

Abstract
Core‐substituted naphthalenediimides (NDIs) attract increasing attention to bind, transport, and transform electrons, anions, anionic intermediates, and anionic transition states, and to shine as most colorful rainbow fluorophores. The energy level of their lowest unoccupied molecular orbital (LUMO) is decisive for many of these applications. Here, differential pulse voltammetry (DPV) measurements for a consistent series of NDIs are reported to extract exact LUMO levels under identical conditions. The influence of primary and secondary substituents in the core and on the primary imides is compared with general trends for the reliable prediction of LUMO levels in functional systems. Emphasis is on sulfur redox switches in the NDI core because of their frequent use as isostructural probes for π acidity. The same sulfur redox chemistry is expanded to perylenediimides (PDIs), and LUMO engineering is discussed in a broader context, including also fullerenes, aminonaphthalimides (ANIs), and aminoperyleneimides (APIs). The result is a comprehensive reference table that graphically maps out the LUMO space covered by the leading families of electronaccepting aromatics. This graphical summary of general trends in the π‐acidic space is expected to be both inspiring and quite useful in practice.
Keywords: anion-π interactions, LUMO, naphthalenediimides, perylenediimides, redox potentials, secondary substituents, sulfur chemistry
Introduction
Core‐substituted naphthalenediimides (NDIs)1 are emerging as unique, most compact, and most versatile components in functional systems.2, 3, 4, 5, 6, 7, 8, 9, 10, 11 The intrinsically low energy of their lowest unoccupied molecular orbitals (LUMOs) makes them attractive to assemble into complex architectures2 and to transport electrons along face‐to‐face stacks.3 Related to these low LUMOs, their large intrinsic positive quadrupole moment4 makes them attractive to elaborate on anion‐π interactions.5 Realized examples with NDIs include anion binding,6, 7 transport across lipid bilayer membranes,4 and catalysis of reactions with anionic transition states,8 including enolate chemistry9 and enamine chemistry.10
The introduction of substituents in their core provides access to an exceptionally rich diversity.1 Withdrawing substituents afford excellent n‐semiconductors on the one hand3 and anion‐π transporters4 and catalysts8, 9, 10 on the other. With electron‐donating substituents in the core, NDI chemistry becomes exceptionally colorful.1, 11 Exchange of only atoms suffices to cover the primary colors.11, 12 Most of these colorful core‐substituted NDIs show symmetry‐breaking charge separation, making them some of the most compact chlorophyll mimics available.13
Most of these decisive contributions of NDIs to functional systems originate from the change of the energy level of HOMO and LUMO with substituents added in the core and to the imides. Particularly, the LUMO levels are important with regard to applications reaching from optoelectronic devices to anion‐π catalysis. As such, the determination of these LUMO levels from cyclic voltammetry (CV) or differential pulse voltammetry (DPV) combined with absorption spectroscopy is straightforward. However, differences in data analysis have led to different values appearing in the literature. This is particularly true for CV data, which can come from very broad peaks or barely visible shoulders, and have been taken either at the onset of the wave, at the maximum on one wave, or the midpoint between reduction and oxidation wave. Moreover, the resulting values have been calibrated against different values, for example, −4.8 eV or, more recently, −5.1 eV for the Fc+/Fc couple against vacuum.1, 14 Most importantly, contributions from secondary substituents on the intrinsic imide and primary substituents in the core have been largely neglected.1, 15
Considering this situation, we decided to prepare a focused collection of NDIs and determine their LUMO levels from the unambiguous maximum in their DPV under identical, clearly defined conditions. As a result, we here provide the pertinent data set needed to dissect individual contributions from substituents to the LUMO levels of NDIs. Moreover, we add an extension to sulfur chemistry in the core of perylenediimides (PDIs)16, 17, 18 to construct a comparative reference table that outlines general trends in the LUMO space covered by leading families of electron acceptors, that is, NDIs, PDIs, and fullerenes.
Results and Discussion
Secondary substituents on imides
In assessments of the LUMO level of core‐substituted NDIs in functional systems, contributions from the secondary substituents of the intrinsic primary imides remain underappreciated.1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15 This seemed justified because these substituents were expected to be largely decoupled at the nitrogen atom. In functional systems, the main difference at the imide periphery concerns aromatic versus aliphatic substituents.6, 7, 8, 9, 10 To determine the impact of these secondary substituents on the LUMO levels precisely, model systems 1–5 with unsubstituted NDIs were prepared (Figure 1). Their synthesis was very straightforward; details can be found in the Supporting Information.19
Figure 1.
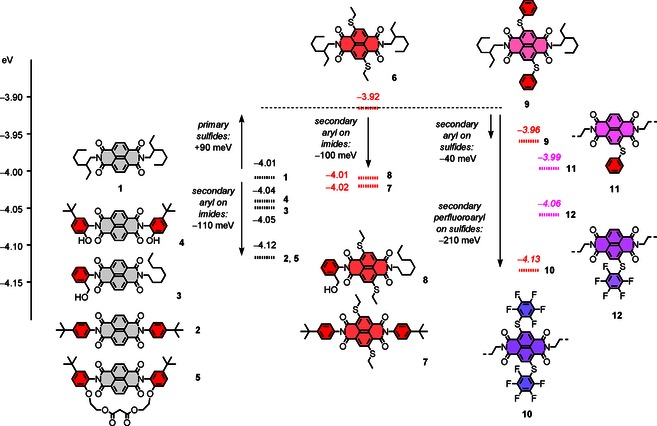
Selected contributions from substituents in NDI core and periphery to the energy of their LUMOs. Energies E LUMO were obtained by differential pulse voltammetry (DPV) in CH2Cl2 and are reported in eV relative to −5.10 eV for Fc+/Fc. Values given in italics are taken from literature16 and are shown for comparison and completion (9–12).
