

Abstract
Many alkaloids containing a tetrahydro‐β‐carboline skeleton have well‐known therapeutic effects, leading to increased interest in the synthesis of these natural products. Enantiomers of N‐Boc‐protected 1‐hydroxymethyl‐1,2,3,4‐tetrahydro‐β‐carboline [(±)‐7], 1‐hydroxymethyl‐6‐methoxy‐1,2,3,4‐tetrahydro‐β‐carboline [(±)‐8], and 1‐hydroxymethyl‐6‐fluoro‐1,2,3,4‐tetrahydro‐β‐carboline [(±)‐9] were prepared through enzymecatalyzed asymmetric acylation of their primary hydroxyl group. The preliminary experiments were performed in a continuous‐flow system, while the preparative‐scale resolutions were done as batch reactions. Excellent enantioselectivities (E>200) were obtained with Candida antarctica lipase B (CAL‐B) and acetic anhydride in toluene at 60 °C. The recovered alcohols and the produced esters were obtained with high enantiomeric excess values (ee≥96 %). The O‐acylated enantiomers [(S)‐10–(S)‐12)] were transformed into the corresponding amino alcohols [(S)‐7–(S)‐9)] with methanolysis. Microwave‐assisted Boc removals were also performed and resulted in the corresponding compounds (R)‐4–(R)‐6 and (S)‐4–(S)‐6 without a drop in the enantiomeric excess values (ee≥96 %).
Keywords: acylation, Candida antarctica lipase B, continuous-flow system, enzyme catalysis, tetrahydro-β-carboline
Introduction
Many alkaloids containing a tetrahydro‐β‐carboline skeleton have been isolated from natural sources. Several of them have well‐known pharmaceutical effects and are used in therapy. As examples, reserpine displays antihypertensive activity,1 while vincristine and vinblastine exhibit cytotoxic activity.2 In view of their potential pharmaceutical activity, tetrahydro‐β‐carboline alkaloids are currently at the forefront of research. New alkaloids have recently been isolated from Vinca major, including vincamajorines A and B3 and vinmajines A–I.4 Terpenoid indole alkaloids, mappiodines A–C, and mappiodosides A–G are found in the stems of Mappianthus iodoides.5 Harmicine, extracted in optically pure form from Kopsia griffithii, has antileishmanial6 and antinociceptive effects.7 The antiproliferative activity of arborescidine alkaloids and their derivatives has been evaluated in vitro in human tumor cell lines.8 A number of studies have reported antimalarial effects of tetrahydro‐β‐carbolines such as (+)‐7‐bromotrypargine, which was extracted from an Australian marine sponge,9 and some pyridoxal β‐carbolines derivatives.10 Trujillo and co‐workers investigated tetrahydro‐β‐carboline‐1‐carboxylic acids and their analogs, such as (±)‐5, as inhibitors of mitogen‐activated protein kinaseactivated protein kinase 2.11 Syntheses of β‐carboline alkaloids, such as henrycinol A and B12 or Eg5, an inhibitor of hydantoin hybrids, have also been reported.13 Several routes for the synthesis of pharmacologically important natural products have been reviewed.14,15
Continuous‐flow techniques are increasingly more often used in lipase‐catalyzed transformations, for example acylation reactions16, 17, 18 or esterifications, such as the resolution of flurbiprofen19 and sugar ester synthesis.20 Most of the lipase‐catalyzed reactions involving the use of continuous‐flow techniques have been reviewed, for example, the compilation by Itabaiana and co‐workers.21
On the basis of earlier excellent results on the enzymatic preparation of various N‐Boc‐protected tetrahydroisoquinolines, intermediates for the preparation of crispine A,22 homocalycotomine,23 or calycotomine,24 we set out to develop a new enzymatic method for the resolution of new tetrahydro‐β‐carboline derivatives: 1‐hydroxymethyl‐1,2,3,4‐tetrahydro‐β‐carboline [(±)‐4], 1‐hydroxymethyl‐6‐methoxy‐1,2,3,4‐tetrahydro‐β‐carboline [(±)‐5], and 1‐hydroxymethyl‐6‐fluoro‐1,2,3,4‐tetrahydro‐β‐carboline [(±)‐6]. We planned to carry out the enantioselective O‐acylation of Boc‐protected derivatives of the above‐mentioned compounds [(±)‐7, (±)‐8, and (±)‐9].
Results and Discussion
The starting compounds [(±)‐7, (±)‐8, and (±)‐9] were synthetized through Pictet–Spengler cyclization of the corresponding tryptamine hydrochloride derivatives [1, 2, and 3] and glycolaldehyde, by a method from the literature.25 Finally, to ensure the acylation exclusively at the OH function, the nitrogen at position 2 was Boc protected (Scheme 1).
Scheme 1.
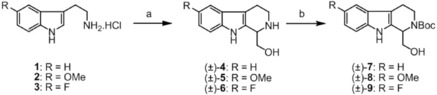
Synthesis of starting compounds (±)‐7, (±)‐8, and (±)‐9. Reagents and conditions: a) 1) water, HCl, glycolaldehyde dimer, 0 °C then 90 °C, 4 h, 2) NaOH, 87 % [(±)‐4], 51 % [(±)‐5], 91 % [(±)‐6]; b) 1,4 dioxane, water, NaOH, (Boc)2O, 0 °C for 1 h then rt for 24 h, 81 % [(±)‐7], 87 % [(±)‐8], 73 % [(±)‐9].
A number of preliminary experiments were performed in order to determine the optimal conditions for the enzymatic acylation of (±)‐7 (Scheme 2). These preliminary reactions were carried out in a continuous‐flow system, using an H‐Cube,24 considering the advantages ensured by this system vs. batch reactions, such as facile automation, reproducibility, constant reaction parameters, and rapid implementation of the reactions (Figure 1).26 The substrate and the acyl donor were dissolved in the solvent, and the solution was pumped through a 70 mm‐long heat‐ and pressure‐resistant CatCart filled with enzyme. We investigated how the enzyme, the acyl donor, the solvent, temperature, and pressure influenced the enantioselectivity and the reaction rate.
Scheme 2.
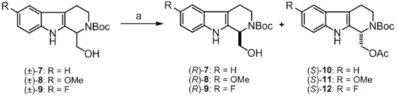
Enzymatic resolution of (±)‐7, (±)‐8, and (±)‐9. Reagents and conditions: a) lipase CAL‐B, acetic anhydride, toluene, 60 °C, 1.5 h: 47 % [(R)‐7], 46 % [(S)‐10], 2.5 h: 47 % [(R)‐8], 43 % [(S)‐11], 2 h: 47 % [(R)‐9], 45 % [(S)‐12].
Figure 1.
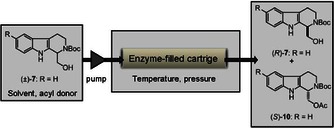
Enzyme‐catalyzed resolution of (±)‐7 in a continuous‐flow system.
In an earlier study on the synthesis of N‐Boc‐protected calycotomine enantiomers, the CAL‐B (Candida antarctica lipase B)‐catalyzed enantioselective acylation (E>200) was performed with vinyl acetate in toluene, with a flow rate of 0.1 mL min−1 in a continuous‐flow system.24 We therefore started the acylation of model compound (±)‐7 under similar conditions (Table 1, entry 1). Poppe and co‐workers16 described the preparative‐scale resolution of different racemic secondary alcohols by using a continuous‐flow system and also at a flow rate of 0.1 mL min−1. CAL‐B catalyzed the reaction with excellent enantioselectivity (E>200), but the conversion (conv.=4 %) was very low after one cycle. Next, several other enzymes, such as PS‐IM (Burkholderia cepacia lipase), CAL‐A (Candida antarctica lipase A) and AK (Pseudomonas fruorescens lipase), were tested under the same conditions (entries 2–4). Lipase PS‐IM catalyzed the reaction with excellent E (entry 2), but with an even lower reaction rate than for CAL‐B (entry 1). CAL‐A displayed moderate reactivity and low E (entry 4), while lipase AK practically did not catalyze the reaction (no product was detected after one cycle) (entry 3). In view of these results, CAL‐B was chosen for further optimization.
Table 1.
Enzyme screening for the acylation of (±)‐7 [a].
| Entry | Enzyme | ee s [b] [%] | ee p [b] [%] | Conv. [%] | E |
|---|---|---|---|---|---|
| 1 | CAL‐B | 4 | 99 | 4 | >200 |
| 2 | PS‐IM | 1.5 | 99 | 1.5 | >200 |
| 3 | AK | No reaction | |||
| 4 | CAL‐A | 3 | 24 | 11 | 1.6 |
[a] Substrate (0.0125 mmol, 3.7 mg); CAL‐B (230 mg), PS‐IM (248 mg), AK (338 mg), CAL‐A (231 mg, 70 mm cartridge); toluene (1 mL); 1.1 equiv vinyl acetate (1.2 μL); 45 °C; 0.1 mL min−1 flow rate; 1 bar; 1 cycle. [b] According to HPLC.
In an attempt to increase the reaction rate, the enzymatic acylation of (±)‐7 was performed with other acyl donors (Table 2). Ethyl acetate and isopropenyl acetate did not react (entries 1 and 2). Although it is known that acylation with an anhydride acyl donor may lead to ‘chemical esterification’ besides enzymatic acylation, thereby causing a decrease in the product enantiomeric excess,27 two anhydride acyl donors, butyric anhydride (entry 4) and acetic anhydride,28,29 (entry 5), were also tested. When butyric anhydride was used, a low E and a relatively good conversion were observed (entry 4). Under the same reaction conditions, acylation with acetic anhydride proceeded in a relatively fast reaction (conversion=17 %) with excellent enantioselectivity (E>200). Consequently, acetic anhydride was chosen as acyl donor in further reactions.
Table 2.
Acyl donor screening for the acylation of (±)‐7 [a].
| Entry | Acyl donor | ee s [b] [%] | ee p [b] [%] | Conv. [%] | E |
|---|---|---|---|---|---|
| 1 | ethyl acetate | No reaction | |||
| 2 | isopropenyl acetate | No reaction | |||
| 3 | vinyl acetate | 4 | 99 | 4 | >200 |
| 4 | butyric anhydride | 31 | 63 | 32 | 6 |
| 5 | acetic anhydride | 20 | 99 | 17 | >200 |
| 6 | 2,2,2‐trifluoroethyl butyrate | 12 | 42 | 22 | 2.7 |
[a] Substrate (0.0125 mmol, 3.7 mg); CAL‐B (230 mg, 70 mm cartridge); toluene (1 mL); 1.1 equiv acyl donor; 60 °C, 0.1 mL min−1 flow rate; 1 bar; 1 cycle. [b] According to HPLC.
We next investigated the acylation of (±)‐7 at different temperatures (Table 3). When the temperature was increased from 60 °C (entry 1) to 70 °C (entry 2) and then to 80 °C (entry 3), the reaction rate increased, but at the same time, E decreased.
Table 3.
Effects of temperature on E and the conversion in the acylation of (±)‐7 [a].
| Entry | Temperature [°C] | ee s [b] [%] | ee p [b] [%] | Conv. [%] | E |
|---|---|---|---|---|---|
| 1 | 60 | 20 | 99 | 17 | >200 |
| 2 | 70 | 23 | 98 | 19 | 124 |
| 3 | 80 | 35 | 97 | 26 | 92 |
[a] Substrate (0.0125 mmol, 3.7 mg); CAL‐B (230 mg, 70 mm cartridge); toluene (1 mL); 1.1 equiv acetic anhydride (1.2 μL); 0.1 mL min−1 flow rate; 1 bar; 1 cycle. [b] According to HPLC.
In an effort to increase the reaction rate without a loss in enantioselectivity, a set of experiments were performed in different solvents, such as toluene, methyl tert‐butyl ether, acetonitrile, diisopropyl ether, chloroform, and 1,4‐dioxane (Table 4). The results demonstrated excellent E (>200) in methyl tert‐butyl ether and 1,4‐dioxane (entries 2 and 6), but the conversions were very low (conv.≤4 %). Excellent E (>200) and relatively good reaction rates were observed in toluene and diisopropyl ether (entries 1 and 4). Finally, diisopropyl ether was chosen for further reactions.
Table 4.
Solvent screening for the acylation of (±)‐7 [a].
| Entry | Solvent | ee s [b] [%] | ee p [b] [%] | Conv. [%] | E |
|---|---|---|---|---|---|
| 1 | toluene | 20 | 99 | 17 | >200 |
| 2 | methyl tert‐butyl ether | 4 | 99 | 4 | >200 |
| 3 | acetonitrile | 1 | 95 | 1 | 39 |
| 4 | diisopropyl ether | 20 | 99 | 17 | >200 |
| 5 | chloroform | 2 | 73 | 3 | 7 |
| 6 | 1,4‐dioxane | 3 | 99 | 3 | >200 |
[a] Substrate (0.0125 mmol, 3.7 mg); CAL‐B (230 mg, 70 mm cartridge); solvent (1 mL); 1.1 equiv acetic anhydride (1.2 μL); 60 °C; 0.1 mL min−1 flow rate; 1 bar; 1 cycle. [b] According to HPLC.
The pressure of the reactions performed in the continuous‐flow system was also examined (Table 5). It was interesting to observe that at about 60 bar, the reaction rate reached a maximum (conversion=32 % after one cycle, entry 4), and further increase of the pressure resulted in a decrease in the conversion.
Table 5.
