

Abstract
A series of tritylated and dimethoxytritylated analogues of selected pyrimidine and purine nucleosides were synthesized and evaluated for their in vitro inhibitory activity against two important members of the genus Flavivirus in the Flaviviridae family, the yellow fever (YFV) and dengue viruses (DENV). Among all compounds tested, the 5′‐O‐tritylated and the 5′‐O‐dimethoxytritylated 5‐fluorouridine derivatives exerted potency against YFV. Interestingly in the series of purine analogues, the 5′O, N‐bis‐tritylated fludarabine derivative revealed strong inhibitory activity against DENV at μm concentrations, however significantly weaker potency against YFV.
Keywords: 4,4′-dimetoxytrityl; antiviral; dengue; flavivirus; nucleoside analogues; trityl; yellow fever
Introduction
Medicinal chemists have directed significant efforts to the synthesis of nucleoside and nucleotide analogues endowed with antiviral and anticancer activities.1 Their synthesis is not always straightforward and often it is complicated by several issues, such us their poor solubility in organic solvents, the presence of several reactive functional groups, and the susceptibility of the glycosidic bond to undergo hydrolytic cleavage. To overcome all these problems, different chemical strategies have been elaborated. Among them, chemical manipulation by means of protecting groups remains a fundamental synthetic approach to prepare nucleoside analogues. Different protecting groups are commonly employed in nucleoside and nucleotide chemistry including both base and acid‐labile. The triphenyl methyl group, most commonly indicated as the trityl group is typically used to protect selectively primary, over secondary hydroxyl functions.2 The positive charge on the alpha carbon, stabilized by the resonance effect of three aromatic rings makes trityl ethers acid‐labile. Since its discovery one hundred years ago, the trityl group has become a key protective group, widely used in nucleoside, oligonucleotide, peptide, carbohydrate chemistry, and indeed in almost all other fields of organic and bioorganic chemistry.3 Typically, the trityl group is introduced prior to manipulation of the structure and then removed when appropriate during the synthesis. However some recent studies indicated that trityl‐containing compounds themselves possess a certain biological activity. To some degree, such discoveries may have been serendipitous. To the best of our knowledge the first account reporting on a trityl compound biologically active was in 2002 from Hernandez et al.4 In their studies, 5′‐O‐tritylthymidine (1, Figure 1) emerges as a novel inhibitor of human mitochondrial thymidine kinase. After that, several other studies described the biological potential of nucleoside analogues bearing the trityl group.5, 6
Figure 1.
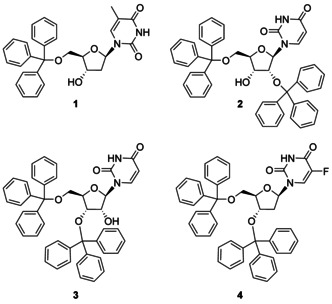
Examples of tritylated nucleosides reported as flavivirus inhibitors.
In particular in 2013, De Burghgraeve et al.7 during the screening of a large compound library aimed at recognizing hit molecules, identified 2′,5′‐bis‐O‐tritylated uridine (2) as a flavivirus inhibitor. As a consequence, several other trityl uridine nucleosides, including different regioisomers, were prepared and evaluated in vitro against two important members of Flaviviridae family, yellow fever (YFV) and dengue (DENV) viruses.8 Among the different compounds 3′,5′‐bis‐O‐tritylated uridine (3) was endowed with the best inhibitory properties (YFV IC50=1.0 μm; DENV IC50=1.75 μm).9 The finding of this lipophilic structure being endowed with high antiviral activity for flaviviruses, encouraged the same authors to undertake further research. However, modifications of the trityl groups, combined with minor variations of the heterocyclic base generally led only to weak in vitro flavivirus inhibition and in some cases to marked in vitro cytotoxicity. From these studies, only 3′,5′‐bis‐O‐tritylated 5‐fluoro‐2′‐deoxyuridine (4) showed no cellular toxicity up to a concentration of 25 μm, while having EC50 values for YFV and DENV respectively of 1.05 μg mL−1 and 1.2 μg mL−1.6a
In the attempt to explain the mode of action of this class of compounds, De Burghgraeve et al. undertook further investigations.7 Their preliminary data suggested that the antiviral activity of this class of compounds might be due to an inhibition of the viral RNA‐dependent RNA polymerase rather than to a mechanism interfering during an early or a late stage of the viral life cycle such as virus entry, assembly, or release.
Despite being under partial control, yellow fever case numbers are now increasing globally, posing a serious risk of local epidemic outbreaks. The global incidence of dengue has also grown dramatically in recent decades. Both viruses are now considered global threats for the entire word population. Although a safe and effective vaccine against YFV exists, there is currently no vaccine available for dengue virus and its development has proven to be very difficult because of the existence of multiple serotypes. With no therapeutic agent available to treat and/or prevent severe epidemics of either yellow fever or dengue virus infections, prevention is currently limited to vector (mosquites) control measures.10 Therefore, there is an obvious and urgent need for the development of effective anti‐flavivirus drugs.11
Based on these considerations and the interesting data reported in the literature for the tritylated nucleosides, we became interested in assessing the impact of the trityl group on several pyrimidine nucleosides not included in the previous studies against both YFV and DENV.
Taking into account that in different studies, hindered nucleoside analogues (NAs) including 3′,5′‐O‐trityl derivatives of adenine nucleoside analogues were found to elicit anti‐flavivirus activity to some extent,[6 g, 12] we decided to include also a few selected purine analogues in our investigations. The structures of the pyrimidine and purine nucleosides used in our studies are shown in Figure 2.
Figure 2.
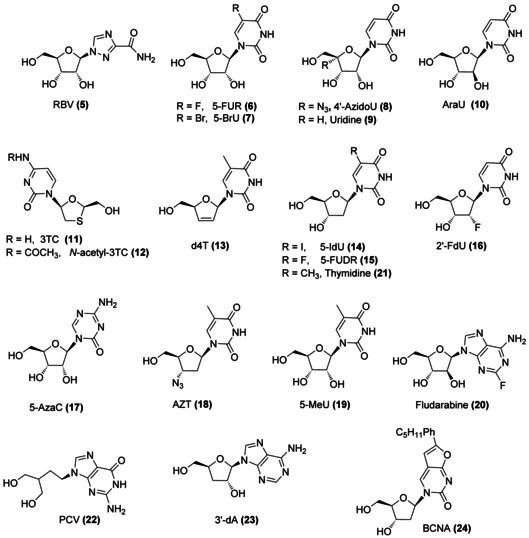
Nucleoside analogues (NAs) considered for this study. RBV: ribavirin, 5‐FUR: 5‐fluorouridine, 5‐BrU: 5‐bromouridine, 4′‐AzidoU: 4′‐azidouridine, AraU: 2′‐β‐d‐arabinouridine, 3TC: lamivudine, d4T: stavudine, 5‐IdU: 5‐iodo‐2′‐deoxyuridine, 5‐FUDR: 5‐fluoro‐2′‐deoxyuridine, 2′‐FdU: 2′‐fluoro‐2′‐deoxyuridine, 5‐AzaC: 5‐azacytidine, AZT: 4′‐azidothymidine, 5‐MeU: 5‐methyluridine, PCV: penciclovir, 3′‐dA: 3′‐deoxyadenosine, BCNA: bicyclic nucleoside analogue.
Results and Discussion
Chemistry
Tritylation of nucleoside analogues has always been a relatively straightforward procedure. Generally, it is sufficient to stir the selected nucleosides with an excess of trityl chloride at an elevated temperature (110 °C).13 However some reports have shown that the temperature can be as low as 80–90 °C. For our synthesis, we followed the milder approach reported by Van Aerschot.8 Briefly, the selected nucleoside was treated in anhydrous pyridine, with an excess of trityl chloride (2.8 equivalents), and the resulting mixture heated at 80 °C for 18 h. After aqueous work‐up, the desired tritylated compounds were isolated by column chromatography on silica gel. In order to avoid decomposition of the trityl moiety, 0.5 % of triethylamine was added to the chromatographic eluent in order to neutralize the silica gel acidity. Although this was not strictly necessary for trityl‐bearing derivatives A–D (provided that their purification was performed in a reasonable time), we found its use imperative during the purification of 4,4′‐dimethoxytrityl derivatives (E–H). The reaction always returned compounds with a diverse degree of tritylation (5′‐, 3′‐, and 2′‐) depending upon the reactivity of the nucleoside. The different regioisomers were separated by column chromatography. The general synthetic procedure for the preparation of the trityl ethers of these compounds is summarized in Scheme 1, and complete compound information can be found in Table 1.
Scheme 1.
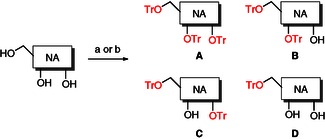
General synthetic methods to trityl compounds 5–24 A–D. Reagents and conditions: a) trityl chloride (2.8 equiv), pyridine (4.5 mL mmol−1), 80 °C, 18 h; b) trityl chloride, (2.8 equiv), DMAP (2.8 equiv), pyridine (4.5 mL mmol−1), 80 °C, 18 h.