DPV measurements were done under standard conditions in dichloromethane with the Fc+/Fc couple as internal standard. From the DPV maxima, LUMO levels were calculated assuming E LUMO=−5.10 eV for Fc+/Fc against vacuum. For unsubstituted NDI 1 with two secondary alkyl substituents on the primary imides, an E LUMO=−4.01 eV was found (Figures 1 and 2). Replacement of these two alkyl by phenyl substituents with tert‐butyl solubilizers in para position in NDI 2 gave E LUMO=−4.12 eV. Replacement of two secondary alkyls by two aryls on the primary imides thus caused a decrease of ΔE LUMO=−110 meV. This decrease, clearly, is not negligible. Only one aryl substituent in NDI 3 gave an intermediate E LUMO=−4.05 eV. Introduction of two most powerful phenol donors in ortho position gave with E LUMO=−4.04 eV a LUMO level for NDI 4 that was still clearly below that of NDI 1 with two secondary alkyls. Conversion of these phenol donors into weaker ethers and bridging of the core of NDI 5 with a malonate dilactone7 returned the LUMO levels to the original E LUMO=−4.12 eV of NDI 2. Although the variation of too many parameters prevented strong conclusions, it could be said that neither bridging nor tertiary alkoxy substituents on the secondary phenyls have an important impact on the LUMO level of the central NDI 5.
Figure 2.
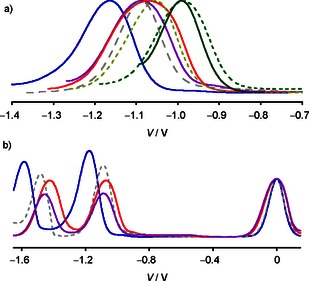
a) Normalized first reduction peaks in the differential pulse voltammogram (DPV) of NDIs 6 (blue), 1 (grey, dashed), 8 (purple), 7 (red), 3 (light green, dashed), 2 (dark green, dashed), and 5 (dark green) in CH2Cl2. b) Original DPV data for 6 (blue), 1 (grey, dashed) and 8 (purple), and 7 (red), normalized at the internal standard Fc+/Fc.
Secondary substituents with sulfides in the NDI core
For NDI 6 with two alkyl substituents at the primary imides, the introduction of two alkyl sulfides in the core, an E LUMO=−3.92 eV was measured by DPV (Figures 1 and 2). Compared with the native NDI 1 at E LUMO=−4.01 eV, this calculated to an increase of ΔE LUMO=+90 meV caused by two alkyl sulfide donors in the core. This increase demonstrated that ethyl sulfides inject electron density into the naphthalene core. This role of ethyl sulfides as electron donors was in conflict with their slightly positive Hammett σp=+0.03. However, this value refers to benzoic acids.20 Recent studies with twisted mechanophores confirmed predictions from theory21 (including σp +=−0.60)20 that with decreasing electron density in the aromatic system, sulfides transform from weak electron acceptors to quite strong electron donors.22 The ΔE LUMO=+90 meV in response to the addition of two ethyl sulfides in the π‐acidic core of NDI 6 was thus in full agreement with this concept of turn‐on sulfide donors (see below).
Replacement of the secondary alkyl by aryl substituents on the primary imides of NDI 6 with two sulfides in the core reproduced the trends found with the native NDI 1. A ΔE LUMO=−100 meV was found for NDI 7 (Figures 1 and 2). This comparison nicely illustrated that contributions from secondary imide substituents in the periphery are not negligible and deserve full attention for the interpretation of results with functional systems. The ΔE LUMO=−90 meV found with only one aryl substituent in NDI 8 exceeded the ΔE LUMO=−40 meV of homolog 3 without primary sulfides clearly. However, the central lesson learned was not affected by this quite puzzling but overall minor inconsistency: The ΔE LUMO∼‐100 meV for substitution of two secondary alkyls by two aryls on the primary imide is roughly independent of the presence of other primary substituents in the NDI core.
LUMO levels of NDIs 9–12 have been previously reported as part of a comprehensive series on aryl sulfides in the NDI core.15 The E LUMO =−3.96 meV of phenyl sulfide 9 was found ΔE LUMO =−40 meV below the E LUMO =−3.92 eV of alkyl sulfide 6. This change with secondary phenyls on primary sulfides in the core was clearly less pronounced than the ΔE LUMO ∼−100 meV with secondary phenyls on primary imides in NDI 7. Secondary pentafluorophenyl substituents on the primary sulfides in NDI 10 lowered to LUMO level to E LUMO=−4.13 eV. Compared with alkyl sulfides in NDI 6, this decrease with secondary pentafluorophenyl substituents in NDI 10 calculated to ΔE LUMO=−210 meV.
Comparison with unsubstituted alkyl NDI 1 at E LUMO=−4.01 eV demonstrated that, consistent with the concept of turn‐on sulfides,22 phenyl sulfides in 9 remain electron donors. Compared with the ethylsulfides in 6 with ΔE LUMO=+90 meV and σp=+0.03, the ΔE LUMO=+50 meV for phenyl sulfides with σp=+0.07 in 9 was very reasonable within the context of the concept of turn‐on sulfide donors. In clear contrast, pentafluorophenyl sulfides in NDI 10 functioned as electron acceptors. The ΔE LUMO=−120 meV below 1 suggested the withdrawing effect from the secondary pentafluorophenyl substituents in NDI 10 overcompensates the donating effect of primary turn‐on sulfides.
To complete the picture, the previously reported15 LUMO levels for NDIs 11 and 12 with only one arylsulfide in the core were added to the graphical summary in Figure 1. Viewed from unsubstituted NDI 1, the contributions were nearly additive. The ΔE LUMO=+20 meV of NDI 11 with one was almost halfway the ΔE LUMO=+50 meV of NDI 9 with two phenyl sulfide donors in the core. The same was true in the pentafluorophenyl series with ΔE LUMO=−50 meV for NDI 12 and ΔE LUMO=−120 meV for NDI 10.