Effects of pressure on E and the conversion in the acylation of (±)‐7 [a].
| Entry | Pressure [bar] | ee s [b] [%] | ee p [b] [%] | Conv. [%] | E |
|---|---|---|---|---|---|
| 1 | 1 | 18 | 99 | 15 | >200 |
| 2 | 20 | 34 | 99 | 25 | >200 |
| 3 | 40 | 35 | 99 | 26 | >200 |
| 4 | 60 | 48 | 99 | 32 | >200 |
| 5 | 80 | 34 | 99 | 25 | >200 |
| 6 | 100 | 21 | 99 | 18 | >200 |
[a] Substrate (0.0125 mmol, 3.7 mg); CAL‐B (230 mg, 70 mm cartridge); diisopropyl ether (1 mL); 1.1 equiv acetic anhydride (1.2 μL); 60 °C; 0.1 mL min−1 flow rate; 1 cycle. [b] According to HPLC.
The CAL‐B‐catalyzed acylation of (±)‐7 was next carried out in an incubator shaker, under the optimized reaction conditions for the H‐Cube (CAL‐B, diisopropyl ether, acetic anhydride, 60 °C). The reaction performed in batch mode reached a conversion of 46 % after 3 h, but the enantioselectivity was relatively low (E=36). Consecutively, the diisopropyl ether was replaced by toluene, which also ensured good results when the acylation of (±)‐7 was carried out in the H‐Cube (Table 4, entry 1 vs. 4). The batch reaction in toluene gave excellent enantioselectivity (E>200) and a conversion of 48 % after 3 h. When the amount of acetic anhydride was increased from 1.1 equiv to 2 equiv, a higher reaction rate was observed (E>200, conversion=50 % after 3 h).
In the small‐scale acylations of (±)‐8 and (±)‐9 under the conditions optimized for (±)‐7 (CAL‐B, 2 equiv acetic anhydride, toluene, 60 °C), lower reaction rates and enantioselectivities were observed (Table 6). The results revealed that the enzymatic acylations slowed down or even stopped after a while, and ee p decreased considerably, as a consequence of chemical esterifications (entries 2 and 5). When the amount of acetic anhydride was increased from 2 equiv to 6 and then 8 equiv, the reactions became faster, and when 50 % conversions were achieved, the enantioselectivities were excellent (>200) (entries 3 and 6).
Table 6.
CAL‐B‐catalyzed O‐acylation of (±)‐8 and (±)‐9 with acetic anhydride.[a]
| Entry | Substrate | Acetic anhydride [equiv] | Time [h] | ee s [b] [%] | ee p [b] [%] | Conv. [%] | E |
|---|---|---|---|---|---|---|---|
| 1 | (±)‐8 | 2 | 3 | 69 | 99 | 41 | >200 |
| 2 | (±)‐8 | 2 | 7 | 70 | 95 | 42 | 82 |
| 3 | (±)‐8 | 8 | 2.5 | 98 | 98 | 50 | >200 |
| 4 | (±)‐9 | 2 | 3 | 68 | 97 | 41 | 134 |
| 5 | (±)‐9 | 2 | 7 | 75 | 89 | 45 | 39 |
| 6 | (±)‐9 | 6 | 2 | 97 | 96 | 50 | >200 |
[a] Substrate (0.0125 mmol); CAL‐B (30 mg); toluene (1 mL); 60 °C. [b] According to HPLC.
On the basis of the above results, the preparative‐scale enzymatic resolutions of (±)‐7–(±)‐9 were performed in toluene, with CAL‐B, acetic anhydride [2 equiv for (±)‐7, 8 equiv for (±)‐8, 6 equiv for (±)‐9], at 60 °C. The results are presented in Table 7 and the Experimental Section.
Table 7.
CAL‐B‐catalyzed preparative‐scale resolution of (±)‐7–(±)‐9 [a].
| Alcohol recovered | Ester produced | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| [(R)‐7–(R)‐9] | [(S)‐10–(S)‐12] | ||||||||
| Entry | Substrate | Time [h] | Conv. [%] | Yield [%] | ee [b] [%] | [α]D 25 | Yield [%] | ee [b] [%] | [α]D 25 |
| 1 | (±)‐7 | 1.5 | 50 | 47 | 98 | +107.5[c] | 46 | 98 | −102.2[d] |
| 2 | (±)‐8 | 2.5 | 50 | 47 | 98 | +82[e] | 43 | 98 | −92.3[f] |
| 3 | (±)‐9 | 2 | 49 | 47 | 96 | +97[g] | 45 | 98 | −131.8[h] |
[a] Toluene, with acetic anhydride, at 60 °C. [b] According to HPLC. [c] c=0.34 in EtOH. [d] c=0.32 in EtOH. [e] c=0.23 in EtOH. [f] c=0.61 in EtOH. [g] c=0.21 in EtOH. [h] c=0.38 in EtOH.
Further transformations
The O‐acylated enantiomers [(S)‐10–(S)‐12] were transformed via methanolysis into the corresponding amino alcohols [(S)‐7–(S)‐9] in K2CO3/MeOH at 60 °C without a loss in ee values (98 %) (Scheme 3). When the protecting Boc in (R)‐8 and (S)‐11 was removed with 18 % HCl at 80 °C, a considerable decrease in ee (≤89 %) was observed. Since methods of Boc deprotection, including catalyst‐free water‐mediated,30 and microwave (MW)‐assisted methods31 are known in the literature, we performed MW‐assisted Boc group removal for (R)‐7–(R)‐9 and (S)‐10–(S)‐12, in water at 100 °C.32 This strategy resulted in the desired products [(R)‐4–(R)‐6 and (S)‐4–(S)‐6] with high ee (≥96 %).
Scheme 3.
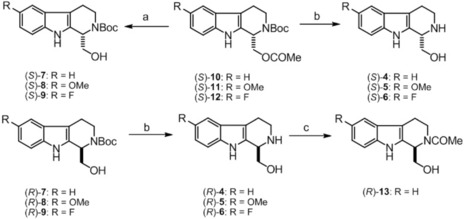
Further transformations. Reagents and conditions: a) MeOH, K2CO3, 60 °C, 10 min, 90 % [(S)‐7], 88 % [(S)‐8], 75 % [(S)‐9]; b) MW, water, 100 °C, 1 h, 52 % [(S)‐4], 38 % [(S)‐5], 28 % [(S)‐6], 79 % [(R)‐4], 38 % [(R)‐5], 52 % [(R)‐6]; c) CH2Cl2, water, acetic anhydride, NaOH, rt, 48 h, 49 %.
For determination of the absolute configuration, amino alcohol 4 was transformed to its N‐acetyl analog (13) by a known literature method (Scheme 3).33
Determination of absolute configuration
The specific rotation earlier reported for (R)‐13 (ee=98 %) was [α]D 25=+17.3 (c=0.2 in EtOH),34 whereas the enantiomeric 13 that we prepared (see Experimental Section) gave [α]D 25=+164 (c=0.2 in EtOH), with the same sign, but with a higher order of magnitude, although the 1H NMR spectroscopic data for our (R)‐13 were similar to those given in the literature.[34][35] Taking into account our earlier observations with regard to the enantioselectivity in the CAL‐B‐catalyzed O‐acylation of related amino alcohols,24 (S) selectivity was accepted in the CAL‐B‐catalyzed O‐acylation of (±)‐7.
Conclusion
An effective enzymatic method was developed for the enantioselective O‐acylation of the primary hydroxyl group of tetrahydro‐β‐carbolines (±)‐7, (±)‐8, and (±)‐9. Taking advantage of the continuous‐flow system, we carried out the preliminary experiments in a continuous‐flow system, while the preparative‐scale resolutions were performed as batch reactions (incubator shaker). Excellent E values (>200) were observed when CAL‐B and acetic anhydride were used in toluene at 60 °C. Enantiomeric N‐Boc‐protected amino alcohols [(R)‐7–(R)‐9], and amino esters [(S)‐10–(S)‐12] were obtained with high ee (≥96 %) in good yields (≥43 %). The transformations of (R)‐7–(R)‐9 and (S)‐10–(S)‐12 with MW‐assisted Boc deprotection resulted in the desired tetrahydro‐β‐carboline amino alcohols without a drop in the ee values (≥96 %).
Experimental Section
Materials and methods
CAL‐B (lipase B from Candida antarctica, Catalog No. L4777, specification:≥5000 U g−1) was purchased from Sigma–Aldrich; lipase AK (Pseudomonas fruorescens) was from Amano Pharmaceuticals. Lipase PS‐IM (Burkholderia cepacia immobilized on diatomaceous earth) was from Amano Enzyme Europe Ltd. Chyrazyme l‐5 (lipase A from Candida antarctica) was from Novo Nordisk. Reactions in the continuous‐flow system were carried out in the H‐Cube from ThalesNano Inc (Budapest, Hungary). The stainless‐steel cartridges used (70 mm in length, 4 mm in internal diameter and 0.75 mL in volume), were also from ThalesNano Inc. With CAL‐B (230 mg) as enzyme charge and a flow rate of 0.1 mL min−1 (toluene), the experimentally determined (staining procedure) residence time within the packed bed of the reactor was 7 min and 40 s. The H‐cube was used in “no H2” mode. The 1H NMR and 13C NMR spectra were recorded with a Bruker Avance DRX 400 instrument (Billerica, MA, USA). Elemental analyses were performed with a PerkinElmer CHNS‐2400 Ser II Elemental Analyzer (Waltham, MA, USA). Optical rotations were measured with a PerkinElmer 341 polarimeter. Microwave (MW) reactions were performed in a CEM Discover MW reactor (Matthews, NC, USA). Melting points were determined on a Kofler apparatus.
The ee values of the N‐Boc‐protected amino alcohols [(R)‐7–(R)‐9) and (S)‐7–(S)‐9] and amino esters [(S)‐10–(S)‐12)] were determined directly, while those of the deprotected enantiomers [(R)‐4–(R)‐6 and (S)‐4–(S)‐6] were determined after derivatization with Boc2O by using high‐performance liquid chromatography (HPLC) with a Chiralpak OD‐H column (4.6 mm×250 mm), eluent: n‐hexane:isopropyl alcohol (93:7), flow rate: 0.5 mL min−1, detection at 260 nm, at rt. Retention times (min): for (R)‐7: 27.7, (S)‐7: 16.6, (R)‐8: 38.6, (S)‐8: 21.9, (R)‐9: 28.8, (S)‐9: 15.6, (S)‐10: 13.8, (R)‐10: 18.8, (S)‐11: 17.0, (R)‐11: 25.3, (S)‐12: 12.6, and (R)‐12: 19.9. The ee value of N‐acetyl amino alcohol (R)‐13 was determined with a Chiralpak IA column (4.6 mm×250 mm), eluent: n‐hexane:isopropyl alcohol (95:5), flow rate: 0.5 mL min−1, detection at 210 nm, at rt. Retention times (min): for (R)‐13: 92.8 and (S)‐13: 88.2.
Small‐scale enzymatic resolutions
Small‐scale experiments in the continuous‐flow system: the racemic substrate [(±)‐7, 0.0125 mmol] and the acyl donor (1.1 equiv) were dissolved in the solvent (1 mL), and the mixtures were pumped with an HPLC pump through the heated (45 °C, 60 °C, 70 °C, and 80 °C) and compressed (1 bar, 20 bar, 40 bar, 60 bar, 80 bar, and 100 bar) cartridge filled with enzyme (flow rate: 0.1 mL min−1).
Small‐scale experiments in batch mode: the racemic compound [(±)‐7, (±)‐8, or (±)‐9, 0.0125 mmol] was dissolved in the solvent (1 mL), and the enzyme (30 mg mL−1) and acyl donor (2, 6, or 8 equiv of acetic anhydride) were then added. The mixture was shaken at 60 °C.
Syntheses
Synthesis of racemic N‐Boc‐protected 1‐hydroxymethyl‐1,2,3,4‐tetrahydro‐β‐carboline [(±)‐7]
Tryptamine hydrochloride (1, 5.9 g, 0.03 mol) was dissolved in a mixture of water and 2 n HCl (15 mL). The solution was cooled to 0 °C, and a solution of glycolaldehyde dimer (2.3 g, 0.02 mol, dissolved in 5 mL water) was added. The reaction mixture was stirred at 90 °C for 4 h. The cooled solution was treated with activated carbon, and then extracted with diethyl ether. To the aqueous layer, 20 % NaOH was added until pH 10, and the mixture was then extracted with EtOAc (3×30 mL). The organic layer was dried on anhydrous Na2SO4 and evaporated. The product (±)‐4 was purified by column chromatography (5.2 g, yield: 87 %, m.p.=146‐147 °C, light‐yellow crystals, R f=0.27, eluent: MeOH). Alcohol (±)‐4 (3.0 g, 0.015 mol) was dissolved in 80 mL 1,4‐dioxane and cooled to 0 °C, and a solution of NaOH (0.62 g, 0.016 mol, in 5 mL water) and then a solution of di‐tert‐butyl dicarbonate (3.56 g, 0.016 mol, in 10 mL 1,4‐dioxane) were added. The reaction was carried out at 1 h under ice‐cooling, and then at room temperature for 24 h. The reaction mixture was extracted with dichloromethane (3×30 mL) and the extract was dried on anhydrous Na2SO4 and evaporated. The resulting N‐Boc‐protected amino alcohol (±)‐7 (3.6 g, yield: 81 %, m.p.=115‐117 °C, light‐yellow crystals from with diethyl ether.) was purified by column chromatography [R f=0.23, eluent: n‐hexane:EtOAc (2:1)].