Table 1.
Summary of all synthesized tritylated compounds.
| Cmpd | Nucleoside | 5′ | 3′ | 2′ |
|---|---|---|---|---|
| 5 A | RBV (5) | OTr | OTr | OTr |
| 5 B | RBV (5) | OTr | OTr | OH |
| 5 C | RBV (5) | OTr | OH | OTr |
| 6 B | 5‐FUR (6) | OTr | OTr | OH |
| 6 C | 5‐FUR (6) | OTr | OH | OTr |
| 6 D | 5‐FUR (6) | OTr | OH | OH |
| 7 B | 5‐BrU (7) | OTr | OTr | OH |
| 7 C | 5‐BrU (7) | OTr | OH | OTr |
| 8 B | 4′‐AzidoU (8) | OTr | OTr | OH |
| 8 C | 4′‐AzidoU (8) | OTr | OH | OTr |
| 8 D | 4′‐AzidoU (8) | OTr | OH | OH |
| 3 | Uridine (9) | OTr | OTr | OH |
| 2 | Uridine (9) | OTr | OH | OTr |
| 10 B | AraU (10) | OTr | OTr | OH |
| 10 C | AraU (10) | OTr | OH | OTr |
| 10 D | AraU (10) | OTr | OH | OH |
| 11 D‐NHTr | 3TC (11) | OTr | – | – |
| 12 D | N‐acetyl‐3TC (12) | OTr | – | – |
| 13 D | d4T (13) | OTr | – | – |
| 14 D | 5‐IdU (14) | OTr | OH | – |
| 4 | 5‐FUDR (15) | OTr | OTr | – |
| 15 D | 5‐FUDR (15) | OTr | OH | – |
| 16 B | 2′‐FdU (16) | OTr | OTr | – |
| 16 D | 2′‐FdU (16) | OTr | OH | – |
| 17 D | 5‐AzaC (17) | OTr | OH | OH |
| 18 D | AZT (18) | OTr | – | – |
| 19 B | 5‐MeU (19) | OTr | OTr | OH |
| 19 C | 5‐MeU (19) | OTr | OH | OTr |
| 19 D | 5‐MeU (19) | OTr | OH | OH |
| 20 B | Fludarabine (20) | OTr | OTr | OH |
| 20 D‐NHTr | Fludarabine(20) | OTr | OH | OH |
| 21 B | Thymidine (21) | OTr | OTr | – |
| 1 | Thymidine (21) | OTr | OH | – |
| 22 B‐NHTr | PCV (22) | OTr | OTr | – |
| 22 B | PCV (22) | OTr | OTr | – |
| 23 C | 3′‐dA (23) | OTr | – | OTr |
| 24 B | BCNA (24) | OTr | OTr | – |
| 24 D | BCNA (24) | OTr | OH | – |
RBV: ribavirin, 5‐FUR: 5‐fluorouridine, 5‐BrU: 5‐bromouridine, 4′‐AzidoU: 4′‐azidouridine, AraU: 2′‐β‐d‐arabinouridine, 3TC: lamivudine, d4T: stavudine, 5‐IdU: 5‐iodo‐2′‐deoxyuridine, 5‐FUDR: 5‐fluoro‐2′‐deoxyuridine, 2′‐FdU: 2′‐fluoro‐2′‐deoxyuridine, 5‐AzaC: 5‐azacytidine, AZT: 4′‐azidothymidine, 5‐MeU: 5‐methyluridine, PCV: penciclovir, 3′‐dA: 3′‐deoxyadenosine, BCNA: bicyclic nucleoside analogue, OTr: O‐trityl.
In order to expand the structure–activity relationship (SAR), and considering that flavivirus inhibition profile has been linked to the critical requirement of bulkiness in the region between the 3’ and 5’ positions14 we decided to synthesize also the 4,4′‐dimethoxytrityl derivatives (5–20 E–H, Scheme 2, Table 2) of nucleosides 5–20.
Scheme 2.
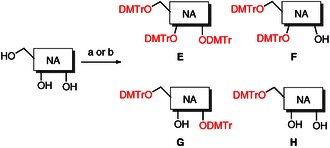
General synthetic method to 4,4′‐dimethoxytrityl compounds 5–20 E–H. Reagents and conditions: a) 4,4′‐dimethoxytrityl chloride, (2.8 equiv), pyridine (4.5 mL mmol−1), 80 °C, 18 h; b) 4,4′‐dimethoxytrityl chloride, (2.8 equiv), DMAP (2.8 equiv), pyridine (4.5 mL mmol−1), 80 °C, 18 h.
Table 2.
Summary of all synthesized 4,4′‐dimethoxytritylated compounds.
| Cmpd | Nucleoside | 5′ | 3′ | 2′ |
|---|---|---|---|---|
| 5 E | RBV (5) | ODMTr | ODMTr | ODMTr |
| 5 G | RBV (5) | ODMTr | OH | ODMTr |
| 6 F | 5‐FUR (6) | ODMTr | ODMTr | OH |
| 6 G | 5‐FUR (6) | ODMTr | OH | ODMTr |
| 6 H | 5‐FUR (6) | ODMTr | OH | OH |
| 8 G | 4′‐AzidoU (8) | ODMTr | OH | ODMTr |
| 9 G | Uridine (9) | ODMTr | OH | ODMTr |
| 9 H | Uridine (9) | ODMTr | OH | OH |
| 10 E | AraU (10) | ODMTr | ODMTr | ODMTr |
| 10 F | AraU (10) | ODMTr | ODMTr | OH |
| 12 H | N‐acetyl‐3TC (12) | ODMTr | – | – |
| 15 F | 5‐FUDR (15) | ODMTr | ODMTr | – |
| 15 H | 5‐FUDR (15) | ODMTr | OH | – |
| 16 F | 2′‐FdU (16) | ODMTr | ODMTr | – |
| 16 H | 2′‐FdU (16) | ODMTr | OH | – |
| 20 H | Fludarabine (20) | ODMTr | OH | OH |
RBV: ribavirin, 5‐FUR: 5‐fluorouridine, 4′‐AzidoU: 4’‐azidouridine, AraU: 2′‐β‐d‐arabinouridine, 3TC: lamivudine, 5‐FUDR: 5‐fluoro‐2′‐deoxyuridine, 2′‐FdU: 2′‐fluoro‐2′‐deoxyuridine, ODMTr: O‐4,4′‐dimethoxytrityl.
5′‐2′‐O‐Bis tritylated regioisomers were distinguished from those 5′‐3′‐O‐bis tritylated analogues using 2D NMR experiments. Correlation spectroscopy (COSY) analysis allowed the correct assignment of each proton for all the compounds. Coupling of the OH signal to either the signal of 2’ or 3’ indicated which position is unalkylated.
Biological evaluation
The antiviral effect of all the parent nucleosides and their corresponding tritylated and 4,4′‐dimethoxytritylated analogues was assessed using a cell viability assay in which Vero cells were infected with either yellow fever (YFV) or dengue viruses (DENV, serotype D2) and further treated with the tested compounds at three different concentrations (5, 10, and 20 μm). Cell viability was determined by measurement of the level of ATP present seven days post infection when compared to vehicle only (DMSO) controls.15 The results are reported as percentage of viable cells left after the assay. Tables 3 and 4 gathered only compounds exhibiting an antiviral activity and include also their CC50 values, defined as the concentration required to decrease cell growth by 50%. The toxicity of all the tested compounds on uninfected cells was also determined in parallel.
Table 3.
Antiviral activity and toxicity of selected tritylated compounds.[a]
| YFV inhibition [% Cells alive] | D2 inhibition [% Cells alive] | Toxicity CC50 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Cmpd | Nucleoside | 5′ | 3′ | 2′ | 5 μm | 10 μm | 20 μm | 5 μm | 10 μm | 20 μm | [μm] |
| 5 B | RBV (5) | OTr | OTr | OH | 0 | 0 | 0 | 0 | 0 | 16.85±1.51 | >20 |
| 6 D | 5‐FUR (6) | OTr | OH | OH | 0 | 0 | 31.24±2.34 | 0 | 0 | 0 | >20 |
| 14 D | 5‐IdU (14) | OTr | OH | – | 0 | 0 | 0 | 0 | 0 | 2.47±0.06 | >20 |
| 16 D | 2′‐FdU (16) | OTr | OH | – | 0 | 0 | 0 | 0 | 0 | 10.88±1.44 | >20 |
| 19 D | 5‐MeU (19) | OTr | OH | OH | 0 | 0 | 8.59±0.85 | 0 | 0 | 0 | >20 |
| 20 D‐NHTr | Fludarabine (20) | OTr | OH | OH | 0 | 0.28 | 0.84 | 71.98±5.75 | 100±1.48 | 100±7.83 | >20 |
| 21 B | Thymidine (21) | OTr | OTr | – | 0 | 0 | 0.95±0.17 | 0 | 0 | 0 | >20 |
| 1 | Thymidine (21) | OTr | OH | – | 0 | 1.25±0.03 | 25.77±1.03 | 0 | 0 | 0 | >20 |
| 22 B‐NHTr | PCV (22) | OTr | OTr | – | 0 | 0 | 8.09±0.04 | 0 | 0 | 0 | >20 |
[a] Determined as an inhibition percentage of yellow fever (YFV, 17 D strain of ASIBI) and dengue viruses (DENV, serotype D2). Values are given as means±SD for n=3. RBV: ribavirin, 5‐FUR: 5‐fluorouridine, 5‐IdU: 5‐iodo‐2′‐deoxyuridine, 2′‐FdU: 2′‐fluoro‐2′‐deoxyuridine, 5‐MeU: 5‐methyluridine, PCV: penciclovir, OTr: O‐trityl.
Table 4.
Antiviral activity and toxicity of selected 4,4′‐dimethoxytritylated compounds.[a]
| YFV inhibition [% Cells alive] | D2 inhibition [% Cells alive] | Toxicity CC50 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Cmpd | Nucleoside | 5′ | 3′ | 2′ | 5 μm | 10 μm | 20 μm | 5 μm | 10 μm | 20 μm | [μm] |
| 6 H | 5‐FUR (6) | ODMTr | OH | OH | 9.22±0.46 | 30.89±1.85 | 100±8.68 | 0 | 0 | 0 | >20 |
| 9 H | Uridine (9) | ODMTr | OH | OH | 1.06±0.27 | 7.00±0.98 | 100±5.23 | 0 | 0 | 0 | >20 |
| 12 H | N‐acetyl‐3TC (12) | ODMTr | – | – | 0 | 0 | 70.45±18.31 | 0 | 0 | 0 | >20 |
| 15 H | 5‐FUDR (15) | ODMTr | OH | – | 0 | 95.17±10.4 | 100±5.4 | 0 | 0 | 0 | >20 |
| 16 H | 2′‐FdU (16) | ODMTr | OH | – | 0 | 0 | 77.99±14.81 | 0 | 0 | 0 | >20 |
[a] Determined as an inhibition percentage of yellow fever (YFV, 17D strain of ASIBI) and dengue viruses (DENV, serotype D2). Values are given as means±SD for n=3. 5‐FUR: 5‐fluorouridine, 3TC: lamivudine, 5‐FUDR: 5‐fluoro‐2′‐deoxyuridine, 2′‐FdU: 2′‐fluoro‐2′‐deoxyuridine.
As expected, the parent nucleosides did not show any antiviral activity versus both the flaviviruses, considered in this study.
In the series of the tritylated nucleoside analogues two compounds: the 5′‐O‐tritylated 5‐FUR derivative (6 D) and the 5′‐O‐tritylated thymidine derivative (1) showed a similar protective effect on the target cells, infected with YFV, when tested at 20 μm, with respectively 31 % and 26 % of the cells maintaining full metabolic activity. Only a low percentage (8.6 %) of YFV inhibition was achieved by the 5′‐O‐tritylated‐5‐Me‐uridine derivative 19 D, at the highest concentration tested. On the other hand, no inhibitory properties were observed for all these compounds (6 D, 1, and 19 D) in the DENV (D2) inhibition assay at any of the concentrations tested.
The 5′,3′‐bis‐O‐tritylated ribavirin (RBV) analogue (5 B) and the 5′‐O‐tritylated 2′‐FdU analogue (16 D) displayed some interesting inhibitory activity against DENV with respectively 17 % and only 11 % viable cells counted at 20 μm.
However, among all the trityl analogues, the 5′‐bis‐O,N‐tritylated derivative (20 D‐NHTr) of the anticancer agent fludarabine (20) displayed the strongest anti‐DENV inhibitory activity with 72 % of viable cells counted at 5 μm and 100 % at both 10 μm and 20 μm. Interestingly, this compound, devoid of any significant anti‐YFV inhibitory effect, seems to selectively target DENV. Despite being the analogue of an anticancer agent, compound 20 D showed no cytotoxicity in our assays, up to a 20 μm.
This noncytotoxic profile was observed also for all the other analogues considered in this work with no evidence of toxicity at concentration up to 20 μm.
Next, the antiviral effect of dimethoxytritylation of selected nucleoside analogues was evaluated similarly to the tritylated congeners against YF and DEN viruses. Table 4 collects the result for the compounds of this series showing some antiviral activity. In particular the 5′‐O‐DMTr‐FUR derivative (6 H), and, the 5′‐O‐DMTr‐5‐FUDR derivative (15 H) emerged as the most active compounds against YFV with 100 % inhibition at 20 μm. Compound 15 H was found to retain high inhibition also at 10 μm with 95 % of viable cells counted. Differently from the 5′‐O‐tritylated‐2′‐deoxy‐2′‐fluorouridine analogue 16 D, its 5′‐O‐dimethoxytrityl congener 16 H had pronounced inhibitory activity but surprisingly only against YFV (78 % at 20 μm).
Intriguing results were also found for the 5′‐O‐dimethoxytritylated‐uridine analogue 9 H, which showed inhibitory activity only against YFV with an inhibition of 1 % at 5 μm; 7 % at 10 μm; and 100 % at 20 μm.
All 4,4′‐dimethoxytritylated compounds tested were devoid of anti‐DENV activity and were found to be not toxic at concentrations up to 20 μm.
As mentioned already, compounds 3 and 2 have been previously reported as relatively potent anti‐YFV and anti DENV agents.6 In contrast, our assays revealed no potency either as anti‐YFV or anti‐DENV agents. Explanation of the differing results from those reported could be found in the differing detection methods used.7 We assayed the metabolic activity of cells, quantitating ATP, which is rapidly degraded in dying cells. Using such an assay indicating active intracellular processes, it is possible to dissect toxic and viral effects that lead to cells with intact membranes but no metabolic activity; that latter method (methylene blue dye exclusion) was used in the prior reports. The ATP assay used here is more sensitive, indicates both toxic and viral effects leading to the loss of metabolic activity, and is the assay of choice to identify compounds, which lead to increased cell survival.14, 15
Conclusion
We herein report the synthesis of tritylated and 4,4′‐dimethoxytritylated pyrimidine and purine nucleoside analogues and the evaluation of their inhibitory activity against YF and DEN viruses. The activity was reported as % of viable cells resulting from treatment with our compounds after exposure to the viruses. In the series of 38 tritylated analogues, two compounds, 6 D and 1, showed selective and moderate YFV inhibition (31 % and 26 %, respectively) at 20 μm concentration. In the DENV inhibition assay, the tritylated fludarabine derivative 20 D‐NHTr displayed pronounced anti‐DENV inhibitory activity, reaching 100 % of virus inhibition at a concentration of 10 μm. Within the panel of 16 4,4′‐dimethoxytritylated compounds, five of the monosubstituted derivatives (6 H, 9 H, 12 H, 15 H, and 16 H) revealed selective anti‐YFV inhibitory activity ranging from 7 to 95 % at 10 μm and from 70 to 100 % at 20 μm. All the tested compounds proved to be noncytotoxic at concentrations up to 20 μm.
In conclusion, our preliminary biological data support an anti‐flavivirus activity for tritylated/4,4′‐dimethoxytritylated nucleoside analogues. Previous studies have suggested that these drugs might act as potential inhibitors of the DENV viral RNA‐dependent RNA polymerase (RdRp).7 However, evidence to unravel their mechanism of action have not been provided yet. In fact, their structure–activity relationship remains unclear and requires additional studies and therefore it is not possible for us here to speculate on a possible mechanism. It might be however that the prior identified lead impacted on cellular metabolic processes rather than a viral target.
Experimental Section
General experimental
All solvents and reagents were used as obtained from commercial sources unless otherwise indicated. All reactions were performed under an argon atmosphere. The 1H and 13C NMR spectra were recorded on a Bruker spectrometer (Billerica, MA, USA) operating at 500 MHz for 1H and 125 MHz for 13C. CDCl3 was used as the solvent for NMR experiments, unless otherwise stated. 1H chemical shift values (δ) are referenced to the residual nondeuterated components of the NMR solvents (δ=7.26 ppm for CHCl3, etc.). The 13C chemical shifts (δ) are referenced to CDCl3 (central peak, δ=77.0 ppm). Fluorine chemical shifts are referenced to CFCl3. Mass spectra (MS) were measured in positive‐mode electrospray ionization (ESI). Thin‐layer chromatography (TLC) was performed on silica gel 60 F254 plastic sheets. Column chromatography was performed using silica gel (35–75 mesh) or on an Isolera Biotage system (Uppsala, Sweden). Purity of prepared compounds was determined to be>95 % by high‐performance liquid chromatography (HPLC)–UV analysis (Thermo HPLC connected with UV detector; Varian Pursuit XS, 4.6 mm×150 mm, 5.0 μm, Palo Alto, CA, USA).
Chemistry
General Procedure 1. A mixture of a nucleoside (1.0 eq mol−1), an appropriate trityl chloride (2.2 eq mol−1), and 4‐dimethylaminopyridine (DMAP, 2.8 mol eq−1, unless stated otherwise) in anhydrous pyridine (4.5 mL mmol−1) was heated at 80 °C under an argon atmosphere for 18 h. The reaction was quenched by addition of MeOH (2 mL mmol−1) at rt, and kept stirring at rt for 30 min. The solution was then concentrated and diluted in CH2Cl2. The organic solution was washed with saturated solution of NaHCO3 (3×20 mL), and the combined aqueous layers were extracted with CH2Cl2. Combined organic layers were dried over Na2SO4, filtered, and concentrated under vacuum. The residue was purified by column chromatography on silica gel (eluent system gradient MeOH in CH2Cl2=1–3 % containing 0.5 % triethylamine).
Experimental data for all active compounds are reported below and for inactive compounds in the Supporting Information.
Tritylated derivatives of ribavirin (5) were prepared according to the general procedure 1 from 5 (0.500 g, 2.05 mmol) and trityl chloride (1.6 g, 5.74 mmol). Column purification with a gradient of MeOH/triethylamine (1:0.5 % to 2:0.5 %) in CH2Cl2 as eluent yielded 5′,3′,2′‐tri‐O‐tritylribavirin (5 A), 5′,3′‐bis‐O‐tritylribavirin (5 B) and 5′,2′‐bis‐O‐tritylribavirin (5 C).
5′,3′‐Bis‐O‐tritylribavirin (5 B) was obtained as a white solid (0.268 g, 18 %); 1H NMR (500 MHz, 25 °C, CDCl3): δ =8.21 (s, 1 H, H‐3), 7.43–7.38 (m, 6 H, H‐Ph), 7.34–7.18 (m, 24 H, H‐Ph), 6.64 (br s, 1 H, NH 2), 5.89 (d, J=3.08 Hz, 1 H, H‐1′), 5.66 (br s, 1 H, NH 2), 4.43–4.39 (m, 1 H, H‐3′), 4.19–4.14 (m, 1 H, H‐4′), 3.61–3.56 (m, 1 H, H‐2′), 3.47–3.43 (m, 1 H, H‐5′), 3.00 (dd, J=10.9, 5.5 Hz, 1 H, H‐5′), 2.85 ppm (d, J=4.5 Hz, 1 H, OH‐2′); 13C NMR (125 MHz, 25 °C, CDCl3): δ=160.4 (C=O), 156.9 (C‐5), 144.1 (C‐3), 143.5, 143.3 (C‐Ph), 128.7, 128.6, 128.3, 127.8, 127.1 (CH‐Ph), 92.4 (C‐1′), 88.1, 87.1 (C(Ph)3), 83.5 (C‐4′), 74.3 (C‐2′), 74.1 (C‐3′), 64.0 ppm (C‐5′); MS (ES+) found: m/z 751.3 [M+Na]+, calcd for [C46H40N4O5]: m/z 728.83 [M]; Reverse‐phase HPLC (H2O/CH3CN from 90:10 to 0:100 in 30 min), flow=1 mL min−1, λ=254 nm, t R=28.10 min.
Tritylated derivatives of 5‐fluorouridine (6) were prepared according to the general procedure 1 from 6 (0.30 g, 1.14 mmol) and trityl chloride (1.02 g, 3.66 mmol) in pyridine (6 mL). Column chromatography purification using a gradient of MeOH/triethylamine (1:0.5 % to 2:0.5 %) in CH2Cl2 as an eluent yielded 5′,3′‐bis‐O‐trityl‐5‐fluorouridine (6 B), 5′,2′‐bis‐O‐trityl‐5‐fluorouridine (6 C), and 5′‐O‐trityl‐5‐fluorouridine (6 D).
5′‐O‐Trityl‐5‐fluorouridine (6 D) was obtained as a white solid (0.183 g, 32 %); 1H NMR (500 MHz, 25 °C, CDCl3,): δ=7.42 (d, J H−F=6.0 Hz, 1 H, H‐6), 7.22–7.20 (m, 6 H, H‐Ph), 7.05–7.02 (m, 6 H, H‐Ph), 6.98–6.95 (m, 6 H, H‐Ph), 5.72 (dd, J=4.5, 1.5 Hz, 1 H, H‐1′), 4.04 (t, J=5.0 Hz, 1 H, H‐3′), 3.96 (t, J=5.0 Hz, 1 H, H‐2′), 3.90–3.87 (m, 1 H, H‐4′), 3.30 (dd, J=11.0, 2.5 Hz, 1 H, H‐5′), 3.25 ppm (dd, J=11.0, 2.5 Hz, 1 H, H‐5′); 13C NMR (125 MHz, 25 °C, [D6]DMSO,): δ=161.0 (d, J C−F=22.5 Hz, C‐2), 152.7 (C‐4), 144.4 (d, J C−F=241.5 Hz, C‐5), 143.3 (C‐Ph), 128.6, 128.3, 127.3 (CH‐Ph), 123.4 (d, J C−F=33.75 Hz, C‐6), 90.1 (C‐1′), 87.5 (C(Ph)3), 84.4 (C‐4′), 75.8 (C‐2′), 71.0 (C‐3′), 63.2 ppm (C‐5′); MS (ES+) found: m/z 527.10 [M+Na]+, calcd for [C28H25FN2O6]: m/z 504.50 [M]; Reverse‐phase HPLC (H2O/CH3CN from 90:10 to 0:50 in 30 min), flow=1 mL min−1, λ=245 nm, t R=17.52 min.
Dimethoxytritylated derivatives of 5‐fluorouridine (6) were prepared according to the general procedure 1 from 6 (0.30 g, 1.14 mmol) and 4,4′‐dimethoxytrityl chloride (1.24 g, 3.66 mmol, 3.2 equiv/mol) in pyridine (6 mL). Column chromatography purification using a gradient of MeOH/triethylamine (1:0.5 % to 2:0.5 %) in CH2Cl2 as an eluent yielded three products, which were further repurified by preparative TLC using MeOH/triethylamine (2:0.5 %) in CH2Cl2 to yield: 5′,3′‐bis‐O‐dimethoxytrityl‐5fluorouridine (6 F), 5′,2′‐bis‐O‐dimethoxytrityl‐5‐fluorouridine (6 G), and 5′‐O‐dimethoxytrityl‐5‐fluorouridine (6 H).
5′‐O‐Dimethoxytrityl‐5‐fluorouridine (6 H) was obtained as a yellowish solid (0.17 g, 26 %); 1H NMR (500 MHz, 25 °C, [D6]DMSO,): δ=7.91 (d, J H−F=7.0 Hz, 1 H, H‐6), 7.40 (dd, J=8.5, 2.5 Hz, 2 H, H‐Ph), 7.33–7.23 (m, 7 H, H‐Ph), 6.90 (apparent d, J=8.5 Hz, 4 H, H‐Ph), 5.73 (dd, J=4.0, 1.5 Hz, 1 H, H‐1′), 5.48 (d, J=5.0 Hz, 1 H, C‐2′‐OH), 5.14 (d, J=5.0 Hz, 1 H, C‐3′‐OH), 4.14 (apparent q, J=5.0 Hz, 1 H, H‐2′), 4.08 (apparent q, J=5.0 Hz, 1 H, H‐3′), 3.98–3.96 (m, 1 H, H‐4′), 3.75 (s, 6 H, 2 x OCH 3), 3.29 (dd, J=11.0, 3.5 Hz, 1 H, H‐5′), 3.19 ppm (dd, J=11.0, 3.5 Hz, 1 H, H‐5′); 13C NMR (125 MHz, 25 °C, [D6]DMSO,): δ=158.2, 158.1 (CH3O‐C‐Ph), 156.9 (d, 2 J C−F=26.0 Hz, C‐4), 149.8 (C‐2), 144.7 (C‐Ph), 139.9 (d, 1 J C−F=230.5 Hz, C‐5), 135.4, 135.2 (C‐Ph), 129.7, 128.9, 128.2, 127.9, 127.6, 126.7, 125.3 (CH‐Ph), 124.6 (d, 2 J C−F=33.8 Hz, C‐6), 113.3, 113.2 (CH‐Ph), 89.0 (C‐1′), 85.8 (C(Ph)3), 82.6 (C‐4′), 73.2 (C‐2′), 69.5 (C‐3′), 63.1 (C‐5′), 55.0 ppm (OCH3); 19F NMR (470 MHz, 25 °C, [D6]DMSO,): δ=−167.25 ppm; MS (ES+) found: m/z 587 [M+Na]+, calcd for [C30H29FN2O8]: m/z 564.55 [M]; Reverse‐phase HPLC (H2O/CH3CN 90:10 to 0:50 in 30 min), flow=1 mL min−1, λ=245 nm, t R=14.85 min.
Dimethoxytritylated derivatives of uridine (9) were prepared according to the general procedure 1 from 9 (0.150 g, 0.61 mmol), 4,4′‐dimethoxytrityl chloride (0.578 g, 1.71 mmol), and DMAP (0.209 g, 1.71 mmol) in anhydrous pyridine (10 mL). Column chromatography eluting with a gradient of MeOH/triethylamine (1:0.5 % to 3.5:0.5 %) in CH2Cl2 as an eluent gave 5′,2′‐bis‐Odimethoxytrityluridine (9 G) and 5′‐dimethoxytrityluridine (9 H).
5′‐O‐Dimethoxytrityluridine (9 H) was obtained as a yellowish solid (0.16 g, 48 %); 1H NMR (500 MHz, 25 °C, CDCl3): δ=8.01 (d, J=8.0 Hz, H‐6), 7.43–7.38 (m, 2 H, H‐Ph), 7.33–7.28 (m, 6 H, H‐Ph), 7.26–7.22 (m, 1 H, H‐Ph), 6.89–6.82 (m, 4 H, H‐Ph), 5.92 (d, J=2.5 Hz, H‐1′), 5.38 (d, J=8.0 Hz, H‐5), 4.44 (dd, J=6.0, 5.5 Hz, H‐3′), 4.36 (dd, J=5.0, 2.5 Hz, H‐2′), 4.22–4.18 (m, 1 H, H‐4′), 3.80 (s, 3 H, OCH 3), 3.79 (s, 3 H, OCH 3), 3.55 (dd, J=11.0, 2.5 Hz, 1 H, H‐5′), 3.50 ppm (dd, J=11.0, 2.5 Hz, 1 H, H‐5′); 13C NMR (125 MHz, 25 °C, CDCl3): δ=163.7 (C‐2), 158.7, 158.6 (CH3O‐C‐Ph) 151.1 (C‐4), 144.4 (C‐Ph), 140.3 (C‐6), 135.5 (C‐Ph), 135.2 (C‐Ph), 130.3, 130.2, 130.1, 129.2, 128.1, 127.9, 127.8, 127.1, 113.4, 113.3, 113.2 (CH‐Ph), 102.3 (C‐5), 90.6 (C‐1′), 83.8 (C‐4′), 87.0 (C(Ph)3), 75.5 (C‐2′), 69.7 (C‐3′), 61.9 (C‐5′), 55.2 ppm (OCH3); MS (ES+) found: m/z 569.2 [M+Na]+, calcd for [C30H30N2O8]: m/z 546.57 [M]; Reverse‐phase HPLC (H2O/CH3CN from 80:20 to 10:90 in 15 min, then to CH3CN 100 % in 25 min), flow=1 mL min−1, λ=245 nm, t R=10.56 min.
5′‐O‐Dimetoxytrityl‐N‐acetyl‐lamivudine (12 H) was prepared according to the general procedure 1 from N‐acetyl‐lamivudine (12) (0.80 g, 2.95 mmol), 4,4′‐dimethoxytrityl chloride (1.2 g, 2.95 mmol), and DMAP (0.036 g, 0.295 mmol) in anhydrous pyridine (5 mL). Column chromatography with a gradient of MeOH/triethylamine (1:0.5 % to 2:0.5 %) in CH2Cl2 as an eluent yielded 12 H as a white solid (0.55 g, 33 %); 1H NMR (500 MHz, 25 °C, CDCl3): δ= 9.73 (bs, 1 H, NH), 8.43 (d, J=7.5 Hz, 1 H, H‐6), 7.49–7.47 (m, 2 H, H‐Ph), 7.38–7.29 (m, 7 H, H‐Ph), 7.21 (d, J=7.5 Hz, H‐5), 6.90–6.88 (m, 4 H, H‐Ph), 6.37 (dd, J=5.5, 2.0 Hz, 1 H, H‐1′), 5.34 (t, J=5.0 Hz, 1 H, H‐4′), 3.84 (s, 6 H, OCH 3), 3.68 (dd, J=12.5, 2.5 Hz, 1 H, H‐5′a), 3.64–3.60 (m, 2 H, H‐5′b and H‐2′a), 3.29 (d, J=12.5 Hz, H‐2′b), 2.27 ppm (s, 3 H, CH 3); 13C NMR (125 MHz, 25 °C, CDCl,): δ=170.5 (COCH3), 163.0 (C‐2), 158.7 (CH3O‐C‐Ph), 154.9 (C‐4), 145.7 (C‐5), 145.3 (C‐Ph), 135.3, 135.1 (C‐Ph), 128.4, 128.1, 113.7, (CH‐Ph), 87.9 (C‐5), 87.6 (CH‐1′), 87.3 (C(Ph)3), 63.5 (CH2‐5′), 55.3 (OCH3), 39.7 (CH2‐2′), 24.9 ppm (COCH3); MS (ES+) found: m/z 596.2 [M+Na]+, calcd for [C31H31N3O6S]: m/z 573.1974 [M]; Reverse‐phase HPLC (H2O/CH3CN from 80:20 to 0:100 in 30 min), flow=1 mL min−1, λ=254 nm, t R=21.59 min.
5′‐O‐Trityl‐5‐iodo‐2′‐deoxyuridine (14 D) was prepared according to the general procedure 1 from 5‐iodo‐2′‐deoxyuridine (14) (0.5 g, 1.41 mmol), trityl chloride (1.26 g, 4.52 mmol), and DMAP (0.172 g, 1.41 mmol) in anhydrous pyridine (10 mL). Column chromatography eluting with a gradient of MeOH (1 % to 4 %) in CH2Cl2 gave 5′‐O‐trityl‐5‐iodo‐2′‐deoxyuridine (14 D) as a white solid (0.46 g, 55 %); 1H NMR (500 MHz, 25 °C, CDCl3): δ=9.34 (bs, 1 H, NH), 8.16 (s, 1 H, H‐6), 7.47–7.46 (m, 6 H, H‐Ph), 7.36–7.33 (m, 6 H, H‐Ph), 7.29–7.26 (m, 3 H, H‐Ph), 6.34 (dd, J=6.0, 8.0 Hz, 1 H, H‐1′), 4.58–4.57 (m, 1 H, H‐3′), 4.15–4.13 (m, 1 H, H‐4′), 3.45–3.39 (m, 2 H, H‐5′), 2.56–2.52 (m, 1 H, H‐2′a), 2.32–2.28 ppm (m, 1 H, H‐2′b); 13C NMR (125 MHz, 25 °C, CDCl3): δ=160.0 (C‐2), 149.7 (C‐4), 144.2 (C‐6), 143.3 (C‐Ph), 128.6, 128.1, 127.4 (CH‐Ph), 88.3 (C‐5), 87.7 (C(Ph)3), 86.3 (C‐4′), 85.5 (C‐1′), 72.3 (C‐3′), 63.6 (C‐5′), 49.5 ppm (C‐2′); MS (ES+) found: m/z 619.2 [M+Na]+, calcd for [C28H25IN2O5] m/z 596.4130 [M]; Reverse‐phase HPLC (H2O/CH3CN from 80:20 to 0:100 in 30 min), flow=1 mL min−1, λ=254 nm, t R=20.71 min.
Dimethoxytritylated derivatives of 5‐fluoro‐2′deoxyuridine (15) were prepared according to the general procedure 1 from 15 (0.30 g, 1.22 mmol) and 4,4′‐dimethoxytrityl chloride (0.90 g, 2.68 mmol) in pyridine (6 mL). Column chromatography purification using gradient of MeOH/triethylamine (1:0.5 % to 2:0.5 %) in CH2Cl2 as an eluent followed by preparative TLC purification with MeOH/triethylamine (2:0.5 %) in CH2Cl2 yielded 5′,3′‐bis‐O‐dimethoxytrityl‐5‐fluoro‐2′‐deoxyuridine (15 F) and 5′‐O‐dimethoxytrityl‐5‐fluoro‐2′‐deoxyuridine (15 H).
5′‐O‐Dimethoxytrityl‐5‐fluoro‐2′‐deoxyuridine (15 H) was obtained as a yellowish solid (0.135 g, 20 %); 1H NMR (500 MHz, 25 °C, MeOD): δ=7.92 (d, J H−F=6.5 Hz, 1 H, H‐6), 7.45 (dd, J=9.0, 1.5 Hz, 2 H, H‐Ph), 7.35–7.29 (m, 6 H, H‐Ph), 7.25–7.23 (m, 1 H, H‐Ph), 6.89–6.87 (m, 4 H, H‐Ph), 6.25–6.22 (m, 1 H, H‐1′), 4.52–4.49 (m, 1 H, H‐3′), 4.05–4.03 (m, 1 H, H‐4′), 3.80 (s, 6 H, 2 x OCH 3), 3.42 (dd, J=10.5, 4.0 Hz, 1 H, H‐5′), 3.35 (dd, J=10.5, 4.0 Hz, 1 H, H‐5′), 2.43–2.38 (m, 1 H, H‐2′), 2.35–2.29 ppm (m, 1 H, H‐2′); 13C NMR (125 MHz, 25 °C, CDCl3): δ=160.6 (C‐2), 158.7 (CH3O‐C‐Ph), 156.7 (C‐4), 144.8 (C‐Ph), 143.4 (d, 1 J C−F=239.5 Hz, C‐5), 129.8, 128.8, (CH‐Ph), 124.3 (d, 2 J C−F=32.8 Hz, C‐6), 113.7 (CH‐Ph), 88.7 (C(Ph)3), 87.8 (C‐1′), 87.0 (C‐4′), 72.3 (C‐3′), 63.9 (C‐5′), 55.3 (OCH3), 40.7 ppm (C‐2′); MS (ES+) found: m/z 571.6 [M+Na]+, calcd for [C30H29FN2O7]: m/z 548.55 [M]; Reverse‐phase HPLC (H2O/CH3CN from 90:10 to 0:50 in 30 min), flow=1 mL min−1, λ=245 nm, t R=18.44 min.
Tritylated derivatives of 2′‐fluoro‐2′‐deoxyuridine (16) were prepared according to the general procedure 1 from 16 (0.50 g, 2.03 mmol) and trityl chloride (1.60 g, 5.69 mmol) in anhydrous pyridine (10 mL). After work‐up, the crude sample was purified by column chromatography using gradient of MeOH/triethylamine (1:0.5 % to 2.5:0.5 %) in CH2Cl2 to give 5′,3′‐bis‐O‐trityl‐2′‐fluoro‐2′‐deoxyuridine 16 B and 5′‐O‐trityl‐2′‐fluoro‐2′‐deoxyuridine 16 D.
5′‐O‐Trityl‐2′‐fluoro‐2′‐deoxyuridine (16 D) was obtained as a white solid (0.79 g, 80 %); 1H NMR (500 MHz, 25 °C, [D6]DMSO): δ=11.43 (bs, 1 H, NH), 7.77 (d, J=8.1 Hz, 1 H, H‐6), 7.41–7.29 (m, 15 H, H‐Ph), 5.91–5.87 (m, 1 H, H‐1′), 5.70–5.69 (m, 1 H, OH‐3′), 5.31 (d, J=8.1 Hz, 1 H, H‐5), 5.18–5.07 (m, 1 H, H‐2′), 4.41–4.32 (m, 1 H, H‐3′), 4.03–4.00 (m, 1 H, H‐4′), 3.34–3.28 ppm (m, 2 H, H‐5′); 13C NMR (125 MHz, 25 °C, [D6]DMSO): δ=163.1 (C‐4), 150.0 (C‐2), 143.3 (‘ipso’ C‐Ph), 140.8 (C‐6), 128.3, 127.9, 127.2 (CH‐Ph), 101.4 (C‐5), 93.4 (d, J C‐F=184.5 Hz, C‐2′), 88.6 (d, J C‐F=35.7 Hz, C‐1′), 86.3 (C(Ph)3), 80.8 (C‐4′), 67.9 (d, J C‐F=16.6 Hz, C‐3′), 62.2 ppm (C‐5′); 19F NMR ([D6]DMSO, 470 MHz): δ=−199.47 ppm; MS (ES+) found: m/z 511.14 [M+Na]+, calcd for [C28H25FN2O5]: m/z 488.51 [M]; Reverse‐phase HPLC (H2O/CH3CN from 50:50 to 0:100 in 30 min), flow=1 mL min−1, λ=254 nm, t R=6.35 min.
Dimethoxytritylated derivatives of 2′‐fluoro‐2′‐deoxyuridine (16) were prepared according to the general procedure 1 from 16 (0.200 g, 0.81 mmol), 4,4′‐dimethoxytrityl chloride (0.769 g, 2.27 mmol), and DMAP (0.278 g, 2.27 mmol) in anhydrous pyridine (10 mL). After work‐up, the crude sample was purified by column chromatography using gradient of MeOH/triethylamine (1:0.5 % to 2.5:0.5 %) in CH2Cl2 to give 5′,3′‐bis‐O‐dimethoxytrityl‐2′‐fluoro‐2′‐deoxyuridine (16 F) and 5′‐O‐dimethoxytrityl‐2′‐fluoro‐2′‐deoxyuridine (16 H).
5′‐O‐Dimethoxytrityl‐2′‐fluoro‐2′‐deoxyuridine (16 H) was obtained as a white solid (0.053 g, 12 %); 1H NMR (500 MHz, 25 °C, CDCl3): δ =7.91 (d, J=8.5 Hz, 1 H, H‐6), 7.41–7.38 (m, 2 H, Ar), 7.35–7.25 (m, 8 H, H‐Ph), 6.89–6.85 (m, 3 H, H‐Ph), 6.09 (dd, J=16.5, 1.5 Hz, 1 H, H‐1′), 5.36 (d, J=8.5 Hz, 1 H, H‐5), 5.05 (dddd, J=52.5, 4.5, 1.5 Hz, 1 H, H‐2′), 4.55 (dddd, J=20.0, 8.0, 4.0, Hz, 1 H, H‐3′), 4.13–4.08 (m, 1 H, H‐4′), 3.83 (s, 3 H, OCH 3), 3.82 (s, 3 H, OCH 3), 3.66 (dd, J=11.5, 2.5 Hz, 1 H, H‐5′), 3.55 ppm (dd, J=11.5, 2.5 Hz, 1 H, H‐5′); 13C NMR (125 MHz, 25 °C, CDCl3): δ=162.4 (C‐2), 158.8, 158.7 (CH3O‐C‐Ph), 149.7 (C‐4), 144.2 (C‐Ph), 139.8 (C‐6), 135.1, 135.0 (C‐Ph), 130.2, 130.1, 128.2, 128.1, 127.2, 113.4 (CH‐Ph), 102.5 (C‐5), 93.9 (d, 1 J CF=185.7 Hz, C‐2′), 87.6 (d, 2 J CF=33.6 Hz, C‐1′), 82.3 (C‐4′), 87.0 (C(Ph)3), 69.1 (d, 2 J CF=17.0 Hz, C‐3′), 61.1 (C‐5′), 55.3 ppm (OCH3); MS (ES+) found: m/z: 571.2 [M+Na]+, calcd for [C30H29FN2O7]: m/z 548.56 [M]; Reverse‐phase HPLC (H2O/CH3CN from 80:20 to 10:90 in 15 min, then to CH3CN 100 % in 25 min, flow=1 mL min−1, λ=245 nm, t R=11.64 min.
Tritylated derivatives of 5‐methyluridine (19) were prepared according to general procedure 1 from 19 (0.50 g, 1.94 mmol) and trityl chloride (1.50 g, 5.42 mmol) in anhydrous pyridine (8.0 mL). Column chromatography using a gradient of MeOH/triethylamine (1:0.5 % to 2:0.5 %) in CH2Cl2 as eluent yielded 5′,3′‐bis‐O‐trityl‐5‐methyluridine (19 B), 5′,2′‐bis‐O‐trityl‐5‐methyluridine (19 C) and 5′‐O‐trityl‐5‐methyluridine (19 D).
5′‐O‐Trityl‐5‐methyl‐uridine (19 D) was obtained as a white solid (0.36 g, 37 %); 1H NMR (500 MHz, 25 °C, [D6]DMSO): δ=11.38 (bs, 1 H, NH), 7.50–7.27 (m, 16 H, H‐Ph, H‐6), 5.81 (d, J=5.2 Hz, 1 H, H‐1′), 5.49 (d, J=5.7 Hz, 1 H, OH‐2′), 5.19 (d, J=5.6 Hz, 1 H, OH‐3′), 4.21–4.17 (m, 1 H, H‐2′), 4.14–4.11 (m, 1 H, H‐3′), 3.98–3.96 (m, 1 H, H‐4′), 3.28–3.16 (m, 2 H, H‐5′), 1.44 ppm (d, J=1.1 Hz, 3 H, CH 3); 13C NMR (125 MHz, 25 °C, [D6]DMSO): δ=163.6 (C‐4), 150.6 (C‐2), 143.4 (‘ipso’ C‐Ph), 135.8 (C‐6), 128.2, 128.0, 127.1 (CH‐Ph), 109.4 (C‐5), 88.0 (C‐1′), 86.4 (C(Ph)3), 82.7 (C‐4′), 73.0 (C‐2′), 70.0 (C‐3′), 63.7 (C‐5′), 11.6 ppm (CH3); MS (ES+) found: m/z 523.16 [M+Na]+, calcd for [C29H28N2O6]: m/z 500.54 [M]; Reverse‐phase HPLC (H2O/CH3CN from 95:5 to 0:100 in 30 min), flow=1 mL min−1, λ=254 nm, t R=20.36 min.
Tritylated derivatives of fludarabine (20) were prepared according to the standard procedure 1 from 20 (0.30 g, 1.05 mmol) and trityl chloride (0.64 g, 2.31 mmol) in pyridine (6 mL). Column chromatography purification using a gradient of MeOH/triethylamine (1:0.5 % to 2:0.5 %) in CH2Cl2 as eluent gave two products which were further repurified by preparative TLC with MeOH/triethylamine (2:0.5 %) in CH2Cl2 to yield 5′, 3′‐bis‐O‐trityl‐fludarabine (20 B) and 5′‐O,N‐trityl‐fludarabine (20 D‐NHTr).
5′‐Bis‐O,N ‐trityl‐fludarabine (20 D‐NHTr) was obtained as a white solid (0.05 g, 6 %); 1H NMR (500 MHz, 25 °C, CDCl3): δ=8.16 (s, 1 H, H‐8), 7.45–7.33 (m, 8 H, H‐Ph), 7.38–7.36 (m, 8 H, H‐Ph), 7.38–7.36 (m, 14 H, H‐Ph), 6.18 (d, J=3.5 Hz, 1 H, H‐1′), 4.36 (apparent broad d, J=2.5 Hz, 1 H, H‐3′), 4.25–4.20 (m, 2 H, H‐2′, NH), 4.05 (q, J=3.5 Hz, 1 H, H‐4′), 3.89 (bs, 1 H, 3′‐OH), 3.62 (dd, J=11.0, 3.0 Hz, 1 H, H‐5′), 3.42 ppm (dd, J=11.0, 3.0 Hz, 1 H, H‐5′); 13C NMR (125 MHz, 25 °C, CDCl3): δ=159.9 (1 J C−F=209.0 Hz, C‐2), 155.7 (3 J C−F=20.8 Hz, C‐6), 149.3 (3 J C−F=18.0 Hz, C‐4), 144.2, 142.9 (C‐Ph), 140.1 (CH‐8), 129.0, 128.6, 128.1, 127.9, 127.5, 127.1 (CH‐Ph), 118.8 (4 J C−F=3.8 Hz, C‐5), 88.2, 88.1 (C(Ph)3), 85.1 (C‐1′), 83.0 (C‐4′), 76.7 (C‐3′), 76.6 (C‐2′), 63.6 ppm (C‐5′); 19F NMR (470 MHz, 25 °C, CDCl3): δ=−49.84; MS (ES+) found: m/z 792.3 [M+Na]+, calcd for [C48H40FN5O4]: m/z 769.86 [M]; Reverse‐phase HPLC (H2O/CH3CN from 90:10 to 0:50 in 30 min), flow=1 mL min−1, λ=245 nm, t R=27.40 min.
Tritylated derivatives of thymidine (21) were prepared according to the general procedure 1 from 21 (0.30 g, 1.24 mmol) and trityl chloride (0.75 g, 2.72 mmol) in pyridine (6 mL). Column chromatography using a gradient of MeOH/triethylamine (1:0.5 % to 3:0.5 %) in CH2Cl2 as an eluent gave 5′,3′‐bis‐O‐trityl‐thymidine (21 B) and 5′‐O‐trityl‐thymidine (1).
5′,3′‐Bis‐O‐trityl‐thymidine (21 B) was obtained as a white solid (0.147 g, 16 %); 1H NMR (500 MHz, 25 °C, CDCl3): δ=7.53 (d, J=1.0 Hz, 1 H, H‐6), 7.41–7.39 (m, 6 H, H‐Ph), 7.27–7.23 (m, 24 H, H‐Ph), 6.47 (t, J=7.5 Hz, 1 H, H‐1′), 4.45–4.42 (m, 1 H, H‐3′), 3.87–3.85 (m, 1 H, H‐4′), 3.24 (dd, J=11.0, 2.5 Hz, 1 H, H‐5′), 2.86 (dd, J=11.0, 2.5 Hz, 1 H, H‐5′), 1.97–1.94 (m, 2 H, H‐2′), 1.41 ppm (d, J=1.0 Hz, 3 H, CH 3); 13C NMR (125 MHz, 25 °C, CDCl3): δ=163.3 (C=O, C‐2), 150.1 (C=O, C‐4), 144.0, 143.3 (C‐Ph), 135.8 (C‐6), 129.0, 128.7, 128.5, 128.2, 128.0, 127.9, 127.4, 127.3, 125.3 (CH‐Ph), 111.0 (C‐5), 87.8, 87.4 (C(Ph)3), 85.4 (C‐4′), 84.9 (C‐1′), 75.4 (C‐3′), 63.8 (C‐5′), 39.7 (C‐2′), 11.7 ppm (CH3); MS (ES+) found: m/z 749.38 [M+Na]+, calcd for [C48H42N2O5]: m/z 726.85 [M]; Reverse‐phase HPLC (H2O/CH3CN from 90:10 to 0:50 in 30 min), flow=1 mL min−1, λ=245 nm, t R=24.56 min.
5′‐O‐Trityl‐thymidine (1) was obtained as a white solid (0.19 g, 33 %); 1H NMR (500 MHz, 25 °C, [D6]DMSO): δ=7.50 (d, J=1.0 Hz, 1 H, H‐6), 7.41–7.38 (m, 6 H, H‐Ph), 7.36–7.33 (m, 6 H, H‐Ph), 7.30–7.26 (m, 3 H, H‐Ph), 6.21 (t, J=6.5 Hz, 1 H, H‐1′), 5.30 (d, J=4.5 Hz, C‐3′‐OH), 4.35–4.31 (m, 1 H, H‐3′), 3.90–3.88 (m, 1 H, H‐4′), 3.24 (dd, J=10.5, 3.0 Hz, 1 H, H‐5′), 3.17 (dd, J=10.5, 3.0 Hz, 1 H, H‐5′), 2.29‐ 2.23 (m, 1 H, H‐2′), 2.18–2.13 (m, 1 H, H‐2′), 1.47 ppm (d, J=1.0 Hz, 3 H, CH 3); 13C NMR (125 MHz, 25 °C, [D6]DMSO): δ=163.6 (C=O, C‐2), 150.3 (C=O, C‐4), 143.4, (C‐Ph), 135.6 (C‐6), 128.2, 127.9, 127.1 (CH‐Ph), 109.5 (C‐5), 86.3 (C(Ph)3), 85.3 (C‐4′), 83.6 (C‐1′), 70.4 (C‐3′), 63.9 (C‐5′), 40.0 (C‐2′), 11.7 ppm (CH3); MS (ES+) found: m/z 507.54 [M+Na]+, calcd for [C29H28N2O5]: m/z 484.54 [M]; Reverse‐phase HPLC (H2O/CH3CN 90:10 to 0:50 in 30 min), flow=1 mL min−1, λ=245 nm, t R=6.29 min.
Tritylated derivatives of penciclovir (22) were prepared according to general procedure 1 from 22 (0.57 g, 2.25 mmol) and trityl chloride (1.86 g, 6.75 mmol) in anhydrous pyridine (10 mL). Column chromatography eluting using a gradient of MeOH/triethylamine (1:0.5 % to 3:0.5 %) in CH2Cl2 yielded 5′,3′‐bis‐O‐trityl‐penciclovir (22 B) and 2‐N,5′,3′‐O‐tri‐trityl penciclovir (22 B‐NHTr).
2‐N‐5′,3′‐O‐Tri‐trityl penciclovir (22 B‐NHTr) was obtained as a white solid (0.47 g, 21 %); 1H NMR (500 MHz, 25 °C, [D6]DMSO): δ=10.51 (bs, 1 H, NH), 7.26 (s, 1 H, H‐8), 7.31–7.28 (m, 31 H, H‐Ph, NH), 7.19–7.17 (m, 6 H, H‐Ph), 7.07–7.04 (m, 6 H, H‐Ph), 6.97–6.96 (m, 3 H, H‐Ph), 3.24–3.21 (m, 2 H, H‐1′), 2.91–2.85 (m, 4 H, H‐4′, H‐5′), 1.62–1.58 (m, 1 H, H‐3′), 1.13–1.09 ppm (m, 2 H, H‐2′); 13C NMR (125 MHz, 25 °C, [D6]DMSO): δ=156.5 (C‐6), 150.3 (C‐2), 149.3 (C‐4), 144.6, 143.8 (‘ipso’ C‐Ph), 137.2 (C‐8), 128.7, 128.4, 128.1, 127.8, 127.4, 126.9, 126.3 (CH‐Ph), 117.0 (C‐5), 85.8 (C(Ph)3), 62.7 (C‐4′, C‐5′), 41.3 (C‐1′), 37.3 (C‐3′), 28.4 ppm (C‐2′); MS (ES+) found: m/z 1003.39 [M+Na]+, calcd for [C67H57N5O3] m/z: 980.20 [M]; Reverse‐phase HPLC (H2O/CH3CN from 50:50 to 0:100 in 40 min), flow=1 mL min−1, λ=254 nm, t R=36.64 min.
Biological assays
Antiviral assay: Trityl compounds were prepared as stock solution of 100 mm in dimethyl sulfoxide (DMSO) and then were diluted to a final assay concentration of 20, 10, 5 μm. Dilutions were made by adding the appropriate amount of DMSO to lyophilized compound to achieve stock concentrations of 10 to 100 mm. Dissolved compounds were stored at −20 °C and brought to rt prior to assay. All compounds were well dissolved in DMSO, and precipitates were not observed upon thawing.
Cell culture: Vero cells were grown in Dulbecco′s modified Eagle′s medium (DMEM, Sigma‐Aldrich) with 10 % FBS (Sigma‐Aldrich). Cells were plated into 96‐well plates at a density of 103 cells per well.
Virus culture: Cells were then infected with dengue virus (serotype D2 (strain New Guinea C), ATCC Number: VR‐1584), and yellow fever vaccine (17 D strain Asibi; grown from a dose of Sanofi Pasteur STAMARIL (1000IU from embryonated chicken eggs) and adapted to Vero cells (Farleigh and Bugert, 2014, unpublished results) at a moi of 1, or left uninfected to determine compound toxicity.
Cell viability was determined after seven days using CellTiterGlo PROMEGA, following the manufacturer's instructions. All experiments were performed in triplicate.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
The authors would like to thank Andrew Gibbs from the School of Pharmacy and Pharmaceutical Sciences, Cardiff University, Cardiff, King Edward VII Avenue, Cardiff CF10 3NB, United Kingdom, for excellent technical assistance.
C. McGuigan, M. Serpi, M. Slusarczyk, V. Ferrari, F. Pertusati, S. Meneghesso, M. Derudas, L. Farleigh, P. Zanetta, J. Bugert, ChemistryOpen 2016, 5, 227.
References
- 1.
- 1a. Jordheim L. P., Durantel D., Zoulim F., Dumontet C., Nat. Rev. Drug Discovery 2013, 12, 447–464; [DOI] [PubMed] [Google Scholar]
- 1b. Galmarini C. M., Mackey J. R., Dumontet C., Lancet Oncol. 2002, 3, 415–424; [DOI] [PubMed] [Google Scholar]
- 1c. De Clercq E., J. Clin. Virol. 2004, 30, 115–133. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Greene T., Wuts P., Protective Groups in Organic Synthesis 3rd ed., New York, 1999; [Google Scholar]
- 2b. Pathak A. K., Pathak V., Seitz L. E., Tiwari K. N., Akhtar M. S., Reynolds R. C., Tetrahedron Lett. 2001, 42, 7755–7757. [Google Scholar]
- 3.
- 3a. Kohli V., Blöcker H., Köster H., Tetrahedron Lett. 1980, 21, 2683–2686; [Google Scholar]
- 3b. Beigelman L. N., Mikhailov S. N., Carbohydr. Res. 1990, 203, 324–329; [Google Scholar]
- 3c. de La Torre B., Marcos M. A., Eritja R., Albericio F., Lett. Pept. Sci. 2002, 8, 331–338; [Google Scholar]
- 3d. Hough L., Salam M. A., Tarelli E., Carbohydr. Res. 1977, 57, 97–101; [Google Scholar]
- 3e. Helferich B. in Adv. Carbohydr. Chem. Vol. 3 (Eds.: M. L. W. W. W. Pigman, P. Stanley), Academic Press, 1948, pp. 79–111. [Google Scholar]
- 4. Hernández A.-I., Balzarini J., Karlsson A., Camarasa M.-J., Pérez-Pérez M.-J., J. Med. Chem. 2002, 45, 4254–4263. [DOI] [PubMed] [Google Scholar]
- 5. Balzarini J., Hernández A.-I., Roche P., Esnouf R., Karlsson A., Camarasa M.-J., Pérez-Pérez M.-J., Mol. Pharmacol. 2003, 63, 263–270. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Pałasz A., Cież D., Eur. J. Med. Chem. 2015, 97, 582–611; [DOI] [PubMed] [Google Scholar]
- 6b. Liekens S., Hernández A.-I., Ribatti D., De Clercq E., Camarasa M.-J., Pérez-Pérez M.-J., Balzarini J., J. Biol. Chem. 2004, 279, 29598–29605; [DOI] [PubMed] [Google Scholar]
- 6c. Liekens S., Bronckaers A., Hernández A.-I., Priego E.-M., Casanova E., Camarasa M.-J., Pérez-Pérez M.-J., Balzarini J., Mol. Pharmacol. 2006, 70, 501–509; [DOI] [PubMed] [Google Scholar]
- 6d. Casanova E., Hernández A.-I., Priego E.-M., Liekens S., Camarasa M.-J., Balzarini J., Pérez-Pérez M.-J., J. Med. Chem. 2006, 49, 5562–5570; [DOI] [PubMed] [Google Scholar]
- 6e. Ruda G. F., Nguyen C., Ziemkowski P., Felczak K., Kasinathan G., Musso-Buendia A., Sund C., Zhou X. X., Kaiser M., Ruiz-Pérez L. M., Brun R., Kulikowski T., Johansson N. G., González-Pacanowska D., Gilbert I. H., ChemMedChem 2011, 6, 309–320; [DOI] [PubMed] [Google Scholar]
- 6f. Whittingham J. L., Leal I., Nguyen C., Kasinathan G., Bell E., Jones A. F., Berry C., Benito A., Turkenburg J. P., Dodson E. J., Perez L. M. R., Wilkinson A. J., Johansson N. G., Brun R., Gilbert I. H., Pacanowska D. G., Wilson K. S., Structure 2005, 13, 329–338; [DOI] [PubMed] [Google Scholar]
- 6g. Angusti A., Manfredini S., Durini E., Ciliberti N., Vertuani S., Solaroli N., Pricl S., Ferrone M., Fermeglia M., Loddo R., Secci B., Visioli A., Sanna T., Collu G., Pezzullo M., La Colla P., Chem. Pharm. Bull. 2008, 56, 423–432. [DOI] [PubMed] [Google Scholar]
- 7. De Burghgraeve T., Selisko B., Kaptein S., Chatelain G., Leyssen P., Debing Y., Jacobs M., Van Aerschot A., Canard B., Neyts J., Antiviral Res. 2013, 98, 242–247. [DOI] [PubMed] [Google Scholar]
- 8. Chatelain G., Debing Y., De Burghgraeve T., Zmurko J., Saudi M., Rozenski J., Neyts J., Van Aerschot A., Eur. J. Med. Chem. 2013, 65, 249–255. [DOI] [PubMed] [Google Scholar]
- 9. Saudi M., Zmurko J., Kaptein S., Rozenski J., Neyts J., Van Aerschot A., Eur. J. Med. Chem. 2014, 76, 98–109. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Leyssen P., De Clercq E., Neyts J., Clin. Microbiol. Rev. 2000, 13, 67–82; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10b. Simmons C. P., Farrar J. J., van Vinh Chau N., Wills B., New Engl. J. Med. 2012, 366, 1423–1432. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Guzman M. G., Halstead S. B., Artsob H., Buchy P., Farrar J., Gubler D. J., Hunsperger E., Kroeger A., Margolis H. S., Martinez E., Nathan M. B., Pelegrino J. L., Simmons C., Yoksan S., Peeling R. W., Nat. Rev. Microbiol. 2010, 8, S7–16; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11b. Abd Kadir S. L., Yaakob H., Mohamed Zulkifli R., J. Nat. Med. 2013, 67, 677–689; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11c. Rothan H. A., Bahrani H., Abd Rahman N., Yusof R., BMC Microbiol. 2014, 14, 140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.S. Pricl, M. Ferrone, M. Paneni, A. Angusti, M. S, P. La Colla, M. Mura, in Convegno GRICU Nuove frontiere di applicazione delle metodologie dell′ingegneria chimica, 2004, 115–118.
- 13.
- 13a. Yung N. C., Fox J. J., J. Am. Chem. Soc. 1961, 83, 3060–3066; [Google Scholar]
- 13b. Žemlička J., Collect. Czech. Chem. Commun. 1964, 29, 1734–1735. [Google Scholar]
- 14. Vernekar S. K. V., Qiu L., Zhang J., Kankanala J., Li H., Geraghty R. J., Wang Z., J. Med. Chem. 2015, 58, 4016–4028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.
- 15a. Kangas L., Grönroos M., Nieminen A., Med. Biol. 1984, 62, 338–343; [PubMed] [Google Scholar]
- 15b. Niles A. L., Moravec R. A., Riss T. L., Curr. Chem. Genomics 2009, 3, 33–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary