

Abstract
By increasing the strength of the negative dielectrophoresis force, we demonstrated a significantly improved electrokinetic actuation and switching microsystem that can be used to elute specifically bound beads from the surface. In this work using atomic layer deposition we deposited a pinhole free nanometer-scale thin film oxide as a protective layer to prevent electrodes from corrosion, when applying high voltages (>20 Vpp) at the electrodes. Then, by exciting the electrodes at high frequency, we capacitively coupled the electrodes to the buffer in order to avoid electric field degradation and, hence, reduction in dielectrophoresis force due to the presence of the insulating oxide layer. To illustrate the functionality of our system, we demonstrated 100% detachment of anti-IgG and IgG bound beads (which is on the same order of magnitude in strength as typical antibody–antigen interactions) from the surface, upon applying the improved negative dielectrophoresis force. The significantly enhanced switching performance presented in this work shows orders of magnitude of improvement in on-to-off ratio and switching response time, without any need for chemical eluting agents, as compared to the previous work. The promising results from this work vindicates that the functionality of this singleplexed platform can be extended to perform a multiplexed bead-based assay where in a single channel an array of proteins are patterned each targeting a different antigen or protein.
Graphical Abstract

The monitoring of genetic and protein biomarkers is of utmost necessity to complete the demands of personalized healthcare.1–8 Protein biomarkers have the potential to be more powerful than genetic biomarkers as the expressed gene products that result from several layers of regulation built into a cell’s machinery. However, protein biomarkers currently have limited utility in the clinical setting due to various limitations of current proteomic technologies.9–11 One of the most pertinent problems limiting the utility of proteomic technologies in the clinical setting is the low throughput nature of these assays, especially since single biomarkers often prove to have low specificity.12 In addition to protein biomarker detection, the quantification of protein–protein interactions is of utmost importance in studying various inter- and intracellular molecular pathways and also in drug screening (small molecule protein–protein interaction inhibitors).13–22 At present, the most routinely used protein assay to analyze clinical samples is that of ELISA, which is performed in a 96 well plate, thus not suited for high-throughput multiplexing. In the research laboratory setting, fluorescence based techniques such as protein/antibody array technology and also mass spectrometry have demonstrated utility in high-throughput proteomic analysis. While high in throughput, the disadvantage of the two mentioned techniques is the high cost associated with the instrumentation. Mass spectrometry requires expensive apparatus and has high maintenance costs as well. Microarray technology requires expensive fluorescent detection apparatus, including an excitation laser, a photodetector, scanning apparatus, resulting in a bulky optical setup.23–27 The bulkiness results from the challenge of imaging fluorescence signal over a relatively wide area (greater than 1 cm2) which necessitates either the whole surface to be scanned or a wide field image sensor. Various efforts are being made in developing miniaturized wide field optical cell imaging systems which have proved successful in particular work done by Ozcan et al.;28–37 however, a solution which would not require wide field analysis where detection at a single point would suffice would result in significantly lower cost. In order to achieve this effectively while keeping costs low and minimizing the footprint of the device, one would require an automated method for actuating either fluids or bioparticles preferably without any physically moving parts, thus making electronic bioactuation the ideal solution. To this end, we set out to develop an electronically actuated smart surface. Figure 1 shows a schematic of the electrokinetically actuated bead based assay we envision. In the case of analyzing protein–protein interactions, we pattern an array of receptor proteins along a single channel, where below each element of our array, we have a pair of addressable interdigitated electrodes. The channel is then loaded with beads which have the protein of interest conjugated to the bead surface. After a series of wash steps to remove the unbound and loosely bound beads, the number of beads binding to each element of the array will depend on the strength of the protein–protein interactions between the protein on the bead and that on the surface of the channel. In addition to the drag force being applied to the beads (resulting from the pressure driven flow), an electrokinetic force is applied to the beads at each element of the array sequentially to detach the beads from each region one by one. The detached beads will then be transported with the flow downstream where they can be quantified. The advantage of this type of approach is that the cost decreases significantly due to the need for detection only at a single point, rather than across the whole array of proteins.
Figure 1.
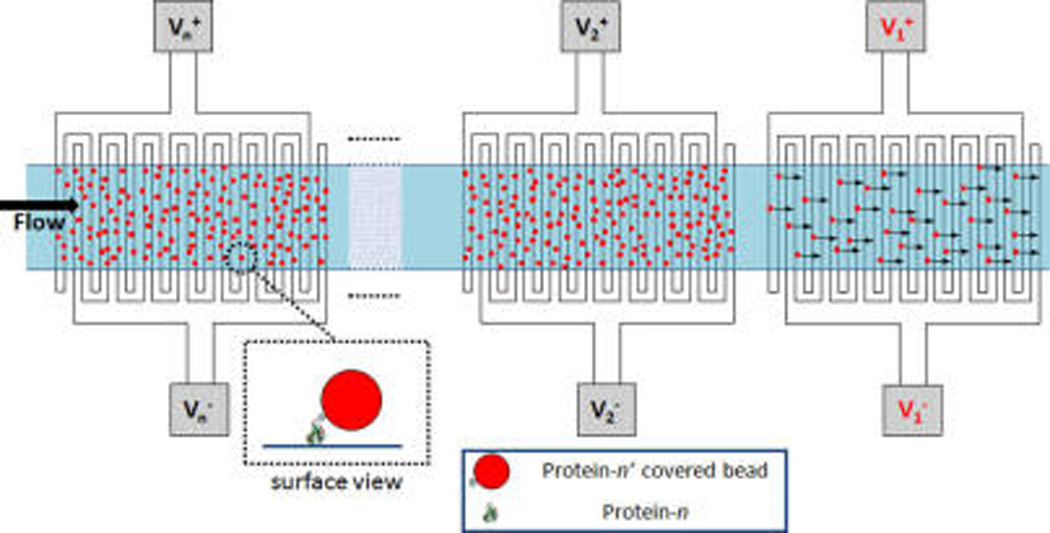
Bead-based multiplexed assay. Each element of an array in the capture region is immobilized with a different protein each targeting a specific protein that is coated on the micrometer-sized beads. Unbound beads are washed out of the channel. Specifically bound beads on each element of the array are eluted one-by-one from the array and are quantified downstream as they pass through. Here, applying voltage V1 turns nDEP on, resulting in elution of specifically bound beads from the surface of the 1st set of interdigitated electrodes.
Given that the beads are held down on the surface via protein–protein interactions, which can typically be on the order of nanonewtons of force,38 a relatively strong method of bead actuation is necessary. Optical tweezers have previously been shown to provide on the order of 0.1–100 pN of force.39 Magnetic tweezers have also shown the capability to achieve 1 fN to 100 pN of force.39 Both optical and magnetic tweezers have advantages of their own, such as matrix independence, but are more bulky than electrokinetic based techniques like negative dielectrophoresis which boasts the advantage of ease of integration and miniaturization with microfluidics; however, similar to optical and magnetic tweezers, conventional DEP provides on the order of 10 pN of force,40,41 due to the fact that bare electrodes begin to corrode beyond low threshold voltages. Relatively thick layers of oxide have been shown to allow higher voltages to be applied; however, with thicker oxides, the voltage drop is primarily across the oxide rather than the bulk solution.42
Previously, with the aid of negative dielectrophoresis (nDEP) force in conjunction with shear force, and at an optimal sodium hydroxide concentration (NaOH), we demonstrated a switch-like functionality to elute specifically bound beads from the surface. The nDEP force was established by applying ac voltage at the interdigitated electrode pair. The role of the NaOH (at an optimal concentration) was to sufficiently weaken the binding such that the nDEP force would be able to push the specifically bound beads off the surface, yet not too weak, so beads would remain firmly bound to the surface at the flow rate of operation when nDEP was off. While the use of NaOH allowed for establishing the switching functionality of nDEP as a proof of concept, its use is not desirable for the purpose of performing multiplexed assay along a single channel. In the multiplexed setting, the optimal NaOH concentration for each interaction may be different and there may be no overlap of the practical range of NaOH concentrations for all interactions. For instance, the lowest NaOH concentration needed to help nDEP with detaching the bound beads for one interaction may already be too strong for another interaction along the array and may undesirably result in detachment of those bound beads with nDEP off. To resolve this issue and eliminate the need for NaOH, we have to significantly increase the nDEP force. The increase in the strength of the nDEP force would enhance the switching performance of the device. In the previous work, a rather low switching on-to-off ratio of 3.6 was achieved (90% and 25% detachment when nDEP was on and off, respectively), while it took a rather long time of about 4 min to detach the majority of the beads (response time). The improved switch should have significantly shorter response time and higher proportional detachment of beads when “on” (due to the increase in nDEP force) and lower proportional detachment when “off” (mainly due to elimination of NaOH, the weakly bound beads no longer detach from the surface at the flow rate of operation with nDEP off).
The most effective way of improving the DEP force in our context is through increasing the gradient of the electric field, which can be achieved by increasing the applied voltage at the electrodes. However, upon applying high voltages (greater than 20 Vpp), the electrodes corrode due to onset of electrochemical reactions at the interface of the electrodes and the solution buffer. To resolve the corrosion issue, for this work, we planned on depositing a thin layer of oxide using atomic layer deposition (ALD) to effectively create a physical barrier between the electrodes and the solution buffer in order to prevent corrosive electrochemical reactions. Deposition of thin film oxide layer on the electrodes imposes four major challenges. First, as a part of the device fabrication procedure, a microfluidic channel embedded in PDMS (polydimethylsiloxane) often needs to be covalently bonded (using oxygen plasma treatment) to the surface of the glass chip on which the metal electrodes are patterned. However, the deposition of the oxide layer modifies the surface of the chip, and hence, the bonding of PDMS to the oxide-deposited glass chip is no longer guaranteed and the bonding of PDMS to a number of different oxide films needed to be investigated. The second challenge is degradation of the electric field in the solution buffer and hence reduction in the dielectrophoresis force, as a result of the undesired voltage drop across the insulating deposited oxide. To compensate for the voltage drop across the oxide film, one may simply increase the applied voltage at the electrodes, but that leads to the third challenge, the oxide breakdown of the deposited thin oxide film upon application of high voltages. As a result, to address the latter two challenges, a careful circuit analysis was required to systematically identify the relevant parameters and the trade-offs involved in our proposed system, as discussed in the following section. A fourth challenge exists, and that is the need for a pinhole free insulative film in order to prevent corrosion. With techniques like plasma enhanced chemical vapor deposition, this is impossible with films less than 50 nm in thickness. This necessitates the use of atomic layer deposition to achieve pinhole free films with thicknesses below 20 nm.
THEORETICAL BACKGROUND
Circuit Model and Analysis
Figure 2a represents the simplified circuit model of interface of the two neighboring electrodes in our interdigitated electrode pair with the solution buffer. The conducting medium, i.e., the solution buffer can simply be modeled as resistance. The parasitic capacitance represents the direct coupling between the two neighboring electrodes. The interface between the oxide-deposited electrodes and solution buffer is effectively an insulator in between two conductors (electrode and solution buffer) and can be modeled as capacitance. In our case, the double-layer capacitance is significantly larger than the thin film deposited oxide capacitance due to double-layer capacitance’s smaller thickness and higher effective dielectric constant. Therefore, as it is effectively in series with the oxide capacitance, the double-layer capacitance can be neglected. With the voltage applied at the two electrodes, the oxide capacitances at each terminal form a voltage divider with the solution buffer resistance. Consequently, there would be an unwanted voltage drop across the oxide films at each end, leading to electric field degradation inside the solution buffer and, hence, significant reduction in the available DEP force. From a circuit analysis point of view, this issue can be resolved by configuring the system such that the impedance of the oxide capacitance is significantly smaller than that of the solution buffer. This can be done through a number of complementary approaches. The first is to use a low conductivity buffer, such as deionized (DI) water or diluted phosphate-buffered saline (PBS), in the wash step. This increases the resistance of the buffer and hence minimizes the voltage drop across the oxide film. Moreover, we need to excite the electrodes at high frequency of operation to effectively reduce the impedance of the oxide film as compared to the buffer resistance. Fortunately, in our case this is aligned with the requirement for operating at high frequencies to accomplish nDEP.43 Finally, the reduction in the oxide film impedance can also be accomplished through minimizing the thickness of the deposited oxide. However, this may result in oxide-breakdown. We simulated the circuit model of our network for different values of oxide thicknesses based on the first-order estimated component values. For this simulation, it was assumed the solution buffer was DI water, the deposited oxide was silicon dioxide (SiO2), and that 100 V is applied at the electrodes. Furthermore, the dimensions of the electrodes and channel in the simulation were chosen the same as that of the fabricated device. Figure 2b,c illustrates the corresponding simulated voltage drop and electric field spectrum across the oxide capacitance at each electrode-buffer interface. As can be seen from Figure 2b, at 1 MHz there is 20% voltage drop across the 50 nm-thick oxide film. The voltage drop can be minimized further (below 5%) when using the oxide with 10 nm thickness. The simulation results show that at about a frequency of 1 MHz and beyond the electric field characteristics across the oxide films of varying thicknesses converge and becomes independent of the thickness of the oxide (Figure 2c). This can be best explained analytically as following:
| (1) |
where VC is the voltage drop across the oxide capacitance Cox at each end of the electrode, R is the resistance of the solution buffer between the two neighboring electrodes, and Vapp is the applied voltage with the excitation frequency f. Here, the oxide capacitance Cox to the first order can be modeled as a parallel plate capacitance, with permittivity ε, thickness t, and area A (equal to the area bound by the width of the channel and an electrode). Therefore, the electric field EC across the oxide at high frequencies becomes
| (2) |
Figure 2.
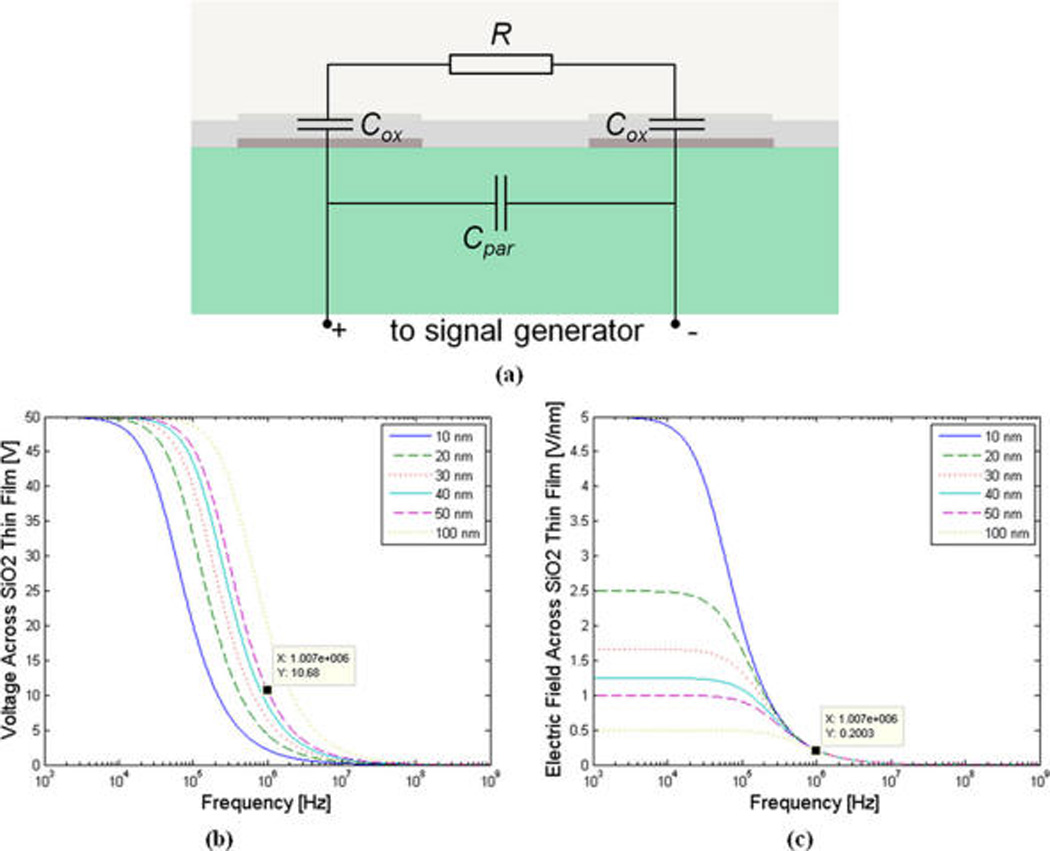
(a) Simplified equivalent circuit model of the two neighboring electrodes in the interdigitated electrode pair. R refers to the resistance of the channel. Cox is the capacitance of the deposited oxide layer for each electrode-oxide-electrolyte interface, and Cpar is the parasitic capacitance. (b,c) Simulated voltage drop spectrum (b) and electric field spectrum (c) across the oxide capacitance at each electrode-buffer interface for various oxide thicknesses.
As can be seen from eq 2, once operating at sufficiently high frequencies (primarily to minimize the undesired voltage drop across the oxide), EC becomes independent of the thickness of the oxide. Hence, oxide-breakdown would not directly set the lower limit on the required thickness. According to our simulation results, at 1 MHz, the electric field across the oxide would be 0.2 V/nm which is well below the ALD SiO2 breakdown field of 1 V/nm.44
Relevant Forces
As discussed in the previous work,43 in addition to the DEP force, the particles that are bound to the surface experience gravitational force and the hydrodynamic drag force. Moreover, for our improved DEP platform, since the applied voltage is increased the electro-thermal effects may become apparent.
The gravitational (sedimentation) force that acts on the particle is governed by the following equation:
| (3) |
where g is the gravitation constant, ρb is the bead density, ρm is the medium density, and r is the particle radius. For a 2.8 µm diameter polystyrene-magnetic bead, this comes to approximately 10 fN, which is orders of magnitude smaller than the other forces in our system.
The hydrodynamic drag force on a bead attached to the surface of the channel can be approximated with the following equation:45
| (4) |
where r is the bead radius, k is a nondimensional factor accounting for wall effects (k ≈ 1.7), η is the dynamic medium viscosity, and νf is the fluid velocity. On the basis of the results from our previous work on quantification of biomolecular interactions,46 flow rates ranging from 10 nL/min to 10 µL/min provide drag forces between 0.2 pN and 200 pN on a 2.8 µm diameter bead.
In our high voltage system, the other relevant force is the electrothermal body force acting on the fluid. This electrothermal effect is due to spatial variations in the electric field that causes nonuniform Joule heating and spatially varying permittivity and conductivity fields in the fluid. In the presence of an externally applied electric field, this results in Coulomb and dielectric body forces and induces microscale fluid motion. Following the approach presented by Meinhart and colleagues,47 the temperature profile of the system was simulated using COMSOL (COMSOL, Stockholm, Sweden) and the fluid velocity field resulting from the electrothermal effect was determined. On the basis of our simulation results, for our electrode and microfluidic channel configurations with a low-conductive buffer solution and applied voltage of 50–100 Vpp, the equivalent hydrodynamic drag force exerted on the beads due to induced fluid motion was on the order of 6–60 pN.
Dielectrophoresis is a force applied to particles in a nonuniform electric field as a result of differences in polarizability of the particle and the fluid medium. The first order time-average DEP force acting on a dielectric sphere is given by
| (5) |
where εm is the relative permittivity of the surrounding medium, r is the particle radius, and ERMS is the root-mean-square value of the electric field. fCM in the above equation is the Clausius-Massotti factor which is related to the effective polarizability of the particle with respect to that of the medium and is of the form
| (6) |
where εp* and εm* are the relative complex permittivities of the particle and the medium, respectively.
The sign of the real part of the Clausius-Massotti factor determines whether the particle is attracted to (positive DEP) or repelled from (nDEP) a region of high electric field strength. On the basis of our previous results,43 in order to achieve DEP in its negative form for the medium conductivity range of interest we need to operate at the frequencies of above 1 MHz. This is aligned with the frequency requirement imposed by our circuit analysis. As one of the pioneers of the field, Voldman et al. through comparison of results from modeling and measurements demonstrated subpiconewton forces in dielectrophoresis-based single-particle traps.48 According to our work and previous work shown in the literature, using micrometer sized interdigitated electrodes, it is possible to provide DEP forces ranging from 0.1 to 10 pN when applying 1–10 Vpp at the electrodes.45 A typical antigen–antibody interaction is roughly on the order of 400 pN. As FDEP is proportional to the square of the applied voltage, in this work by increasing the capability of applying higher voltages at the electrodes we were able to increase the DEP significantly beyond this value to comfortably detach the bound beads from the surface.
RESULTS AND DISCUSSIONS
Applying the standard oxygen plasma treatment process at the PDMS bonding step was only successful for the SiO2 coated film. It formed a strong bond with the same order of magnitude in strength as PDMS-glass bonding which is amenable to microfluidic pressure driven applications. The same treatment processes did not result in a strong bond for the cases of the Al2O3 and HfO2 coated thin films. At this step since we achieved a well-sealed and practical device with the pinhole-free SiO2 coated chip (no outside channel leakage at the flow rates of interest and beyond), we proceeded with our experiments using the SiO2-coated devices.
To validate the developed circuit model of the device, we measured the impedance spectrum between the two interdigitated electrodes. Deionized (DI) water with measured conductivity of 1.2 mS/m was used as the solution buffer. Figure 3a illustrates the measured impedance spectrum between the two interdigitated electrodes. In this graph, the curve-fitted spectrum based on the equivalent circuit model is also overlaid. The close agreement between the two graphs not only supports our developed circuit model for the device but also allows us to characterize and extract the resistance value of the buffer as well as the oxide and parasitic capacitance values. Upon validation of the circuit model, the characterized values from the measured impedance spectrum were used to plot the voltage drop and the electric field characteristic across the deposited oxide film for the frequency range of interest. Figure 3b,c illustrate these characteristics as well as the corresponding originally simulated voltage drop and electric field spectrum, based on our first-order estimate of the resistance and capacitance values. As can be seen from these figures, our simulation and measurement results are close. The slight deviation between them in fact implies that the simulation results originally led us to a more conservative conclusion by projecting a higher voltage drop for a given frequency. On the basis of the above results, and the available margin in design space specifications, we can also conclude that using a more conductive solution such as diluted PBS as our buffer would still be practical when demonstrating the improved switching capability of nDEP (as we will see shortly).
Figure 3.
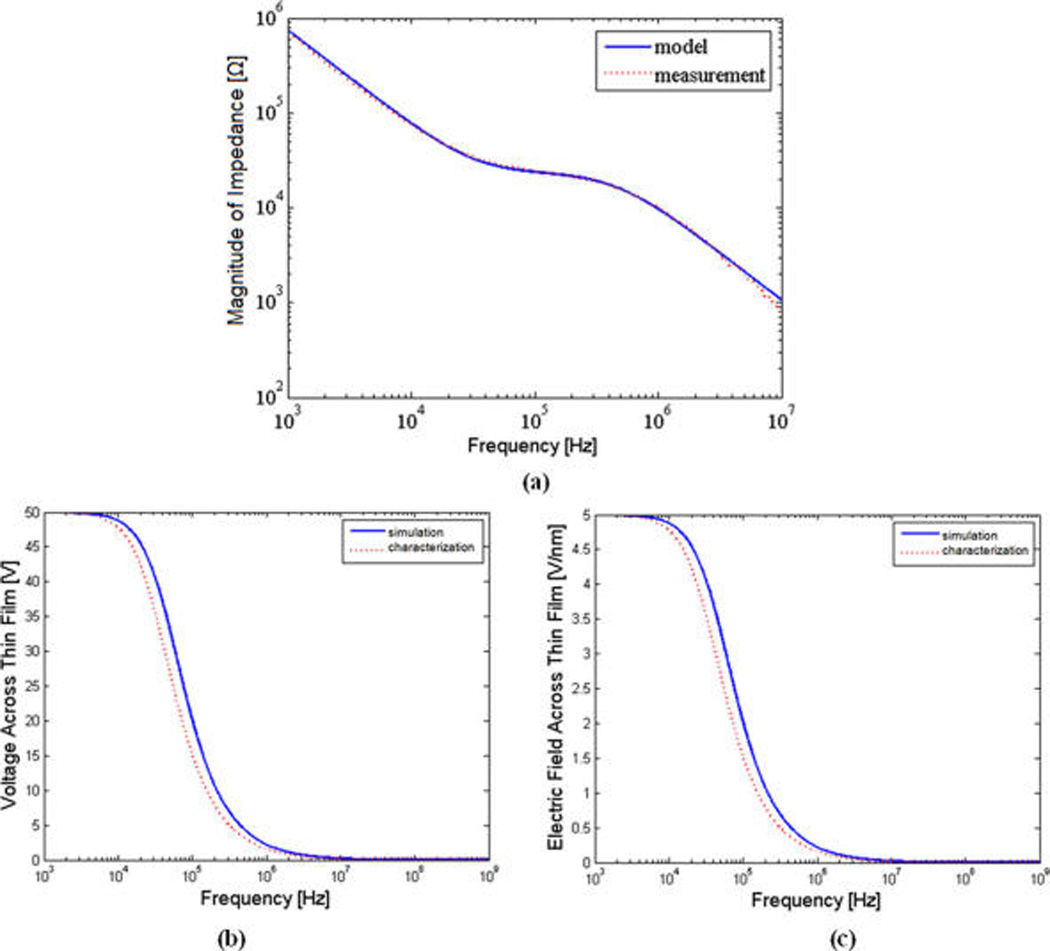
(a) Impedance spectrum (measured vs curve-fitted model) between the two neighboring electrodes in the interdigitated electrode pair. (b,c) Characterized vs simulated voltage drop spectrum (b) and electric field spectrum (c) across the deposited 10-nm SiO2 layer on the electrodes.
Next, we characterized the voltage tolerance of the fabricated device by increasing the applied voltage in small steps and scanning the impedance spectrum at each point to see whether or not the device parameters have stayed the same or significantly changed due to oxide breakdown or other undesired irreversible effects. Our voltage tolerance characterization process indicated that our device can practically provide up to 100 Vpp of voltage which practically provides nDEP on the order of nanoNewtons above the oxide. As was visually evident and confirmed through comparison of impedance spectra of the device captured at each incremental voltage step, there was no sign of damage and the device parameters stayed the same. However, increasing the applied voltage to 120 Vpp resulted in bubble formation. We speculate that the bubble formation can be attributed to the generated heat in the channel resulting in outgassing or evaporation phenomena, with temperature approaching the boiling point (Figure 4). To ensure that the device integrity was preserved after the bubble formation, the channel was flushed and filled with buffer. Then, by capturing the impedance spectrum we verified that the device parameters stayed the same. We repeated the same procedure for three other devices to verify the bubble formation at high voltages (about 120 Vpp), while subsequently validating the preserved device integrity after flushing and filling the channel. It is worth noting that here we demonstrated a design where about 100 Vpp of voltage is effectively dropped across the buffer with minimal voltage and field degradation across the deposited oxide. This implies voltage tolerance improvement of a factor of 5 is achieved, resulting in 25-fold stronger nDEP for the same interdigitated electrode configuration. Moreover, with our fabricated device, the upper bound on the electrode’s voltage tolerance is no longer imposed by fabrication limitations (such as electrode corrosion in previous work or other irreversible phenomena such as possible oxide breakdown, etc.) but in fact is restricted by physical phenomena affecting the buffer.
Figure 4.

Upper bound on the voltage tolerance of the improved DEP device is limited by the bubble formation inside the channel due to the generated heat. The device integrity was preserved throughout the above procedure.
Once the circuit model and voltage tolerance of the device were characterized, we demonstrated the core functionality of the device. We first flushed the channel with diluted PBS to eliminate the air bubbles. Next, the prepared sample beads were injected into the channel by directly pipetting them into the inlet port. The beads were allowed to settle for 15 min so that they had sufficient time to bind to the surface. Negative pressure was applied to the syringe, which was connected to the outlet well through tygon tubing, to pull the solution. The flow rate was controlled with the syringe pump. All experiments were video recorded using a digital microscope camera ProgRes MF (Jenoptik, Germany) that was mounted on a microscope (Nikon, Japan). For each experiment, the device on the imaging stage was positioned such that the imaging camera’s field of view covered the portion of the channel with the interdigitated electrodes as well as the neighboring region along the channel with no electrodes. By applying voltage at the electrodes, we turned nDEP on. The upward nDEP force caused the specifically bound beads to be pushed away from the surface of the interdigitated electrodes, without disturbing the bound beads on the neighboring no-electrode zone along the channel. Then, to detach the remaining bound beads, the flow rate and hence drag force was increased gradually until all eluted. At each flow rate, we quantified the percentage of detached beads relative to the total number of initially captured beads (at rest). To quantify the beads accurately and efficiently we used a custom-made automated video and image processing software. Using this software, and by comparing the consecutive captured frames, we were able to distinguish between the stationary beads that were actually bound and the moving beads that were not originally bound. If analyzing only a single frame captured at low flow rate, the moving beads might have appeared and mistakenly quantified as stationary bound beads. Overall, this procedure was repeated on three different devices where we analyzed the total of 9000 beads. The collective result for the anti-IgG and IgG interaction is shown in Figure 5a for the cases of nDEP-off (the regions outside the interdigitated electrodes) and nDEP-on (applying 50 Vpp at 1 MHz). Upon turning nDEP on, almost all (99.8%) of the bound beads on the interdigitated electrodes region got eluted without disturbing the bound beads on the neighboring no-electrode zone along the channel. The remaining bound beads outside the interdigitated electrodes region (represented as nDEP-off curve in Figure 5a) got detached as we increased the flow rate gradually. For this case, no beads got detached up to 0.15 µL min−1, and the flow rate had to be increased to 0.33 µL min−1, 2.40 µL min−1, and 15.1 µL min−1 (equivalent drag forces of about 7 pN, 50 pN, and 300 pN) in order to remove 10%, 50%, and 90% of the bound beads respectively.
Figure 5.
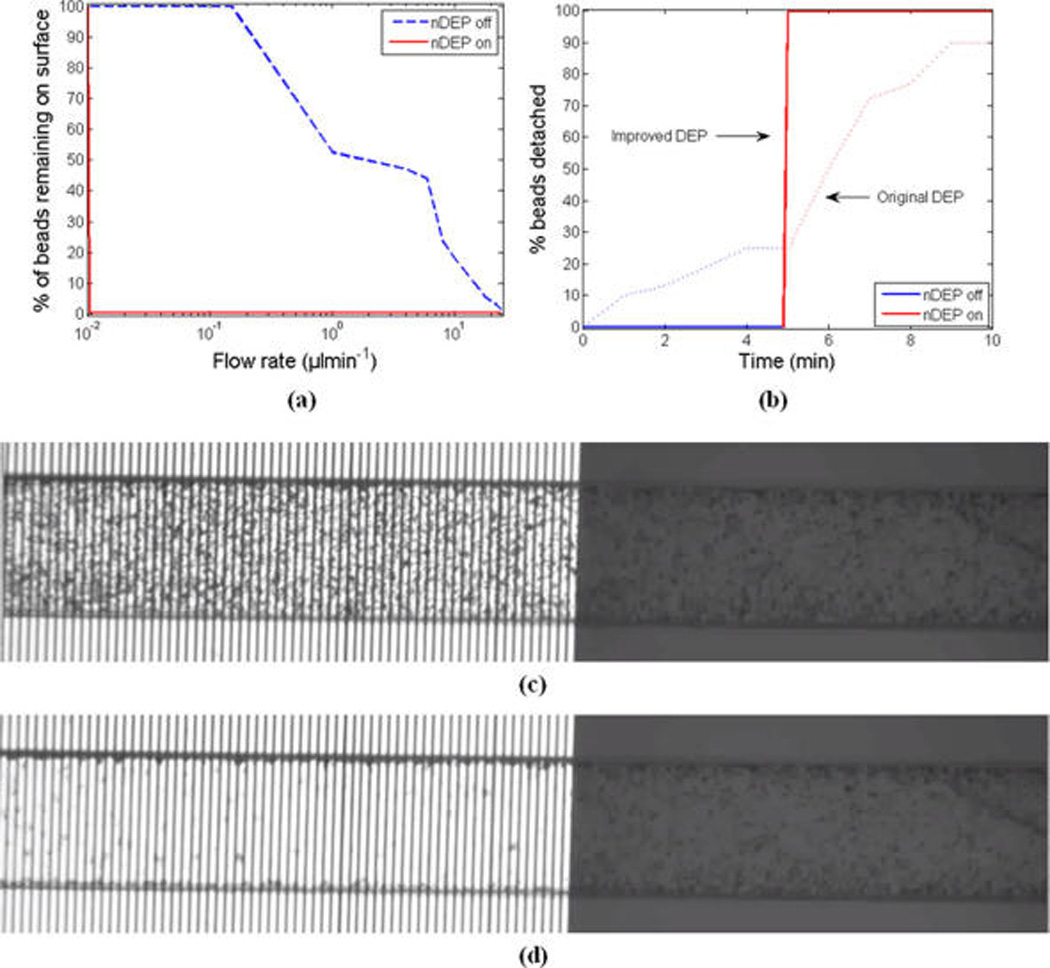
(a) Percentage of the beads remaining on the surface. (b) Bead detachment time profile at a flow rate of 0.15 µL min−1 using the improved vs original DEP device. (c,d) The corresponding raw video snapshots of the beads distribution (c) before (d) after turning nDEP on. The remaining beads on the electrode region are actually detached and are passing through. Similarly, the small difference between the two nonelectrode regions in parts c and d are mainly due to the moving beads passing by (but appearing as stationary in a single frame) as verified by the image-processing software.
Finally, we further demonstrated our device’s much improved functionality as a switch. The relevant performance measures in our context are ideally zero-switching response time upon turning the switch on, and 0 and 100% bead detachment when the switch is off and on, respectively. In this experiment, after sample and surface chemistry preparation and allowing for the injected beads to settle and bind to the surface, the flow rate was set to 0.15 µL min−1 and allowed to run for a total of 10 min. The nDEP was turned on at the end of the fifth minute. Figure 5b illustrates the bead detachment time profile for this process as well as the detachment profile for the previous work with nonimproved DEP device. As can be seen from this figure, at 0.15 µL min−1, a very small fraction of beads (about 0.3%) beads got detached in the first 5 min while nDEP was off. At the end of the fifth minute, once the nDEP was turned on and as expected 99.8% of beads immediately got eluted, resulting in achieving an on-to-off ratio of 333 which is about 2 orders of magnitude improvement in this performance measure as compared to the previous work, where the on-to-off ratio of 3.6 was demonstrated. Moreover, the elution of beads took place almost instantly, unlike the previous work where it took at least 4 min to detach the majority (90%) of beads. Specifically, upon applying the nDEP and within one frame of the captured video (with frame-capture interval of 0.4 s), the beads were fully detached. This shows improvement in switching response time of at least 600-fold. Representative captured snapshots from the video recorded experiment are shown by Figure 5c,d.
Together our results demonstrate that by improving the strength of nDEP we successfully developed a robust microfluidic switch to elute specifically bound beads. The enhanced nDEP switch not only eliminated the need for eluting agents but also shows about 2 orders of magnitude of improvement in the switching on-to-off ratio as well as at least 600-fold reduction in the switching response time. Having achieved an electrokinetic switch capable of eluting 100% of immunobound beads instantaneously, future efforts will be focused on implementation of a high throughput multiplexing system by fabricating an array of electrode pairs along a single channel and integrating a bead detector downstream. In addition to this we will use this device to quantify and compare various protein–protein interactions and also validate new protein biomarkers in biological samples.
METHODS
Fabrication
The microchannel with 200 µm width, 50 µm height, and 1 cm length was fabricated in PDMS. The master mold for the microchannel was patterned onto a silicon substrate using SU-8 photoresist. PDMS (10:1 prepolymer/curing agent) was poured onto the master mold and allowed to cure at 80 °C overnight. Once the PDMS channel was formed, it was peeled off from the mold. Then, two holes of diameter 3 mm were punched, one at each end, to create the channel’s inlet and outlet ports.
To fabricate the electrodes, standard evaporation and lift-off processing techniques were used. An array of interdigitated Au/Cr electrode pair was fabricated on a glass substrate with electrode width and spacing of 7 µm each. Then, we used atomic layer deposition (ALD) Fiji F202 system from Cambridge Nanotech to deposit a 10 nm oxide film on the surface of the device. For this purpose, with the wafers transferred to the reaction chamber, high purity Ar was used as a carrier gas. Through a repeated cycle, pulses of tris-[dimethylamino]silane and O2 plasma were generated, for a duration of 0.3 and 20 s (at 300 W), respectively, resulting in an effective deposition rate of 0.7 Å/cycle at 200 °C. Next, we selectively oxide-etched (dry etched) the surface to expose and access the electrical bonding pads. The oxide-deposited electrode chip and the PDMS microchannel were then aligned and bonded together after standard oxygen plasma treatment.49 All three available oxide films (SiO2, Al2 O3, and HfO2) were used on different devices to test for PDMS bonding using our standard plasma treatment process.
Sample Preparation and Surface Chemistry
To demonstrate the switch-like functionality in eluting specifically bound beads from the surface, we chose anti-IgG and IgG interactions that are in the same order of magnitude in strength as typical antibody–antigen interactions. For this purpose, 2.8 µm-diameter goat antimouse IgG covered beads (initially in 0.5% w/v suspension) were used. A volume of 250 µL of this solution was washed with PBS (containing 1% BSA and 0.05% Tween) and resuspended in 50 µL of 30×-diluted PBS.
To prepare the channel surface for this interaction, mouse IgG (originally 2 mg/mL, diluted by ×100) was pipetted into the channel and incubated for 20 min so that the IgG molecules became physically adsorbed on the channel surface. This was followed by introducing 1 mg/mL bovine serum albumin (BSA) in the channel to eliminate nonspecific binding. The channel was then flushed and filled with diluted PBS. As a control experiment to test for binding specificity, on a separate chip, the above steps were performed minus the addition of mouse IgG, thus minimizing attachment of beads due to nonspecific binding.
Experiment Setup
We used an impedance spectroscope (Zurich Instruments HF2IS, Switzerland) and a transimpedance amplifier (Zurich Instruments HF2TA, Switzerland) to capture the impedance spectrum of the device in order to validate and characterize the equivalent circuit model. This impedance spectroscope was also used as a signal generator in conjunction with a high voltage 50 V/V amplifier (Trek 2100HF) to excite the electrodes with a sine wave at 2 MHz. Furthermore, a syringe pump (Harvard Apparatus) was used to control the flow rate through the device.
CONCLUSIONS
In conclusion, we have achieved two significant results in this paper. First and foremost, using ALD to fabricate a pinhole free thin film layer, we have demonstrated the ability to generate nanoNewton DEP forces. The pinhole free thin film prevents corrosion, but at the same time because of the film thickness (<20 nm) and also the high frequency applied, the electric field fully couples capacitively to the bulk electrolyte rather than being localized primarily across the film also resulting in film breakdown. The fact that we have the ability to apply DEP forces on this order of magnitude opens up the possibility of a broad range of applications. Examples include actuation of cells at higher flow rates during cell sorting and other various cell handling applications which up until now where DEP was limited in utility because of its relatively weak nature.
In addition to the several order of magnitude improvement in DEP, for the first time, we have demonstrated the ability to instantaneously detach specific protein–protein bound beads using electronic actuation without the need for any special buffer modification. The conditions we applied resulted in 100% bead detachment in the case where DEP was applied, whereas no bead detachment occurs where DEP was off. This localized electronic actuator will open up many possibilities for performing rapid and inexpensive multiplexed protein biomarker detection. The ability to inexpensively detect protein biomarkers in high throughput will undoubtedly have a large impact on clinical medicine by equipping clinicians with the tools necessary to provide more complete personalized healthcare.
Acknowledgments
The authors would like to thank M. Bendernagel for her significant contribution in quantifying and verifying the experiment results through developing an automated image processing software which will also be utilized for the future extension of the project. Furthermore, the authors would like to thank J. Provine for the training, expertise, and help with atomic layer deposition. This work was supported by the National Institutes of Health through Grant PO1HG000205 and Natural Sciences and Engineering Research Council of Canada Scholarship through graduate tuition support.
Footnotes
Author Contributions
All authors contributed to the manuscript preparation. S. Emaminejad and M. Javanmard set up the simulation platform, fabricated the microfluidic devices, and performed the experiments.
The authors declare no competing financial interest.
REFERENCES
- 1.Hamburg MA, Collins FS. N. Engl. J. Med. 2010;363(4):301–304. doi: 10.1056/NEJMp1006304. [DOI] [PubMed] [Google Scholar]
- 2.Harvey A, Brand A, Holgate ST, Kristiansen LV, Lehrach H, Palotie A, Prainsack B. New Biotechnol. 2012;29(6):625–633. doi: 10.1016/j.nbt.2012.03.009. [DOI] [PubMed] [Google Scholar]
- 3.Wei DQ, Chen Q. Curr. Drug Metab. 2012;13(7):951–951. doi: 10.2174/138920012802138651. [DOI] [PubMed] [Google Scholar]
- 4.Souied EH, Leveziel N. Am. J. Ophthalmol. 2012;154(3):427–428. doi: 10.1016/j.ajo.2012.05.022. [DOI] [PubMed] [Google Scholar]
- 5.Iacobuzio-Donahue CA. J. Gastrointest. Surg. 2012;16(9):1651–1652. doi: 10.1007/s11605-012-1943-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sadeghi S, Gilardi G. FEBS J. 2012;279:345–345. [Google Scholar]
- 7.O’Brien CP. Alcohol.: Clin. Exp. Res. 2012;36:12a–12a. [Google Scholar]
- 8.Burckart GJ, Green DJ. Pediatr. Transplant. 2012;16(6):530–532. doi: 10.1111/j.1399-3046.2011.01638.x. [DOI] [PubMed] [Google Scholar]
- 9.Garbis S, Lubec G, Fountoulakis M. J. Chromatogr., A. 2005;1077(1):1–18. doi: 10.1016/j.chroma.2005.04.059. [DOI] [PubMed] [Google Scholar]
- 10.Ray S, Reddy PJ, Jain R, Gollapalli K, Moiyadi A, Srivastava S. Proteomics. 2011;11(11):2139–2161. doi: 10.1002/pmic.201000460. [DOI] [PubMed] [Google Scholar]
- 11.Yarmush ML, Jayaraman A. Annu. Rev. Biomed. Eng. 2002;4:349–373. doi: 10.1146/annurev.bioeng.4.020702.153443. [DOI] [PubMed] [Google Scholar]
- 12.MacBeath G. Nat. Genet. 2002;32:526–532. doi: 10.1038/ng1037. [DOI] [PubMed] [Google Scholar]
- 13.Schweitzer B, Predki P, Snyder M. Proteomics. 2003;3(11):2190–2199. doi: 10.1002/pmic.200300610. [DOI] [PubMed] [Google Scholar]
- 14.Price JV, Tangsombatvisit S, Xu GY, Yu JT, Levy D, Baechler EC, Gozani O, Varma M, Utz PJ, Liu CL. Nat. Med. 2012;18(9):1434–U190. doi: 10.1038/nm.2913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gemovic B, Perovic V, Glisic S, Veljkovic N. FEBS J. 2012;279:295–295. [Google Scholar]
- 16.Lalli D, De Pablo R, Del Conte R, Turano P. FEBS J. 2012;279:437–437. [Google Scholar]
- 17.Mamitsuka H. Wiley Interdiscip. Rev. Data Min. Knowledge Discovery. 2012;2(5):400–410. [Google Scholar]
- 18.Takayama Y, Clore GM. J. Biol. Chem. 2012;287(32):26962–26970. doi: 10.1074/jbc.M112.382960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pierrot C, Freville A, Olivier C, Souplet V, Khalife J. Curr. Pharm. Des. 2012;18(24):3522–3530. [PubMed] [Google Scholar]
- 20.Smith MC, Gestwicki JE. Expert Rev. Mol. Med. 2012:14. doi: 10.1017/erm.2012.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen C, Zhao JF, Huang Q, Wang RS, Zhang XS. BMC Syst. Biol. 2012:6. doi: 10.1186/1752-0509-6-S1-S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Salimi-Moosavi H, Rathanaswami P, Rajendran S, Toupikov M, Hill J. Anal. Biochem. 2012;426(2):134–141. doi: 10.1016/j.ab.2012.04.023. [DOI] [PubMed] [Google Scholar]
- 23.Iwamoto S, Iwamoto AT, Cha J, Clark T, Lu M, Breen C, Bhawan J, Falanga V. Am. J. Dermatopathol. 2009;31(3):218–222. doi: 10.1097/DAD.0b013e31818ffde1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Satterfield MB, Lippa K, Lu ZQ, Salit ML. J. Res. Natl. Inst. Stand. Technol. 2008;113(3):157–174. doi: 10.6028/jres.113.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Microarray scanner. Scientist. 2005;19(16):30–31. [Google Scholar]
- 26.Shi LM, Tong WD, Su ZQ, Han T, Han J, Puri RK, Fang H, Frueh FW, Goodsaid FM, Guo L, Branham WS, Chen JJ, Xu ZA, Harris SC, Hong HX, Xie Q, Perkins RG, Fuscoe JC. BMC Bioinf. 2005:6. [Google Scholar]
- 27.Sinclair MB, Timlin JA, Haaland DM, Werner-Washburne M. Appl. Opt. 2004;43(10):2079–2088. doi: 10.1364/ao.43.002079. [DOI] [PubMed] [Google Scholar]
- 28.Biener G, Greenbaum A, Isikman SO, Lee K, Tseng D, Ozcan A. Lab Chip. 2011;11(16):2738–2743. doi: 10.1039/c1lc20169g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bishara W, Isikman SO, Ozcan A. Ann. Biomed. Eng. 2012;40(2):251–262. doi: 10.1007/s10439-011-0385-3. [DOI] [PubMed] [Google Scholar]
- 30.Bishara W, Zhu HY, Ozcan A. Opt. Express. 2010;18(26):27499–27510. doi: 10.1364/OE.18.027499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Coskun AF, Sencan I, Su TW, Ozcan A. Lensfree Fluorescent On-Chip Imaging of Transgenic Caenorhabditis elegans Over an Ultra-Wide Field-of-View. PLoS One. 2011;6(1) doi: 10.1371/journal.pone.0015955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Isikman SO, Bishara W, Mavandadi S, Yu FW, Feng S, Lau R, Ozcan A. Proc. Natl. Acad. Sci. U.S.A. 2011;108(18):7296–7301. doi: 10.1073/pnas.1015638108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Isikman SO, Bishara W, Mudanyali O, Sencan I, Su TW, Tseng DK, Yaglidere O, Sikora U, Ozcan A. IEEE J. Sel. Top. Quantum Electron. 2012;18(3):1059–1072. doi: 10.1109/JSTQE.2011.2161460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Isikman SO, Bishara W, Zhu HY, Ozcan A. Optofluidic Tomography on a Chip. Appl. Phys. Lett. 2011;98(16) doi: 10.1063/1.3548564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Isikman SO, Greenbaum A, Lee M, Bishara W, Mudanyali O, Su TW, Ozcan A. Anal. Cell. Pathol. 2012;35(4):229–247. doi: 10.3233/ACP-2012-0057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tseng D, Mudanyali O, Oztoprak C, Isikman SO, Sencan I, Yaglidere O, Ozcan A. Lab Chip. 2010;10(14):1787–1792. doi: 10.1039/c003477k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhu HY, Mavandadi S, Coskun AF, Yaglidere O, Ozcan A. Anal. Chem. 2011;83(17):6641–6647. doi: 10.1021/ac201587a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Javanmard M, Babrzadeh F, Davis RW. Microfluidic force spectroscopy for characterization of biomolecular interactions with piconewton resolution. Appl. Phys. Lett. 2010;97(17) doi: 10.1063/1.3491547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Neuman KC, Nagy A. Nat. Methods. 2008;5(6):491–505. doi: 10.1038/nmeth.1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Auerswald J, Widmer D, de Rooij NF, Sigrist A, Staubli T, Stokli T, Knapp HF. Electrophoresis. 2005;26(19):3697–3705. doi: 10.1002/elps.200500401. [DOI] [PubMed] [Google Scholar]
- 41.Li HB, Zheng YN, Akin D, Bashir R. J. Microelectromech. Syst. 2005;14(1):103–112. [Google Scholar]
- 42.Park IS, Eom K, Son J, Chang WJ, Park K, Kwon T, Yoon DS, Bashir R, Lee SW. ACS Nano. 2012;6(10):8665–8673. doi: 10.1021/nn302202t. [DOI] [PubMed] [Google Scholar]
- 43.Javanmard M, Emaminejad S, Dutton RW, Davis RW. Anal. Chem. 2012;84(3):1432–1438. doi: 10.1021/ac202508u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Seok-Jun W, Sungin S, Myung Soo H, Hyeong Joon K. Electron Device Lett., IEEE. 2010;31(8):857–859. [Google Scholar]
- 45.Li H, Yanan Z, Akin D, Bashir R. J. Microelectromech. Syst. 2005;14(1):103–112. [Google Scholar]
- 46.Javanmard M, Babrzadeh F, Davis RW. Appl. Phys. Lett. 2010;97:173704. doi: 10.1063/1.3491547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Meinhart C, Wang D, Turner K. Biomed. Microdevices. 2003;5(2):139–145. [Google Scholar]
- 48.Voldman J, Braff RA, Toner M, Gray ML, Schmidt MA. Biophys. J. 2001;80(1):531–542. doi: 10.1016/S0006-3495(01)76035-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McDonald JC, Duffy DC, Anderson JR, Chiu DT, Wu H, Schueller OJA, Whitesides GM. Electrophoresis. 2000;21(1):27–40. doi: 10.1002/(SICI)1522-2683(20000101)21:1<27::AID-ELPS27>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]