1H NMR (400 MHz, CDCl3) for (±)‐4: δ=8.12–8.26 (br s, 1 H, NH), 7.49–7.56 (d, J=7.69 Hz, 1 H, Ar–H), 7.32–7.39 (d, J=8.14 Hz, 1 H, Ar–H), 7.10–7.23 (m, 2 H, Ar–H), 4.18–4.26 (t, J=2.30 Hz, 1 H, CH), 3.77–3.96 (m, 2 H, CH2), 3.11–3.36 (m, 2 H, CH2), 2.71–2.87 ppm (m, 2 H, CH2); 13C NMR (400 MHz, [D4]MeOH) for (±)‐4: δ=135.35, 131.43, 125.98, 119.67, 117.22, 116.13, 109.46, 107.18, 62.11, 53.19, 40.17, 20.35 ppm; Anal. calcd. for C12H14N2O: C 71.26, H 6.98, N 13.85, found: C 71.21, H 6.93, N 13.79.
1H NMR (400 MHz, DMSO) for (±)‐7: δ=10.66–10.87 (br s, 1 H, NH), 7.35–7.43 (d, J=7.6 Hz, 1 H, Ar–H), 7.28–7.35 (d, J=8.0 Hz, 1 H, Ar–H), 7.01–7.09 (t, J=7.3 Hz, 1 H, Ar–H), 6.91–6.99 (t, J=7.2 Hz, 1 H, Ar–H),), 4.88–5.24 (m, 2 H, CH2), 4.10–4.43 (m, 1 H, CH), 3.69–3.84 (m, 2 H, CH2), 2.57–2.74 (m, 2 H, CH2), 1.44 ppm (s, 9 H, C(CH3)3); 13C NMR (400 MHz, CDCl3) for (±)‐7: δ=136.68, 132.30, 127.01, 122.24, 119.76, 118.50, 111.56, 81.10, 64.62, 53.32, 28.94, 21.91 ppm; Anal. calcd. for C17H22N2O3: C 67.53, H 7.33, N 9.26, found: C 67.43, H 7.39, N 9.22.
Synthesis of racemic N‐Boc‐protected 1‐hydroxymethyl‐6methoxy‐1,2,3,4‐tetrahydro‐β‐carboline [(±)‐8]
With the procedure described above [5‐methoxytryptamine hydrochloride (2, 1.0 g, 4.4 mmol), water (40 mL), 2 n HCl (2.8 mL), glycolaldehyde dimer (0.52 g, 4.3 mmol)], the reaction resulted in (±)‐5 [0.52 g, yield: 51 %, m.p.=152–153 °C, R f=0.25, eluent: MeOH] as yellow crystals. To a solution of (±)‐5 (0.52 g, 2.26 mmol) in 1,4‐dioxane (35 mL), NaOH (0.09 g, 2.25 mmol) in water (5 mL) and di‐tert‐butyl dicarbonate (0.54 g, 2.47 mmol) in 1,4‐dioxane (5 mL) were added. The method was as described above. (±)‐8 [0.65 g, yield: 87 %, m.p.=155–156 °C from n‐hexane, R f=0.34, eluent: n‐hexane:EtOAc (2:1)] was obtained as light‐yellow crystals.
1H NMR (400 MHz, CDCl3) for (±)‐5: δ=8.08–8.24 (br s, 1 H, NH), 7.22–7.24 (d, 1 H, J=8.80 Hz, Ar–H), 6.95 (s, 1 H, Ar–H), 6.78–6.88 (d, J=8 Hz, 1 H, Ar–H), 4.13–4.26 (m, 1 H, CH), 3.75–3.97 (m, 2 H, CH2 overlapping with s, 3 H, CH3), 3.04–3.37 (m, 2 H, CH2), 2.63–2.88 ppm (m, 2 H, CH2); 13C NMR (400 MHz, [D4]MeOH) for (±)‐5: δ=152.55, 132.38, 130.56, 126.30, 110.07, 109.48, 107.05, 98.70, 62.12, 53.90, 53.27, 40.23, 20.42 ppm; Anal. calcd. for C13H16N2O2: C 67.22, H 6.94, N 12.06, found: C 67.20, H 6.88, N 12.14.
1H NMR (400 MHz, DMSO) for (±)‐8: δ=10.49‐10.72 (br s, 1 H, NH), 7.16–7.31 (d, 1 H, J=8.48 Hz, Ar–H), 6.88 (s, 1 H, Ar‐H), 6.61–6.77 (dd, J=2.2 Hz, 8.7 Hz, 1 H, Ar–H), 4.90–5.23 (m, 2 H, CH2), 4.08–4.43 (m, 1 H, CH), 3.70–3.85 (m, 2 H, CH2 overlapping with s, 3 H, CH3), 2.51–2.61 (m, 2 H, CH2), 1.44 ppm (s, 9 H, C(CH3)3); 13C NMR (400 MHz, CDCl3) for (±)‐8: δ=154.45, 133.12, 131.80, 127.37, 112.18, 100.91, 81.05, 64.68, 56.42, 53.32, 28.90, 21.92 ppm; Anal. calcd. for C18H24N2O4: C 65.04, H 7.28, N 8.43, found: C 65.07, H 7.19, N 8.49.
Synthesis of racemic N‐Boc‐protected 1‐hydroxymethyl‐6‐fluoro‐1,2,3,4‐tetrahydro‐β‐carboline, (±)‐9
With the procedure described above [5‐fluorotryptamine hydrochloride (3, 1.0 g, 4.6 mmol), water (40 mL), 2 n HCl (2.5 mL), glycolaldehyde dimer (0.55 g, 4.6 mmol)], the reaction resulted in (±)‐6 [0.93 g, yield: 91 %, m.p.=138–141 °C, R f=0.15, eluent: toluene:MeOH (1:1)] as yellow crystals. To a solution of (±)‐6 (0.83 g, 3.77 mmol) in 1,4‐dioxane (30 mL), NaOH (0.15 g, 3.75 mmol) in water (5 mL) and di‐tert‐butyl dicarbonate (0.91 g, 4.17 mmol) in 1,4‐dioxane (5 mL) were added. The method was as described above. The product (±)‐9 [0.88 g, yield: 73 %, m.p.=124–125 °C from n‐hexane, R f=0.26, eluent: n‐hexane:EtOAc (2:1)] was obtained as light‐yellow crystals.
1H NMR (400 MHz, [D4]MeOH) for (±)‐6: δ=7.16–7.28 (q, J=4.44 Hz, 1 H, Ar–H), 7.00–7.08 (dd, J=2.48 Hz, 9.68 Hz, 1 H, Ar–H), 6.72–6.85 (dt, J=2.44 Hz, 9.16 Hz, 1 H, Ar–H), 4.03–4.15 (m, 1 H, CH), 3.78–3.92 (m, 1 H, CH2), 3.56–3.66 (m, 1 H, CH2), 3.25–3.32 (m, 1 H, CH2), 2.93–3.03 (m, 1 H, CH2), 2.62–2.78 ppm (m, 2 H, CH2); 13C NMR: (400 MHz, [D4]MeOH) for (±)‐6: δ=157.62, 155.31, 133.72, 131.84, 126.26, 110.07, 107.34, 100.86, 62.02, 47.39, 40.16, 20.28 ppm; Anal. calcd. for C12H13FN2O: C 65.44, H 5.95, N 12.72, found: C 65.27, H 5.99, N 12.85.
1H NMR (400 MHz, DMSO) for (±)‐9: δ=10.78–10.92 (br s, 1 H, NH), 7.25–7.34 (q, J=8.7 Hz, 1 H, Ar–H), 7.09–7.18 (dd, J=2.4 Hz, 9.8 Hz, 1 H, Ar–H), 6.83–6.91 (dt, J=2.8 Hz, 9.4 Hz, 1 H, Ar–H), 4.95–5.19 (m, 2 H, CH2), 4.11–4.39 (m, 1 H, CH), 3.69–3.81 (m, 2 H, CH2), 2.56–2.69 (m, 2 H, CH2), 1.44 ppm (s, 9 H, C(CH3)3); 13C NMR: (400 MHz, CDCl3) for (±)‐9: δ=159.37, 157.04, 134.25, 133.13, 127.32, 112.03 110.33, 103.54, 81.22, 64.49, 53.24, 40.36, 28.89, 21.85 ppm; Anal. calcd. for C17H21FN2O3: C 63.74, H 6.61, N 8.74, found: C 63.70, H 6.71, N 8.68.
Enzymatic resolutions
Enzymatic resolution of (±)‐7
To (±)‐7 (0.5 g, 1.66 mmol) in toluene (30 mL), lipase CAL‐B (900 mg) and acetic anhydride (2 equiv, 310 μL) were added, and the reaction mixture was shaken in an incubator shaker at 60 °C for 1.5 h. The reaction was stopped at 50 % conversion (ee=98 %) by filtering off the enzyme, and the solvent was then evaporated off. The products were separated by column chromatography on silica [eluent: n‐hexane:EtOAc (2:1)], resulting in the unreacted amino alcohol (R)‐7 as light‐yellow crystals [235 mg, yield: 47 %, [α]D 25=+107.5 (c=0.34 in EtOH), m.p.=136–137 °C, R f=0.24] and the product amino ester (S)‐10 as white crystals [263 mg, yield: 46 %, [α]D 25=−102.2 (c=0.32 in EtOH), m.p.=124–125 °C, R f=0.76].
The 1H NMR (400 MHz, DMSO) spectroscopic data for (R)‐7 were similar to those for (±)‐7. 1H NMR (400 MHz, CDCl3) for (S)‐10: δ=7.98–8.15 (br s, 1 H, NH), 7.52–7.54 (d, 1 H, J=8.0 Hz, Ar–H), 7.37–7.39 (d, 1 H, J=7.6 Hz, Ar–H), 7.2–7.26 (t, J=7.5 Hz, 1 H, Ar–H), 7.12–7.18 (t, J=7.2 Hz, 1 H, Ar–H), 5.31–5.69 (m, 1 H, CH), 4.29–4.47 (m, 1 H, CH2 overlapping with m, 2 H, CH2), 3.47–3.56 (m, 1 H, CH2), 2.71–2.96 (m, 2 H, CH2), 2.15 (s, 3 H, CH3), 1.46 ppm (s, 9 H, C(CH3)3); 13C NMR: (400 MHz, CDCl3) for (S)‐10: δ=171.30, 136.70, 130.76, 127.06, 122.57, 120.00, 118.66, 111.47, 80.81, 65.16, 50.09, 39.90 28.87, 21.90, 21.37 ppm; Anal. calcd. for C19H24N2O4: C 66.26, H 7.02, N 8.13, found: C 66.29, H 7.12, N 8.04.
Enzymatic resolution of (±)‐8
With the procedure described above, the reaction of (±)‐8 (200 mg, 0.6 mmol), in toluene (25 mL), CAL‐B (750 mg) and acetic anhydride (8 equiv, 466 μL) after 2.5 h resulted in (R)‐8 as white crystals [93 mg, yield: 47 %, [α]D 25=+82 (c=0.23 in EtOH), m.p.=196–198 °C, ee=98 %, R f=0.20, eluent: n‐hexane:EtOAc (2:1)] and (S)‐11 as a yellow oil [98 mg, yield: 43 %, [α]D 25=−92.3 (c=0.61 in EtOH), ee=98 %, R f=0.63, eluent: n‐hexane:EtOAc (2:1)].
The 1H NMR (400 MHz, DMSO) spectroscopic data for (R)‐8 were similar to those for (±)‐8. 1H NMR (400 MHz, DMSO) for (S)‐11: δ=10.72–10.88 (d, 1H J=14.04 Hz, NH), 7.18–7.25 (d, 1 H, J=8.4 Hz, Ar–H), 6.89–6.94 (d, 1 H, J=2.12 Hz, Ar–H), 6.69–6.75 (dd, 1 H, J=2.34 Hz, 8.84 Hz, Ar–H), 5.28–5.47 (m, 1 H, CH), 4.10‐4.50 (m, 2 H, CH2 overlapping with m, 2 H, CH2), 3.75 (s, 3 H, CH3), 2.55–2.73 (m, 2 H, CH2), 1.95–2.10 (m, 3 H, CH3), 1.44 ppm (s, 9 H, C(CH3)3); 13C NMR: (400 MHz, CDCl3) for (S)‐11: δ=171.36, 154.56, 131.84, 127.45, 112.19, 100.96, 80.78, 65.07, 56.39, 50.17, 28.87, 21.93, 21.35 ppm; Anal. calcd. for C20H26N2O5: C 64.15, H 7.00, N 7.48, found: C 64.26, H 7.02, N 7.39.
Enzymatic resolution of (±)‐9
Similarly, the preparative‐scale reaction of (±)‐9 (500 mg, 1.56 mmol) in toluene (50 mL), CAL‐B (1500 mg) and acetic anhydride (6 equiv, 885 μL) resulted after 2 h in (R)‐9 as light‐yellow crystals [234 mg, isolated yield: 47 %, [α]D 25=+97 (c=0.21 in EtOH), m.p.=99–100 °C, ee=96 %, R f=0.12, eluent: n‐hexane:EtOAc (3:1)] and (S)‐12 as white crystals [255 mg, isolated yield: 45 %, [α]D 25=−131.8 (c=0.38 in EtOH), m.p.=143–145 °C, ee=98 %, R f=0.49, eluent: n‐hexane:EtOAc (3:1)].
The 1H NMR (400 MHz, DMSO) spectroscopic data for (R)‐9 were similar to those for (±)‐9. 1H NMR (400 MHz, DMSO) for (S)‐12: δ=10.99–11.21 (d, 1 H, J=16 Hz, NH), 7.27–7.35 (2d, J=4.6 Hz, 1 H, Ar–H), 7.15–7.21 (dd, 1 H, J=2.4 Hz, 9.82 Hz, Ar–H), 6.87–6.96 (dt, J=2.5 Hz, 9.4 Hz, 1 H, Ar–H), 5.31–5.53 (d, 1 H, J=27.4 Hz, CH), 4.10–4.59 (m, 2 H, CH2 overlapping with m, 2 H, CH2), 2.55–2.70 (m, 2 H, CH2), 1.94–2.12 (m, 3 H, CH3), 1.44 ppm (s, 9 H, C(CH3)3); 13C NMR: (400 MHz, CDCl3) for (S)‐12: δ=171.31, 159.46, 157.12, 133.17, 132.64, 127.45, 111.98, 110.79, 103.73, 80.94, 65.07, 50.13, 28.85, 21.82, 21.35 ppm; Anal. calcd. for C19H23N2O4: C 62.97, H 6.40, N 7.73, found: C 62.89, H 6.44, N 7.78.
Deacylation of (S)‐10, (S)‐11, and (S)‐12
Enantiomeric (S)‐10 (30 mg, 0.09 mmol), (S)‐11 (40 mg, 0.11 mmol), or (S)‐12 (50 mg, 0.14 mmol) was dissolved in MeOH (10 mL). K2CO3 (50 mg, 0.36 mmol) was added, and the reaction mixture was shaken at 60 °C for 10 min. The following products were obtained as white crystals: (S)‐7 [24 mg, yield: 90 %, [α]D 25=−108.8 (c=0.33 in EtOH), m.p.=135–137 °C, ee=98 %, R f=0.48, eluent: n‐hexane:EtOAc (1:1)], (S)‐8 [31 mg, yield: 88 %, [α]D 25=−81 (c=0.12 in EtOH), m.p.=195–197 °C, ee=98 %, R f=0.47, eluent: n‐hexane: EtOAc (1:1)], or (S)‐9 [33 mg, yield: 75 %, [α]D 25=−105 (c=0.195 in EtOH), m.p.=98–100 °C, ee=98 %, R f=0.20, eluent: n‐hexane: EtOAc (3:1)].
Deprotection of (R)‐7, (R)‐8, (R)‐9, (S)‐10, (S)‐11, and (S)‐12
(R)‐7 (50 mg, 0.16 mmol), (R)‐8 (30 mg, 0.09 mmol), (R)‐9 (30 mg, 0.09 mmol), (S)‐10 (50 mg, 0.15 mmol), (S)‐11 (40 mg, 0.1 mmol), or (S)‐12 (30 mg, 0.08 mmol) was suspended in water (5 mL) in a tube, and stirred at 100 °C for 1 h under maximum MW irradiation of 150 W. The solvent was evaporated off, and the residue was purified by column chromatography with toluene:MeOH (1:1) as eluent. The following products were crystallized from n‐hexane: (R)‐4 as yellow crystals [26 mg, yield: 79 %, [α]D 25=−37.8 (c=0.41 in EtOH), m.p.=145–147 °C, ee=98 %, R f=0.15], (S)‐4 as yellow crystals [15 mg, yield: 52 %, [α]D 25=+36.1 (c=0.4 in EtOH), m.p.=146–147 °C, ee=98 %, R f=0.16], (R)‐5 as light‐yellow crystals [8 mg, yield: 38 %, [α]D 25=−22.0 (c=0.4 in EtOH), m.p.=167–168 °C, ee=98 %, R f=0.18], (S)‐5 as light‐yellow crystals [9 mg, yield: 38 %, [α]D 25=+21.8 (c=0.45 in EtOH), m.p.=168‐170 °C, ee=98 %, R f=0.21], (R)‐6 as light‐yellow crystals [11 mg, yield: 52 %, [α]D 25=−29 (c=0.155 in EtOH), m.p.=96‐98 °C, ee=96 %, R f=0.14], or (S)‐6 as light‐yellow crystals [5 mg, yield: 28 %, [α]D 25=+29 (c=0.25 in EtOH), m.p.=99‐101 °C, ee=98 %, R f=0.18].
Synthesis of racemic and enantiomeric 1‐hydroxymethyl‐2‐acetyl‐1,2,3,4‐tetrahydro‐β‐carboline [(±)‐13 and (R)‐13]
A mixture of (±)‐4 (30 mg, 0.13 mmol), 6 equiv acetic anhydride (75 μL, 0.8 mmol) and NaOH (100 mg, 2.5 mmol) in CH2Cl2 (20 mL) and water (20 mL) was stirred for 48 h at rt. The reaction mixture was then extracted with CH2Cl2 (3×15 mL). The organic phase was dried on anhydrous Na2SO4 and evaporated. The resulting dark‐yellow oil was purified by column chromatography with CH2Cl2:MeOH (30:1) as eluent. The product (±)‐13 (18 mg, yield: 49 %, m.p.=191–192 °C, R f=0.10) was obtained as white crystals from EtOH and diisopropyl ether (1:9).
1H NMR (400 MHz, CDCl3) for (±)‐13: δ=8.58–8.71 (br s, 1 H, NH), 7.44–7.49 (d, J=7.72 Hz, 1 H, Ar–H), 7.31–7.35 (d, J=8.36 Hz, 1 H, Ar–H), 7.13–7.20 (dt, J=1 Hz, 7.02 Hz, 1 H, Ar–H), 7.06–7.13 (t, J=7.2 Hz, 1 H, Ar–H), 5.74–5.82 (t, J=6.56 Hz, 1 H, CH), 3.96–4.11 (m, 2 H, CH2), 3.84–3.93 (m, 1 H, CH2), 3.43–3.55 (m, 1 H, CH2), 2.79–2.96 (m, 2 H, CH2), 2.29 ppm (s, 3 H, CH3).
With the above procedure, the reaction of (R)‐4 (26 mg, 0.11 mmol) resulted in the desired product (R)‐13 as white crystals [22 mg, yield: 70 %, [α]D 25=+164 (c=0.2 in EtOH), m.p.=201–203 °C, ee=98 %]. The 1H NMR (400 MHz, CDCl3) spectroscopic data for (R)‐13 were similar to those for (±)‐13.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
The authors are grateful to the Hungarian Research Foundation (OTKA No. K108943 and K115731) for financial support.
R. Megyesi, E. Forró, F. Fülöp, ChemistryOpen 2016, 5, 254.
References
- 1. Garrett L., O. Carrier Jr. , Douglas B. H., Eur. J. Pharmacol. 1967, 2, 236–238. [DOI] [PubMed] [Google Scholar]
- 2. Creasey W. A., Markiw M. E., Biochim. Biophys. Acta 1964, 87, 601–609. [DOI] [PubMed] [Google Scholar]
- 3. Zhang Z.-J., Yang J., He J., Wu X.-D., Shao L.-D., Li Y., Huang S.-X., Li R.-T., Zhao Q.-S., Tetrahedron Lett. 2014, 55, 6490–6494. [Google Scholar]
- 4. Cheng G.-G., Zhao Y.-L., Zhang Y., Lunga P.-K., Hu D.-B., Li Y., Gu J., Song C.-W., Sun W.-B., Liu Y.-P., Luo X.-D., Tetrahedron 2014, 70, 8723–8729. [Google Scholar]
- 5. Cong H.-J., Zhao Q., Zhang S.-W., Wei J.-J., Wang W.-Q., Xuan L.-J., Phytochemistry 2014, 100, 76–85. [DOI] [PubMed] [Google Scholar]
- 6. Kam T.-S., Sim K.-M., Phytochemistry 1998, 47, 145–147. [Google Scholar]
- 7. Spindola H. M., Vendramini-Costa D. B., M. T. Rodrigues Jr. , Foglio M. A., Pilli R. A., Carvalho J. E., Pharmacol. Biochem. Behav. 2012, 102, 133–138. [DOI] [PubMed] [Google Scholar]
- 8. Santos L. S., Theoduloz C., Pilli R. A., Rodriguez J., Eur. J. Med. Chem. 2009, 44, 3810–3815. [DOI] [PubMed] [Google Scholar]
- 9. Davis R. A., Duffy S., Avery V. M., Camp D., Hooper J. N. A., Quinn R. J., Tetrahedron Lett. 2010, 51, 583–585. [Google Scholar]
- 10. Brokamp R., Bergmann B., Müller I. B., Bienz S., Bioorg. Med. Chem. 2014, 22, 1832–1837. [DOI] [PubMed] [Google Scholar]
- 11. Trujillo J. I., Meyers M. J., Anderson D. R., Hegde S., Mahoney M. W., Vernier W. F., Buchler I. P., Wu K. K., Yang S., Hartmann S. J., Reitz D. B., Bioorg. Med. Chem. Lett. 2007, 17, 4657–4663. [DOI] [PubMed] [Google Scholar]
- 12. Prasad K. R., Nidhiry J. E., Sridharan M., Tetrahedron 2014, 70, 4611–4616. [Google Scholar]
- 13. Shankaraiah N., Nekkanti S., Chudasama K. J., Senwar K. R., Sharma P., Jeengar M. K., Naidu V. G. M., Srinivasulu V., Srinivasulu G., Kamal A., Bioorg. Med. Chem. Lett. 2014, 24, 5413–5417. [DOI] [PubMed] [Google Scholar]
- 14. Roszkowski P., Czarnocki S., Maurin J. K., Siwicka A., Zawadzka A., Szawkało J., Leniewski A., Czarnocki Z., Int. Congr. Ser. 2007, 1304, 46–59. [Google Scholar]
- 15. Chakraborty I., Jana S., Synthesis 2013, 45, 3325–3331. [Google Scholar]
- 16. Csajági Cs., Szatzker G., Tőke E. R., Ürge L., Darvas F., Poppe L., Tetrahedron: Asymmetry 2008, 19, 237–246. [Google Scholar]
- 17. Tomin A., Hornyánszky G., Kupai K., Dorkó Zs., Ürge L., Darvas F., Poppe L., Process Biochem. 2010, 45, 859–865. [Google Scholar]
- 18. De Miranda A. S., Gomes J. C., M. T. Rodrigues Jr. , Costa I. C. R., Almeida W. P., Lopes R. de O., Miranda L. S. M., Coelho F., de Souza R. O. M. A., J. Mol. Catal. B 2013, 91, 77–80. [Google Scholar]
- 19. Tamborini L., Romano D., Pinto A., Bertolani A., Molinari F., Conti P., J. Mol. Catal. B 2012, 84, 78–82. [Google Scholar]
- 20. Sutili F. K., Ruela H. S., Leite S. G. F., Miranda L. S. de M., Leal I. C. R., de Souza R. O. M. A., J. Mol. Catal. B 2013, 85–86, 37–42. [Google Scholar]
- 21. I. Itabaiana Jr. , Miranda L. S. de M., de Souza R. O. M. A., J. Mol. Catal. B 2013, 85–86, 1–9. [Google Scholar]
- 22. Forró E., Schönstein L., Fülöp F., Tetrahedron: Asymmetry 2011, 22, 1255–1260. [Google Scholar]
- 23. Schönstein L., Forró E., Fülöp F., Tetrahedron: Asymmetry 2013, 24, 1059–1062. [Google Scholar]
- 24. Schönstein L., Forró E., Fülöp F., Tetrahedron: Asymmetry 2013, 24, 202–206. [Google Scholar]
- 25. Spenser I. D., Can. J. Chem. 1959, 37, 1851–1858. [Google Scholar]
- 26. Wegner J., Ceylan S., Kirschning A., Chem. Commun. 2011, 47, 4583–4592. [DOI] [PubMed] [Google Scholar]
- 27. Forró E., Lundell K., Fülöp F., Kanerva L. T., Tetrahedron: Asymmetry 1997, 8, 3095–3099. [Google Scholar]
- 28. Simas A. B. C., da Silva A. A. T., Cunha A. G., Assumpção R. S., Hoelz L. V. B., Neves B. C., Galvão T. C., Almeida R. V., Albuquerque M. G., Freire D. M. G., de Alencastro R. B., J. Mol. Catal. B 2011, 70, 32–40. [Google Scholar]
- 29. Miyazawa T., Kaito E., Yukawa T., Murashima T., Yamada T., J. Mol. Catal. B 2005, 37, 63–67. [Google Scholar]
- 30. Wang G., Li C., Li J., Jia X., Tetrahedron Lett. 2009, 50, 1438–1440. [Google Scholar]
- 31. Dandepally S. R., Williams A. L., Tetrahedron Lett. 2009, 50, 1071–1074. [Google Scholar]
- 32. Benedek G., Palkó M., Wéber E., Martinek T. A., Forró E., Fülöp F., Tetrahedron: Asymmetry 2009, 20, 2220–2225. [Google Scholar]
- 33. Araźny Z., Czarnocki Z., Wojtasiewicz K., Maurin J. K., Tetrahedron: Asymmetry 2000, 11, 1623–1628. [Google Scholar]
- 34. Araźny Z., Czarnocki Z., Wojtasiewicz K., Maurin J. K., Tetrahedron: Asymmetry 2000, 11, 2793–2800. [Google Scholar]
- 35.Apparently the value of the specific rotation of compound questioned was incorrectly given in the text in our publication (Ref. [28]) due to an error in decimal point placement (Czarnocki, Z., personal communication).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary