recycling markers in the AmVs, avoidance of lysosomal fusion, and maintenance of neutral pH.
To determine whether active A. marginale protein synthesis is required to maintain the AmVs, we treated infected cells with tetracycline to inhibit bacterial protein production. Inhibition of bacterial protein synthesis significantly reduced the labeling of AmVs with Rab5, Rab4, and Rab11 (Fig. 2, 3, 4, and 10) from averages of 85%, 76%, and 79% in untreated controls to averages of 12%, 14%, and 9%, respectively, in tetracycline-treated infected cells (P < 0.001 for all pairwise comparisons between treated and untreated). Conversely, labeling with the lysosomal marker LAMP1 and cathepsin L increased significantly from 16% and 14% in untreated controls to 93% and 95% in tetracycline-treated infected cells, respectively (Fig. 7, 8, and 10) (P < 0.001 for all pairwise comparisons between treated and untreated). There was no significant alteration in AmV labeling with Rab7 between tetracycline-treated AmVs (94%) and controls (86%). Thus, maintenance of the AmV and the avoidance of fusion with the lysosome required active protein synthesis from A. marginale. Additionally, inhibition of bacterial protein synthesis resulted in the failure of AmVs to maintain a neutral pH (Fig. 9A and B, bottom). Significantly more AmVs in treated cells accumulated the acidic pH dye DND-189 (76%), and less accumulated the neutral dye DND-153 (16%) compared to untreated controls in which 12% and 62% of infected cells accumulated the acidic and neutral dyes on average, respectively (P < 0.001). In electron micrographs of tetracycline-treated infected cells, AmVs were often found within large multimembrane vacuoles with organelles (Fig. 11A). This, together with the acidification and labeling with lysosomal markers, is consistent with normal maturation of the vacuoles toward phagolysosomes in the absence of active bacterial protein synthesis.
FIG 10.
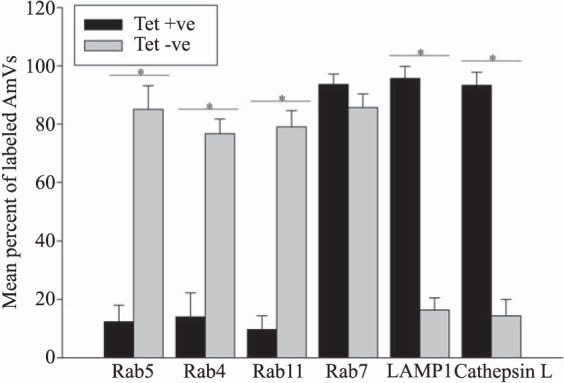
Retention of the early and recycling markers on the AmVs and avoidance of lysosomal fusion require bacterial protein synthesis. Mean of the percentages (± SD) of infected D. andersoni AmVs labeled with a specified marker. Cells that were synchronously infected with A. marginale were untreated (Tet −ve) or treated (Tet +ve) with tetracycline for 1 h. Asterisks denote statistically significantly different means with a P value of <0.001.
FIG 11.

The AmVs are associated with the rough endoplasmic reticulum and mature to phagolysosomes in the absence of bacterial protein synthesis. D. andersoni cells that were infected with A. marginale were evaluated by transmission electron microscopy. (A) In tetracycline-treated infected D. andersoni cells (tetracycline treated, enlarged), colonies of A. marginale are often within multimembranous phagolysosomes with other organelles (white arrow). An untreated control (Am-iDAE untreated control) demonstrates AmVs unfused to other cellular vesicles and closely associated with recruited RER (black arrow). (B) The rough endoplasmic reticulum is closely applied to the outer aspect of the AmV and is devoid of ribosomes on the contact side (black arrow) in Am-iDAE untreated and at a higher magnification (RER enlarged). Long strands of RER are adjacent to the nucleus in the uninfected control.
AmVs associate with vacuoles from the rough endoplasmic reticulum and Golgi apparatus.
Bacterial survival in intracellular vacuoles requires a mechanism for nutrient acquisition. Association with the nutrient-rich secretory vesicles of the host is a common mechanism used by intracellular bacteria in mammalian host cells (16). To investigate whether this mechanism was employed in tick cells, we determined whether markers of the endoplasmic reticulum or Golgi apparatus were associated with the AmVs. A low percentage of AmVs (4%) were labeled with the secretory markers as early as 1 h postinfection (Fig. 8A and B), indicating early recruitment of secretory markers. Recruitment of the markers of the endoplasmic reticulum, calnexin (60%), and calreticulin (63%) (28) and of the 58K Golgi protein (65%) increased significantly by 5 days postinoculation in pairwise comparisons with all earlier times for the respective markers and with all sampling times of LAMP1 and cathepsin L (P < 0.001 in each pairwise comparison). The percentage of AmVs positively labeled with secretory markers continued to increase from 5 days to 10 days postinoculation, with little change between day 10 and day 21 (Fig. 8A, 12, 13, and 14). As reported previously for the AmV in ISE6 cells, ultrastructural evaluation (Fig. 11B) confirmed that rough endoplasmic reticulum (RER) that was devoid of ribosomes on the contact side segmentally covered the outer aspect of the AmVs (29).
FIG 13.

AmVs accumulate and retain the endoplasmic reticulum marker calreticulin. Indirect immunofluorescence localization of calreticulin using an anti-calreticulin antibody (red) in D. andersoni cells that were synchronously infected with A. marginale labeled with anti-Msp2 antibodies (green) or mock infected with medium (uninfected). Calreticulin accumulates around AmVs 5 days postinoculation and is retained at 10 days postinoculation. The uninfected control shows the pattern of staining of calreticulin D. andersoni cells. The box in the Merge+DAPI image outlines the area of magnification in the Enlarged column. Scale bars represent 10 μm on a 63×/1.20 objective. The white arrowhead in the first column indicates the location of the AmV and the area enlarged in the inset. All images are at the same magnification, with the exception of the insets and last column, which are enlarged 3-fold.
FIG 14.

AmVs accumulate the 58K Golgi protein and retain it throughout infection. Indirect immunofluorescence localization of Golgi proteins using an anti-58K Golgi protein antibody (red) in D. andersoni cells that were synchronously infected with A. marginale labeled with anti-Msp2 antibodies (green) or mock infected with medium (uninfected). AmVs associate with the Golgi marker by 5 days postinoculation, and it is retained at 10 days postinfection. The uninfected control shows the pattern of staining of the 58K Golgi protein in D. andersoni cells. The box in the Merge+DAPI image outlines the area of magnification in the Enlarged column. Scale bars represent 10 μm on a 63×/1.20 objective. The white arrowhead in the first column indicates the location of the AmV and the area enlarged in the inset. All images are at the same magnification, with the exception of the insets and last column, which are enlarged 3-fold.
In D. andersoni ticks, AmVs were associated with early, recycling, and late endosome markers, the endoplasmic reticulum, and the Golgi apparatus but not with lysosomal markers.
To confirm that the features of AmVs identified in cultured tick cells are representative of those expressed in vivo, we evaluated the association of the endosome and organelle markers with established A. marginale colonies in adult male D. andersoni midguts. Similar to our findings in vitro, established AmVs retained the early endosome marker Rab5, the recycling endosome marker Rab11, and the late endosome marker Rab7. The labeling was punctate and subtle (Fig. 15A to C). Also, similar to findings in cultured cells, the lysosome marker LAMP1 did not associate with the AmVs (Fig. 15D). In the midgut, the colocalization of the AmV with the endoplasmic reticulum marker calnexin and the Golgi marker 58K Golgi protein (Fig. 15E and F) also occurred, adding support to the in vitro observations.
DISCUSSION
We accept the hypothesis that A. marginale actively modulates its vacuole and maintains this modulation over the time required for onward transmission. This conclusion is supported by four lines of evidence. (i) The AmV phenotype is aberrant and has features of the early, recycling, and late endosome with close association with the ER and Golgi apparatus. (ii) The AmV fails to acidify and excludes lysosomal enzymes. (iii) This aberrant phenotype is stably maintained for a minimum of 21 days, a period sufficient for transmission. (iv) In the absence of A. marginale protein synthesis, the AmV loses the markers of the early and recycling endosome, acidifies, and accumulates lysosomal enzymes as modeled in Fig. 16.
FIG 16.

Model summarizing the interaction of the AmV with proteins from the endocytic and secretory pathways. The early endosome marker Rab5, the recycling endosome markers Rab4 and Rab11, the late endosome marker Rab7, the rough endoplasmic reticulum and endoplasmic reticulum markers calnexin and calreticulin, and the Golgi marker 58K Golgi protein were recruited to a neutral pH AmV, while LAMP1 and cathepsin were excluded. The inset at the lower right shows the maturation of the AmV into an acidic, multiorganelle phagolysosome that were labeled with LAMP1, Rab7, and cathepsin L following inhibition of A. marginale protein synthesis. Details of the endocytic and secretory pathways are adapted from references 12, 13, 15, 44, and 45. AmV, Anaplasma marginale-infected tick vacuole; Am, Anaplasma marginale; cG, cis-Golgi; mG, medial-Golgi; tG, trans-Golgi; 58kD GP, 58K Golgi protein; ERC, endocytic recycling center; SG, secretory granule; SRE, slow recycling endosome; RRE, rapid recycling endosome; EE, early endosome; RER, rough endoplasmic reticulum; LE, late endosome; MVB, multivesicular body.
A. marginale in mammals resides in mature erythrocytes, which do not have a nucleus and lack regulated vesicular transport, precluding the establishment of parallels to our findings in the arthropod vector and mammalian host. Little is known about the tick-pathogen interface for other members of the Rickettsiales. However, the Anaplasma phagocytophilum-infected vacuole (ApV) in mammalian cells is well characterized. The ApV failed to associate with Rab5 and Rab7 (30, 31), while the AmVs in tick cells accumulated both. Rab5 and Rab7 are of particular interest, as they, along with associated effector molecules, largely mediate the critical transition from the early to late endosome (32). Subversion of the normal Rab5 to Rab7 transition by intracellular bacterial pathogens is often central to the establishment and maintenance of the replicative niche (33, 34). For example, Listeria monocytogenes and Mycobacterium tuberculosis isolates reside in a modified Rab5 endocytic compartment (16). The Coxiella burnetii-containing vacuole exchanges Rab5 for Rab7. Rab7 remains on the vacuole for the duration of infection. Few pathogens occupy vacuoles that are labeled simultaneously with Rab5 and Rab7 through time. Tropheryma whipplei, the cause of Whipple's disease, and now A. marginale are the recently described exceptions (32). The mechanisms by which this Rab5- and Rab7-positive state is established and maintained are unknown for either pathogen. Since the conserved transport functions of Rab7 include moving vesicles from the early endosome compartment to the late endosome, lysosome biogenesis, and phagosome maturation (12), the maintenance of Rab7 on the AmV without normal maturation suggests interference with or disruption of the normal function of Rab7, its specific guanine nucleotide exchange factors, or GTPase-activating proteins. The restoration of normal maturation of the AmV with the disruption of A. marginale protein synthesis indicates that the Rab7 loss of function is mediated by A. marginale. The maintenance of Rab5 on the AmV and its loss once bacterial protein synthesis is inhibited suggest that its continued presence on the membrane is important in the failure of vacuolar maturation.
Similar to the ApV and Chlamydia sp. inclusions in mammalian cells, the AmV was labeled with Rab4 and Rab11, which regulate recycling endosomes (16, 35). The recycling endosome returns endocytosed cargo to the plasma membrane via the rapid or slow recycling endosome (12, 34, 36). The significance of the recycling endosome to the function and integrity of the pathogen-containing vacuole is unclear, but it may help to stall progression of the vacuole toward a late endosome. Also, similar to most pathogen-containing vacuoles, including the ApV, the AmV fails to acidify, is nonfusogenic with lysosomes, and fails to acquire LAMP1 (30, 31, 37). Failure of the AmVs to acidify likely interferes not only with the activation of the proteases but also with the fusogenicity of the vacuole.
Similar to A. phagocytophilum, the AmVs engaged the ER and the Golgi apparatus early in infection and maintained association throughout the course of infection (29, 38, 39). The ER and the Golgi apparatus are nutrient-rich compartments of the secretory pathway and likely serve as a rich source of glycoprotein and lipids, which can be degraded to supply amino acids, fatty acids, nitrogen, carbon, and energy to auxotrophic A. marginale (40, 41). The dynamics of transport in the secretory pathway are complex and involve many molecules that are targeted by organisms that parasitize these nutrient-rich compartments. These molecules include Rab10 targeted by A. phagocytophilum for hijacking secretory vesicles from the trans-Golgi network (38) and Rab1 used by Legionella pneumophila to engage traffic from the ER (16).
Establishing that A. marginale actively modulates the tick cell raises the question of which specific bacterial effectors and cellular targets are required to establish this vacuolar replicative niche. Additionally, does expression of these bacterial effectors differ among the strains of A. marginale and determine their ability to colonize and be successfully transmitted by specific genera of ticks? This knowledge gap is specifically relevant to understanding the epidemiology of tick-borne A. marginale transmission and requires evaluation of more strains of A. marginale. The ability of genetically distinct A. marginale strains to colonize the tick midgut has been identified as a critical determinant of strain-specific transmission efficiency (5). For example, the infection rate of the St. Maries strain is consistently 100%, while the Mississippi strain is unable to colonize the midgut. A. marginale subsp. centrale has an intermediate phenotype with lower midgut infection rates and levels than the St. Maries strain (5, 42). There are likely many molecules and complex interactions and mechanisms that determine these strain differences, and the characterization of the replicative niche of strains of A. marginale within the tick, as reported here, is the first step in identifying these molecules. Equally notable, Scoles et al. identified significant differences in the susceptibility of geographically and phenotypically distinct populations of D. andersoni to midgut colonization with a single strain of A. marginale (43). Whether this reflects variation in the establishment of the replication permissive intracellular vacuole is a compelling question and highlights the need to understand the bacterial-tick interactions at a molecular level.
ACKNOWLEDGMENTS
We thank Guy Palmer for critical review of the manuscript. We also appreciate the intellectual guidance provided by Kelly Brayton, Glen Scoles, and Viveka Vadyvaloo. We also thank Debby Alperin, Jessie Ujczo, Ralph Horn, James Allison, Kathy Mason, Christine Mary Davitt, Daniel Mullendore, Valerie Lynch-Holm, and Lee Deobald for their expert technical assistance and Ulrike G. Munderloh (Department of Entomology, University of Minnesota) and Anthony Barbet (University of Florida) for cell lines and bacterial strains used in this study.
Mass spectrometry was performed at the University of Idaho Mass Spectrometry Core INBRE under NIH/NIGMS grant P20GM103408. This work was supported by the United States Department of Agriculture under Agricultural Research Service project 5348-32000-033-00D and by the National Institutes of Health under grant R37 AI44005.
Mention of trade names or commercial products in this article is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the U.S. Department of Agriculture.
F. Magunda and S. M. Noh were involved in the design, execution, analysis, and interpretation of all experiments and in the writing of the manuscript. C. W. Thompson and D. Schneider contributed significantly to the design and execution of immunofluorescence labeling experiments and to the review of manuscript drafts.
Funding Statement
The funding agencies had no role in the design, execution, interpretation, and decision to submit the study.
REFERENCES
- 1.Jongejan F, Uilenberg G. 2004. The global importance of ticks. Parasitology 129(Suppl):S3–S14. [DOI] [PubMed] [Google Scholar]
- 2.Ge NL, Kocan KM, Blouin EF, Murphy GL. 1996. Developmental studies of Anaplasma marginale (Rickettsiales:Anaplasmataceae) in male Dermacentor andersoni (Acari:Ixodidae) infected as adults by using nonradioactive in situ hybridization and microscopy. J Med Entomol 33:911–920. doi: 10.1093/jmedent/33.6.911. [DOI] [PubMed] [Google Scholar]
- 3.Kocan KM, Goff WL, Stiller D, Claypool PL, Edwards W, Ewing SA, Hair JA, Barron SJ. 1992. Persistence of Anaplasma marginale (Rickettsiales: Anaplasmataceae) in male Dermacentor andersoni (Acari: Ixodidae) transferred successively from infected to susceptible calves. J Med Entomol 29:657–668. doi: 10.1093/jmedent/29.4.657. [DOI] [PubMed] [Google Scholar]
- 4.Kocan KM, Stiller D, Goff WL, Claypool PL, Edwards W, Ewing SA, McGuire TC, Hair JA, Barron SJ. 1992. Development of Anaplasma marginale in male Dermacentor andersoni transferred from parasitemic to susceptible cattle. Am J Vet Res 53:499–507. [PubMed] [Google Scholar]
- 5.Ueti MW, Reagan JO Jr, Knowles DP Jr, Scoles GA, Shkap V, Palmer GH. 2007. Identification of midgut and salivary glands as specific and distinct barriers to efficient tick-borne transmission of Anaplasma marginale. Infect Immun 75:2959–2964. doi: 10.1128/IAI.00284-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sojka D, Franta Z, Frantova H, Bartosova P, Horn M, Vachova J, O'Donoghue AJ, Eroy-Reveles AA, Craik CS, Knudsen GM, Caffrey CR, McKerrow JH, Mares M, Kopacek P. 2012. Characterization of gut-associated cathepsin D hemoglobinase from tick Ixodes ricinus (IrCD1). J Biol Chem 287:21152–21163. doi: 10.1074/jbc.M112.347922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Horn M, Nussbaumerova M, Sanda M, Kovarova Z, Srba J, Franta Z, Sojka D, Bogyo M, Caffrey CR, Kopacek P, Mares M. 2009. Hemoglobin digestion in blood-feeding ticks: mapping a multipeptidase pathway by functional proteomics. Chem Biol 16:1053–1063. doi: 10.1016/j.chembiol.2009.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sojka D, Franta Z, Horn M, Caffrey CR, Mares M, Kopacek P. 2013. New insights into the machinery of blood digestion by ticks. Trends Parasitol 29:276–285. doi: 10.1016/j.pt.2013.04.002. [DOI] [PubMed] [Google Scholar]
- 9.Lara FA, Lins U, Bechara GH, Oliveira PL. 2005. Tracing heme in a living cell: hemoglobin degradation and heme traffic in digest cells of the cattle tick Boophilus microplus. J Exp Biol 208:3093–3101. doi: 10.1242/jeb.01749. [DOI] [PubMed] [Google Scholar]
- 10.Kocan KM, Holbert D, Ewing SA, Hair JA, Barron SJ. 1983. Development of colonies of Anaplasma marginale in the gut of incubated Dermacentor andersoni. Am J Vet Res 44:1617–1620. [PubMed] [Google Scholar]
- 11.Stenmark H, Olkkonen VM. 2001. The Rab GTPase family. Genome Biol 2:3007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brumell JH, Scidmore MA. 2007. Manipulation of Rab GTPase function by intracellular bacterial pathogens. Microbiol Mol Biol Rev 71:636–652. doi: 10.1128/MMBR.00023-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fischer JA, Eun SH, Doolan BT. 2006. Endocytosis, endosome trafficking, and the regulation of Drosophila development. Annu Rev Cell Dev Biol 22:181–206. doi: 10.1146/annurev.cellbio.22.010605.093205. [DOI] [PubMed] [Google Scholar]
- 14.Satoh AK, Tokunaga F, Ozaki K. 1997. Rab proteins of Drosophila melanogaster: novel members of the Rab-protein family. FEBS Lett 404:65–69. doi: 10.1016/S0014-5793(97)00094-X. [DOI] [PubMed] [Google Scholar]
- 15.Stenmark H. 2009. Rab GTPases as coordinators of vesicle traffic. Nat Rev Mol Cell Biol 10:513–525. doi: 10.1038/nrm2728. [DOI] [PubMed] [Google Scholar]
- 16.Sherwood RK, Roy CR. 2013. A Rab-centric perspective of bacterial pathogen-occupied vacuoles. Cell Host Microbe 14:256–268. doi: 10.1016/j.chom.2013.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Munderloh UG, Kurtti TJ. 1989. Formulation of medium for tick cell culture. Exp Appl Acarol 7:219–229. doi: 10.1007/BF01194061. [DOI] [PubMed] [Google Scholar]
- 18.Munderloh UG, Liu Y, Wang M, Chen C, Kurtti TJ. 1994. Establishment, maintenance and description of cell lines from the tick Ixodes scapularis. J Parasitol 80:533–543. doi: 10.2307/3283188. [DOI] [PubMed] [Google Scholar]
- 19.Eriks IS, Stiller D, Goff WL, Panton M, Parish SM, McElwain TF, Palmer GH. 1994. Molecular and biological characterization of a newly isolated Anaplasma marginale strain. J Vet Diagn Invest 6:435–441. doi: 10.1177/104063879400600406. [DOI] [PubMed] [Google Scholar]
- 20.Crosby FL, Brayton KA, Magunda F, Munderloh UG, Kelley KL, Barbet AF. 2015. Reduced infectivity in cattle for an outer membrane protein mutant of Anaplasma marginale. Appl Environ Microbiol 81:2206–2214. doi: 10.1128/AEM.03241-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Borjesson DL. 2008. Culture, isolation, and labeling of Anaplasma phagocytophilum for subsequent infection of human neutrophils. Methods Mol Biol 431:159–171. [DOI] [PubMed] [Google Scholar]
- 22.Visser ES, McGuire TC, Palmer GH, Davis WC, Shkap V, Pipano E, Knowles DP Jr. 1992. The Anaplasma marginale msp5 gene encodes a 19-kilodalton protein conserved in all recognized Anaplasma species. Infect Immun 60:5139–5144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Scoles GA, Ueti MW, Noh SM, Knowles DP, Palmer GH. 2007. Conservation of transmission phenotype of Anaplasma marginale (Rickettsiales: Anaplasmataceae) strains among Dermacentor and Rhipicephalus ticks (Acari: Ixodidae). J Med Entomol 44:484–491. doi: 10.1093/jmedent/44.3.484. [DOI] [PubMed] [Google Scholar]
- 24.Ducken DR, Brown WC, Alperin DC, Brayton KA, Reif KE, Turse JE, Palmer GH, Noh SM. 2015. Subdominant outer membrane antigens in Anaplasma marginale: conservation, antigenicity, and protective capacity using recombinant protein. PLoS One 10:e0129309. doi: 10.1371/journal.pone.0129309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lopez JE, Siems WF, Palmer GH, Brayton KA, McGuire TC, Norimine J, Brown WC. 2005. Identification of novel antigenic proteins in a complex Anaplasma marginale outer membrane immunogen by mass spectrometry and genomic mapping. Infect Immun 73:8109–8118. doi: 10.1128/IAI.73.12.8109-8118.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Noh SM, Brayton KA, Brown WC, Norimine J, Munske GR, Davitt CM, Palmer GH. 2008. Composition of the surface proteome of Anaplasma marginale and its role in protective immunity induced by outer membrane immunization. Infect Immun 76:2219–2226. doi: 10.1128/IAI.00008-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shevchenko A, Wilm M, Vorm O, Mann M. 1996. Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal Chem 68:850–858. doi: 10.1021/ac950914h. [DOI] [PubMed] [Google Scholar]
- 28.Benham AM. 2012. Protein secretion and the endoplasmic reticulum. Cold Spring Harb Perspect Biol 4:a012872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Truchan HK, Cockburn CL, Hebert KS, Magunda F, Noh SM, Carlyon JA. 2016. The pathogen-occupied vacuoles of Anaplasma phagocytophilum and Anaplasma marginale interact with the endoplasmic reticulum. Front Cell Infect Microbiol 6:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Webster P, Ijdo JW, Chicoine LM, Fikrig E. 1998. The agent of human granulocytic ehrlichiosis resides in an endosomal compartment. J Clin Invest 101:1932–1941. doi: 10.1172/JCI1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mott J, Barnewall RE, Rikihisa Y. 1999. Human granulocytic ehrlichiosis agent and Ehrlichia chaffeensis reside in different cytoplasmic compartments in HL-60 cells. Infect Immun 67:1368–1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mottola G. 2014. The complexity of Rab5 to Rab7 transition guarantees specificity of pathogen subversion mechanisms. Front Cell Infect Microbiol 4:180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rink J, Ghigo E, Kalaidzidis Y, Zerial M. 2005. Rab conversion as a mechanism of progression from early to late endosomes. Cell 122:735–749. doi: 10.1016/j.cell.2005.06.043. [DOI] [PubMed] [Google Scholar]
- 34.Segev N. 2011. Coordination of intracellular transport steps by GTPases. Semin Cell Dev Biol 22:33–38. doi: 10.1016/j.semcdb.2010.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang B, Hubber A, McDonough JA, Roy CR, Scidmore MA, Carlyon JA. 2010. The Anaplasma phagocytophilum-occupied vacuole selectively recruits Rab-GTPases that are predominantly associated with recycling endosomes. Cell Microbiol 12:1292–1307. doi: 10.1111/j.1462-5822.2010.01468.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kelly EE, Horgan CP, Adams C, Patzer TM, Ni Shuilleabhain DM, Norman JC, McCaffrey MW. 2010. Class I Rab11-family interacting proteins are binding targets for the Rab14 GTPase. Biol Cell 102:51–62. doi: 10.1042/BC20090068. [DOI] [PubMed] [Google Scholar]
- 37.Carlyon JA, Abdel-Latif D, Pypaert M, Lacy P, Fikrig E. 2004. Anaplasma phagocytophilum utilizes multiple host evasion mechanisms to thwart NADPH oxidase-mediated killing during neutrophil infection. Infect Immun 72:4772–4783. doi: 10.1128/IAI.72.8.4772-4783.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Truchan HK, VieBrock L, Cockburn CL, Ojogun N, Griffin BP, Wijesinghe DS, Chalfant CE, Carlyon JA. 2016. Anaplasma phagocytophilum Rab10-dependent parasitism of the trans-Golgi network is critical for completion of the infection cycle. Cell Microbiol 18:260–281. doi: 10.1111/cmi.12500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Capmany A, Damiani MT. 2010. Chlamydia trachomatis intercepts Golgi-derived sphingolipids through a Rab14-mediated transport required for bacterial development and replication. PLoS One 5(11):e14084. doi: 10.1371/journal.pone.0014084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rikihisa Y. 2011. Mechanisms of obligatory intracellular infection with Anaplasma phagocytophilum. Clin Microbiol Rev 24:469–489. doi: 10.1128/CMR.00064-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brayton KA, Kappmeyer LS, Herndon DR, Dark MJ, Tibbals DL, Palmer GH, McGuire TC, Knowles DP. 2005. Complete genome sequencing of Anaplasma marginale reveals that the surface is skewed to two superfamilies of outer membrane proteins. Proc Natl Acad Sci U S A 102:844–849. doi: 10.1073/pnas.0406656102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ueti MW, Knowles DP, Davitt CM, Scoles GA, Baszler TV, Palmer GH. 2009. Quantitative differences in salivary pathogen load during tick transmission underlie strain-specific variation in transmission efficiency of Anaplasma marginale. Infect Immun 77:70–75. doi: 10.1128/IAI.01164-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Scoles GA, Ueti MW, Palmer GH. 2005. Variation among geographically separated populations of Dermacentor andersoni (Acari: Ixodidae) in midgut susceptibility to Anaplasma marginale (Rickettsiales: Anaplasmataceae). J Med Entomol 42:153–162. doi: 10.1093/jmedent/42.2.153. [DOI] [PubMed] [Google Scholar]
- 44.Grant BD, Donaldson JG. 2009. Pathways and mechanisms of endocytic recycling. Nat Rev Mol Cell Biol 10:597–608. doi: 10.1038/nrm2755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Doherty GJ, McMahon HT. 2009. Mechanisms of endocytosis. Annu Rev Biochem 78:857–902. doi: 10.1146/annurev.biochem.78.081307.110540. [DOI] [PubMed] [Google Scholar]