

ABSTRACT
The σE envelope stress response is an essential signal transduction pathway which detects and removes mistargeted outer membrane (OM) β-barrel proteins (OMPs) in the periplasm of Escherichia coli. It relies on σE, an alternative sigma factor encoded by the rpoE gene. Here we report a novel mutation, a nucleotide change of C to A in the third base of the second codon, which increases levels of σE (rpoE_S2R). The rpoE_S2R mutation does not lead to the induction of the stress response during normal growth but instead changes the dynamics of induction upon periplasmic stress, resulting in a faster and more robust response. This allows cells to adapt faster to the periplasmic stress, avoiding lethal accumulation of unfolded OMPs in the periplasm caused by severe defects in the OMP assembly pathway.
IMPORTANCE Survival of bacteria under conditions of external or internal stresses depends on timely induction of stress response signaling pathways to regulate expression of appropriate genes that function to maintain cellular homeostasis. Previous studies have shown that strong preinduction of envelope stress responses can allow bacteria to survive a number of lethal genetic perturbations. In our paper, we describe a unique mutation that enhances kinetics of the σE envelope stress response pathway rather than preinducing the response. This allows bacteria to quickly adapt to sudden and severe periplasmic stress.
INTRODUCTION
Maintaining the integrity of the cell envelope is an essential function in bacteria. In the case of Gram-negative bacteria, such as Escherichia coli, the inner membrane (IM) and outer membrane (OM) delimit the periplasm, an extracytoplasmic compartment that is not accessible to cytoplasmic protein quality control mechanisms (1). Accordingly, detection and removal of mistargeted OM β-barrel proteins (OMPs) in the periplasm rely on a dedicated signal transduction system known as the σE envelope stress response (2). At the center of this response is σE (σ24 or RpoE), an alternative sigma factor encoded by the rpoE gene, which is cotranscribed with rseABC from a σ70 (RpoD)-dependent promoter (Fig. 1) (3). The rseA gene encodes an anti-sigma factor, which binds to and inhibits RpoE activity during normal growth (4, 5).
FIG 1.
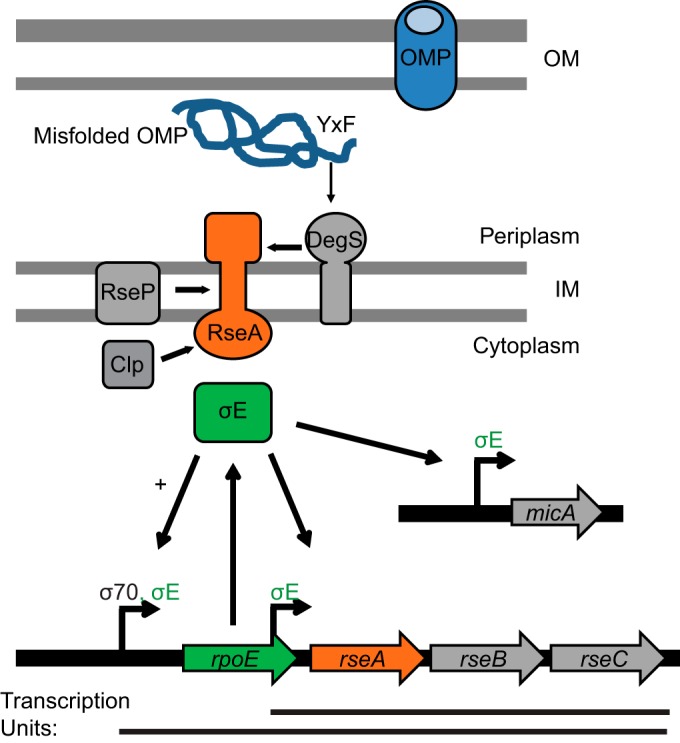
σE stress response pathway. σE is encoded by the rpoE gene, which is transcribed with rseABC from a σ70 promoter. RseA is an IM protein, which binds and inhibits σE. When the OMP assembly pathway is compromised, OMPs misfold in the periplasm and a YXF motif activates the DegS protease. DegS initiates the proteolytic pathway, and RseA is further degraded by the RseP and Clp proteases. σE is released and activates expression from σE-dependent promoters, e.g., those of the micA gene and the rpoE-rse and rseABC operons.
Induction of σE activity in response to stress is regulated at the level of RseA degradation and has become a paradigm for regulated intramembrane proteolysis (Fig. 1) (2). RseA is an IM protein with a cytoplasmic domain that is responsible for binding and inhibiting σE (4, 5). When the OMP assembly pathway is compromised, misfolded OMPs accumulate in the periplasm and expose their C-terminal YXF motif to the IM protease DegS (6). Binding of these motifs to a periplasmic PDZ domain of DegS activates its proteolytic activity and results in the first cut of RseA (6–8). This event induces a further proteolytic cascade, including a cut within the RseA transmembrane domain by the IM protease RseP (8, 9) and degradation of the remaining RseA cytoplasmic domain by cytoplasmic proteases, such as Clp (10). When σE is released, it directs RNA polymerase to transcribe a specific subset of genes: the well-characterized σE regulon (11). Expression of small RNAs (sRNA), such as MicA and RybB, is induced, resulting in translational downregulation of OMP synthesis (12, 13). This reduces the load on the OMP assembly pathway and minimizes further accumulation of unfolded OMPs in the periplasm. Moreover, σE also upregulates the expression of the OM biogenesis machines and periplasmic chaperones to promote OMP folding as well as periplasmic proteases to degrade misfolded OMPs (11).
In addition, σE increases transcription of its own operon from a σE-dependent promoter, as well as transcription of rseABC from an additional σE-dependent promoter located within rpoE (Fig. 1) (11). This creates a positive-feedback loop that persists as long as the stress continues and RseA is cleaved. When periplasmic stress is removed, RseA degradation is inhibited, and the accumulated anti-sigma factor binds and inhibits the activity of σE. Although rpoE and rseA lie in the same operon, recent studies performed using ribosome profiling have shown that RseA is made in excess of σE (14), ensuring that σE activity is tightly inhibited in the absence of periplasmic stress.
In this paper, we report a novel suppressor mutation in rpoE that allows cells to survive lethal periplasmic stress. The suppressor mutation increases production of σE and RseA, thereby priming cells for a faster and more potent envelope stress response.
MATERIALS AND METHODS
All strains used in this study are listed in Table S1 in the supplemental material.
Suppressor analysis of the OMP assembly double mutants.
A spontaneous suppressor of the synthetic lethal phenotype of the surA degP double mutant was isolated through selection for growth at 37°C (rpoE_S2R). The mutation was mapped by identifying linked Tn10 insertions and sequence analysis of suspected target genes. rpoE_S2R linked to nadB::Tn10 was then transduced into PBAD-surA or PBAD-bamE depletion strains, carrying additional degP, skp, or bamB mutations. Viability of these strains was tested by an efficiency-of-plating assay. For this assay, strains were grown overnight in LB supplemented with 0.2% arabinose and washed in LB to remove arabinose, and 10-fold serial dilutions were plated on the LB agar with or without arabinose and incubated at 37°C overnight.
β-Galactosidase assay.
Strains were grown at 37°C unless otherwise indicated. A 100-μl volume of mid-log culture (optical density at 600 nm [OD600], 0.5 to 0.7) was taken and added directly to 900 μl of Z buffer (60 mM Na2HPO4, 40 mM NaH2PO4, 10 mM KCl, 1 mM MgSO4, 50 mM β-mercaptoethanol, 0.03% SDS). A 50-μl volume of chloroform was added to stop growth, and the reaction mixture was mixed vigorously by pipetting. A 100-μl volume of each cell lysate was mixed with 100 μl of 4 mg/ml ONPG (O-nitrophenyl-β-d-galactopyranoside) solution in Z buffer. β-Galactosidase activity was analyzed by a kinetic measurement of the OD420 in a BioTek Synergy 1 plate reader, and Vmax was determined using Gen5 software. The Vmax value was normalized by OD600. Experiments were performed in three biological replicates, and mean values ± standard errors of the means (SEM) of the results were plotted. Graphs were built using GraphPad Prism 6 software.
Immunoblot analysis.
Strains were grown at 37°C until mid-log phase (OD600, 0.5 to 0.7). Samples were normalized by determination of OD600. Anti-RpoE antibodies (Neoclone) were used for immunoblotting. RpoE levels were normalized to the loading control and quantified relative to the wild type (WT) using ImageJ software.
Determination of the rate of translation initiation of the reporter fusions.
Overnight cultures of corresponding strains containing reporter plasmids or empty vector controls were diluted 1:500 in LB supplemented with 0.2% arabinose and ampicillin. Strains were grown in a 200-μl volume in 96-well black plates with clear bottoms (Costar) at 37°C and OD600, and green fluorescence (excitation, 481 nm; emission, 507 nm) and red fluorescence (excitation, 580 nm; emission, 610 nm) were monitored every 20 min using a BioTek Synergy 1 plate reader. Each strain was grown in six wells to avoid plate effects, and mean values for these replicates were calculated. The background fluorescence of the empty vector control was subtracted from the fluorescence of strains containing reporter fusions. This specific fluorescence was normalized for OD600 and plotted as a function of time. Experiments were performed three independent times, and a plot of the mean values ± SEM is shown in Fig. S1 in the supplemental material.
Linear regression analysis was performed on the graphs within the linear range of 300 to 480 min, with R2 values of 0.98 ± 0.07 (standard deviation [SD]) for green fluorescent protein (GFP) fluorescence graphs and 0.97 ± 0.02 (SD) for mCherry fluorescence graphs. gfp or mCherry translation rates were taken as the slope of the line and calculated for each of the biological replicates separately. The ratio between the mCherry and gfp translation rates was determined and calculated as the fold change from the rate determined for the WT-gfp of WT-mCherry construct. These values are plotted as a bar graph with mean values ± SEM. Significance analysis was performed using an unpaired t test, and differences were considered to be significant with P values of ≤0.05. Exact P values for significantly different pairs are shown on the graph (see Fig. 3C).
FIG 3.
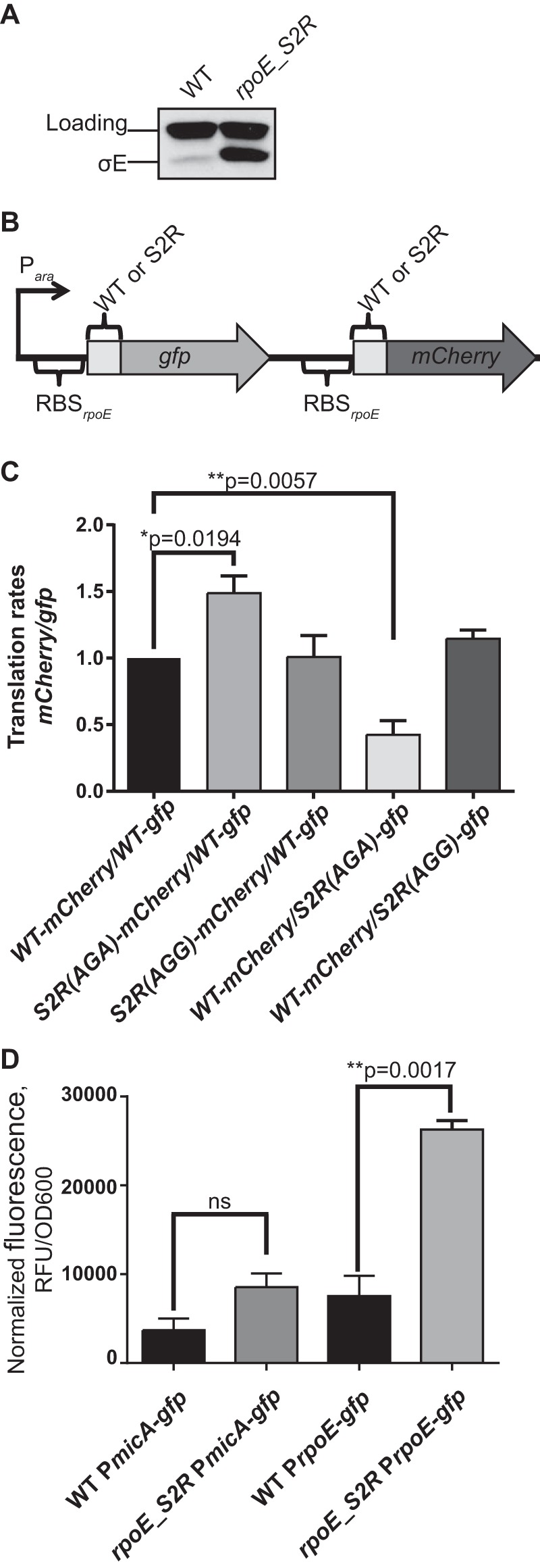
rpoE_S2R upregulates expression of rpoE. (A) Levels of the σE protein were significantly increased in an rpoE_S2R mutant during steady-state growth. (B) Organization of translational reporter construct. gfp and mCherry genes were translationally fused with the first five codons of rpoE. The expression of both genes was controlled by an arabinose-inducible promoter and the native rpoE RBS. S2R mutations (AGA or AGG codons) were introduced in either the gfp or mCherry gene fusions. (C) The S2R(AGA) mutation significantly increased the translation initiation rate. This graph represents the ratio of the translation rates of mCherry and gfp, normalized to the WT-gfp/WT-mCherry construct. Means ± SEM are indicated with P values for the constructs with significant changes in ratios as determined using a t test. (D) PrpoE but not PmicA promoter activity was increased in the rpoE_S2R strain. This graph represents the normalized fluorescence of GFP expressed from PrpoE or PmicA promoters during mid-logarithmic growth of the WT or rpoE_S2R strains. Means ± SEM are indicated with P values for the constructs with significant changes in GFP expression as determined using a t test. RFU, relative fluorescence units.
Determination of promoter activity.
GFP fluorescence normalized to the OD600 during steady-state growth was used to test differences in expression of the PrpoE-gfp and PmicA-gfp reporter fusions in rpoE_S2R strains versus WT strains. Strains were grown to mid-log phase (OD600 of 0.5), and GFP fluorescence (excitation, 481 nm; emission, 507 nm) was measured in a 200-μl volume using a BioTek Synergy 1 plate reader and blank normalized against the strain lacking GFP reporters. This experiment was performed three independent times, and a plot of mean values ± SEM is shown (see Fig. 3D). Significance analysis was performed pairwise using an unpaired t test, and differences were considered to be significant with P values of ≤0.05. Exact P values for significantly different pairs are shown on the graph (see Fig. 3D).
To determine the kinetics of σE induction in response to OmpC_YYF peptide expression, the corresponding strains were grown overnight in LB supplemented with corresponding antibiotics and 0.2% fucose to avoid the expression of the OmpC_YYF peptide. Cells were washed in LB and diluted 1:500 in LB supplemented with corresponding antibiotics, 0.5% glucose, and 0.2% arabinose for autoinduction. Strains were grown in a 200-μl volume in 96-well black plates with clear bottoms (Costar) at 37°C and OD600, and green fluorescence was monitored every 20 min using a BioTek Synergy 1 plate reader. Each strain was grown in 12 wells to avoid plate effects, and mean values for these replicates were calculated. The background fluorescence of the “no-gfp” control was subtracted from the fluorescence of strains containing reporter fusions. The increase of the specific fluorescence was plotted as a function of growth (OD600), resulting in a differential plot within the window of exponential growth (see Fig. S2 in the supplemental material). The experiment was performed three independent times; a plot of mean values ± SEM is shown (see Fig. 4), and corresponding growth curves (mean OD600 ± SEM over time) are shown (see Fig. S2). Note that the OD600 values are those obtained from microwell reads and have not been path length corrected for a 1-cm-path-length cuvette.
FIG 4.

Kinetics of σE induction monitored by GFP expression from PrpoE- or PmicA-gfp promoter fusions. Strains were grown in LB with glucose and arabinose in order to create autoinduction of OmpC_YYF peptide expression (where applicable). Graphs represent a differential plot of the increase of fluorescence as a function of growth (means ± SEM). Corresponding growth curves are included as described for Fig. S2 in the supplemental material. Induction of OmpC_YYF peptide at an OD600 of 0.5 led to the induction of expression of the PrpoE- or PmicA-gfp transcriptional reporters. The upper graphs show induction in the WT background. The lower panels show induction in rpoE_S2R compared to the WT results (note scale difference).
RESULTS
rpoE_S2R is a powerful suppressor of a number of synthetically lethal combinations.
Simultaneous loss of the major periplasmic chaperone SurA and the periplasmic chaperone/protease DegP confers a strong synthetic phenotype caused by a severe OMP assembly defect, evidenced by the accumulation of unfolded OMPs in the periplasm (15, 16). The surA degP double mutant is viable only at 24°C. A search for suppressors that would allow growth of the surA degP double mutant at 37°C yielded null mutations in rseA. This class of suppressors allows survival because the σE response is highly induced in this strain (Fig. 2A), resulting in the downregulation of OMP synthesis and upregulation of alternative chaperones and the OM biogenesis machines. This minimizes the periplasmic stress caused by the shift to the nonpermissive temperature.
FIG 2.
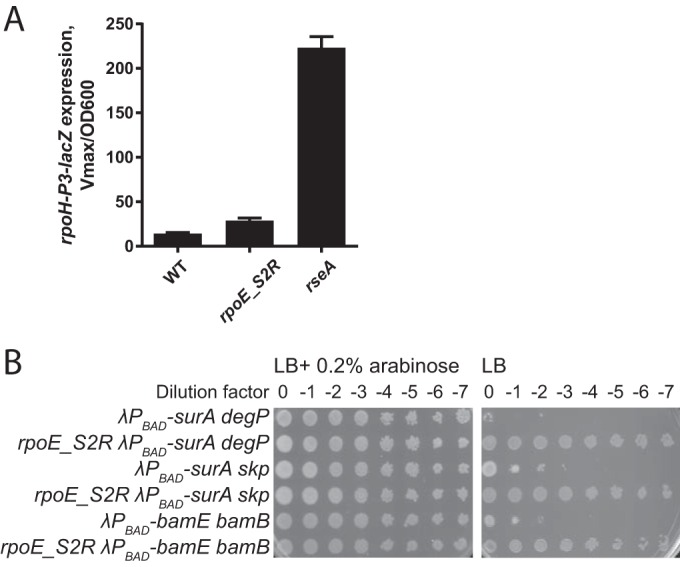
rpoE_S2R is a suppressor of synthetic double mutants in OMP assembly pathway. (A) Null mutations in rseA but not rpoE_S2R increase basal activity of σE based on expression of the rpoH-P3–lacZ reporter. Graphs represent mean β-galactosidase activity ± SEM; n = 3. (B) Chromosomal arabinose-dependent depletion constructs of surA or bamE were combined with null mutations in the degP, skp, or bamB genes. Strain viability was assayed by plating 10-fold serial dilutions of overnight cultures grown in LB with 0.2% arabinose and washed to remove arabinose before plating. rpoE_S2R fully rescued growth of the strains in the absence of arabinose.
We also found a mutation in the open reading frame (ORF) of rpoE, which changes the second codon from Ser (AGC) to Arg (AGA). Unlike in rseA null strains, steady-state levels of the σE response were not increased (Fig. 2A). Despite this, rpoE_S2R is a powerful suppressor and fully restores normal growth at 37°C of the surA degP strain, as well as of other strains with synthetically lethal mutations, such as surA skp and bamB bamE (Fig. 2B).
Intrigued by the finding that the σE response was not preinduced in the rpoE_S2R strain, we chose this mutant for further study in order to understand the mechanism of suppression.
rpoE_S2R upregulates expression of rpoE without induction of the σE response.
Levels of σE are quite low during normal growth in the WT background; however, levels were increased 8-fold in the rpoE_S2R mutant (Fig. 3A). rpoE_S2R has a nucleotide change of C to A in the third base of the second codon. The nucleotide sequence of the second codon has been shown to be important for translation initiation (17–19). In E. coli, all codons have been previously tested for their effect on translation initiation based on reporter assays (19). Interestingly, the Arg codon AGA was found to be one of the best-performing codons, conferring increased translation rates compared to the Ser AGC codon (19).
To test whether the rpoE_S2R mutation resulted in translational upregulation of rpoE, we generated reporter plasmids that encode rpoE-translational fusions to gfp and mCherry genes that are under the control of arabinose-inducible PBAD promoter. Translation of both gfp and mCherry fusions is initiated by the native ribosome-binding site (RBS) of rpoE, and each of the genes is translationally fused to the five initial codons of WT rpoE, resulting in WT-gfp and WT-mCherry fusions. We generated the second-codon mutations in either rpoE-gfp or rpoE-mCherry and then plotted the increase of red or green fluorescence normalized to the OD600 as a function of time (see Fig. S1 in the supplemental material). gfp or mCherry translation rates were determined by calculating the slope of the line in these plots. The ratio between the corresponding mCherry and gfp translation rates was set to 1 in the reporter strain containing WT-gfp and WT-mCherry fusions. The effects of the various second-codon mutations are expressed as the fold change of this ratio and are plotted as a bar graph (Fig. 3C).
Figure 3C shows that the translation rates of S2R(AGA) were 1.5-fold to 2-fold higher than those of the WT, regardless of whether the S2R(AGA) mutation was introduced into the gfp or mCherry reporter construct, and the results showed an increase of about 2-fold to 3-fold in final fluorescence (see Fig. S1A and B in the supplemental material). To test, whether it was the nucleotide change rather than the amino acid change that was responsible for this effect, we used an alternative Arg codon, AGG, which occurs at the same frequency in the E. coli genome as AGA. The S2R(AGG) mutation did not cause the same increase in the translation rate as S2R(AGA). We therefore concluded that it was the C6A nucleotide change and not the S2R amino acid change that was responsible for translational upregulation.
To determine if the increased rpoE translation rate caused by the rpoE_S2R mutation causes induction of the σE response, we monitored the expression of two previously published transcriptional reporters, PrpoE-gfp and PmicA-gfp (Fig. 3D). The PrpoE-gfp fusion is the most sensitive reporter for a change in σE protein levels (20). This fusion does not contain the native rpoE RBS or part of the rpoE ORF and therefore reports exclusively on the transcription. The micA gene is a member of the σE regulon; it encodes a small RNA that functions to downregulate OMP synthesis. PmicA-gfp is the most sensitive promoter reporter for the σE regulon (20).
During logarithmic growth, the expression of PrpoE-gfp was about 3-fold higher than that of the WT (Fig. 3D). This increased PrpoE promoter activity also contributed to increased σE levels. However, increased transcription from this promoter also resulted in increased levels of RseA, which would keep σE inhibited. Consistent with this increase in RseA, the σE response was not induced, as judged by lack of significant increase in the activity of the PmicA promoter (Fig. 3D), and was in line with what we observed using the rpoH-P3–lacZ reporter (Fig. 2A).
rpoE_S2R changes the kinetics of the response to periplasmic stress.
Our genetic analysis suggested that preinduction of the σE response cannot explain the suppression caused by rpoE_S2R. For example, the presence of rpoE_S2R does not lead to increased σE activity in the degP mutant (see Fig. S2A in the supplemental material); however, the surA mutation can be introduced only in the degP rpoE_S2R strain and not in the degP strain at 37°C. When grown at room temperature, the degP surA strain induces σE activity very strongly (even more strongly than the degP surA rpoE_S2R strain). This suggest that rpoE_S2R allows the degP surA strain to grow and that the growth is not due to higher levels of initial or endpoint σE activity but is perhaps due to the fact that the σE is induced more rapidly. We reasoned that the increased levels of σE might be responsible for suppression because they would change the kinetics of the response to periplasmic stress through an enhanced positive-feedback loop.
During steady-state growth, the mutants described above were adapted to chronic periplasmic stress, by readjusting the gene expression of OMPs as well as of the biogenesis machines. Therefore, we sought an experimental approach in which we could transiently generate periplasmic stress in an otherwise-WT background and then follow the kinetics of σE induction in the rpoE_S2R strain versus the WT. For this, we used an OmpC-derived C-terminal peptide, which acts as an inducing signal for DegS (6). For this assay, we generated a plasmid that directs periplasmic expression of the OmpC_YYF peptide under the control of the arabinose-inducible PBAD promoter according to the method described in reference 6. This plasmid was introduced into the WT or rpoE_S2R strain containing either the PrpoE-gfp or the PmicA-gfp reporter. We then monitored induction of the σE response by following the increased production of GFP as a function of growth. To induce expression of the OmpC_YYF peptide, we used an autoinduction approach that was based on catabolite repression. We grew strains in LB containing glucose and arabinose. In the presence of glucose, expression of PBAD is tightly repressed (21). This avoids any leaky production of the OmpC_YYF peptide and therefore avoids any preinduction of the σE response. Upon glucose depletion through the cell growth, arabinose should rapidly induce expression of the PBAD promoter, resulting in the production of the OmpC-YYF peptide. First, we tested this autoinduction approach in a WT background (Fig. 4, upper panels). In the presence of glucose, at a lower OD600, reporter activity was present at similarly low levels, regardless of the presence or absence of the OmpC_YYF construct. Once cultures reached an OD600 of 0.5 and glucose levels became depleted, the activity of both the PrpoE promoter and the PmicA promoter increased as expected in an OmpC_YYF-dependent manner, indicating that the σE response was induced under those conditions.
We next compared the kinetics of σE induction in an rpoE_S2R strain to the kinetics in the WT. Figure 4 (lower panels) shows that once cultures reached an OD600 of 0.5, both reporters were induced more rapidly and also to much higher levels in the rpoE_S2R strain than in the WT background (note the scale difference between the upper and lower panels of Fig. 4). Therefore, we concluded that the rpoE_S2R stain responds more quickly and more robustly to periplasmic stress, allowing cells to combat the resulting damage before it reaches lethal levels.
DISCUSSION
In this paper, we report a novel mutation, rpoE_S2R, that suppresses the synthetic lethality observed with double mutant strains lacking components of the OMP assembly pathway. The rpoE_S2R mutation increases the levels of σE by increasing the translation rate. Strikingly, however, the rpoE_S2R mutation does not increase the activity of σE but instead changes the dynamics of σE induction upon periplasmic stress, resulting in a faster and more robust response. When a cell encounters stress, in order to survive, it must mount an effective defense that prevents damage from reaching lethal levels. The faster and more robust σE response caused by the rpoE_S2R suppressor mutation allows cells to win the race against the periplasmic stress caused by the crippled OMP assembly pathways.
How does rpoE_S2R change the dynamics of the σE stress response? In WT cells, σE is maintained at very low levels during vegetative growth. Although rpoE and rseA are located in the same operon, RseA is made in excess of σE (14), likely because rpoE possesses a weak RBS that is suboptimally positioned relative to the translation start site. Because a positive-feedback loop is incorporated into the σE pathway, an excess of RseA makes the pathway noise resistant. We found that levels of σE are significantly increased in the rpoE_S2R strain for two reasons. First, the S2R mutation directly increases RpoE levels by enhancing translation. Using a translational reporter, we showed that the nucleotide change C6A in rpoE results in an increased translation rate. We show that this increase is due to the nucleotide change and not the amino acid change, because another Arg codon failed to support increased translation. Although we did not address the mechanisms for translation enhancement, it has been previously shown that the nucleotide sequence of the second codon affects translation initiation and that this is especially important in cases of leaderless mRNAs or genes with weak RBSs (17–19). Second, the rpoE_S2R mutation indirectly increases RpoE levels by increasing transcription of the rpoE-rseA operon from the σE-dependent promoter. This increased transcription from the PrpoE promoter (Fig. 1) would also increase RseA levels, and this, in turn, would inhibit σE, preventing the induction of the stress response. Although we have not determined the ratio of RseA to σE in the rpoE_S2R strain, we think that it is likely that the levels of σE relative to RseA are somewhat increased because of the increased transcription from PrpoE. However, this increase is apparently not sufficient to support an increase in the expression of regulon genes, as evidenced by the lack of a significant increase in the activity of the PmicA or rpoH-P3 promoters.
We demonstrated that the increased levels of σE dramatically affect the kinetics of the stress response. Unlike in WT cells, RseA is fully saturated with σE in the rpoE_S2R mutant, due to the increase in σE levels. When cells experience periplasmic stress, similar degradation rates of RseA would release more σE molecules in the rpoE_S2R strain than in the WT. Because the activity of σE is directly linked to the number of free (uninhibited by RseA) molecules, the release of more molecules of σE results in faster activation of the response. In addition, the increased translation rate of rpoE_S2R would result in faster signal amplification through the positive-feedback loop, which would result in a response much stronger than that seen in the WT.
Mutations that increase the basal activity of the envelope stress responses are able to suppress a number of lethal genetic permutations. For example, a null mutation in rseA, which leads to full activation of the σE pathway during steady-state growth, can also suppress the surA degP, surA skp, bamE bamB (our work), and degP bamB (22) synthetically lethal double mutants. This is because OMP expression is significantly reduced in this strain, bypassing the requirement for certain nonessential components in the OMP pathway that function to increase the efficiency of the assembly reaction. Consistent with this, mutations in the EnvZ/OmpR two-component signal transduction pathway, which also lead to decreased expression of abundant OMPs, also suppress degP bamB synthetic lethality (23). Similarly, mutations such as cpxA*, which causes constitutively high activity of the Cpx envelope stress response, can suppress lethality caused by toxic LamB-LacZ fusions or misfolded P pilus subunits (24, 25). In this respect, the rpoE_S2R mutation is unique, because the mechanism of suppression does not depend on the preinduction of the stress response but instead relies on enhanced kinetics, which allows the σE response to win the race against increasing periplasmic stress.
If the rpoE_S2R mutation creates a faster, more robust stress response, why then has evolution apparently selected against it? On the basis of the results of our experiments with the OmpC_YYF peptide, we suggest that the rpoE_S2R strain overreacts to small stresses. This may come with a fitness cost, because high-level induction of the σE pathway can be detrimental to cells (26–28). These harmful effects are caused at least in part by destabilization of the OM due to decreased OMP levels (29). In addition, decreased OMP levels result in increased sensitivities to detergents and antibiotics (30, 31). Because of these defects in OM integrity, it is likely that the rpoE_S2R mutation would be detrimental to the survival of E. coli in its natural environment.
Supplementary Material
ACKNOWLEDGMENT
We thank Carol A. Gross (UCSF) for providing PrpoE- and PmicA-gfp reporter plasmids.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00340-16.
REFERENCES
- 1.Silhavy TJ, Kahne D, Walker S. 2010. The bacterial cell envelope. Cold Spring Harb Perspect Biol 2:a000414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barchinger SE, Ades SE. 2013. Regulated proteolysis: control of the Escherichia coli sigma(E)-dependent cell envelope stress response. Subcell Biochem 66:129–160. doi: 10.1007/978-94-007-5940-4_6. [DOI] [PubMed] [Google Scholar]
- 3.Raina S, Missiakas D, Georgopoulos C. 1995. The rpoE gene encoding the sigma E (sigma 24) heat shock sigma factor of Escherichia coli. EMBO J 14:1043–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.De Las Peñas A, Connolly L, Gross CA. 1997. The sigmaE-mediated response to extracytoplasmic stress in Escherichia coli is transduced by RseA and RseB, two negative regulators of sigmaE. Mol Microbiol 24:373–385. doi: 10.1046/j.1365-2958.1997.3611718.x. [DOI] [PubMed] [Google Scholar]
- 5.Missiakas D, Mayer MP, Lemaire M, Georgopoulos C, Raina S. 1997. Modulation of the Escherichia coli sigmaE (RpoE) heat-shock transcription-factor activity by the RseA, RseB and RseC proteins. Mol Microbiol 24:355–371. doi: 10.1046/j.1365-2958.1997.3601713.x. [DOI] [PubMed] [Google Scholar]
- 6.Walsh NP, Alba BM, Bose B, Gross CA, Sauer RT. 2003. OMP peptide signals initiate the envelope-stress response by activating DegS protease via relief of inhibition mediated by its PDZ domain. Cell 113:61–71. doi: 10.1016/S0092-8674(03)00203-4. [DOI] [PubMed] [Google Scholar]
- 7.Kanehara K, Ito K, Akiyama Y. 2003. YaeL proteolysis of RseA is controlled by the PDZ domain of YaeL and a Gln-rich region of RseA. EMBO J 22:6389–6398. doi: 10.1093/emboj/cdg602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alba BM, Leeds JA, Onufryk C, Lu CZ, Gross CA. 2002. DegS and YaeL participate sequentially in the cleavage of RseA to activate the sigma(E)-dependent extracytoplasmic stress response. Genes Dev 16:2156–2168. doi: 10.1101/gad.1008902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kanehara K, Ito K, Akiyama Y. 2002. YaeL (EcfE) activates the sigma(E) pathway of stress response through a site-2 cleavage of anti-sigma(E), RseA. Genes Dev 16:2147–2155. doi: 10.1101/gad.1002302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Flynn JM, Levchenko I, Sauer RT, Baker TA. 2004. Modulating substrate choice: the SspB adaptor delivers a regulator of the extracytoplasmic-stress response to the AAA+ protease ClpXP for degradation. Genes Dev 18:2292–2301. doi: 10.1101/gad.1240104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rhodius VA, Suh WC, Nonaka G, West J, Gross CA. 2006. Conserved and variable functions of the sigmaE stress response in related genomes. PLoS Biol 4:e2. doi: 10.1371/journal.pbio.0040002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Johansen J, Rasmussen AA, Overgaard M, Valentin-Hansen P. 2006. Conserved small non-coding RNAs that belong to the sigmaE regulon: role in down-regulation of outer membrane proteins. J Mol Biol 364:1–8. doi: 10.1016/j.jmb.2006.09.004. [DOI] [PubMed] [Google Scholar]
- 13.Udekwu KI, Wagner EG. 2007. Sigma E controls biogenesis of the antisense RNA MicA. Nucleic Acids Res 35:1279–1288. doi: 10.1093/nar/gkl1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li GW, Burkhardt D, Gross C, Weissman JS. 2014. Quantifying absolute protein synthesis rates reveals principles underlying allocation of cellular resources. Cell 157:624–635. doi: 10.1016/j.cell.2014.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sklar JG, Wu T, Kahne D, Silhavy TJ. 2007. Defining the roles of the periplasmic chaperones SurA, Skp, and DegP in Escherichia coli. Genes Dev 21:2473–2484. doi: 10.1101/gad.1581007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rizzitello AE, Harper JR, Silhavy TJ. 2001. Genetic evidence for parallel pathways of chaperone activity in the periplasm of Escherichia coli. J Bacteriol 183:6794–6800. doi: 10.1128/JB.183.23.6794-6800.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ringquist S, Shinedling S, Barrick D, Green L, Binkley J, Stormo GD, Gold L. 1992. Translation initiation in Escherichia coli: sequences within the ribosome-binding site. Mol Microbiol 6:1219–1229. doi: 10.1111/j.1365-2958.1992.tb01561.x. [DOI] [PubMed] [Google Scholar]
- 18.Looman AC, Bodlaender J, Comstock LJ, Eaton D, Jhurani P, de Boer HA, van Knippenberg PH. 1987. Influence of the codon following the AUG initiation codon on the expression of a modified lacZ gene in Escherichia coli. EMBO J 6:2489–2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stenström CM, Jin H, Major LL, Tate WP, Isaksson LA. 2001. Codon bias at the 3′-side of the initiation codon is correlated with translation initiation efficiency in Escherichia coli. Gene 263:273–284. doi: 10.1016/S0378-1119(00)00550-3. [DOI] [PubMed] [Google Scholar]
- 20.Mutalik VK, Nonaka G, Ades SE, Rhodius VA, Gross CA. 2009. Promoter strength properties of the complete sigma E regulon of Escherichia coli and Salmonella enterica. J Bacteriol 191:7279–7287. doi: 10.1128/JB.01047-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guzman LM, Belin D, Carson MJ, Beckwith J. 1995. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J Bacteriol 177:4121–4130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leiser OP, Charlson ES, Gerken H, Misra R. 2012. Reversal of the DeltadegP phenotypes by a novel rpoE allele of Escherichia coli. PLoS One 7:e33979. doi: 10.1371/journal.pone.0033979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gerken H, Charlson ES, Cicirelli EM, Kenney LJ, Misra R. 2009. MzrA: a novel modulator of the EnvZ/OmpR two-component regulon. Mol Microbiol 72:1408–1422. doi: 10.1111/j.1365-2958.2009.06728.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cosma CL, Danese PN, Carlson JH, Silhavy TJ, Snyder WB. 1995. Mutational activation of the Cpx signal transduction pathway of Escherichia coli suppresses the toxicity conferred by certain envelope-associated stresses. Mol Microbiol 18:491–505. doi: 10.1111/j.1365-2958.1995.mmi_18030491.x. [DOI] [PubMed] [Google Scholar]
- 25.Isaac DD, Pinkner JS, Hultgren SJ, Silhavy TJ. 2005. The extracytoplasmic adaptor protein CpxP is degraded with substrate by DegP. Proc Natl Acad Sci U S A 102:17775–17779. doi: 10.1073/pnas.0508936102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nitta T, Nagamitsu H, Murata M, Izu H, Yamada M. 2000. Function of the sigma(E) regulon in dead-cell lysis in stationary-phase Escherichia coli. J Bacteriol 182:5231–5237. doi: 10.1128/JB.182.18.5231-5237.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kabir MS, Yamashita D, Koyama S, Oshima T, Kurokawa K, Maeda M, Tsunedomi R, Murata M, Wada C, Mori H, Yamada M. 2005. Cell lysis directed by sigmaE in early stationary phase and effect of induction of the rpoE gene on global gene expression in Escherichia coli. Microbiology 151:2721–2735. doi: 10.1099/mic.0.28004-0. [DOI] [PubMed] [Google Scholar]
- 28.Noor R, Murata M, Nagamitsu H, Klein G, Raina S, Yamada M. 2009. Dissection of sigma(E)-dependent cell lysis in Escherichia coli: roles of RpoE regulators RseA, RseB and periplasmic folding catalyst PpiD. Genes Cells 14:885–899. doi: 10.1111/j.1365-2443.2009.01318.x. [DOI] [PubMed] [Google Scholar]
- 29.Murata M, Noor R, Nagamitsu H, Tanaka S, Yamada M. 2012. Novel pathway directed by sigma E to cause cell lysis in Escherichia coli. Genes Cells 17:234–247. doi: 10.1111/j.1365-2443.2012.01585.x. [DOI] [PubMed] [Google Scholar]
- 30.Liu A, Tran L, Becket E, Lee K, Chinn L, Park E, Tran K, Miller JH. 2010. Antibiotic sensitivity profiles determined with an Escherichia coli gene knockout collection: generating an antibiotic bar code. Antimicrob Agents Chemother 54:1393–1403. doi: 10.1128/AAC.00906-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nichols RJ, Sen S, Choo YJ, Beltrao P, Zietek M, Chaba R, Lee S, Kazmierczak KM, Lee KJ, Wong A, Shales M, Lovett S, Winkler ME, Krogan NJ, Typas A, Gross CA. 2011. Phenotypic landscape of a bacterial cell. Cell 144:143–156. doi: 10.1016/j.cell.2010.11.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.