Sulfur chemistry in the NDI core
Oxidation of the two sulfides in the core of NDI 6 gradually converted the turn‐on donors (σp=+0.03) into strong sulfoxide (σp=+0.48) and even stronger sulfone acceptors (σp=+0.78). This redox switch is of highest importance in functional systems because π acidity6, 7, 8, 9, 10 or macrodipoles22, 23 can be changed without global structural changes. According to their DPV maxima, the LUMO levels dropped correspondingly from NDI 13 with one sulfoxide in the core at E LUMO=−4.09 eV to NDI 14 with two sulfoxides at E LUMO=−4.31 eV (Figures 3 and 4). The E LUMO=−4.09 eV of NDI 13 with one sulfoxide and one sulfide in the core was clearly, that is ΔE LUMO =−80 meV, below the E LUMO=−4.01 eV of unsubstituted NDI 1. This finding demonstrated that turn‐on sulfide donors are less effective than conventional sulfoxide acceptors. The LUMO level of mixed sulfide/sulfoxide NDI 13 was ΔE LUMO =−170 meV below that of NDI 6 with sulfides and ΔE LUMO =+220 meV above that of NDI 14 with two sulfoxides in the core. These differences showed that the sulfide‐to‐sulfoxide substitution in the NDI core is a) most significant and b) slightly overadditive.
Figure 3.
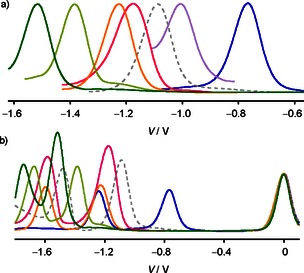
a) Normalized first reduction peaks in the differential pulse voltammogram (DPV) of NDIs 19 (dark green), 20 (light green), 18 (orange), 6 (red), 1 (grey, dashed), 13 (purple), and 14 (blue) in CH2Cl2. b) Original DPV data for 1, 14, and 18–20 normalized at the internal standard Fc+/Fc (colors as in a).
Figure 4.
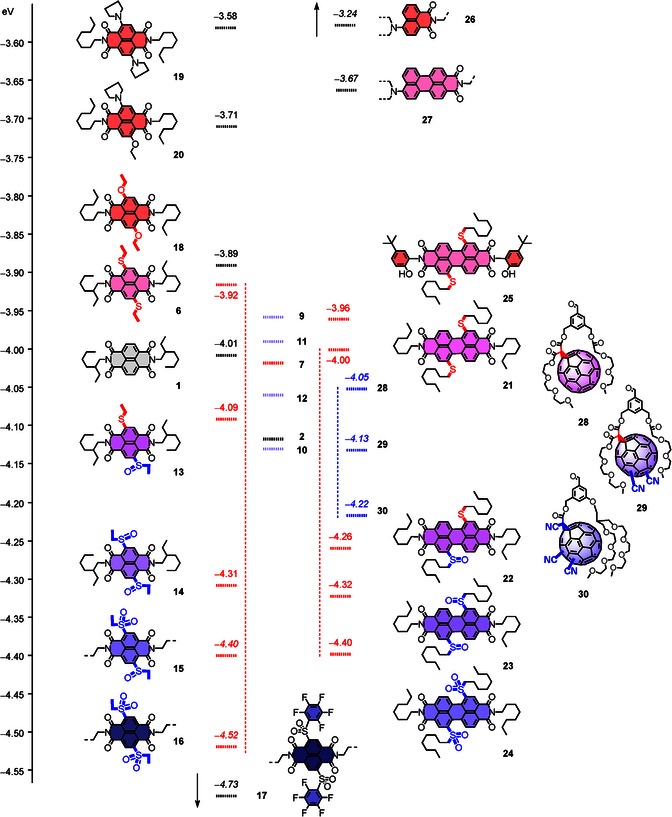
Reference table for LUMO space covered by leading electron acceptors: NDIs (left), PDIs (middle), and fullerenes (right), compared also with ANIs and APIs (top right). Representative data from Figure 1 (1–12) are transcribed here to show the contribution of secondary aromatic NDI substituents on sulfides (blue) and imides (red, black). LUMO levels were obtained by differential pulse voltammetry (DPV) in CH2Cl2 and are reported in eV relative to −5.10 eV for Fc+/Fc. Values given in italics are extrapolated from literature15, 26, 28 and are shown for comparison and completion (15–17, 26–30).
Less important for this study focusing on precise fine‐tuning around unsubstituted NDIs, values extrapolated from the literature for the more extreme LUMOs of mixed sulfoxide/sulfone NDI 15 (E LUMO=−4.40 eV), NDI 16 with two ethyl sulfone (E LUMO=−4.52 eV) and NDI 17 with two pentafluorophenyl sulfones (E LUMO=−4.73 eV) were added in Figure 4 for comparison and completion.15
Other donors in the NDI core
The increase of LUMO levels with stronger donors in the core is understood.1 In the current series, two ethoxy donors with σp=−0.24 in the core of NDI 18 gave E LUMO =−3.89 eV, two stronger pyrrolidyl donors in the core of NDI 19 gave E LUMO =−3.58 eV, and the mixed NDI 20 was with E LUMO =−3.71 eV quite exactly in between (Figures 3 and 4). Most important for this study on small changes, ethoxy NDI 18 with σp=−0.24 could be firmly placed ΔE LUMO =+30 meV above NDI 6 with the turn‐on ethyl sulfide donors (σp=+0.03, Figure 3 a). Compared with unsubstitued NDI 1, the increase with two turn‐on sulfides (ΔE LUMO =+90 meV) was not so much weaker than that with strong ethoxy donors (ΔE LUMO =+120 meV), that is, +45 mV against +60 mV per donor.
NDI 6 with turn‐on sulfide donors is red (λ max=528 nm), whereas NDI 18 with ethoxy donors is yellow (λ max=469 nm).1, 11, 12, 13, 14 The red color of turn‐on NDI 6 is similar to that of mixed NDI 20 with one amine and one ether in the core (λ max=552 nm), whereas diamino NDI 19 is blue (λ max=620 nm).1, 11, 12, 13, 14 The red color of sulfide NDI 6 compared with the yellow color of ethoxy NDI 18 suggested that the turn‐on anomaly of sulfide substituents22 is more pronounced on the HOMO than on the LUMO level. Estimated from the intercept of absorption and emission spectra,1, 12, 13, 14, 15 the HOMO level of NDI 6 with turn‐on donors (σp=+0.03) approximated to quite remarkable ΔE HOMO=+270 mV above the HOMO level of NDI 18 with permanent ethoxy donors (σp=−0.24). A HOMO far above and a LUMO slightly below the conventional alkoxy standards supported that turn‐on sulfide donors22 operate more on the HOMO than on the LUMO level. Contributions from 1,5 O−S interactions24 to the imide carbonyls are possible.25
Sulfur chemistry in the PDI core
Contrary to the situation with NDIs, sulfur chemistry in the core of PDIs has not received much attention.17 To put the results with NDIs into context, we prepared PDI 21 with two octylsulfides in the core (Figure 4). Long alkyl chains were needed also on the primary sulfides to assure good solubility. Controlled oxidation with meta‐chloroperbenzoic acid (mCPBA) and BF3 .Et2O at 0 °C afforded the PDI 22 with one sulfide and one chiral sulfoxide and 23 with two chiral sulfoxides in the core. The two diastereomers of PDI 23 showed very different retentions on thin‐layer chromatography (TLC) and could be easily separated by column chromatography. The pairs of enantiomers in 22 and 23 were not separated. Further oxidation with excess mCPBA at room temperature yielded the highly fluorescent PDI 24 with two sulfones in the core.
DPV revealed an E LUMO =−4.00 eV for PDI 21 with two sulfide donors (Figures 4 and 5). With increasingly strong primary π acceptors in the core, their LUMO levels decreased from E LUMO =−4.26 eV for the mixed PDI 22 to E LUMO =−4.32 eV for sulfoxide PDI 23 and E LUMO =−4.40 eV for sulfone PDI 24 (Figures 4 and 5). For completion, PDI 25 with powerful secondary phenol donors on the primary imides was prepared as well. Compared with the standard alkyl substituents, secondary phenol donors on the imides raised the LUMO level of PDI 25 by ΔE LUMO =+40 meV to E LUMO =−3.96 eV (Figures 4 and 5).
Figure 5.
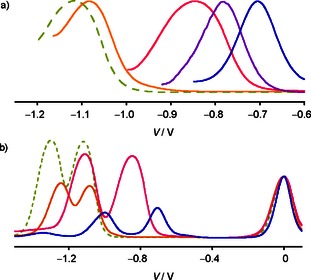
a) Normalized first reduction peaks in the differential pulse voltammogram (DPV) of PDIs 25 (light green, dashed), 21 (orange), 22 ( red), 23 (purple), and 24 (blue) in CH2Cl2. b) Original DPV data for 25 (light green, dashed), 21 (orange), 22 (red), and 24 (blue), normalized at the internal standard Fc+/Fc.
The big picture
The E LUMO =−4.00 eV of PDI 21 with two turn‐on sulfide donors in the core was found ΔE LUMO =−80 meV below the E LUMO =−3.92 eV of the homologous NDI 6 (Figure 4). This ΔE LUMO =−80 meV was clearly significant yet relatively small considering the structural modifications involved. Already the replacement of normal alkyl by aryl substituents at the imine periphery of NDI 7 was sufficient to lower the LUMO below this level. The E LUMO =−4.00 eV of PDI 21 with two primary sulfides was roughly identical with that of unsubstituted NDI 1. Interestingly, the most powerful secondary phenol donors on the imides raised the LUMO from PDI 21 to PDI 25 by ΔE LUMO =+40 meV, whereas the same secondary phenols lowered the LUMO of the original NDI 1 to NDI 4 by ΔE LUMO =−30 meV (Figure 4).
The E LUMO =−4.26 eV of the mixed sulfide/sulfoxide PDI 22 was ΔE LUMO =−170 meV below the E LUMO =−4.09 eV of the homologous NDI 13 (Figure 4). Further oxidation to sulfoxides strongly decreased this difference to nearly negligible ΔE LUMO =−10 meV for PDI 23 below NDI 14. Complete oxidation to sulfones made PDI 24 appear ΔE LUMO =+120 meV above NDI 16. Overall, the expansion of the aromatic system from naphthalene to perylene decreased the responsiveness to oxidation of turn‐on sulfide donors to sulfoxide and sulfone acceptors down to 67 %.
For completion, we point out that similar trends can be extracted for strong donors. NDI 19 with two amines in the core reaches E LUMO =−3.58 eV. The formal removal of one imide acceptor and one amine donor leads to push–pull amino‐naphthalimide (ANI) 26 at E LUMO =−3.24 eV (Figure 4).26, 27 The homologous amino peryleneimide (API) 27 appears at E LUMO =−3.67 eV.26 Compared with the standard NDI 6 (ΔE LUMO =+720 meV) and PDI 21 (ΔE LUMO =−330 meV), this calculated to a decrease in the responsiveness to 50 % for APIs compared with ANIs. This observation was intriguing because compared with the ideal NDIs, the reduced responsiveness of PDIs to primary acceptors might question their usefulness to integrate anion‐π interactions into functional systems.4, 5, 6, 7, 8, 9, 10 With APIs and ANIs compared with PDIs and NDIs, however, the reversed conclusion applies: an even more decreased responsiveness to the primary donor could eventually increase the appeal of APIs for studies on ionpair‐π interactions.26, 27
To provide a general overview for LUMO engineering with the arguably most popular families of electron‐accepting aromatics, selected data from the recently reported nine‐component gradient of fullerenes were added to Figure 4 as well.28 These data were obtained under identical conditions, that is, from DPV maxima calibrated against Fc+/Fc at −5.10 eV. Fullerene 28 with one cyclopropane substituent, that is, the classical motif obtained by fullerene modification with the Bingel reaction, was found at E LUMO =−4.05 eV. This is just below unsubstituted NDI 1 with alkyl substituents on the imides (E LUMO =−4.01 eV) and PDI 21 with turn‐on sulfides in the core (E LUMO =−4.00 eV).
The addition of two cyano acceptors as primary substituents lowers the LUMO level of 28 by ΔE LUMO =−80 meV to E LUMO =−4.13 eV for 29. With ΔE LUMO =−90 meV, the impact of one secondary cyano acceptor in 30 exceeded the moderate ΔE LUMO =−80 meV from two primary acceptors in 29 slightly. The ΔE LUMO =−80 meV of fullerenes in response to two strong primary acceptors with σp=+0.66 was small compared with NDIs. Two strong primary acceptors with σp=+0.48 gave an ΔE LUMO =−300 meV, primary sulfone acceptors with σp=+0.77 an ΔE LUMO =−510 meV. The responsiveness of fullerenes to primary substituents thus calculated to ∼20 % of that of NDIs. The response of comparable NDIs to two primary cyano groups is even more impressive: E LUMO =−4.80 eV, ΔE LUMO =−280 meV below disulfone 16, −790 meV below unsubstituted 1 has been estimated under similar conditions.1 Compared with these dicyano NDIs, the responsiveness of fullerenes drops to ∼10 %. In any case, the poor responsiveness of fullerenes to primary acceptors was also clearly below the 67 % observed for PDIs.
For completion, all newly determined LUMO levels were listed together with the measured first reduction potentials in DPV (Table 1). Although less important for most applications, the second reduction potentials were added as well. The influence of primary and secondary substituents on first and second reduction potentials was similar overall.
Table 1.
Summary of newly measured differential pulse voltammetry (DPV) data.[a]
| Entry | Compound[a] | E LUMO [eV][b] | E red1 [V][c] | E red2 [V][d] |
|---|---|---|---|---|
| 1 | 1 | −4.01 | −1.09 | −1.48 |
| 2 | 2 | −4.12 | −0.98 | −1.40 |
| 3 | 3 | −4.05 | −1.05 | −1.37 |
| 4 | 4 | −4.04 | −1.06 | −1.37 |
| 5 | 5 | −4.12 | −0.98 | −1.54 |
| 6 | 6 | −3.92 | −1.18 | −1.58 |
| 7 | 7 | −4.02 | −1.08 | −1.46 |
| 8 | 8 | −4.01 | −1.09 | −1.43 |
| 9 | 13 | −4.09 | −1.01 | −1.46 |
| 10 | 14 | −4.31 | −0.79 | −1.37 |
| 11 | 18 | −3.89 | −1.21 | −1.59 |
| 12 | 20 | −3.71 | −1.39 | −1.69 |
| 13 | 19 | −3.58 | −1.52 | −1.75 |
| 14 | 21 | −4.00 | −1.10 | −1.24 |
| 15 | 22 | −4.26 | −0.84 | −1.01 |
| 16 | 23 | −4.32 | −0.78 | −1.09 |
| 17 | 24 | −4.40 | −0.70 | −1.00 |
Conclusion
The objective of this study was to produce a comprehensive reference table for quantitative LUMO engineering of NDIs, in comparison with PDIs, fullerenes, as well as ANIs and APIs. The responsiveness of NDIs to primary substituents is shown to exceed that of PDIs (67 %) and fullerenes (∼20 %) by far. These trends confirm the most responsive NDIs as ideal to integrate anion‐π interactions into functional systems,4, 5, 6, 7, 8, 9, 10 whereas the less responsive APIs could be of interest to explore ionpair‐π interactions.25, 26
With primary substituents in the NDI core, the comparison of sulfides and ethers deserves particular attention. With electron‐rich aromatics, ethyl sulfides are weak acceptors (σp=+0.03), but they transform into quite strong donors with electron‐poor aromatics such as NDIs.21, 22 In contrast, ethoxy substituents are conventional donors (σp=−0.24). The LUMO level of NDIs with ethyl sulfides in the core (E LUMO =−3.92 eV) could be firmly localized right below that of NDIs with alkoxy substituents (E LUMO =−3.89 eV) but clearly above that of unsubstituted NDIs (E LUMO =−4.01 eV). However, their HOMO levels show inversion of this order: The red NDIs with ethyl sulfides in the core appear far above the yellow NDIs with ethoxy substituents (ΔE HOMO=+135 meV per donor). This particular impact of turn‐on sulfides22 on the HOMO level invites for computational analysis21, 24 and could be of use to elaborate on the nature of anion‐π interactions.5
Secondary aryl in place of alkyl substituents for the intrinsic primary imide substituents of NDIs lowers their LUMO levels by ΔE LUMO∼−100 meV. Aryl in place of alkyl substituents for primary turn‐on sulfides in the NDI core lowers their LUMO levels by ΔE LUMO=−40 meV (perfluorophenyl: ΔE LUMO=−210 meV). As a result, NDIs with primary sulfides in the core can be more or less π acidic than unsubstituted NDIs, depending on the secondary substituents on imides and sulfides. The availability of comparable values for different primary and secondary substituents is thus most important for design and analysis of functional systems. For example, the answer to the question whether the dependence of anion‐π catalysts on their π acidity is linear or exponential can change, depending on the LUMO energies used.7, 9, 29, 30
The graphical summary of general trends with primary and secondary substituent effects for NDIs, PDIs, and fullerenes provided in Figure 4 is obviously far from complete. Nearly endless possibilities exist to refine the collection with more, or less, subtle structural modifications. However, the data set provided in Figure 4 identifies the relevant trends needed to clarify open questions and map out the LUMO space for electron‐accepting aromatics with appreciable certainty and completeness. It could be quite useful.
Experimental Section
For details of the materials, methods and synthetic procedures, and characterization data for all new compounds, see the Supporting Information.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
The authors thank Y. Zhao, S. Benz, A. Bolag, N. Sakai, and D.‐H. Tran (University of Geneva) for their contribution to synthesis, the NMR and the Sciences Mass Spectrometry (SMS) platforms for services, and the University of Geneva, the European Research Council (ERC Advanced Investigator Grant), the National Centre of Competence in Research (NCCR) Molecular Systems Engineering, the NCCR Chemical Biology, and the Swiss National Science Foundation (NSF) for financial support.
F. N. Miros, S. Matile, ChemistryOpen 2016, 5, 219.
References
- 1. Sakai N., Mareda J., Vauthey E., Matile S., Chem. Commun. 2010, 46, 4225–4237. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Suraru S.-L., Würthner F., Angew. Chem. Int. Ed. 2014, 53, 7428–7448; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 7558–7578; [Google Scholar]
- 2b. Bhosale S. V., Jani C. H., Langford S. J., Chem. Soc. Rev. 2008, 37, 331–342; [DOI] [PubMed] [Google Scholar]
- 2c. Sikder A., Das A., Ghosh S., Angew. Chem. Int. Ed. 2015, 54, 6755–6760; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 6859–6864; [Google Scholar]
- 2d. Das A., Ghosh S., Angew. Chem. Int. Ed. 2014, 53, 2038–2054; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 2068–2084; [Google Scholar]
- 2e. Narayan B., Bejagam K. K., Balasubramanian S., George S. J., Angew. Chem. Int. Ed. 2015, 54, 13053–13057; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 13245–13249; [Google Scholar]
- 2f. Avinash M. B., Samanta P. K., Sandeepa K. V., Pati S. K., Govindaraju T., Eur. J. Org. Chem. 2013, 5838–5847; [Google Scholar]
- 2g. Ponnuswamy N., Pantoş G. D., Smulders M. M. J., Sanders J. M. K., J. Am. Chem. Soc. 2012, 134, 566–573; [DOI] [PubMed] [Google Scholar]
- 2h. Hagihara S., Tanaka H., Matile S., J. Am. Chem. Soc. 2008, 130, 5656–5657; [DOI] [PubMed] [Google Scholar]
- 2i. Talukdar P., Bollot G., Mareda J., Sakai N., Matile S., Chem. Eur. J. 2005, 11, 6525–6532; [DOI] [PubMed] [Google Scholar]
- 2j. Gabriel G. J., Iverson B. L., J. Am. Chem. Soc. 2002, 124, 15174–15175; [DOI] [PubMed] [Google Scholar]
- 2k. Chen W., Zhang J., Long G., Liu Y., Zhang Q., J. Mater. Chem. C 2015, 3, 8219–8224. [Google Scholar]
- 3.
- 3a. Zhang K.-D., Matile S., Angew. Chem. Int. Ed. 2015, 54, 8980–8983; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 9108–9111; [Google Scholar]
- 3b. Berezin A. A., Sciutto A., Demitri N., Bonifazi D., Org. Lett. 2015, 17, 1870–1873; [DOI] [PubMed] [Google Scholar]
- 3c. Rocard L., Berezin A., De Leo F., Bonifazi D., Angew. Chem. Int. Ed. 2015, 54, 15739–15743; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 15965–15969; [Google Scholar]
- 3d. Sakai N., Charbonnaz P., Ward S., Matile S., J. Am. Chem. Soc. 2014, 136, 5575–5578; [DOI] [PubMed] [Google Scholar]
- 3e. Zhang F., Hu Y., Schuettfort T., Di C.-A., Gao X., McNeill C. R., Thomsen L., Mannsfeld S. C. B., Yuan W., Sirringhaus H., Zhu D., J. Am. Chem. Soc. 2013, 135, 2338–2349; [DOI] [PubMed] [Google Scholar]
- 3f. Chang J., Ye Q., Huang K.-W., Zhang J., Chen Z.-K., Wu J., Chi C., Org. Lett. 2012, 14, 2964–2967; [DOI] [PubMed] [Google Scholar]
- 3g. Würthner F., Stolte M., Chem. Commun. 2011, 47, 5109–5115; [DOI] [PubMed] [Google Scholar]
- 3h. Lista M., Areephong J., Sakai N., Matile S., J. Am. Chem. Soc. 2011, 133, 15228–15231; [DOI] [PubMed] [Google Scholar]
- 3i. Zhan X., Facchetti A., Barlow S., Marks T. J., Ratner M. A., Wasielewski M. R., Marder S. R., Adv. Mater. 2011, 23, 268–284. [DOI] [PubMed] [Google Scholar]
- 4. Gorteau V., Bollot G., Mareda J., Perez-Velasco A., Matile S., J. Am. Chem. Soc. 2006, 128, 14788–14789. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Giese M., Albrecht M., Rissanen K., Chem. Rev. 2015, 115, 8867–8895; [DOI] [PubMed] [Google Scholar]
- 5b. He Q., Ao Y. F., Huang Z. T., Wang D.-X., Angew. Chem. Int. Ed. 2015, 54, 11785–11790; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 11951–11956; [Google Scholar]
- 5c. Wang D.-X., Wang M.-X., J. Am. Chem. Soc. 2013, 135, 892–897; [DOI] [PubMed] [Google Scholar]
- 5d. Chifotides H. T., Dunbar K. R., Acc. Chem. Res. 2013, 46, 894–906; [DOI] [PubMed] [Google Scholar]
- 5e. Watt M. M., Zakharov L. N., Haley M. M., Johnson D. W., Angew. Chem. Int. Ed. 2013, 52, 10275–10280; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 10465–10470; [Google Scholar]
- 5f. Vargas Jentzsch A., Matile S., J. Am. Chem. Soc. 2013, 135, 5302–5303; [DOI] [PubMed] [Google Scholar]
- 5g. Ballester P., Acc. Chem. Res. 2013, 46, 874–884; [DOI] [PubMed] [Google Scholar]
- 5h. Bretschneider A., Andrada D. M., Dechert S., Meyer S., Mata R. A., Meyer F., Chem. Eur. J. 2013, 19, 16988–17000; [DOI] [PubMed] [Google Scholar]
- 5i. Frontera A., Gamez P., Mascal M., Mooibroek T. J., Reedijk J., Angew. Chem. Int. Ed. 2011, 50, 9564–9583; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 9736–9756; [Google Scholar]
- 5j. Chudzinski M. G., McClary C. A., Taylor M. S., J. Am. Chem. Soc. 2011, 133, 10559–10567; [DOI] [PubMed] [Google Scholar]
- 5k. Estarellas C., Frontera A., Quiñonero D., Deyà P. M., Angew. Chem. Int. Ed. 2011, 50, 415–418; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 435–438. [Google Scholar]
- 6.
- 6a. Kumar S., Ajayakumar M. R., Hundal G., Mukhopadhyay P., J. Am. Chem. Soc. 2014, 136, 12004–12010; [DOI] [PubMed] [Google Scholar]
- 6b. Schneebeli S. T., Frasconi M., Liu Z., Wu Y., Gardner D. M., Strutt N. L., Cheng C., Carmieli R., Wasielewski M. R., Stoddart J. F., Angew. Chem. Int. Ed. 2013, 52, 13100–13104; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 13338–13342. [Google Scholar]
- 7.F. N. Miros, Y. Zhao, G. Sargsyan, M. Pupier, C. Besnard, C. Beuchat, J. Mareda, N. Sakai, S. Matile, Chem. Eur. J 2016, DOI: 10.1002/chem.201504008. [DOI] [PubMed]
- 8.
- 8a. Zhao Y., Domoto Y., Orentas E., Beuchat C., Emery D., Mareda J., Sakai N., Matile S., Angew. Chem. Int. Ed. 2013, 52, 9940–9943; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 10124–10127; [Google Scholar]
- 8b. Zhao Y., Beuchat C., Mareda J., Domoto Y., Gajewy J., Wilson A., Sakai N., Matile S., J. Am. Chem. Soc. 2014, 136, 2101–2111. [DOI] [PubMed] [Google Scholar]
- 9. Zhao Y., Benz S., Sakai N., Matile S., Chem. Sci. 2015, 6, 6219–6223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.
- 10a. Zhao Y., Cotelle Y., Avestro A.-J., Sakai N., Matile S., J. Am. Chem. Soc. 2015, 137, 11582–11585; [DOI] [PubMed] [Google Scholar]
- 10b. Lee K. S., Parquette J. R., Chem. Commun. 2015, 51, 15653–15656. [DOI] [PubMed] [Google Scholar]
- 11. Würthner F., Ahmed S., Thalacker C., Debaerdemaeker T., Chem. Eur. J. 2002, 8, 4742–4750. [DOI] [PubMed] [Google Scholar]
- 12.
- 12a. Kishore R. S. K., Kel O., Banerji N., Emery D., Bollot G., Mareda J., Gomez-Casado A., Jonkheijm P., Huskens J., Maroni P., Borkovec M., Vauthey E., Sakai N., Matile S., J. Am. Chem. Soc. 2009, 131, 11106–11116; [DOI] [PubMed] [Google Scholar]
- 12b. Maniam S., Cox R. P., Langford S. J., Bell T. D., Chem. Eur. J. 2015, 21, 4133–4140. [DOI] [PubMed] [Google Scholar]
- 13.
- 13a. Bhosale S., Sisson A. L., Talukdar P., Fürstenberg A., Banerji N., Vauthey E., Bollot G., Mareda J., Röger C., Würthner F., Sakai N., Matile S., Science 2006, 313, 84–86; [DOI] [PubMed] [Google Scholar]
- 13b. Sakai N., Lista M., Kel O., Sakurai S.-i., Emery D., Mareda J., Vauthey E., Matile S., J. Am. Chem. Soc. 2011, 133, 15224–15227. [DOI] [PubMed] [Google Scholar]
- 14. Míšek J., Vargas Jentzsch A., Sakurai S., Emery D., Mareda J., Matile S., Angew. Chem. Int. Ed. 2010, 49, 7680–7683; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 7846–7849. [Google Scholar]
- 15. Zhao Y., Huang G., Besnard C., Mareda J., Sakai N., Matile S., Chem. Eur. J. 2015, 21, 6202–6207. [DOI] [PubMed] [Google Scholar]
- 16.
- 16a. Würthner F., Chem. Commun. 2004, 40, 1564–1579; [DOI] [PubMed] [Google Scholar]
- 16b.F. Würthner, C. Saha-Möller, B. Fimmel, S. Ogi, P. Leowanawat, D. Schmidt, Chem. Rev 2016, DOI: 10.1021/acs.chemrev.5b00188. [DOI] [PubMed]
- 17.
- 17a. Blas-Ferrando V. M., Ortiz J., Ohkubo K., Fukuzumi S., Fernandez-Lazaro F., Sastre-Santos A., Chem. Sci. 2014, 5, 4785–4793; [Google Scholar]
- 17b. Langbein S., Wadepohl H., Gade L. H., J. Org. Chem. 2015, 80, 12620–12626; [DOI] [PubMed] [Google Scholar]
- 17c. Räisänen M. T., Slater A. G., Champness N. R., Buck M., Chem. Sci. 2012, 3, 84–92. [Google Scholar]
- 18.
- 18a. Marty R., Nigon R., Leite D., Frauenrath H., J. Am. Chem. Soc. 2014, 136, 3919–3927; [DOI] [PubMed] [Google Scholar]
- 18b. Charbonnaz P., Zhao Y., Turdean R., Lascano S., Sakai N., Matile S., Chem. Eur. J. 2014, 20, 17143–17151; [DOI] [PubMed] [Google Scholar]
- 18c. Probst M., Wenger D., Biner S. M., Häner R., Org. Biomol. Chem. 2012, 10, 755–759; [DOI] [PubMed] [Google Scholar]
- 18d. Li W.-S., Saeki A., Yamamoto Y., Fukushima T., Seki S., Ishii N., Kato K., Takata M., Aida T., Chem. Asian J. 2010, 5, 1566–1572; [DOI] [PubMed] [Google Scholar]
- 18e. Wasielewski M. R., Acc. Chem. Res. 2009, 42, 1910–1921; [DOI] [PubMed] [Google Scholar]
- 18f. Foster S., Finlayson C. E., Keivanidis P. E., Huang Y.-S., Hwang I., Friend R. H., Otten M. B. J., Lu L.-P., Schwartz E., Nolte R. J. M., Rowan A. E., Macromolecules 2009, 42, 2023–2030; [Google Scholar]
- 18g. Baumstark D., Wagenknecht H.-A., Angew. Chem. Int. Ed. 2008, 47, 2612–2614; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 2652–2654; [Google Scholar]
- 18h. Shaller A. D., Wang W., Gan H., Li A. D. Q., Angew. Chem. Int. Ed. 2008, 47, 7705–7709; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 7819–7823; [Google Scholar]
- 18i. Würthner F., Chen Z., Hoeben F. J. M., Osswald P., You C.-C., Jonkheijm P., van Herrikhuyzen J., Schenning A. P. H. J., van der Schoot P. P. A. M., Meijer E. W., Beckers E. H. A., Meskers S. C. J., Janssen R. A. J., J. Am. Chem. Soc. 2004, 126, 10611–10618; [DOI] [PubMed] [Google Scholar]
- 18j. Chen W., Yang X., Long G., Wan X., Chen Y., Zhang Q., J. Mater. Chem. C 2015, 3, 4698–4705. [Google Scholar]
- 19.see Supporting Information.
- 20. Hansch C., Leo A., Taft R. W., Chem. Rev. 1991, 91, 165–195. [Google Scholar]
- 21. Bernardi F., Mangini A., Epiotis N. D., Larson J. R., Shaik S., J. Am. Chem. Soc. 1977, 99, 7465–7470. [Google Scholar]
- 22. Verolet Q., Rosspeintner A., Soleimanpour S., Sakai N., Vauthey E., Matile S., J. Am. Chem. Soc. 2015, 137, 15644–15647. [DOI] [PubMed] [Google Scholar]
- 23.
- 23a. Dal Molin M., Verolet Q., Colom A., Letrun R., Derivery E., Gonzalez-Gaitan M., Vauthey E., Roux A., Sakai N., Matile S., J. Am. Chem. Soc. 2015, 137, 568–571; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23b. Kramer J. R., Deming T. J., J. Am. Chem. Soc. 2014, 136, 5547–5550; [DOI] [PubMed] [Google Scholar]
- 23c. Sakai N., Gerard D., Matile S., J. Am. Chem. Soc. 2001, 123, 2517–2524; [DOI] [PubMed] [Google Scholar]
- 23d. Dado G. P., Gellman S. H., J. Am. Chem. Soc. 1993, 115, 12609–12610. [Google Scholar]
- 24.
- 24a. Bauzá A., Mooibroek T. J., Frontera A., ChemPhysChem 2015, 16, 2496–2517; [DOI] [PubMed] [Google Scholar]
- 24b. Beno B. R., Yeung K.-S., Bartberger M. D., Pennington L. D., Meanwell N. A., J. Med. Chem. 2015, 58, 4383–4438. [DOI] [PubMed] [Google Scholar]
- 25. Lin N.-T., Vargas Jentzsch A., Guénée L., Neudörfl J.-M., Aziz S., Berkessel A., Orentas E., Sakai N., Matile S., Chem. Sci. 2012, 3, 1121–1127. [Google Scholar]
- 26.A. Bolag, N. Sakai, S. Matile, unpublished data.
- 27. Fujisawa K., Humbert-Droz M., Letrun R., Vauthey E., Wesolowski T. A., Sakai N., Matile S., J. Am. Chem. Soc. 2015, 137, 11047–11056. [DOI] [PubMed] [Google Scholar]
- 28. López-Andarias J., Bolag A., Nançoz C., Vauthey E., Atienza C., Sakai N., Martín N., Matile S., Chem. Commun. 2015, 51, 7543–7545. [DOI] [PubMed] [Google Scholar]
- 29. Adriaenssens L., Gil-Ramírez G., Frontera A., Quinonero D., Escudero-Adán E. C., Ballester P., J. Am. Chem. Soc. 2014, 136, 3208–3218. [DOI] [PubMed] [Google Scholar]
- 30.Several reports on the complementary cation-π interactions point toward linear dependence:
- 30a. Dougherty D. A., Acc. Chem. Res. 2013, 46, 885–893; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30b. Knowles R. R., Lin S., Jacobsen E. N., J. Am. Chem. Soc. 2010, 132, 5030–5032; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30c. Holland M. C., Paul S., Schweizer W. B., Bergander K., Mück-Lichtenfeld C., Lakhdar S., Mayr H., Gilmour R., Angew. Chem. Int. Ed. 2013, 52, 7967–7971; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 8125–8129; [Google Scholar]
- 30d. Faraldos J. A., Antonczak A. K., Gonzalez V., Fullerton R., Tippmann E. M., Allemann R. K., J. Am. Chem. Soc. 2011, 133, 13906–13909. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary