

ABSTRACT
Varicella-zoster virus (VZV) is an alphaherpesvirus that causes varicella and herpes zoster. Membrane fusion is essential for VZV entry and the distinctive syncytium formation in VZV-infected skin and neuronal tissue. Herpesvirus fusion is mediated by a complex of glycoproteins gB and gH-gL, which are necessary and sufficient for VZV to induce membrane fusion. However, the cellular requirements of fusion are poorly understood. Integrins have been implicated to facilitate entry of several human herpesviruses, but their role in VZV entry has not yet been explored. To determine the involvement of integrins in VZV fusion, a quantitative cell-cell fusion assay was developed using a VZV-permissive melanoma cell line. The cells constitutively expressed a reporter protein and short hairpin RNAs (shRNAs) to knock down the expression of integrin subunits shown to be expressed in these cells by RNA sequencing. The αV integrin subunit was identified as mediating VZV gB/gH-gL fusion, as its knockdown by shRNAs reduced fusion levels to 60% of that of control cells. A comparable reduction in fusion levels was observed when an anti-αV antibody specific to its extracellular domain was tested in the fusion assay, confirming that the domain was important for VZV fusion. In addition, reduced spread was observed in αV knockdown cells infected with the VZV pOka strain relative to that of the control cells. This was demonstrated by reductions in plaque size, replication kinetics, and virion entry in the αV subunit knockdown cells. Thus, the αV integrin subunit is important for VZV gB/gH-gL fusion and infection.
IMPORTANCE Varicella-zoster virus (VZV) is a highly infectious pathogen that causes chickenpox and shingles. A common complication of shingles is the excruciating condition called postherpetic neuralgia, which has proven difficult to treat. While a vaccine is now available, it is not recommended for immunocompromised individuals and its efficacy decreases with the recipient's age. These limitations highlight the need for new therapies. This study examines the role of integrins in membrane fusion mediated by VZV glycoproteins gB and gH-gL, a required process for VZV infection. This knowledge will further the understanding of VZV entry and provide insight into the development of better therapies.
INTRODUCTION
Varicella-zoster virus (VZV) is an alphaherpesvirus and a host-specific human pathogen that causes the diseases varicella and herpes zoster, commonly known as chickenpox and shingles (1). Prior to the approval of attenuated vaccines by the Food and Drug Administration, varicella was endemic in the United States population and it was estimated that one in three individuals would develop zoster in their lifetime (1, 2). The program for universal varicella vaccination of children in the United States has proven to be successful in preventing disease by reducing varicella incidence by 57% to 90% (3). The zoster vaccine has been effective in reducing the zoster incidence by 51.3% (4). Individuals afflicted with zoster risk developing postherpetic neuralgia (PHN), a debilitating, painful condition that can last weeks to months after the acute herpes zoster rash has healed (1). Effective therapies are not currently available to treat PHN, as the cause of pain associated with this condition has not been clarified. While the vaccine can significantly reduce the incidence of herpes zoster and PHN, its effectiveness has been reported to wane over time (5). Critically, the attenuated VZV vaccines are contraindicated for immunocompromised individuals. These limitations highlight the importance of identifying new targets for drug and vaccine development that focus on essential steps in VZV infection.
Herpesvirus membrane fusion is an essential first step of virion entry that allows the nucleocapsid to gain access to the cytoplasm of the host cell (6). The formation of the multinucleated cells called syncytia is a consequence of membrane fusion and is associated with VZV-induced pathology in infected skin and neuronal tissue (7, 8). Fusion is induced by a conserved complex of herpesvirus glycoproteins consisting of gB and the heterodimer gH-gL, which are present on the virion and expressed on the surface of infected cells (9). Expression of VZV gB and gH-gL is necessary and sufficient to induce fusion, in contrast to other herpesviruses which require additional virally encoded accessory proteins, such as gD for herpes simplex virus (HSV), gp42 for Epstein-Barr virus (EBV) for certain cell types, and gO or UL128/UL130/UL131 for human cytomegalovirus (HCMV) (9–13). Efforts to identify cellular components that contribute to VZV gB/gH-gL-mediated fusion activity have been hampered by the highly cell-associated nature of VZV in cell culture. This has made it challenging to study the stages of VZV infection, including fusion, because of the difficulty in generating a purified cell-free virus inoculum with a high titer, which is in contrast to HSV, which releases complete virions into the culture media (14–16).
Three cell proteins, cation-independent mannose 6-phosphate receptor (MPRci), insulin-degrading enzyme (IDE), and myelin-associated glycoprotein (MAG), have been proposed to function as receptors for VZV entry. MPRci, which binds mannose 6-phosphate groups, is required for cell-free VZV infection of MeWo cells (17). However, MPRci is unlikely to be directly involved in fusion because knockdown cells are still susceptible to cell-associated VZV infection and could form syncytia. Furthermore, a direct interaction has not been shown between MPRci and the VZV glycoproteins that contain N-linked complex oligosaccharides with mannose 6-phosphate groups, including gB and gH (18). IDE interacts with VZV gE and has been suggested to contribute to entry (17). However, IDE is unlikely to be an entry receptor, because VZV gE is not required for membrane fusion (12). Furthermore, the disruption of the gE and IDE interaction in a VZV gE mutant virus did not prevent infection of melanoma cells or fibroblasts in vitro or of T cells or neuronal cells in vivo (19, 20). Since VZV does not induce cell-cell fusion between T cells, this cell type was used to document interactions required for VZV entry without possible spread by syncytium formation. MAG associates with gB and contributes to gB/gH-gL-mediated fusion (12), but the fact that its expression is restricted to neuronal tissue suggests that there are additional cellular proteins that contribute to membrane fusion, as VZV can infect other cell types.
Integrins are heterodimeric proteins of noncovalently bound α and β subunits that are expressed on the cell surface (21). In humans, the known 18 α subunits and 8 β subunits can be assembled into 24 unique integrins with various functions, including cell adhesion and mobility (21, 22). Specific integrins have been implicated in having a role in the entry of five of the eight known human herpesviruses. These include HSV-1, HSV-2, EBV, HCMV, and Kaposi's sarcoma-associated herpesvirus (KSHV), suggesting that integrins are a conserved receptor or coreceptor for the entry of all herpesviruses (23–26). The role of integrins in VZV gB/gH-gL-mediated fusion and infection has not been explored to date.
In this study, the role of integrins in VZV gB/gH-gL-mediated fusion and infection was examined in human melanoma cells, which are permissive to VZV infection and model the syncytia that are observed in infected skin (27). To identify the integrin subunits that are expressed, the transcriptome of the melanoma cells was determined using RNA sequencing (RNA-seq). The role of integrins in VZV gB/gH-gL-mediated fusion was assessed using a newly developed cell-cell fusion assay that uses simultaneous and constitutive expression of a reporter protein and small hairpin RNAs (shRNAs) to target the transcripts of integrin subunits for knockdown. Data derived from the newly developed assay were corroborated using infection studies with the VZV pOka strain that demonstrated that the αV integrin subunit is important for VZV gB/gH-gL-mediated fusion.
MATERIALS AND METHODS
Cells and viruses.
Chinese hamster ovary K1 (CHO) cells (CCL-61; ATCC) and CHO-DSP1 cells (generated in this study) were propagated using F-12K nutrient mixture with Kaighn's modification (Invitrogen) supplemented with 10% fetal bovine serum (FBS; Invitrogen) and penicillin (100 U/ml; Invitrogen), with CHO-DSP1 cells maintained under puromycin selection (8 μg/ml; Invitrogen). MeWo (human melanoma) cells (HTB-65; ATCC), Mel-DSP2 cells (generated in this study), and Mel-DSP2 cells expressing shRNAs (generated in this study) were propagated in minimal essential medium (Invitrogen) supplemented with 10% FBS, nonessential amino acids (100 μM; Omega Scientific), and antibiotics (penicillin, 100 U/ml; streptomycin, 100 μg/ml; Invitrogen), with the last two cell lines maintained under puromycin selection (5 μg/ml). The 293T (CRL-3216; ATCC) cells were propagated using Dulbecco's modified Eagle's medium (DMEM) with 4.5 g/liter glucose, l-glutamine, and sodium pyruvate (Invitrogen) supplemented with 10% FBS and penicillin. The parental Oka strain of VZV (pOka) (gift from Klaus Osterrieder, Freie Universität, Berlin, Germany) was derived from a self-excisable bacterial artificial chromosome (BAC) and used throughout the study (28).
Vectors.
pGIPZ-DSP1 and pGIPZ-DSP2 vectors were generated by subcloning the R8(1-8) (DSP1) and R8(9-11) (DSP2) cassettes from a pair of phRL-CMV vectors that expressed the DSP proteins (gift from Zene Matsuda, University of Tokyo, Tokyo, Japan) (29). Briefly, the DSP1 and DSP2 genes were amplified by PCR (AccuPrime Taq; Invitrogen) from pH-RL-CMV-R8(1-8) and pH-RL-CMV-R8(9-11) using primers [P]R8(1-8)-pGIPZ, BsrG1(1-8)-pGIPZ, [P]R8(9-11)-pGIPZ, and BsrG1(9-11)-pGIPZ (Table 1). The PCR fragments were separated on a 1% agarose gel, purified by gel extraction (Qiagen), and digested with restriction enzyme, BsrGI (New England BioLabs). The products were then ligated (T4 ligase; Invitrogen) with an XbaI (New England BioLabs)-digested PCR fragment amplified from the pGIPZ vector using primers pGIPZ_XbaI and pGIPZ_5CMV and a BsrG1- and XbaI-digested pGIPZ vector. TOP10 (Invitrogen) electrocompetent cells were transformed with the ligated products and plated on LB agar plates supplemented with ampicillin (100 μg/ml; Sigma) and Zeocin (25 μg/ml; Invitrogen) antibiotics. Clones were confirmed by restriction enzyme digestion and sequencing.
TABLE 1.
Primers for effector/target cell and viral fusogen vector generation
| Primer name | Purpose | Sequence |
|---|---|---|
| [P]R8(1-8)-pGIPZ | Cloning | ATGGCTTCCAAGGTGTACGACCCCGAGCAACGC |
| BsrG1(1-8)-pGIPZ | Cloning | TACTTGTACATTAGATCACTTGTCGGCGGTGATGCACACG |
| [P]R8(9-11)-pGIPZ | Cloning | ATGCAGAAGAACGGCATCAAGGCCAACTTC |
| BsrG1(9-11)-pGIPZ | Cloning | TACTTGTACATTACTGCTCGTTCTTCAGCACGCG |
| pGIPZ_XbaI | Cloning | TGCTGCAGGTCCGAGGTTCTAGACG |
| pGIPZ_5CMV | Cloning | GGTGGCAGATCCTCTAGTAGAGTCG |
| miR30PCRXHO1F | shRNA adapters | CAGAAGGCTCGAGAAGGTATATTGCTGTTGACAGTGAGCG |
| miR30PCRECOR1R | shRNA adapters | CTAAAGTAGCCCCTTGAATTCCGAGGCAGTAGGCA |
| shINTGAV_1F | shRNA | TGCTGTTGACAGTGAGCGCGTGAGGTCGAAACAGGATAAATAGTGAAGCCACAGATGTA |
| shINTGAV_1R | shRNA | TCCGAGGCAGTAGGCATGTGAGGTCGAAACAGGATAAATACATCTGTGGCTTCACTA |
| shINTGA6_1F | shRNA | TGCTGTTGACAGTGAGCGACGGATCGAGTTTGATAACGATTAGTGAAGCCACAGATGTA |
| shINTGA6_1R | shRNA | TCCGAGGCAGTAGGCAGCGGATCGAGTTTGATAACGATTACATCTGTGGCTTCACTA |
| shINTGB1_1F | shRNA | TGCTGTTGACAGTGAGCGAGCCTTGCATTACTGCTGATATTAGTGAAGCCACAGATGTA |
| shINTGB1_1R | shRNA | TCCGAGGCAGTAGGCAGGCCTTGCATTACTGCTGATATTACATCTGTGGCTTCACTA |
| shINTGB3_1F | shRNA | TGCTGTTGACAGTGAGCGACCACGTCTACCTTCACCAATATAGTGAAGCCACAGATGTA |
| shINTGB3_1R | shRNA | TCCGAGGCAGTAGGCAGCCACGTCTACCTTCACCAATATACATCTGTGGCTTCACTA |
| shINTGB5_2F | shRNA | TGCTGTTGACAGTGAGCGCCTGAGGGCAAACCTTGTCAAATAGTGAAGCCACAGATGTA |
| shINTGB5_2R | shRNA | TCCGAGGCAGTAGGCATCTGAGGGCAAACCTTGTCAAATACATCTGTGGCTTCACTA |
| shINTGB8_3F | shRNA | TGCTGTTGACAGTGAGCGCGCTCAGTTGATTCAATAGAATTAGTGAAGCCACAGATGTA |
| shINTGB8_3R | shRNA | TCCGAGGCAGTAGGCATGCTCAGTTGATTCAATAGAATTACATCTGTGGCTTCACTA |
| shSLC1A5_1F | shRNA | TGCTGTTGACAGTGAGCGCGCCTGAGTTGATACAAGTGAATAGTGAAGCCACAGATGTA |
| shSLC1A5_1R | shRNA | TCCGAGGCAGTAGGCAAGCCTGAGTTGATACAAGTGAATACATCTGTGGCTTCACTA |
| pGIPZ-shRNA-seq001 | Sequencing | TTCACCGTCACCGCCGACGTCG |
Primers to clone shRNAs targeting the integrin subunit and SLC1A5 (solute carrier family 1, member 5) transcripts were designed using the shRNA psm2 design algorithm on the RNAi Central website (http://cancan.cshl.edu/RNAi_central/RNAi.cgi?type=shRNA) (30). The cloning fragment was generated by two PCRs, with the first amplifying a primer dimer containing the shRNA using the primers described above (Table 1) and the second adding on the remaining sequence of the miR30 cassette using primers miR30PCRXHO1F and miR30PCRECOR1R. The final PCR product was resolved on a 4% agarose gel, purified by gel extraction (Qiagen), and digested with EcoRI and XhoI restriction enzymes (New England Biolab). The pGIPZ-DSP2 vector was also digested with EcoRI and XhoI, dephosphorylated with antarctic phosphatase (Invitrogen), separated on a 0.8% agarose gel, and purified by gel extraction. The purified products were ligated together and then transformed into TOP10 cells. Cells were then plated on LB plates supplemented with the antibiotics ampicillin and Zeocin. Positive clones were confirmed by restriction enzyme digestion using MluI and XhoI (New England BioLabs), followed by sequencing using pGIPZ-shRNA-seq001.
Construction of CHO-DSP1, Mel-DSP2, and Mel-DSP2 expressing shRNA cell lines using lentivirus.
Lentiviruses were generated by transfecting 1.2 × 106 293T cells with 2.5 μg pGIPZ-DSP variant plasmids, 2.5 μg psPAX2, and 1 μg pMD2.G plasmids using Lipofectamine 2000 (Invitrogen). At 24 h posttransduction, virus-containing supernatant was harvested and passed through a 0.45-μm syringe filter. A spinoculation was performed by adding 1 ml of supernatant to 8 × 105 CHO or 1.5 × 106 human melanoma cells along with Polybrene (4 μg/ml; EMD Millipore) and centrifuging the cells at 750 relative centrifugal force (rcf) for 45 min at room temperature to generate CHO-DSP1 and Mel-DSP2, respectively. Melanoma cells constitutively expressing both DSP2 and shRNAs were generated in a similar manner. Puromycin was added at 48 h posttransduction for selection. All cell lines were adherent despite interference with expression of the integrin subunit of interest, and cell proliferation was not altered during passaging of the cell lines.
qPCR.
RNA from the cells was harvested and isolated using the QIAshredder columns (Qiagen) and the RNeasy Plus minikit (Qiagen) in accordance with the manufacturer's instructions. cDNA was generated from the isolated RNA using the SuperScriptIII first-strand synthesis system (Invitrogen) in accordance with the manufacturer's instructions. Quantitative PCR (qPCR) was performed with the cDNA using SsoAdvanced Universal SYBR green supermix (Bio-Rad) and exon-exon junction primers designed using the NCBI/Primer-BLAST tool (http://www.ncbi.nlm.nih.gov/tools/primer-blast/) (Table 2). The reactions were carried out on a CFX384 real-time system mounted on a C1000 thermal cycler (Bio-Rad) in quadruplicate. The results were normalized to Mel-DSP2 using levels of a housekeeping gene, PGM1, as a loading control. Statistical analysis was performed using the CFX Manager (Bio-Rad).
TABLE 2.
Quantitative PCR primers
| Primer name | Sequence |
|---|---|
| INTGAV-F | TTGGAGCATCTGTGAGGTCG |
| INTGAV-R | ACATGGAGCATACTCAACAGTCT |
| INTGA6-F | GACACTCGGGAGGACAACG |
| INTGA6-R | TGGAAGCGCTTCTGCCCGC |
| INTGB1-F | GAGTCGCGGAACAGCAGG |
| INTGB1-R | AGCAAACACACAGCAAACTGA |
| INTGB3-F | CATCACCATCCACGACCGAA |
| INTGB3-R | GTGCCCCGGTACGTGATATT |
| INTGB5-F | TGATACCTGGAACAACGGTGG |
| INTGB5-R | GGCTGATCCCAGACTGACAA |
| INTGB8-F | TTGTCTGCCTGCAAAACGAC |
| INTGB8-R | TGGCACAGGATGCTGCATTT |
| PGM-qPCR03 | CCAGAGTATCATCTCCACCGT |
| PGM-qPCR04 | TGTCCGATAACCAAGCGACC |
| SLC1A5-F | GTCGACCATATCTCCTTGATCCTG |
| SLC1A5-R | CAGCTCACTCTTCACTTGTATCAAC |
VZV gB/gH-gL and syncytin-1 DSP fusion assays.
The DSP fusion assay was adapted from the previously described quantitative cre reporter assay (10). Briefly, 8 × 105 CHO-DSP1 cells were transfected with 1.6 μg each of pCAGGs-gB (gift from Tadahiro Suenaga and Hisashi Arase, Osaka University, Osaka, Japan), pME18s-gH[TL], and pcDNA-gL plasmids or 20 ng of pHCMV-ERVW1Δ16 (gift from George Murphy, Stanford University, Stanford, CA, USA) plasmid with Lipofectamine 2000. Both the gH[TL] plasmid and the ERVW1Δ16 plasmid, which encodes a form of syncytin-1 with a 16-amino-acid truncation from the cytoplasmic domain, were selected over their respective wild type because they induced a hyperfusion phenotype that provided for a stronger readout without compromising the fusion mechanisms involving the ectodomain (27, 31). At 6 h posttransfection, the transfected CHO cells were trypsinized, collected by centrifugation, and resuspended in 1 ml of medium, of which 500 μl of cells was cocultured with either 1.5 × 106 Mel-DSP2 cells or 1.5 × 106 Mel-DSP2 cells expressing shRNAs and seeded in a 6-well plate. The medium was changed at 12 h after coculture, and the cells were harvested after an additional 24 h with trypsin and resuspended in 1 ml of resuspension buffer (phosphate-buffered saline [PBS], 2.5% FBS, 5 mM EDTA). The frequency of green fluorescent protein (GFP)-positive cells in 105 cells was quantified using a FACSCalibur flow cytometer controlled by CellQuest Pro (BD Bioscience) and analyzed with FlowJo (TreeStar). A negative-control experiment was performed with pME18s− and pcDNA− empty vectors to establish background levels of GFP expression, which were subtracted from the results for the test constructs. For the antibody inhibition study, 10 μg/ml of either anti-integrin αV (AV1; EMD Millipore), anti-gI (SG4; Meridian), or anti-gH (206) antibody (gift from Charles Grose, University of Iowa, Iowa City, IA, USA) (32) was added to 250 μl of transfected CHO cells cocultured with 7.5 × 105 Mel-DSP2 cells and then seeded in a 12-well plate. The medium was replaced with medium supplemented with 10 μg/ml of antibody at 24 h after coculture, harvested after an additional 24 h, and resuspended in 0.5 ml of resuspension buffer. Fusion was quantified as described above. Experiments were performed in duplicate at a minimum.
RNA-seq.
RNA was extracted from human melanoma cells using an RNeasy Plus minikit (Qiagen). The RNA-seq libraries were prepared from purified mRNA using a TruSeq RNA sample preparation kit v2 (Illumina), and the libraries were sequenced on a HiSeq2000 platform (Illumina) at the Stanford Stem Cell Institute Genome Center (SCIGC). Reads were aligned with the human genome hg19 sequence using TopHat (https://ccb.jhu.edu/software/tophat/index.shtml), and the values of fragments per kilobase of exon per million fragments mapped (FPKM) were calculated using Cufflinks (http://cole-trapnell-lab.github.io/cufflinks/). An FPKM value of 3 or greater was chosen as the cutoff to determine the expression of mRNA of integrin subunits.
Western blotting.
Protein lysates from transduced and nontransduced melanoma cells were obtained by cell lysate fractionation and chloroform methanol precipitation to enrich the lysate for αV integrins. Briefly, cell pellets were incubated in hypotonic buffer (20 mM HEPES, 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 10% glycerol, 1 mM dithiothreitol [DTT]) with EDTA-free protease inhibitors (Roche) for 15 min on ice. The buffer was then supplemented with 4% NP-40 to a final concentration of 0.2%, and the cells were briefly vortexed. The sample was then centrifuged at 16,200 rcf for 1 min, and the supernatant was transferred to a fresh tube. The supernatant was then treated with methanol, chloroform, and water, with vortexing after the addition of each buffer. Samples were centrifuged at 16,200 rcf for 1 min, and the aqueous layer was then removed. Additional methanol was added, and the sample was vortexed and centrifuged. Methanol was removed and further evaporated using an Eppendorf Vacufuge concentrator. The protein pellet was resuspended in Laemmli sample buffer (Bio-Rad) supplemented with 2-mercaptoethanol, boiled at 95°C for 10 min, and run on a 7.5% Mini-Protean TGX gel (Bio-Rad). Protein transfer was performed using a Transblot SD cell (Bio-Rad), and the membranes were probed for either αV integrin (rabbit polyclonal antibody [PAb] 4711; Cell Signaling) or α-tubulin (clone B-5-1-2 mouse anti-α-tubulin; Sigma). Primary antibodies were detected using secondary horseradish peroxidase-conjugated antibodies to either anti-mouse or anti-rabbit antibody and the ECL Plus detection kit (GE Healthcare Bio-Sciences).
VZV plaque size assay.
Mel-DSP2 cells or Mel-DSP2 cells expressing shRNA were inoculated with 200 PFU of cell-associated pOka or were transfected with 2 μg of pOka-TK GFP BAC (27). For the assay involving pOka infection, the medium was replaced with medium supplemented with puromycin (5 μg/ml) at 24 and 48 h postinfection to remove the puromycin-sensitive cells in the inoculum. The cells were fixed with 4% paraformaldehyde (PFA) at 96 h postinfection. Immunohistochemistry was performed on the fixed cells using an anti-VZV polyclonal serum (C05108MA; Meridian Life Science), and images of randomly selected plaques (n = 40) were taken with an AX10 microscope (Zeiss). The plaques were outlined, and the area was calculated using Image J, as previously described (11).
Replication kinetics and virus titration.
Mel-DSP2 cells or Mel-DSP2 cells expressing shRNA were inoculated with 1,000 PFU of cell-associated parental Oka strain of VZV (pOka). At 6 and 24 h postinfection, medium was replaced with medium supplemented with puromycin (5 μg/ml). Infected cells were harvested at 24-h intervals and titrated on melanoma cells in triplicate to examine the replication kinetics as previously described (33). For the cell-free virus infection, 5.5 × 105 cells were seeded in wells of a 12-well plate. Cells were inoculated with 20 PFU of cell-free pOka, which had been prepared as previously described and contained virions attached to short membrane segments and some cell-free virions (34). Cells were maintained under puromycin selection for 4 days. Cells were fixed at 4 days postinfection, and plaques were visualized by immunohistochemical staining as described above.
Confocal microscopy.
Confocal microscopy of CHO-DSP1 cells transiently expressing viral fusogenic glycoproteins fused with Mel-DSP2 cells was performed as previously described (10). Cells were fixed with 4% PFA, permeabilized with 0.1% Triton-X, and probed with either anti-gB MAb SG2 (mouse; Meridian Life Sciences) or anti-syncytin pAb H-280 (rabbit; Santa Cruz Biotechnology). The primary antibodies were detected with anti-mouse or anti-rabbit secondary antibody, Alexa Fluor 555 (Invitrogen). GFP expression was used as a marker of fused cells. Nuclei were stained with Hoechst 33342 (Invitrogen). Images were captured with a Leica SP2 AOBS confocal laser scanning microscope or an inverted Zeiss LSM 780 multiphoton laser scanning confocal microscope at the Stanford Cell Sciences Imaging Facility. Channel merging and image processing were performed with ImageJ and Photoshop.
Statistical analysis.
All quantitative results were analyzed by either a t test, one-way analysis of variance (ANOVA), or two-way ANOVA to determine the statistical significance using Prism (GraphPad Software).
RESULTS
VZV glycoproteins gB and gH-gL are necessary and sufficient to induce membrane fusion.
To address the limitations of sensitivity and reproducibility associated with current fusion assays, a novel mechanism-of-action assay called the stable reporter fusion assay (SRFA) was developed to quantify VZV gB/gH-gL-mediated fusion. The SRFA combined the dual split protein (DSP) system, which was developed by Ishikawa et al. (29), with a lentivirus system to generate cells that constitutively expressed the two reporter proteins, DSP-1 and DSP-2 (Fig. 1A). A GFP or Renilla luciferase signal is produced only upon reconstitution of the two proteins when effector cells that express DSP-1 and the viral glycoprotein fuse with the target cells that express DSP-2. Fused cells with reconstituted GFP activity and gB expression were observed by confocal microscopy after coculture of effector cells transiently expressing VZV gB/gH-gL and target cells derived from human melanoma cells (Fig. 1B). This was consistent with previous observations that gB and gH-gL are the minimum viral proteins required for fusion (12, 35). Melanoma cells were selected, as VZV replication in these cells induces the cell-cell fusion characteristic of VZV pathogenesis and the cells were expected to express the cellular components necessary for gB/gH-gL-mediated fusion (27). Fused cells were not observed in cocultures using effector cells transfected with the empty vector. The mean frequency of fused cells was >1 × 103 fused cells for every 1 × 105 cells analyzed for GFP activity by fluorescence-activated cell sorting (FACS) (Fig. 1C). In contrast, less than 0.2% of the cells in the coculture containing effector cells transfected with empty vector had GFP activity. Thus, the SRFA was successfully adapted to quantify VZV gB/gH-gL-mediated fusion and demonstrated that the constitutive expression of the DSP proteins provided for low background and a high signal-to-noise ratio.
FIG 1.

Quantifying VZV gB/gH-gL-mediated fusion with the SRFA. (A) Effector cells (square) that constitutively express the DSP1 reporter (black hexagons) and transiently express the fusogen (white circles) are cocultured with target cells (circle) that constitutively express the cellular receptor(s) (orange) or coreceptors needed for membrane fusion and the DSP2 reporter (purple hexagons). Fusion between the effector and target cells (green) results in GFP and Renilla luciferase activity from the reconstituted DSP, allowing for a dual readout of fusion. Fusion events were quantified by flow cytometry. (B) Confocal micrographs of CHO-DSP1 cells expressing VZV gB/gH-gL and Mel-DSP2 cells. Cells were stained for VZV gB (red) and nuclei (Hoechst 33342; blue). Fused cells have GFP activity from reconstituted DSP1 and DSP2 (green). The merged image is on the left. The middle images are of GFP and nuclei (top) and VZV gB and nuclei (bottom). The top right image (*) is a magnification of a representative fused cell. The bottom right image is the vector control. White scale bars, 100 μm. (C) The frequency of GFP-positive cells in effectors cells only (CHO-DSP1), target cells only (Mel-DSP2), cocultured effector and target cells (CHO-DSP1/Mel-DSP2), and cocultured effector and target cells transfected with either empty vector (vector) or plasmids expressing VZV gB and gH-gL (gB/gH-gL). Values were compared to those of the vector control, and statistical analysis was performed using ANOVA (***, P < 0.001). The data shown are the mean results of four independent experiments, with bars indicating the standard error of the mean (SEM).
Validation of SLC1A5 as a required receptor for syncytin-1-mediated fusion using a novel assay that combined the SRFA with shRNA expression.
To reduce experimental variability resulting from the use of small interfering RNA (siRNA) knockdown by transfection to identify cellular components critical for fusion, a new fusion assay was developed involving a single lentiviral vector encoding both the DSP and an shRNA to interfere with cellular protein expression (Fig. 2). This assay utilized transcriptome data of the target cell to infer the expression of relevant proteins that could contribute to fusion. The abundance of mRNAs encoding membrane proteins has been shown to correlate with high levels of protein expression in mammalian cells (36). Target cells that expressed both the DSP and an shRNA to knock down protein expression would be generated and evaluated in the SRFA to determine if the protein had a role in fusion.
FIG 2.

Experimental design to identify cellular proteins that function in viral glycoprotein-mediated fusion using RNA-seq, cell-cell fusion, and shRNA knockdown. Transcripts of candidate cellular receptors (green, orange, and blue sinusoids) expressed by cells permissive to infection by the enveloped virus of interest can be identified by RNA sequencing (RNA-seq). A single lentiviral vector encoding both the DSP2 (purple hexagons) and an shRNA (green, orange, and blue hairpins) targeting the transcript of the candidate receptor is generated. Target cells that stably express the DSP2 and the shRNA that inhibits expression (red line) of the candidate receptor (green, orange, and blue blocks) are generated by lentivirus transduction of cells. Transduced target cells are selected using puromycin. The stable reporter fusion assay is performed by coculturing effector cells that transiently express the fusogen of the enveloped virus of interest (white circles) and constitutively express the DSP1 (black hexagon) with the transduced target cells. Knockdown of a relevant protein will result in a reduction in fusion relative to that of the control cells (blue bar in chart). Receptors determined to be important for fusion will be confirmed in the context of viral infection.
This approach was evaluated first by adapting the SRFA to the fusogenic glycoprotein syncytin-1 that is encoded by human endogenous retrovirus-W that integrated into the germline 25 to 40 million years ago (37, 38). Syncytin-1 is expressed on the surface of placental trophoblasts and induces cell-cell fusion when triggered upon binding to its receptor, solute carrier family 1, member 5 (SLC1A5) (38). Similar to the VZV gB/gH-gL fusion assay, fused cells with GFP activity could be observed by confocal microscopy and quantified by FACS in cocultures of effector cells expressing syncytin-1 and target cells that were confirmed to express SLC1A5 by RNA-seq (Fig. 3A to C). Target cells in which each cell expressed the DSP2 construct and an shRNA that knocks down SLC1A5 were generated using a single lentiviral vector. The knockdown target cells had a 97% reduction in SLC1A5 transcripts and a 95% reduction in fusion events relative to those of the control cells (Fig. 3D). The requirement of SLC1A5 for syncytin-1 fusion was consistent with previous observations of the absence of cell-cell fusion in native CHO cells compared to CHO cells constitutively expressing SLC1A5 (39). The frequency of VZV gB/gH-gL fusion with the SLC1A5 knockdown cells was comparable to that of the control cells, indicating that the knockdown was specific and that expression of shRNAs did not affect expression of the DSP2 reporter protein (Fig. 3E). Thus, these results demonstrate that a single lentiviral vector that expresses both the reporter protein and shRNAs can be used in the SRFA to validate the functional role of cellular receptors for fusogenic proteins.
FIG 3.

Solute carrier family 1, member 5 (SLC1A5), is a required receptor for syncytin-1-mediated fusion. (A) Integrative Genomics Viewer coverage track of SLC1A5 reads derived from RNA-seq data of libraries generated from human melanoma cells. Exons of SLC1A5 are indicated in blue. (B) Confocal micrographs of cocultures of CHO-DSP1 cells expressing syncytin-1 and Mel-DSP2 cells. Cells were stained for syncytin-1 (red) and nuclei (Hoechst 33342; blue). Fused cells have GFP activity from reconstituted DSP1 and DSP2 (green). The merged image is on the left. The middle images are of GFP and nuclei (top) and syncytin-1 and nuclei (bottom). The top right image (*) is a magnification of a representative fused cell. The bottom right image is the vector control. White scale bars, 50 μm. (C) Frequency of GFP-positive cells produced by syncytin-1-induced fusion determined by flow cytometry analysis of 1 × 105 cells. (D) SLC1A5 mRNA (white) and syncytin-1 fusion (black) levels of a Mel-DSP2 knockdown cell line (SLC1A5) determined by qRT-PCR and the fusion assay, respectively. Values were normalized and compared to those of the Mel-DSP2 control (Ctrl). (E) Fusion assay using CHO-DSP1 cells transfected with either empty vector (Vector) or plasmids expressing gB/gH-gL and either Mel-DSP2 (Ctrl; white) or SLC1A5 knockdown target cells (black). All statistical analysis was performed using either a t test (C) or ANOVA (D and E) (***, P < 0.001; ns, not significant). Data shown for all experiments are the mean results of at least two independent experiments, with bars indicating the standard error of the mean (SEM).
VZV-permissive human melanoma cells express integrins.
To determine which of the 24 known human integrins were expressed on the melanoma cells, RNA lysate from the cells was analyzed by RNA-seq. Of the 18 α subunits and 8 β subunits known to be expressed by human cells, the abundance of mRNA transcripts of αV, αE, α1 to α7, β1, β3 to β5, and β8 subunits was determined to be above the expression threshold selected for this study, a value of fragments per kilobase of exon per million fragments mapped (FPKM) of 3 or greater (Fig. 4A). Based upon the transcript levels, 12 integrins were implicated as potential receptors important for VZV fusion, including αVβ1, αVβ3, αVβ5, αVβ8, α1β1, α2β1, α3β1, α4β1, α5β1, α6β1, α6β4, and α7β1. Integrin αEβ7 was excluded from this study, as the FPKM value for the β7 subunit was below the expression threshold.
FIG 4.

Inhibition of integrin subunit expression by shRNA knockdown. (A) FPKM (fragments per kilobase of exon per million fragments mapped) values of integrin subunits (circles) expressed in human melanoma cells determined by RNA sequencing, with darker colors indicating larger values, as indicated. Integrins are represented by the circles linked with a line. (B) Integrin subunit mRNA levels of Mel-DSP2 cell lines expressing shRNAs targeting subunits αV (ITGAV), α6 (ITGA6), β1 (ITGB1), β5 (ITGB5), and β8 (ITGB8) quantified by qRT-PCR. Levels of RNA for shRNA knockdown cells (white) are represented as percentages of those of the control Mel-DSP2 cells (black). Statistical analysis was performed using ANOVA (***, P < 0.001). Data shown are the mean results of at least duplicate experiments, with bars indicating the standard error of the mean (SEM).
Knockdown of αV integrins reduces VZV gB/gH-gL-mediated fusion.
To examine all 12 candidate integrins for their role in gB/gH-gL-mediated fusion, the αV (ITGAV), α6 (ITGA6), and β1 (ITGB1) subunits were individually targeted for knockdown by shRNA, as all of the candidates required at least one of these subunits. Relative to the control cell line (Mel-DSP2), a 95% knockdown of αV in ITGAV cells, a 42% knockdown of α6 in ITGA6 cells, and a 99% knockdown of β1 in ITGB1 cells were measured by quantitative reverse transcription (RT)-PCR (Fig. 4B). All cell lines were adherent despite interference with expression of the integrin subunit of interest, and cell proliferation was not altered during passaging of the cell lines. Expression of the shRNA targeting αV (ITGAV) reduced gB/gH-gL-mediated fusion to 60% of that of the control melanoma cells that did not express an shRNA (Mel-DSP2) (Fig. 5A, Ctrl). In contrast, expression of shRNAs targeting α6 (ITGA6) and β1 (ITGB1) increased fusion to 38% and 30% compared to that of the control melanoma cells, respectively. Knockdown of the αV subunit protein was confirmed by its absence in protein lysates from the ITGAV cell line compared to the control cell line (Fig. 5B). An increase in αV expression for the ITGB1 cells was shown by Western blotting, suggesting that the increase in fusion for ITGB1 cells was a consequence of increased αV integrin expression.
FIG 5.

Inhibition of αV subunit expression reduces VZV gB/gH-gL-mediated fusion. (A) VZV gB/gH-gL (white) and vector control (black) fusion levels with Mel-DSP2 (Ctrl) or Mel-DSP2 cell lines expressing shRNAs targeting subunits αV (ITGAV), α6 (ITGA6), β1 (ITGB1), β5 (ITGB5), and β8 (ITGB8). Values were normalized and compared to that of the Mel-DSP2 cells (Ctrl, gB/gH-gL). Statistical analysis was performed using ANOVA (*, P < 0.05; ***, P < 0.001). Data shown for all experiments are the mean results of at least two independent experiments, with bars indicating the standard error of the mean (SEM). (B) Western blot of αV subunit in membrane and cytoplasmic portions of cell-fractionated lysate from Mel-DSP2 cells (Ctrl) and shRNA knockdown target cells, ITGAV (αV) and ITGB1 (β1). Alpha tubulin (α-Tub) was probed as a loading control. Arrows indicate molecular weight (MW) in thousands for αV and α-tubulin.
To determine which of the αV heterodimers, αVβ3, αVβ5, or αVβ8, were involved in gB/gH-gL-mediated fusion, cell lines designated ITGB3, ITGB5, and ITGB8 were generated by shRNA knockdown of β3, β5, and β8 subunits, respectively. Each of the cell lines had reduced expression of their respective subunits by ≥89% relative to the expression of the control in each line (Fig. 4B). A moderate 15% reduction in fusion was observed with ITGB3 cells relative to the control, while significant differences were not observed with the ITGB5 and ITGB8 cells (Fig. 5A). Thus, the αV integrin subunit has a more significant role in VZV gB/gH-gL-mediated fusion than the β3 subunit.
Anti-αV antibody inhibits VZV gB/gH-gL-mediated fusion.
The αV subunit was postulated to directly participate in VZV gB/gH-gL-mediated fusion because of the marked reduction in fusion levels observed with the ITGAV cells. To determine the relevance of the extracellular domain of the αV subunit to fusion, cocultures of transfected CHO-DSP1 and Mel-DSP2 cells were treated with an antibody to the αV domain. The anti-αV antibody reduced fusion to 63% of that of the untreated cells (Fig. 6, No Ab), which was consistent with the reduction observed in the knockdown cell line, while addition of VZV glycoprotein I (gI) as a control antibody resulted in fusion levels that were similar to those of the untreated cells (Fig. 6). An anti-gH antibody (mAb206) with known neutralizing activity was used as a positive control and reduced fusion to background levels (32, 35). Thus, the reduction in fusion by the antibody to the extracellular domain of the αV subunit confirms the role of the subunit in VZV gB/gH-gL-mediated fusion.
FIG 6.
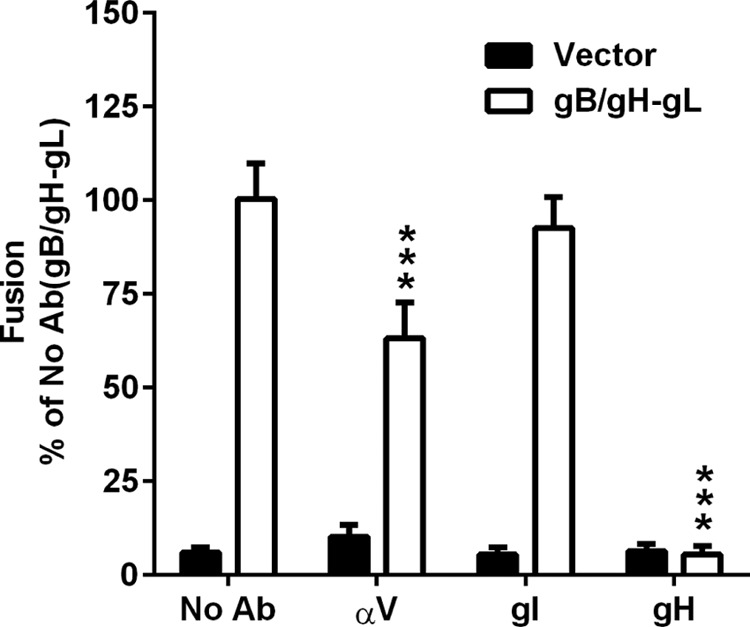
Anti-αV antibody inhibits VZV gB/gH-gL-mediated fusion. VZV gB/gH-gL (white) and vector control (black) fusion levels with 10 μg of anti-αV (αV), -glycoprotein I (gI), or -glycoprotein H (gH) monoclonal antibody added during coculture. Values were normalized and compared to those of the untreated Mel-DSP2 cells (No Ab, gB/gH-gL). Statistical analysis was performed using ANOVA (*, P < 0.05; ***, P < 0.001). Data shown are the mean results of at least two independent experiments, with bars indicating the standard error of the mean (SEM).
Knockdown of the αV subunit reduces VZV infection and spread.
To demonstrate the relevance of the fusion assay for VZV infection, ITGAV, ITGB1, and ITGB3 cells and control melanoma cells were infected with cell-associated VZV pOka and treated with puromycin at 24 h postinfection to remove the inoculum cells. The average size of the plaques, a measurement of VZV spread, in ITGAV cells was reduced by 60% compared to the control cells at 96 h postinfection, indicating that the loss of the αV subunit limited VZV spread (Fig. 7A and B). In contrast, the average plaque sizes in the ITGB1 and ITGB3 cell lines and the SLC1A5 cell line, which served as an shRNA control, were comparable those of the control cells. While inhibition of the β3 subunit reduced fusion events by 15% in the VZV fusion assay, the lack of an effect on VZV plaque size in the ITGB3 cells indicated that this level of fusion impairment was not sufficient to reduce viral spread. A similar reduction in plaque size was also observed in ITGAV cells (50% reduced) that were transfected with a BAC containing the pOka genome compared to the control cells at 96 h posttransfection (Fig. 7C). The reduced spread limited virus propagation, as a statistically significant 0.4- to 1-log10 decrease in viral titers in cell-associated pOka-infected ITGAV cells compared to the control cells between 1 and 3 days postinfection (dpi) was observed (Fig. 7D). Both infected ITGAV and control cells had similar viral titers at 4 dpi, indicating that the initial delay in propagation could be overcome. In contrast, the viral titers in the ITGB1, ITGB3, and SLC1A5 cells were similar to those in the control cells at all time points measured, which corroborated the plaque size assay results. The inoculation of uninfected monolayers with cell-associated VZV exposes the cells to large quantities of virions attached to the surface of each infected cell in the inoculum, increasing infection efficiency. To further determine the importance of the αV subunit for virus infection, the cell lines were infected with cell-free virus, which exposes the target cell to a lower titer of virions. At 4 dpi, a statistically significant reduction in the mean number of plaques was observed in ITGAV (3.5 ± 2.1, P < 0.01) and ITGB3 (6.5 ± 2.1, P < 0.01) cells compared to the control cells (18.25 ± 3.8), supporting the critical role of the αV subunit. Under these low-inoculum conditions, it was possible to detect a modest contribution of the β3 subunit, consistent with observations in the fusion assay. In contrast, the numbers of plaques in ITGB1 and SLC1A5 cells were not significantly different from the number in the control cells. Thus, the αV integrin subunit has an important role in effective VZV infection and spread.
FIG 7.

Knockdown of αV integrin subunit limits pOka infection and spread. (A) Phase-contrast images show representative plaques from a plaque size assay of Mel-DSP2 (Ctrl) and shRNA knockdown cell lines, ITGAV (αV), ITGB1 (β1), ITGB3 (β3), and SLC1A5, at 96 h after VZV pOka infection. Bars, 200 μm. (B) Box-and-whisker plot of plaque sizes, measured (n = 40) in square millimeters and compared to those of Mel-DSP2 cells. (C) Box-and-whisker plot of plaque sizes (n = 40) of Ctrl, ITGAV, and ITGB3 cell lines at 96 h posttransfection with pOka-TK GFP BAC. For the box-and-whisker plots, the boxes represent the 25th to 75th percentile, with the median indicated by the band and the whiskers representing the minimum and maximum values. (D) Replication of kinetics of pOka in ITGAV (red), ITGB1 (blue), ITGB3 (green), SLC1A5 (purple), and Mel-DSP2 (Ctrl; black) cells over 4 days using cell-associated inoculum. Individual points represent the mean of harvested infected cells titrated in triplicate in melanoma cells, including the titrated inoculum at day 0. Titers were measured in log10 of PFU per milliliter (PFU/ml). (E) Plaque assay for shRNA knockdown and Mel-DSP2 (Ctrl) cell lines infected with 20 PFU of cell-free pOka at 4 days postinfection. Statistical differences between the control and other cells were evaluated by one-way ANOVA (B, C, and E) (**, P < 0.01; ***, P < 0.001) or two-way ANOVA (D) (*, P < 0.05; ***, P < 0.001). Data shown are the mean results of at least two independent experiments, with bars in panels D and E indicating the standard error of the mean (SEM).
DISCUSSION
Membrane fusion induced by viral fusogens is a complex mechanism that involves both viral and cellular proteins (40). This study identified the human αV subunit of integrins as having a role in VZV gB/gH-gL-mediated membrane fusion and VZV infection and spread in melanoma cells. Integrin heterodimers that include the αV subunit have also been shown to be important for fusion-mediated entry of other human herpesviruses, including αVβ3, αVβ6, and αVβ8 for HSV-1, αVβ3 for HSV-2, αVβ5, αVβ6, and αVβ8 for EBV, αVβ3 for HCMV, and αVβ3 for KSHV (23, 24, 26, 41–43). In contrast to what has been observed in other herpesviruses, a heterodimer consisting of a specific β subunit together with the αV subunit was not discernibly involved in VZV gB/gH-gL-mediated fusion. Knockdown of the β subunits expressed in melanoma cells did not reduce fusion to levels of the αV knockdown, although a small change was observed when β3 was knocked down. This suggests that the β subunits of the integrin heterodimer are largely interchangeable, with fusion depending predominately on the αV subunit. This redundancy has been observed in studies of the role of integrins in HSV-1 entry and EBV glycoprotein-mediated fusion (23, 44). A similar reduction in viral spread has also been observed in HSV-2-infected human cervical epithelial cells expressing siRNA targeting the αV subunit (24). A slight but significant reduction in plaque number was observed when cells with the β3 subunit knockdown were infected with low-titer cell-free virus, suggesting that the β3 integrin subunit might contribute to VZV infection. Consistent with the limited number of plaques observed, siRNA knockdown of the β3 subunit has been previously reported to reduce HSV-2 spread (24). The presence of the αV integrin subunit on cells known to support VZV replication during natural infection, including T cells, epithelial cells, keratinocytes, and dermal fibroblasts, implicate αV integrin in VZV tropism (45–52). Although the abundantly expressed αV subunit was the emphasis of this study, it is plausible that integrin subunits expressed at low levels might have a contributory role in VZV gB/gH-gL-mediated fusion. Thus, VZV, like other members of the family Herpesviridae, exploits integrins to facilitate entry, suggesting that this capacity was acquired by a common ancestor from which all three human herpesvirus subfamilies were derived.
The reduction in fusion caused by the addition of an anti-αV antibody that binds the extracellular domain suggests that an interaction of either gB or gH-gL with the αV extracellular domain was inhibited. This possibility has precedence because αV integrins have been reported to act as coreceptors and to bind to the canonical, integrin-binding motif, Arg-Gly-Asp (RGD), in the extracellular domains of KSHV gB and HSV-1 gH (42, 53). A disintegrin-like domain (DLD) present in KSHV and HCMV gB has also been shown to function in integrin binding (54, 55). Strikingly, both the RGD and DLD motifs are absent in VZV gB, gH, and gL, suggesting that any potential interaction would occur through an unidentified motif in these glycoproteins. Such an interaction would implicate αV integrins as a coreceptor to facilitate or stabilize an interaction of gB/gH-gL with the currently unknown receptor(s) for VZV that triggers fusion.
The αV integrins might contribute to VZV gB/gH-gL-mediated fusion by facilitating the localization of the fusion-triggering receptor to lipid rafts that promote effective membrane fusion and virion entry. CHO cells expressing αVβ3 integrin have been observed to relocalize nectin-1, a receptor for HSV-1 entry, to lipid rafts that also contain the integrin in the absence of infection (56). Virions of KSHV have also been found to localize to lipid rafts that contain αVβ3 integrin (57). Therefore, lack of αV integrin expression could limit the localization of the fusion-triggering receptor to these lipid rafts and hinder fusion and entry. An interaction of αV integrins to VZV gB/gH-gL might also alter the cellular membrane to increase the susceptibility of the cells to fusion, as binding of cellular ligands to αV integrins can trigger downstream signaling that promotes cell migration, adhesion, and proliferation (58, 59). The binding of HSV-2 gH/gL to αVβ3 has been shown to cause intracellular Ca2+ release and activation of the focal adhesion kinase (24, 60). Both of these events promote actin rearrangement, which has been demonstrated to be important for the membrane fusion step during entry of enveloped viruses, including vaccinia virus and respiratory syncytial virus (61, 62). Actin rearrangement was found to be essential for KSHV entry, but it is not clear whether this process is involved in the fusion step of entry (63). It has also been shown in Drosophila cells that actin-propelled membrane protrusions are necessary and sufficient to promote cell-cell fusion mediated by fusogenic proteins (64). VZV gB/gH-gL might also alter the lipid composition of the cellular membrane via αV integrins to enhance fusion, as has been proposed for the interaction of the alphaherpesvirus equine herpesvirus 1 gH with the α4β1 integrin (65).
This study demonstrated the ability of the stable reporter fusion assay to overcome obstacles to the study of viral and cell proteins involved in VZV entry that result from its highly cell-associated nature. Furthermore, the assay can be used as a quantifiable mechanism-of-action assay in tandem with shRNA knockdown to study VZV gB/gH-gL-mediated fusion. The use of lentiviruses to generate the target cells removes the experimental variability that has plagued previous technologies that are reliant on transient transfection of reporter constructs, leading to heterogeneous levels of reporter expression. The experimental design of this study included RNA sequencing and generation of cells that stably coexpress shRNA and the reporter protein. This provides a platform that can be readily customized to study diverse fusogens and to identify cellular counterparts in different cell types, which was demonstrated by the syncytin-1 fusion assay. This novel assay can be adapted to study membrane fusion in diverse cell types because many human cells are susceptible to lentivirus transduction.
ACKNOWLEDGMENTS
We acknowledge John Mulholland, Kitty Lee, and Cedric Espenel of the Stanford Cell Sciences Imaging Facility and Marvin Sommer for their technical assistance.
Funding Statement
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
REFERENCES
- 1.Arvin AM, GIlden D. 2013. Varicella-zoster virus, p 2015–2057. In Howley P, Knipe DM (ed), Fields virology, 6th ed, vol 2 Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 2.Sampathkumar P, Drage LA, Martin DP. 2009. Herpes zoster (shingles) and postherpetic neuralgia. Mayo Clin Proc 84:274–280. doi: 10.4065/84.3.274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Marin M, Meissner HC, Seward JF. 2008. Varicella prevention in the United States: a review of successes and challenges. Pediatrics 122:e744–51. doi: 10.1542/peds.2008-0567. [DOI] [PubMed] [Google Scholar]
- 4.Oxman MN, Levin MJ, Johnson GR, Schmader KE, Straus SE, Gelb LD, Arbeit RD, Simberkoff MS, Gershon AA, Davis LE, Weinberg A, Boardman KD, Williams HM, Zhang JH, Peduzzi PN, Beisel CE, Morrison VA, Guatelli JC, Brooks PA, Kauffman CA, Pachucki CT, Neuzil KM, Betts RF, Wright PF, Griffin MR, Brunell P, Soto NE, Marques AR, Keay SK, Goodman RP, Cotton DJ, Gnann JW Jr, Loutit J, Holodniy M, Keitel WA, Crawford GE, Yeh SS, Lobo Z, Toney JF, Greenberg RN, Keller PM, Harbecke R, Hayward AR, Irwin MR, Kyriakides TC, Chan CY, Chan IS, Wang WW, Annunziato PW, Silber JL. 2005. A vaccine to prevent herpes zoster and postherpetic neuralgia in older adults. N Engl J Med 352:2271–2284. doi: 10.1056/NEJMoa051016. [DOI] [PubMed] [Google Scholar]
- 5.Tseng HF, Harpaz R, Luo Y, Hales CM, Sy LS, Tartof SY, Bialek S, Hechter RC, Jacobsen SJ. 2016. Declining effectiveness of herpes zoster vaccine in adults aged ≥60 Years. J Infect Dis doi: 10.1093/infdis/jiw047. [DOI] [PubMed] [Google Scholar]
- 6.Connolly SA, Jackson JO, Jardetzky TS, Longnecker R. 2011. Fusing structure and function: a structural view of the herpesvirus entry machinery. Nat Rev Microbiol 9:369–381. doi: 10.1038/nrmicro2548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cheatham WJ, Dolan TF Jr, Dower JC, Weller TH. 1956. Varicella: report of two fatal cases with necropsy, virus isolation, and serologic studies. Am J Pathol 32:1015–1035. [PMC free article] [PubMed] [Google Scholar]
- 8.Esiri MM, Tomlinson AH. 1972. Herpes zoster. Demonstration of virus in trigeminal nerve and ganglion by immunofluorescence and electron microscopy. J Neurol Sci 15:35–48. [DOI] [PubMed] [Google Scholar]
- 9.Eisenberg RJ, Atanasiu D, Cairns TM, Gallagher JR, Krummenacher C, Cohen GH. 2012. Herpes virus fusion and entry: a story with many characters. Viruses 4:800–832. doi: 10.3390/v4050800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vleck SE, Oliver SL, Brady JJ, Blau HM, Rajamani J, Sommer MH, Arvin AM. 2011. Structure-function analysis of varicella-zoster virus glycoprotein H identifies domain-specific roles for fusion and skin tropism. Proc Natl Acad Sci U S A 108:18412–18417. doi: 10.1073/pnas.1111333108.22025718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Oliver SL, Brady JJ, Sommer MH, Reichelt M, Sung P, Blau HM, Arvin AM. 2013. An immunoreceptor tyrosine-based inhibition motif in varicella-zoster virus glycoprotein B regulates cell fusion and skin pathogenesis. Proc Natl Acad Sci U S A 110:1911–1916. doi: 10.1073/pnas.1216985110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Suenaga T, Satoh T, Somboonthum P, Kawaguchi Y, Mori Y, Arase H. 2010. Myelin-associated glycoprotein mediates membrane fusion and entry of neurotropic herpesviruses. Proc Natl Acad Sci U S A 107:866–871. doi: 10.1073/pnas.0913351107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhou M, Lanchy JM, Ryckman BJ. 2015. Human cytomegalovirus gH/gL/gO promotes the fusion step of entry into all cell types, whereas gH/gL/UL128-131 broadens virus tropism through a distinct mechanism. J Virol 89:8999–9009. doi: 10.1128/JVI.01325-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harper DR, Mathieu N, Mullarkey J. 1998. High-titre, cryostable cell-free varicella zoster virus. Arch Virol 143:1163–1170. doi: 10.1007/s007050050364. [DOI] [PubMed] [Google Scholar]
- 15.Carpenter JE, Grose C. 2014. Varicella-zoster virus glycoprotein expression differentially induces the unfolded protein response in infected cells. Front Microbiol 5:322. doi: 10.3389/fmicb.2014.00322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reichelt M, Brady J, Arvin AM. 2009. The replication cycle of varicella-zoster virus: analysis of the kinetics of viral protein expression, genome synthesis, and virion assembly at the single-cell level. J Virol 83:3904–3918. doi: 10.1128/JVI.02137-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen JJ, Zhu Z, Gershon AA, Gershon MD. 2004. Mannose 6-phosphate receptor dependence of varicella zoster virus infection in vitro and in the epidermis during varicella and zoster. Cell 119:915–926. doi: 10.1016/j.cell.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 18.Gabel CA, Dubey L, Steinberg SP, Sherman D, Gershon MD, Gershon AA. 1989. Varicella-zoster virus glycoprotein oligosaccharides are phosphorylated during posttranslational maturation. J Virol 63:4264–4276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Berarducci B, Rajamani J, Zerboni L, Che X, Sommer M, Arvin AM. 2010. Functions of the unique N-terminal region of glycoprotein E in the pathogenesis of varicella-zoster virus infection. Proc Natl Acad Sci U S A 107:282–287. doi: 10.1073/pnas.0912373107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zerboni L, Berarducci B, Rajamani J, Jones CD, Zehnder JL, Arvin A. 2011. Varicella-zoster virus glycoprotein E is a critical determinant of virulence in the SCID mouse-human model of neuropathogenesis. J Virol 85:98–111. doi: 10.1128/JVI.01902-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cox D, Brennan M, Moran N. 2010. Integrins as therapeutic targets: lessons and opportunities. Nat Rev Drug Discov 9:804–820. doi: 10.1038/nrd3266. [DOI] [PubMed] [Google Scholar]
- 22.Wang Z, Symons JM, Goldstein SL, McDonald A, Miner JH, Kreidberg JA. 1999. (Alpha)3(beta)1 integrin regulates epithelial cytoskeletal organization. J Cell Sci 112:2925–2935. [DOI] [PubMed] [Google Scholar]
- 23.Gianni T, Salvioli S, Chesnokova LS, Hutt-Fletcher LM, Campadelli-Fiume G. 2013. αvβ6- and αvβ8-integrins serve as interchangeable receptors for HSV gH/gL to promote endocytosis and activation of membrane fusion. PLoS Pathog 9:e1003806. doi: 10.1371/journal.ppat.1003806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cheshenko N, Trepanier JB, Gonzalez PA, Eugenin EA, Jacobs WR Jr, Herold BC. 2014. Herpes simplex virus type 2 glycoprotein H interacts with integrin αvβ3 to facilitate viral entry and calcium signaling in human genital tract epithelial cells. J Virol 88:10026–10038. doi: 10.1128/JVI.00725-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Akula SM, Pramod NP, Wang FZ, Chandran B. 2002. Integrin alpha3beta1 (CD 49c/29) is a cellular receptor for Kaposi's sarcoma-associated herpesvirus (KSHV/HHV-8) entry into the target cells. Cell 108:407–419. doi: 10.1016/S0092-8674(02)00628-1. [DOI] [PubMed] [Google Scholar]
- 26.Wang X, Huang DY, Huong SM, Huang ES. 2005. Integrin alphavbeta3 is a coreceptor for human cytomegalovirus. Nat Med 11:515–521. doi: 10.1038/nm1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang E, Arvin AM, Oliver SL. 2014. The cytoplasmic domain of varicella-zoster virus glycoprotein H regulates syncytia formation and skin pathogenesis. PLoS Pathog 10:e1004173. doi: 10.1371/journal.ppat.1004173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tischer BK, Kaufer BB, Sommer M, Wussow F, Arvin AM, Osterrieder N. 2007. A self-excisable infectious bacterial artificial chromosome clone of varicella-zoster virus allows analysis of the essential tegument protein encoded by ORF9. J Virol 81:13200–13208. doi: 10.1128/JVI.01148-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ishikawa H, Meng F, Kondo N, Iwamoto A, Matsuda Z. 2012. Generation of a dual-functional split-reporter protein for monitoring membrane fusion using self-associating split GFP. Protein Eng Des Sel 25:813–820. doi: 10.1093/protein/gzs051. [DOI] [PubMed] [Google Scholar]
- 30.Chang K, Elledge SJ, Hannon GJ. 2006. Lessons from nature: microRNA-based shRNA libraries. Nat Methods 3:707–714. doi: 10.1038/nmeth923. [DOI] [PubMed] [Google Scholar]
- 31.Drewlo S, Leyting S, Kokozidou M, Mallet F, Potgens AJ. 2006. C-terminal truncations of syncytin-1 (ERVWE1 envelope) that increase its fusogenicity. Biol Chem 387:1113–1120. [DOI] [PubMed] [Google Scholar]
- 32.Montalvo EA, Grose C. 1986. Neutralization epitope of varicella zoster virus on native viral glycoprotein gp118 (VZV glycoprotein gpIII). Virology 149:230–241. doi: 10.1016/0042-6822(86)90124-8. [DOI] [PubMed] [Google Scholar]
- 33.Oliver SL, Zerboni L, Sommer M, Rajamani J, Arvin AM. 2008. Development of recombinant varicella-zoster viruses expressing luciferase fusion proteins for live in vivo imaging in human skin and dorsal root ganglia xenografts. J Virol Methods 154:182–193. doi: 10.1016/j.jviromet.2008.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Provost PJ, Krah DL, Friedman PA. 1997. Process for attenuated varicella zoster virus vaccine production. US patent 5607852.
- 35.Vleck SE, Oliver SL, Reichelt M, Rajamani J, Zerboni L, Jones C, Zehnder J, Grose C, Arvin AM. 2010. Anti-glycoprotein H antibody impairs the pathogenicity of varicella-zoster virus in skin xenografts in the SCID mouse model. J Virol 84:141–152. doi: 10.1128/JVI.01338-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lundberg E, Fagerberg L, Klevebring D, Matic I, Geiger T, Cox J, Algenas C, Lundeberg J, Mann M, Uhlen M. 2010. Defining the transcriptome and proteome in three functionally different human cell lines. Mol Syst Biol 6:450. doi: 10.1038/msb.2010.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Voisset C, Blancher A, Perron H, Mandrand B, Mallet F, Paranhos-Baccala G. 1999. Phylogeny of a novel family of human endogenous retrovirus sequences, HERV-W, in humans and other primates. AIDS Res Hum Retroviruses 15:1529–1533. doi: 10.1089/088922299309810. [DOI] [PubMed] [Google Scholar]
- 38.Blond JL, Lavillette D, Cheynet V, Bouton O, Oriol G, Chapel-Fernandes S, Mandrand B, Mallet F, Cosset FL. 2000. An envelope glycoprotein of the human endogenous retrovirus HERV-W is expressed in the human placenta and fuses cells expressing the type D mammalian retrovirus receptor. J Virol 74:3321–3329. doi: 10.1128/JVI.74.7.3321-3329.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lavillette D, Marin M, Ruggieri A, Mallet F, Cosset FL, Kabat D. 2002. The envelope glycoprotein of human endogenous retrovirus type W uses a divergent family of amino acid transporters/cell surface receptors. J Virol 76:6442–6452. doi: 10.1128/JVI.76.13.6442-6452.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Podbilewicz B. 2014. Virus and cell fusion mechanisms. Annu Rev Cell Dev Biol 30:111–139. doi: 10.1146/annurev-cellbio-101512-122422. [DOI] [PubMed] [Google Scholar]
- 41.Chesnokova LS, Hutt-Fletcher LM. 2011. Fusion of Epstein-Barr virus with epithelial cells can be triggered by αvβ5 in addition to αvβ6 and αvβ8, and integrin binding triggers a conformational change in glycoproteins gHgL. J Virol 85:13214–13223. doi: 10.1128/JVI.05580-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Garrigues HJ, Rubinchikova YE, Dipersio CM, Rose TM. 2008. Integrin αVβ3 binds to the RGD motif of glycoprotein B of Kaposi's sarcoma-associated herpesvirus and functions as an RGD-dependent entry receptor. J Virol 82:1570–1580. doi: 10.1128/JVI.01673-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gianni T, Gatta V, Campadelli-Fiume G. 2010. αVβ3-integrin routes herpes simplex virus to an entry pathway dependent on cholesterol-rich lipid rafts and dynamin2. Proc Natl Acad Sci U S A 107:22260–22265. doi: 10.1073/pnas.1014923108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chesnokova LS, Nishimura SL, Hutt-Fletcher LM. 2009. Fusion of epithelial cells by Epstein-Barr virus proteins is triggered by binding of viral glycoproteins gHgL to integrins alphavbeta6 or alphavbeta8. Proc Natl Acad Sci U S A 106:20464–20469. doi: 10.1073/pnas.0907508106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sen N, Mukherjee G, Sen A, Bendall SC, Sung P, Nolan GP, Arvin AM. 2014. Single-cell mass cytometry analysis of human tonsil T cell remodeling by varicella zoster virus. Cell Rep 8:633–645. doi: 10.1016/j.celrep.2014.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Overstreet MG, Gaylo A, Angermann BR, Hughson A, Hyun YM, Lambert K, Acharya M, Billroth-Maclurg AC, Rosenberg AF, Topham DJ, Yagita H, Kim M, Lacy-Hulbert A, Meier-Schellersheim M, Fowell DJ. 2013. Inflammation-induced interstitial migration of effector CD4+ T cells is dependent on integrin αV. Nat Immunol 14:949–958. doi: 10.1038/ni.2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schmidt-Chanasit J, Bleymehl K, Rabenau HF, Ulrich RG, Cinatl J Jr, Doerr HW. 2008. In vitro replication of varicella-zoster virus in human retinal pigment epithelial cells. J Clin Microbiol 46:2122–2124. doi: 10.1128/JCM.00122-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Weinacker A, Ferrando R, Elliott M, Hogg J, Balmes J, Sheppard D. 1995. Distribution of integrins alpha v beta 6 and alpha 9 beta 1 and their known ligands, fibronectin and tenascin, in human airways. Am J Respir Cell Mol Biol 12:547–556. doi: 10.1165/ajrcmb.12.5.7537970. [DOI] [PubMed] [Google Scholar]
- 49.Gershon MD, Gershon AA. 2010. VZV infection of keratinocytes: production of cell-free infectious virions in vivo. Curr Top Microbiol Immunol 342:173–188. doi: 10.1007/82_2010_13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Haapasalmi K, Zhang K, Tonnesen M, Olerud J, Sheppard D, Salo T, Kramer R, Clark RA, Uitto VJ, Larjava H. 1996. Keratinocytes in human wounds express alpha v beta 6 integrin. J Invest Dermatol 106:42–48. doi: 10.1111/1523-1747.ep12327199. [DOI] [PubMed] [Google Scholar]
- 51.Shiraki K, Yoshida Y, Asano Y, Yamanishi K, Takahashi M. 2003. Pathogenetic tropism of varicella-zoster virus to primary human hepatocytes and attenuating tropism of Oka varicella vaccine strain to neonatal dermal fibroblasts. J Infect Dis 188:1875–1877. doi: 10.1086/379835. [DOI] [PubMed] [Google Scholar]
- 52.Gailit J, Clark RA. 1996. Studies in vitro on the role of alpha v and beta 1 integrins in the adhesion of human dermal fibroblasts to provisional matrix proteins fibronectin, vitronectin, and fibrinogen. J Invest Dermatol 106:102–108. doi: 10.1111/1523-1747.ep12328177. [DOI] [PubMed] [Google Scholar]
- 53.Parry C, Bell S, Minson T, Browne H. 2005. Herpes simplex virus type 1 glycoprotein H binds to alphavbeta3 integrins. J Gen Virol 86:7–10. doi: 10.1099/vir.0.80567-0. [DOI] [PubMed] [Google Scholar]
- 54.Walker LR, Hussein HA, Akula SM. 2014. Disintegrin-like domain of glycoprotein B regulates Kaposi's sarcoma-associated herpesvirus infection of cells. J Gen Virol 95:1770–1782. doi: 10.1099/vir.0.066829-0. [DOI] [PubMed] [Google Scholar]
- 55.Feire AL, Koss H, Compton T. 2004. Cellular integrins function as entry receptors for human cytomegalovirus via a highly conserved disintegrin-like domain. Proc Natl Acad Sci U S A 101:15470–15475. doi: 10.1073/pnas.0406821101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gianni T, Campadelli-Fiume G. 2012. αVβ3-integrin relocalizes nectin1 and routes herpes simplex virus to lipid rafts. J Virol 86:2850–2855. doi: 10.1128/JVI.06689-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Garrigues HJ, DeMaster LK, Rubinchikova YE, Rose TM. 2014. KSHV attachment and entry are dependent on αVβ3 integrin localized to specific cell surface microdomains and do not correlate with the presence of heparan sulfate. Virology 464-465:118–133. doi: 10.1016/j.virol.2014.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kaido T, Perez B, Yebra M, Hill J, Cirulli V, Hayek A, Montgomery AM. 2004. Alphav-integrin utilization in human beta-cell adhesion, spreading, and motility. J Biol Chem 279:17731–17737. doi: 10.1074/jbc.M308425200. [DOI] [PubMed] [Google Scholar]
- 59.Cruet-Hennequart S, Maubant S, Luis J, Gauduchon P, Staedel C, Dedhar S. 2003. alpha(v) integrins regulate cell proliferation through integrin-linked kinase (ILK) in ovarian cancer cells. Oncogene 22:1688–1702. doi: 10.1038/sj.onc.1206347. [DOI] [PubMed] [Google Scholar]
- 60.Leung CS, Yeung TL, Yip KP, Pradeep S, Balasubramanian L, Liu J, Wong KK, Mangala LS, Armaiz-Pena GN, Lopez-Berestein G, Sood AK, Birrer MJ, Mok SC. 2014. Calcium-dependent FAK/CREB/TNNC1 signalling mediates the effect of stromal MFAP5 on ovarian cancer metastatic potential. Nat Commun 5:5092. doi: 10.1038/ncomms6092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Laliberte JP, Weisberg AS, Moss B. 2011. The membrane fusion step of vaccinia virus entry is cooperatively mediated by multiple viral proteins and host cell components. PLoS Pathog 7:e1002446. doi: 10.1371/journal.ppat.1002446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kallewaard NL, Bowen AL, Crowe JE Jr. 2005. Cooperativity of actin and microtubule elements during replication of respiratory syncytial virus. Virology 331:73–81. doi: 10.1016/j.virol.2004.10.010. [DOI] [PubMed] [Google Scholar]
- 63.Greene W, Gao SJ. 2009. Actin dynamics regulate multiple endosomal steps during Kaposi's sarcoma-associated herpesvirus entry and trafficking in endothelial cells. PLoS Pathog 5:e1000512. doi: 10.1371/journal.ppat.1000512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shilagardi K, Li S, Luo F, Marikar F, Duan R, Jin P, Kim JH, Murnen K, Chen EH. 2013. Actin-propelled invasive membrane protrusions promote fusogenic protein engagement during cell-cell fusion. Science 340:359–363. doi: 10.1126/science.1234781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Azab W, Gramatica A, Herrmann A, Osterrieder N. 2015. Binding of alphaherpesvirus glycoprotein H to surface α4β1-integrins activates calcium-signaling pathways and induces phosphatidylserine exposure on the plasma membrane. mBio 6:e01552-15. doi: 10.1128/mBio.01552-15. [DOI] [PMC free article] [PubMed] [Google Scholar]