

ABSTRACT
Influenza A virus (IAV) infections cause major morbidity and mortality, generating an urgent need for novel antiviral therapeutics. We recently established a dual myxovirus high-throughput screening protocol that combines a fully replication-competent IAV-WSN strain and a respiratory syncytial virus reporter strain for the simultaneous identification of IAV-specific, paramyxovirus-specific, and broad-spectrum inhibitors. In the present study, this protocol was applied to a screening campaign to assess a diverse chemical library with over 142,000 entries. Focusing on IAV-specific hits, we obtained a hit rate of 0.03% after cytotoxicity testing and counterscreening. Three chemically distinct hit classes with nanomolar potency and favorable cytotoxicity profiles were selected. Time-of-addition, minigenome, and viral entry studies demonstrated that these classes block hemagglutinin (HA)-mediated membrane fusion. Antiviral activity extends to an isolate from the 2009 pandemic and, in one case, another group 1 subtype. Target identification through biolayer interferometry confirmed binding of all hit compounds to HA. Resistance profiling revealed two distinct escape mechanisms: primary resistance, associated with reduced compound binding, and secondary resistance, associated with unaltered binding. Secondary resistance was mediated, unusually, through two different pairs of cooperative mutations, each combining a mutation eliminating the membrane-proximal stalk N-glycan with a membrane-distal change in HA1 or HA2. Chemical synthesis of an analog library combined with in silico docking extracted a docking pose for the hit classes. Chemical interrogation spotlights IAV HA as a major druggable target for small-molecule inhibition. Our study identifies novel chemical scaffolds with high developmental potential, outlines diverse routes of IAV escape from entry inhibition, and establishes a path toward structure-aided lead development.
IMPORTANCE This study is one of the first to apply a fully replication-competent third-generation IAV reporter strain to a large-scale high-throughput screen (HTS) drug discovery campaign, allowing multicycle infection and screening in physiologically relevant human respiratory cells. A large number of potential druggable targets was thus chemically interrogated, but mechanistic characterization, positive target identification, and resistance profiling demonstrated that three chemically promising and structurally distinct hit classes selected for further analysis all block HA-mediated membrane fusion. Viral escape from inhibition could be achieved through primary and secondary resistance mechanisms. In silico docking predicted compound binding to a microdomain located at the membrane-distal site of the prefusion HA stalk that was also previously suggested as a target site for chemically unrelated HA inhibitors. This study identifies an unexpected chemodominance of the HA stalk microdomain for small-molecule inhibitors in IAV inhibitor screening campaigns and highlights a novel mechanism of cooperative resistance to IAV entry blockers.
INTRODUCTION
Influenza viruses are members of the Orthomyxoviridae family and are responsible for major morbidity and mortality worldwide (1). Influenza A viruses (IAVs) have caused four recorded pandemics, the most devastating of which was the 1918 Spanish flu (H1N1) outbreak, which resulted in over 40 million deaths (2). This was followed by outbreaks of Asian flu (H2N2) in 1957, Hong Kong flu (H3N2) in 1968, and, most recently, swine-origin H1N1 pandemic flu in 2009 (3–5). Even in nonpandemic years, influenza virus infections are estimated to cause approximately 30,000 deaths in the United States alone (6). Two factors predominantly drive the morbidity and high mortality rates associated with influenza virus infections: first, the effectiveness of current influenza vaccines is moderate and particularly low in the elderly (7), and second, the efficacy of licensed antivirals is controversially discussed (8) and is further compromised by an increasing frequency of preexisting resistance in circulating virus strains (9–11).
For the past 10 influenza seasons, average vaccination effectiveness was calculated to be 40.6% (http://www.cdc.gov/flu/professionals/vaccination/effectiveness-studies.htm) but was substantially lower in years in which the seasonal vaccine did not fully match the circulating virus strains (12). In the case of newly emerging pandemic strains, low production response rates for the egg-grown vaccine stock can further aggravate the problem through temporary vaccine shortages (13).
Two classes of influenza virus-specific antivirals have received FDA approval for clinical use against influenza virus infection in the United States: inhibitors of the viral neuraminidase and blockers of the viral M2 ion channel. In addition, favipiravir (14), a substrate analog polymerase blocker with a broad RNA virus indication spectrum, is approved in Japan for the treatment of influenza when other drugs are ineffective. The neuraminidase inhibitors, such as oseltamivir and zanamivir, prevent release of newly assembled virions from the host cell surface (15, 16), whereas the adamantanes, such as amantadine and rimantadine, prevent uncoating of the incoming virus particle by blocking the M2 ion channel (17). However, clinical use of the adamantanes is no longer recommended due to widespread resistance to M2 channel blockers in circulating virus strains (18, 19). Furthermore, the emergence of preexisting resistance likewise increasingly compromises the efficacy of the neuraminidase inhibitors, as experienced, for instance, during the 2009 swine-origin H1N1 pandemic (11, 20–23). Since these resistant strains do not show significant attenuation (23–25), an urgent and unmet clinical need exists for the development of mechanistically novel classes of influenza virus inhibitors.
Propelled by the advent of modern genomics, proteomics, and bioinformatics, efforts to identify novel medicines since the 1990s have increasingly centered on hypothesis-driven target-based drug discovery (TDD) approaches (26, 27). In the search for novel influenza virus inhibitors, for instance, several genome-wide small interfering RNA (siRNA) screens reported a decade ago aimed to identify host factors required for virus replication as a starting point for TDD campaigns for host-directed antivirals (28). However, despite this shift in the industry toward target-based approaches and the commitment of major resources, the impact of TDD on the identification of first-in-class medicines remains below expectations. The majority of small-molecule drugs approved in recent years originated from empirical phenotypic drug discovery (PDD) campaigns (26, 27), resulting in a PDD renaissance for the development of novel drugs.
PDD anti-influenza virus high-throughput screening (HTS) campaigns have traditionally relied on monitoring virus-induced cytotoxicity as the primary criterion for hit identification. More recently, several generations of reporter systems were developed to assess virus replication, and these provide a quantitative readout and support miniaturization below the level of the 96-well-plate format (29). First-generation reporter assays employed a plasmid-based minigenome luciferase reporter driven by superinfection of transfected cells with influenza virus (30, 31). However, the suitability of this approach for HTS was limited by the labor-intensive transfection of target cells, a narrow range of suitable target cell lines, and a restriction to single-cycle infection at a high multiplicity of infection (MOI) for robust reporter expression, which prevents the discovery of late-acting compounds. Some of these limitations were eliminated by second-generation screens that employed recombinant virions in which one of the viral open reading frames was replaced by a reporter gene (32). Since virus stocks must be generated on stably transfected helper cell lines providing the missing viral protein in trans, however, second-generation assays also demand single-cycle infection or narrow the assay host range to the helper cell line. Recently developed replication-competent third-generation reporter strains integrate the reporter as additional genetic information in one of the viral genome segments (33, 34), which supports multicycle infections and provides target cell flexibility. However, third-generation influenza virus reporter strains have not yet been applied to large-scale HTS campaigns.
We recently developed a novel PDD approach for the identification of myxovirus inhibitors that employs third-generation IAV reporter strain technology. Based on coinfection of cells with independently traceable replication-competent IAV and respiratory syncytial virus (RSV) reporter strains expressing nano and firefly luciferases, respectively, this protocol is designed to simultaneously identify orthomyxovirus-specific, paramyxovirus-specific, and broad-spectrum likely host-directed hit candidates. Full assay validation against a small test set confirmed the suitability of this innovative approach for automated drug discovery (35).
In the present study, we applied the assay to an approximately 142,000-entry open discovery library that covers a diverse spectrum of the drug-like chemical space. Focusing on the IAV-specific primary hit pool, we confirmed candidate compounds through a panel of direct and orthogonal counterscreens and characterized the molecular mechanism of activity (MMoA). This approach yielded three novel and structurally distinct classes of entry inhibitors blocking the IAV hemagglutinin (HA) protein with nanomolar potency. The compounds were subjected to label-free positive target identification, resistance profiling, chemical elaboration, and in silico modeling of a docking pose for structure-aided lead development.
MATERIALS AND METHODS
Cell lines and transfections.
Human bronchial epithelial cells (BEAS-2B; ATCC CRL-9609), human embryotic kidney cells (293T; ATCC CRL-3216), Madin-Darby canine kidney cells (MDCK; ATCC CCL-34), baby hamster kidney cells (BHK-21; ATCC CCL-10) stably expressing T7 polymerase (BSR T7/5), and African green monkey kidney epithelial cells (Vero; ATCC CCL1-81) were maintained at 37°C and 5% CO2 in Dulbecco's modified Eagle's medium (DMEM) supplemented with 7.5% fetal bovine serum (FBS). Pooled human peripheral blood mononuclear cells (PBMCs) were incubated at 37°C and 5% CO2 in Roswell Park Memorial Institute (RPMI) medium supplemented with 10% FBS, 2 mM glutamine, 1% nonessential amino acids, and 1% sodium pyruvate. GeneJuice (Invitrogen) was used for all transient-transfection reactions.
Viruses.
Influenza A/WSN/1933(WSN) (H1N1), influenza A/Panama/2007/99 (H3N2), influenza A/Wisconsin/67/2005 (H3N2), influenza A/Wyoming/03/2003 (H3N2), and influenza A/Aichi/2/1968 (H3N2) viruses were propagated in MDCK cells for 2 days at 37°C. Influenza A/Mallard/Potsdam/83 (H2N2) virus was propagated in 10-day-old embryonated chicken eggs for 2 days at 37°C. Influenza viruses were titrated by a standard plaque assay with MDCK cells. Measles virus was propagated in Vero cells for 2 days at 37°C and titrated by determining the 50% tissue culture infective dose (TCID50) for Vero cells. Respiratory syncytial virus was grown in Hep2 cells and titrated by determining the TCID50 for Hep2 cells. Sindbis virus and vesicular stomatitis virus (VSV) were grown in Vero cells and titrated by determining the TCID50 for Vero cells. Vaccinia virus was grown in Vero cells and titrated by plaque assay.
Purification of virus stock.
Progeny virions were collected from cell culture supernatants (IAV) or released from infected cells through one freeze-thaw cycle (RSV) and subjected to a clearance step (4,000 rpm for 20 min at 4°C). Virions were diluted in DMEM, purified through a 20%-60% one-step sucrose gradient in TNE buffer (1 mM Tris [pH 7.2], 100 mM NaCl, 10 mM EDTA) (30,000 rpm for 120 min at 4°C), and harvested from the gradient intersection. Purified virus stocks were stored in aliquots at −80°C.
Generation of recIAV-NL/09-nanoLuc and recovery of recombinant IAV strains.
The recombinant IAV (recIAV) reporter strain IAV-NL/09-nanoLuc (H1N1) was generated using an 8-plasmid influenza virus rescue system (36). recIAV-NL/09-nanoLuc containing a nano luciferase reporter in the PA segment was generated analogously to a previously reported strategy (33). A cassette consisting of an inactivated 3′ PA packaging signal, the nano luciferase open reading frame (ORF) followed by a KDEL-encoding endoplasmic reticulum retention signal, and a copy of the original PA packaging signal flanked by AseI and ApaI restriction sites was synthesized (GeneWiz) and cloned into the original PA segment-containing plasmid. To recover recombinant IAV-NL and IAV-WSN, 293T cells were transfected with the 8-plasmid rescue system, treated with tosylsulfonyl phenylalanyl chloromethyl ketone (TPCK)-supplemented trypsin, and overlaid onto MDCK cells after 24 h of incubation at 37°C. Recovered recIAV strains were amplified on MDCK cells, and progeny virus was titrated by utilizing nano luciferase reporter activity or through plaque assay.
Compounds.
The screening set was assembled from commercial libraries (ChemBridge [100,000 compounds] and ChemDiv [30,000 compounds], both curated against chemical structures with undesirable reactivity) and proprietary compound collections from the Emory Institute for Drug Development (EIDD) (1,155 compounds) and Kansas University (11,520 compounds). All compounds were dissolved in dimethyl sulfoxide (DMSO) to a concentration of 10 μM and stored at −80°C. The MScreen software package (37) was used for electronic compound management, HTS data storage, and data analysis. To generate a screening set, all compounds were inventoried in MScreen and reformatted into barcoded 384-well daughter plates by use of a Nimbus96 liquid handler (Hamilton Robotics). Thirty-two wells of each 384-well plate were reserved for positive and negative (vehicle) controls, arranged in a checkerboard pattern in the two lateral columns at either side.
Chemical synthesis.
Dry solvents for chemical synthesis were obtained under an inert atmosphere in Sure Seal bottles and used without any further drying. Reactions were monitored by thin-layer chromatography on TLC silica gel 60 F254 or by liquid chromatography-mass spectrometry (LC-MS) with UV detection at 254 nm and low-resonance electrospray ionization (ESI). Purification of title compounds was accomplished on a Teledyne ISCO CombiflashRf+ purification system. Products were analyzed by nuclear magnetic resonance (NMR) analysis on a 400-MHz spectrometer (Varian 400) with deuteriochloroform (CDCl3) solvent, unless otherwise stated. Chemical shifts are reported in parts per million relative to CDCl3, and coupling constants (J) are reported in hertz. Solvents for NMR were CDCl3 (residual shifts of δ 7.27 for 1H and δ 77.23 for 13C). The residual shifts were taken as internal references and reported in parts per million. Title compounds were also analyzed by LC-MS with low-resonance ESI or high-resolution mass spectrometry (HRMS) (samples were dissolved in methanol and ionized by picospray, a form of electrospray). Analysis was done on an LTQ-FTMS instrument at a resolution of 100,000.
Automated HTS protocol in 384-well-plate format.
BEAS-2B cells (8 × 103 cells/well) were injected (30 μl/well) into barcoded white-walled/clear-bottomed 384-well plates by using a MultiFlo automated dispenser (BioTek) equipped with dual 10-μl peristaltic pump manifolds, collected (150 × g for 90 s at 25°C), and incubated for 5 h at 37°C and 5% CO2. Compound was added to a final concentration of 5 μM (20 nl/well) by using a high-density pin tool (V&P Scientific) attached to the pipetting head of the Nimbus liquid handler, followed by coinfection with recRSVA2-L19FD489E-fireSMASh (multiplicity of infection [MOI] = 0.1) and recIAV-nanoLuc (MOI = 0.02) at 10 μl/well by use of a MultiFlo dispenser unit, spin collection (150 × g for 90 s at 25°C), and incubation for 40 h at 37°C and 5% CO2. The final vehicle (DMSO) concentration was 0.05%. Barcodes of source and assay plates were automatically detected and recorded by the Nimbus unit at the time of stamping. Using a stacker unit with an integrated barcode reader attached to an H1 synergy plate reader (BioTek), plates were automatically loaded, nano-Glo substrates (10 μl/well [each]) injected, and bioluminescence recorded after 3 min of lag time for each well and substrate. Readouts were automatically saved by plate barcode.
Data normalization and analysis.
The MScreen package was employed for automated data analysis. Plate reader raw data files together with source and assay plate barcode maps generated by the Nimbus system were directly imported into the package. Normalized relative inhibition values were calculated for each compound by subtracting each value from the average for the plate vehicle controls, followed by dividing the results by the difference between the means for plate vehicle positive controls. Robust Z-scores were calculated as follows: robust Z-score = [Si − median(Sall)]/MAD(Sall) and MAD(Sall) = 1.4826 × median[|Si − median(Sall)|], where Si is an individual compound value and Sall are values of all compounds on a screening plate. Hit candidates were defined as compounds showing ≥75% inhibition of normalized signal intensity against either or both viral targets and having robust Z-scores of ≥3.5. The SciFinder database package (American Chemical Society) was used to query a chemical database with hit candidate structures to evaluate known bioactivity.
Dose-response counterscreens.
Threefold serial dilutions of cherry-picked hit candidates were prepared as three replicates each in 384-well plates by using a Nimbus liquid handler. BEAS-2B cells (8 × 103 cells/well) were plated as described above, serial dilutions transferred using the pin tool, and cells infected with recRSV-L19FD489E-fireSMASh (MOI = 0.1) and IAV-WSN-nanoLuc (MOI = 0.02) or left uninfected for cell viability assessment. Reporter signals were recorded as outlined above. To determine cell viability, PrestoBlue substrate (Life Technologies) was added after 40 h of incubation of the cells at 37°C (5 μl/well), and the top-read fluorescence (excitation at 560 nm, emission at 590 nm, and instrument gain of 85) was recorded after incubation for 45 min at 37°C by using an H1 Synergy plate reader. Raw data for all dose-response screens were analyzed according to the following formula: % inhibition = (XSample − XMin)/(XMax − XMin) × 100, with XMin representing the average for the positive-control wells and XMax the average for the negative-control wells. Four-parameter variable-slope regression was applied to determine 50% effective (EC50) and cytotoxic (CC50) concentrations.
Assay interference screen.
Serial dilutions of cherry-picked hit candidates were tested against a recVSV-nanoLuc strain (MOI = 0.02) generated for the counterscreen and a previously developed recRSV-A2-renilla strain (31, 38). Reporter signals were determined after 24 h (recVSV-nanoLuc) or 48 h (recRSV-A2-renilla) of incubation at 37°C.
Orthogonal counterscreen.
BEAS-2B or MDCK cells (1.5 × 105 cells/well) were seeded in a 12-well plate and then, at approximately 90% confluence, infected with a dilution of IAV-WSN (MOI = 0.02) in the presence of compound. For some viruses, the medium contained 1.0 μg/ml TPCK-supplemented trypsin. At 36 to 48 h postinfection, culture supernatants were harvested and viral titers determined by standard plaque assay on MDCK cells.
Cytotoxicity assay.
MDCK (1.2 × 104 cells/well), BSR T7/5 (1.0 × 104 cells/well), Hep2 (1.0 × 104 cells/well), BEAS-2B (1.0 × 104 cells/well), and Vero (1.0 × 104 cells/well) cells or stimulated human PBMCs were seeded in a 96-well plate. For stimulation, PBMCs were incubated in RPMI medium with 100 ng/ml phorbol myristate acetate (PMA) and 1 μg/ml ionomycin for 6 h at 37°C prior to seeding into microtiter plates and compound addition. Sixteen wells received positive (cycloheximide) and negative (vehicle) controls. Test compounds were added to the remaining wells in three replicates and serial dilutions ranging from 20.0 to 0.009 μM. To determine cell viability, PrestoBlue substrate (Life Technologies) was added at 10 μl/well after 72 h of incubation of cells at 37°C, followed by incubation for 2 h at 37°C and fluorescence reading (excitation at 560 nm, emission at 590 nm, and instrument gain of 85) using an H1 Synergy plate reader.
Statistical analysis.
The Excel and Prism 6 (GraphPad) software packages were used for data analysis. The 50% and 90% effective concentrations (EC50 and EC90) were assessed by four-parameter variable-slope regression modeling. The statistical significance of differences between two sample groups was assessed by two-way analysis of variance (ANOVA; Prism 6) in combination with Sidak's multiple-comparison posttest as specified in the figure legends. Experimental uncertainties are identified by error bars, which show standard deviations (SD).
Hemolysis inhibition assay.
Fresh chicken erythrocytes (red blood cells [RBCs]) were washed thrice with phosphate-buffered saline (PBS) and resuspended to make a 2% (vol/vol) suspension in PBS that was stored at 4°C until use. Concentrated virus was mixed with the same volume of 2% RBCs in PBS. After incubation of the virus-RBC mixture for 30 min on ice, compound was added. Incubation of the mixture for another 10 min on ice was followed by centrifugation at 3,500 rpm for 5 min, disposal of the supernatant, and resuspension in 200 μl PBS, pH 5.1, containing the compound or a control (DMSO or JMN3-003). To trigger hemolysis, the mixture was incubated for 30 min at 37°C. For removal of intact erythrocytes, samples were centrifuged at 3,500 rpm for 5 min, and then 100 μl of supernatant was transferred to a flat-bottomed 96-well plate and the absorbance at 410 nm determined using an H1 Synergy plate reader.
IAV minigenome assay.
For IAV minigenome reporter assays, 293T cells were transfected with 1.0 μg of WSN firefly luciferase reporter plasmid and 0.5 μg each of PB1, PB2, NP, and PA expression plasmids. At 3 h posttransfection, compound was added at the specified concentration range, followed by incubation at 37°C for 30 h, lysis, and assessment of luciferase activity.
Induction of viral resistance.
Cells were infected with IAV-WSN-nanoLuc (MOI = 0.02) in the presence of compound at 10× EC50. Viral growth was determined based on luciferase activity, and compound concentrations were increased to a maximum of 20 μM when robust replication was detected. Once the compound-treated virus showed replication activity equal to that of the DMSO-treated control, virus stocks were amplified in the presence of 5 μM compound, total viral RNA extracted, and cDNA copies of the HA segment subjected to sequence analysis. To confirm resistance, individual mutations were rebuilt in the genome plasmid encoding the IAV-WSN HA segment through site-directed mutagenesis using the QuikChange protocol (Stratagene), and the corresponding recombinants were recovered. After gradient ultrapurification of virus stocks, resistance was confirmed through dose-response testing in a 96-well-plate format, using a compound concentration range of 8.0 to 0.06 μM, and EC90 values were determined.
Surface biotinylation and streptavidin precipitation.
IAV-WSN-encoding expression plasmids were transfected into BSR T7/5 cells. After 36 h at 37°C, cells were washed with PBS and subjected to biotinylation on ice for 20 min, followed by quenching with DMEM supplemented with 10% FBS. Cells were subsequently treated with RIPA buffer for 15 min at 4°C, followed by clearance centrifugation of lysates (14,000 rpm for 30 min at 4°C). Supernatants were mixed with prewashed streptavidin beads and incubated for 2 h at 4°C. After the beads were washed, biotinylated proteins were released from the beads by incubation with urea-dithiothreitol (urea-DTT) (25 min and 50°C with agitation). Where indicated, precipitated biotinylated proteins were subjected to peptide-N-glycosidase F (PNGase F)-mediated deglycosylation prior to gel fractionation. Samples were denatured in the presence of 1× glycoprotein buffer 1 (New England BioLabs) for 20 min at 55°C, followed by treatment with PNGase F in 1× glycoprotein buffer 2 and NP-40 for 40 min at 37°C.
Gel fractionation and immunodetection.
Protein samples were fractionated by SDS-PAGE using 10% Tris-glycine gels. Fractionated proteins were transferred to polyvinylidene difluoride (PVDF) membranes, followed by coating of membranes with 5% nonfat milk in PBS with Tween (PBST) and decoration with anti-IAV HA (H1N1) antiserum (GTX127357; GeneTex) for 1.5 h at room temperature and subsequent staining with anti-TfR antibodies (ab84036; Abcam) for 1.5 h. Decorated membranes were stained with horseradish peroxidase (HRP)-conjugated antiserum and subjected to chemiluminescence detection using a Bio-Rad ChemiDoc imaging unit.
In silico docking.
Docking studies were carried out with MOE 2015.1001, using the Amber10 force field. The A/PR/8/34 (H1N1) (PDB entry 1RU7) and A/chicken/Potsdam/4705/1984 (H2N2) (PDB entry 2WRF) virus HA crystal structures were used for docking studies. The models were loaded into the MOE package (version 2015.1001), and the structure was prepared for docking. After protonation and energy minimization, an induced-fit protocol was used to dock the fusion inhibitors in the HA structure. All atoms were considered possible targets for docking. Once the docking procedure was completed, results were analyzed to determine the most probable binding sites.
Expression and purification of soluble HA proteins.
A DNA fragment corresponding to the ectodomain of HA (residues 412 to 529) fused to a C-terminal BirA biotinylation site (GLNDIFEAQKIEWHE; Avitag), foldon trimerization domain, and His tag was synthesized (GeneWiz). A thrombin cleavage site was inserted between the Avitag and the trimerization domain. This sequence was then inserted into a pCDNA5-HA helper vector that encoded residues 1 to 411 of WSN-HA. The full soluble HA construct (encoding residues 1 to 529, with C-terminal tags) was then cloned into the pCMV-ER/myc vector in frame with an N-terminal signal peptide. Recombinant soluble HA was expressed in 293T cells transiently transfected with the pCMV-ER/myc vector by use of GeneJuice transfection reagent. The medium was changed 12 h after transfection and replaced with serum-free DMEM. At 48 h posttransfection, the medium was harvested and buffered using Tris, pH 8.0. Medium containing secreted HA was flowed over Ni-nitrilotriacetic acid (Ni-NTA) resin. The resin was then washed in buffer containing 20 mM Tris, pH 8, with 250 mM NaCl and 5 mM imidazole. The bound resin was washed with Tris buffer containing 20 mM imidazole. Thrombin (8 U/100 μg HA) was used to elute recombinant HA. Fractions containing purified soluble HA were combined and concentrated to 5 mg/ml. After purification, soluble HA proteins were biotinylated by the addition of 25 μg BirA enzyme/mg total protein in accordance with standard protocols (Avidity). Biotinylated HAs were purified by size-exclusion chromatography (Superdex 200; GE Healthcare) and concentrated to approximately 5 mg/ml.
Label-free compound docking studies using BLI.
Binding assays with purified soluble IAV-WSN HA were conducted by biolayer interferometry (BLI) using the Octet Red 96 system (Fortebio). Purified and biotinylated HA protein was coupled to Super Streptavidin (SSA) high-binding biosensors (Fortebio) in PBS. HA-loaded SSA biosensors were dipped into increasing concentrations of lead compounds in PBS with 0.1% DMSO. Double controls were included in all experiments. Dissociation rates were determined by dipping the sensors back into PBS in the absence of compound. Reference sensors loaded with biotinylated anti-FLAG antibody (M2; Sigma) were used to control for nonspecific binding. Real-time binding kinetics were analyzed and calculated using the Octet Red software package. Briefly, concentrations of 50 nM to 25.6 μM were used to analyze small-molecule binding. Association of small molecules (kon) was measured for 80 to 120 s by dipping the sensors into 8 or 9 concentrations of inhibitor in kinetics buffer. Dissociation of small molecules (koff) was measured for 120 to 180 s in kinetics buffer. Experiments were performed at 30°C with shaking at 1,000 rpm. The ratio of koff to kon was used to determine the KD (equilibrium dissociation constant).
Viral growth curves in cultured cells.
For one-step growth curves, BEAS-2B cells were infected with the respective IAV-WSN-nanoLuc strain (MOI = 3). At the indicated time points postinfection, viral replication was monitored using nano luciferase reporter activity as a surrogate readout. For each strain tested, the number of relative light units (RLUs) at time zero was subtracted from those for all following time points, and all activity values were expressed relative to the maximum signal observed for the standard IAV-WSN-nanoLuc strain.
Cell-based fusion assay.
BSR T7/5 cells were cotransfected with 1.5 μg expression plasmid DNA encoding the respective HA protein and 0.15 μg plasmid DNA encoding enhanced green fluorescent protein (EGFP). After 24 h, cells were treated with 2.5 μg/ml TPCK-supplemented trypsin for 15 min at 37°C, with subsequent incubation in PBS at the specified pH for 5 min. Cells were neutralized with DMEM supplemented with 10% FBS, and syncytium formation was monitored microscopically after 60 min of incubation at 37°C.
RESULTS
Large-scale HTS campaign using fully replication-competent reporter strains to identify novel myxovirus inhibitors.
Using the previously validated dual myxovirus coinfection HTS protocol, we screened a library of over 142,000 compounds that was assembled from different proprietary and commercially available collections and curated against substructures with undesirable properties or known pan-assay interference (PAIN) activity (39). Compounds were tested in 384-well-plate format in single replicates at a final concentration of 5 μM, using human respiratory epithelial BEAS-2B cells as the host cell line, followed by automated dose-response direct and orthogonal counterscreens of hit candidates (Fig. 1A).
FIG 1.

Summary of the HTS campaign implemented. (A) Flow chart of the primary hit discovery and counterscreening strategies used for hit discovery and verification. (B) Primary hit identification strategy. Raw data were analyzed using both control-dependent (% inhibition by plate) and -independent (robust Z-score) approaches. Dashed lines represent hit cutoffs (75% inhibition and a Z-score of ≥3.5, respectively), and red symbols denote hit candidates (643 total) selected for counterscreening. (Inset) Summary of the counterscreening campaign. Forty confirmed IAV-specific (yellow) and two broad-spectrum candidates (green) passed the counterscreens. Three IAV-specific hit candidates (red) were selected for mechanistic characterization and development in this study.
To boost the accuracy of hit identification, we employed two independent statistical approaches: the calculation of % inhibition by plate and robust Z-scores. The former relies on independent positive and negative controls present on each plate, whereas the latter is based on the median values obtained for all library entries on each plate and is therefore control independent (40). Only compounds that adhered to the inclusion criteria of both analyses (robust Z-scores of ≥3.5 and ≥75% inhibition) were considered hit candidates and short-listed for counterscreening (Fig. 1B). All counterscreens were carried out in a 384-well format, using dose-response assays with a concentration range of 20 to 0.009 μM. A total of 643 influenza virus-specific and broad-spectrum hit candidates were admitted to counterscreening (Fig. 1B, inset). Of 319 candidates with activity against both myxovirus reporter strains in primary screening, 126 failed at the cytotoxicity counterscreening stage. In contrast, only 28 of the 324 IAV-specific hit candidates failed toxicity testing. Overall, 40 IAV-specific compounds and 2 broad-spectrum candidates passed all counterscreens, corresponding to a hit rate of approximately 0.03%.
Based on their performances in the primary screen and the panel of subsequent counterscreens (Fig. 2A), we selected three chemically distinct scaffolds for further evaluation in this study. Orthogonal efficacy testing of resynthesized compounds against the standard influenza A/WSN/1933 (H1N1) virus (IAV-WSN), using the virus yield determined by plaque assay as a readout, confirmed the potent inhibition of virus replication, with EC50s ranging from 0.17 to 0.38 μM (Fig. 2B and Table 1). Inhibition was not restricted to the laboratory-adapted IAV-WSN strain, since a luciferase-expressing reporter strain that we generated based on the influenza A/Netherlands/602/2009 (H1N1) virus isolate (IAV-NL-nanoLuc) was blocked at an efficiency similar to that with IAV-WSN-nanoLuc (Fig. 2B and Table 1). Cytotoxicity tests on different established cell lines and stimulated human PBMCs returned robust selectivity indices (SI = CC50/EC50), exceeding 500, 1,100, and 3,300, for the three hit classes (Fig. 2B and Table 2). IAV strains can be divided into group 1 and group 2 subtypes (41). Of the subtypes associated with past pandemics, two (H1N1 and H2N2) belong to group 1, whereas the third (H3N2) is part of group 2. When we tested the hit compounds against representatives of different subtypes, we noted that antiviral activity of GRP-103594 and GRP-115249 was restricted to H1N1 strains, whereas GRP-71271 was also active against an H2N2 test strain (Table 1). None of the compounds blocked group 2 IAV strains or a viral panel representing unrelated negative- and positive-strand RNA and DNA viruses (Table 1).
FIG 2.

Activity profiling of the three hit compounds selected for further characterization. (A) Primary screening and counterscreening results and chemical structures of the hit candidates. (B) Dose-response inhibition curves for the compounds shown in panel A for virus yield assays against IAV-WSN (black) and reporter assays against IAV-NL-nanoLuc (red). Titers and reporter signals were determined at 36 h postinfection. Curves are based on four-parameter variable-slope regression modeling. Blue symbols represent metabolic activities of uninfected host cells after 72 h of exposure to a compound. All symbols represent averages for three independent replicates ± SD.
TABLE 1.
| Viral target | GRP-71271 |
GRP-103594 |
GRP-115249 |
|||
|---|---|---|---|---|---|---|
| EC50 (μM) | EC90 (μM) | EC50 (μM) | EC90 (μM) | EC50 (μM) | EC90 (μM) | |
| IAV-WSN (H1N1)b | 0.17 (0.11–0.25) | 0.34 (0.16–0.73) | 0.28 (0.15–0.51) | 0.36 (0.30–0.43) | 0.38 (0.36–0.39) | 0.84 (0.73–0.97) |
| IAV-WSN-nanoLuc (H1N1)c | 0.05 (0.03–0.08) | 0.310 (0.21–0.45) | 0.02 (0.01–0.04) | 0.321 (0.26–0.38) | 0.06 (0.04–0.09) | 0.46 (0.31–0.69) |
| IAV-NL-nanoLuc (H1N1)c | 0.20 (0.15–0.27) | 0.55 (0.30–1.00) | 0.09 (0.05–0.16) | 0.42 (0.12–1.40) | 0.03 (0.01–0.08) | 0.25 (0.09–0.70) |
| A/Mallard/Potsdam/83 (H2N2)b | 5.36 (3.11–9.30) | 15.5 (3.6–50.5) | >20d | >10e | ||
| A/Panama/2007/99 (H3N2)b,e | >10 | >10 | >10 | |||
| A/Wisconsin/67/2005 (H3N2)b,e | >10 | >10 | >10 | |||
| A/Wyoming/03/2003 (H3N2)b,e | >10 | >10 | >10 | |||
| A/Aichi/2/68 (H3N2)b,e | >10 | >10 | >10 | |||
| Measles viruse,f | >10 | >10 | >10 | |||
| Respiratory syncytial viruse,f | >10 | >10 | >10 | |||
| Sindbis virusb,e | >10 | >10 | >10 | |||
| Vaccinia virusb,e | >10 | >10 | >10 | |||
| Vesicular stomatitis virusc,e | >10 | >10 | >10 | |||
Values represent three independent repeats and were calculated through four-parameter variable-slope regression modeling. Values in parentheses specify 95% confidence intervals.
Data are based on plaque titers.
Data are based on relative RLUs.
The highest concentration assessed was 20 μM.
The highest concentration assessed was 10 μM.
Data are based on TCID50 values.
For IAVs, the CC50 value as assessed on BEAS-2B cells was >100 μM (the highest concentration assessed was 100 μM) for all three compounds, and the SI values were >500, >1,100, and >3,300 for GRP-71271, GRP-103594, and GRP-115249, respectively.
TABLE 2.
Cytotoxicity testing of selected compounds against target cell lines or pooled, stimulated human PBMCs from healthy donorsa
| Target cells | CC50 (μM) |
||
|---|---|---|---|
| GRP-71271 | GRP-103594 | GRP-115249 | |
| MDCK | >100 | >100 | >100 |
| BEAS-2B | >100 | >100 | >100 |
| Vero | >100 | >100 | >100 |
| BSR T7/5 | >100 | >100 | >100 |
| Hep2 | >100 | >100 | >100 |
| Stimulated human PBMCs | >100 | >100 | ND |
The highest concentration assessed was 100 μM. ND, not determined.
Mechanistic characterization of confirmed hits.
To gain insight into the MMoA of the three hit classes, we employed time-of-addition (ToA) studies using IAV-WSN-nanoLuc as the target strain. Pretreatment of either the virus inoculum or the host cells followed by compound washout demonstrated that none of the hits had virucidal activity or induced a host cell antiviral state (Fig. 3A and B). Virion absorption to target cells was likewise unaffected by the presence of compound (Fig. 3C). While compound addition at the time of infection resulted in efficient inhibition, antiviral potency rapidly decreased when compounds were added at different times postinfection (Fig. 3D). These ToA profiles are characteristic for viral entry inhibitors and starkly distinct from those obtained for compound JMN3-003, a blocker of influenza virus RNA-dependent RNA polymerase (RdRp) activity that we previously developed (42, 43) and included in this experiment for reference. Likewise in contrast to JMN3-003, all three hit classes lacked inhibitory activity in a plasmid-based, IAV-WSN-derived minireplicon reporter assay (Fig. 3E). However, we noted dose-dependent inhibition of IAV-WSN-mediated hemolysis by each of the hit compounds (Fig. 3F) and efficient suppression of cell-to-cell fusion induced by transiently expressed IAV-WSN HA (Fig. 3G).
FIG 3.

Mechanistic characterization of selected hits. (A to C) Pretreatment of virus or cells with compounds. IAV-WSN-nanoLuc (A) or BEAS-2B cells (B) were preincubated with 10 μM compound for 2 h, or the virus was adsorbed to cells for 2 h at 4°C in the presence of compound (C). After compound removal and, for panels A and B, infection of cells, luciferase activity was determined after incubation at 37°C for 30 h. Bars represent averages for three independent experiments and SD; one-way ANOVA with Sidak's multiple-comparison posttest was used for statistical analysis (ns, not significant [P ≥ 0.05]). (D) Time-of-addition study. Compounds (10 μM final concentration) or an equivalent volume of vehicle (DMSO) was added at the specified times postinfection to BEAS-2B cells infected with IAV-WSN-nanoLuc. Luciferase activities in all samples were determined after incubation at 37°C for 20 h. Values represent averages for three independent experiments ± SD. (E) IAV-WSN-based minireplicon assay. 293T cells transiently expressing IAV-WSN NA, NP, PB1, and PB2 proteins and transfected with an IAV-WSN-based minigenome reporter containing firefly luciferase (31) were incubated in the presence of different compound concentrations or an equivalent volume of vehicle (DMSO). Luciferase activity was determined after incubation at 37°C for 36 h. Values represent averages for three independent experiments ± SD. (F) Hemolysis inhibition assay. Chicken red blood cells were mixed with purified IAV-WSN and compounds or an equivalent volume of vehicle (DMSO), subjected to pH 5.1, and incubated for 30 min at 37°C. The absorbance of released hemoglobin was measured at 410 nm and corrected for baseline signals of equally treated but mock-infected cells. Values represent averages for three independent experiments ± SD. (G) Cell-to-cell fusion assay. BSR T7/5 cells were transfected with expression plasmids encoding IAV-WSN HA or EGFP at a relative ratio of 10:1, followed by trypsin treatment and exposure to pH 5.1 for 5 min at 37°C in the presence of 10 μM compound or an equivalent volume of vehicle (DMSO). Syncytium formation was assessed microscopically after 30 min of incubation at 37°C. Representative fields of view are shown. Magnification, ×100.
Taken together, these results confirm the identification of three chemically novel IAV entry inhibitor classes. The IAV-specific indication profile and efficient attachment of virions to target cells in the presence of each of the hits indicates that these compounds do not block receptor binding or endocytotic virion uptake but rather interfere with HA protein expression or membrane fusion activity.
Target identification through biolayer interferometry (BLI).
To test whether the hit compounds interfere with HA protein synthesis or intracellular transport, we transiently expressed plasmid-encoded IAV-WSN HA in the presence of compounds or vehicle (equivalent volume of DMSO) and examined plasma membrane steady-state levels by using cell surface biotinylation and precipitation with immobilized streptavidin. As an internal standard, the cellular transferrin receptor was immunodetected in parallel with the HA protein on Western blots (Fig. 4A). Densitometric quantitation of the HA signals demonstrated that none of the tested compounds significantly affected HA surface levels, suggesting direct docking to HA and interference with HA fusion activity.
FIG 4.

Compound target identification. (A) Surface biotinylation of BSR T7/5 cells transiently transfected with expression plasmids encoding IAV-WSN HA and incubated in the presence of 10 μM compound or an equivalent volume of vehicle (DMSO). Streptavidin-precipitated surface proteins were subjected to SDS-PAGE, and blots were decorated with specific antibodies directed against HA or the host cell transferrin receptor (TfR), as an internal standard. Bars show average results and SD for densitometric quantitation of signal intensities of three independent repeats. Control cells received equal amounts of empty vector DNA (mock). One-way ANOVA with Sidak's multiple-comparison posttest was used for statistical analysis (ns, not significant [P ≥ 0.05]). (B) Purification of foldon-stabilized soluble IAV-WSN HA protein. Nickel affinity chromatography-purified soluble HA was biotinylated in vitro at a C-terminally inserted Avitag prior to SDS-PAGE. Gels were either subjected to Western blotting (WB) using specific antibodies directed against IAV HA or HRP-conjugated streptavidin or directly subjected to silver staining (Ag stain). (C) BLI analysis of compound binding to purified soluble IAV-WSN HA protein immobilized on high-density streptavidin-coated sensors. Association and dissociation curves are shown for all hit compounds and an inactive analog of GRP-71271 at different concentrations. Lines represent regression modeling, and numbers represent dissociation values (KD) and the goodness of fit of the underlying regression model (R2).
Based on these results, we employed label-free BLI to explore positive target identification. We generated and expressed a soluble, foldon-stabilized form of the IAV-WSN-derived HA ectodomain (solHA) (Fig. 4B) and subjected purified solHA to specific in vitro biotinylation of a carboxy-terminally inserted Avitag (44). The resulting biotinylated solHA trimers were bound to streptavidin-coated sensors and exposed to different concentrations of each of the three hit compounds or a biologically inactive chemical analog of GRP-71271. We noted dose-dependent docking of all three hit compounds, with the high on- and off-rate profiles characteristic of small-molecule binding (Fig. 4C). In contrast, the inactive analog did not bind to the sensors, confirming the specificity of the assay. Regression modeling returned affinity constants (KD) ranging from 0.54 to 1.8 μM, with an acceptable goodness of curve fit (R2 ≥ 0.84) which is within an approximately 2- to 5-fold range of the corresponding EC50s and thus in good agreement with the measured bioactivities of the different compounds. These results demonstrate specific and direct binding of each of the hit compounds to the HA protein.
Viral adaptation for resistance profiling.
To complement in vitro binding with bioactivity-driven target identification and to gain the first insight into the nature of the candidate target microdomains, we induced resistance to each of the hit classes through passaging of the IAV-WSN-nanoLuc reporter strain in the presence of gradually increasing compound concentrations, starting with 10× EC50 during the first passage. Viruses were subjected to reverse transcription-PCR (RT-PCR) and sequencing of the HA genome segment when efficient replication was detected in the presence of 20 μM compound, which occurred latest after 20 passages.
Among four independent adaptations (one each to compounds GRP-103594 and GRP-115249 and two to compound GRP-71271), we found two adapted strains with single point mutations in the HA protein and one escape strain, each with double and triple point mutations, respectively (Fig. 5A). Two mutations, I257T and V323I, were located in the HA1 subunit, and all others were located in HA2. Localization of the altered residues in a structural model of IAV-WSN HA in prefusion conformation positioned the mutations in three spatial clusters: a membrane-proximal region near the base of the HA stalk, within or near the membrane-distal section of the HA stalk, and at the base of the HA head domain (Fig. 5A). One substitution, I257T, which emerged in the HA with three point mutations, was located outside these clusters, near the top of the HA head domain.
FIG 5.

Resistance profiling. (A) Ribbon model of the prefusion HA trimer (left) and linear schematic of the protein organization (right). HA1 is shown in blue, HA2 is shown in green, and individual domains in HA2 are highlighted. FP, fusion peptide; SH, small α-helix; LH, large α-helix; TM, transmembrane domain. The position of the most membrane-proximal HA stalk N-glycan is shown in the structural model for one monomer only. The positions of the disulfide bridge linking both HA subunits (S-S; black line) and of previously reported resistance mutations of structurally different HA fusion inhibitors (gray lines) are marked. (B to E) Activity testing of recombinant IAV-WSN strains harboring reintroduced specific HA mutations against all three hit compound classes. Luciferase activity was calculated at 40 h postinfection; values represent averages for three independent experiments ± SD. Where justified, four-parameter variable-slope regression modeling was applied to calculate inhibitory concentrations (summarized in Table 3). Mutations that emerged in isolation were tested individually (B and C), and mutations found together were rebuilt individually and in combination (D and E). The gray line in each graph represents reference testing of GRP-71271 against the standard IAV-WSN-nanoLuc strain.
Reinsertion of either of the mutations that emerged in isolation, i.e., HA2 K58Q or HA2 F110L, into IAV-WSN HA and recovery of the corresponding recombinant viruses resulted in complete resistance of the recombinants to the compound used for adaptation and full cross-resistance to the other two compound classes (Fig. 5B and C and Table 3). Two recent studies reported that resistance to two structurally unrelated IAV H1N1 entry inhibitors is mediated by an HA2 M59I or M59L mutation (45, 46). We therefore generated recombinant IAV-WSN HA2-M59I and found that mutation of residue 59 also resulted in broad cross-resistance to our three hit classes (Table 3), suggesting that resistances mediated by mutations at residues 58 and 59 use a common mechanism.
TABLE 3.
Inhibition testing of recombinant IAV-WSN-nanoLuc strains harboring proteins with specific mutations
| IAV-WSN-nanoLuc HA mutation(s) | EC90 (μM)a |
||
|---|---|---|---|
| GRP-71271 | GRP-103594 | GRP-115249 | |
| None (standard HA) | 0.3 (0.20–0.49) | 0.3 (0.23–0.49) | 0.2 (0.22–0.79) |
| I257T | 0.8 (0.62–1.02) | 1.3 (0.83–2.02) | 1.1 (0.80–1.43) |
| V323I | 0.7 (0.27–0.96) | 1.3 (0.83–2.02) | 0.5 (0.24–1.01) |
| T156A | 1.0 (0.55–2.03) | 3.3 (1.35–3.98) | 2.3 (0.98–1.58) |
| D85N | 1.8 (0.92–3.58) | 3.6 (1.23–10.56) | 4.2 (9.61–10.87) |
| N154S | 1.1 (0.75–1.66) | 1.7 (1.31–2.21) | 2.5 (1.11–5.64) |
| V323I/T156A | >8.0 | >8.0 | >8.0 |
| I257T/V323I/T156A | >8.0 | >8.0 | >8.0 |
| D85N/N154S | >8.0 | >8.0 | >8.0 |
| V323I/N154S | >8.0 | >8.0 | >8.0 |
| D85N/T156A | >8.0 | >8.0 | >8.0 |
| K58Q | >8.0 | >8.0 | >8.0 |
| F110L | >8.0 | >8.0 | >8.0 |
| M59I | >8.0 | >8.0 | >8.0 |
Values represent three independent repeats and were calculated through four-parameter variable-slope regression modeling. Numbers in parentheses specify 95% confidence intervals. The highest concentration assessed was 8 μM.
In contrast to the adapted strains with single substitutions in HA, none of the several changes identified in the HA proteins of the remaining two adapted strains conferred robust escape from any of the compounds when rebuilt individually into recombinant IAV-WSN (Fig. 5D and E). However, we found that combinations of two point mutations, i.e., HA1 V323I/HA2 T156A and HA2 D85N/HA2 N154S, which were identified in viruses resistant to GRP-71271 and GRP-115249, respectively, cooperatively mediated high-level resistance (Fig. 5D and E and Table 3). As noted for the individual escape mutations K58Q and F110L, the two sets of combination mutations also mediated escape from inhibition by each of the three compound classes. Addition of the I257T mutation in the HA head to the V323I/T156A mutant, thus fully rebuilding the HA variant with the triple mutation, did not result in noticeably more pronounced resistance.
These results demonstrate that all three hit classes belong to a single cross-resistance group despite their high chemical diversity. Resistance mutations located in the HA2 B-loop domain were previously reported for at least six structurally unrelated classes of small-molecule IAV entry inhibitors (45–47). Remarkably, our adaptations reveal alternative, cooperative escape strategies mediated by substitutions located in distinct domains of the protein. Cooperative resistance is a known phenomenon exemplified, for instance, by HIV escape from antiretrovirals (48). However, interdependent escape mutations located in distal regions of the native influenza virus HA protein, as identified in our study, are unusual and highlight the possibility that mechanistically distinct avenues may be available for IAV escape from entry inhibitors.
Cooperative resistance depends on elimination of an HA stalk N-linked glycan.
Three universally conserved N-linked glycans exist in the IAV HA stem domain (49). We noted that the N154S and T156A mutations, which appeared in the virions with double and triple HA mutations, respectively, are both predicted to eliminate the most membrane proximal of these sites, which is reportedly the only one of these carbohydrates that can be removed without major penalties for HA bioactivity and virus replication in cell culture (49).
Transient expression of all reengineered mutant HAs followed by cell surface biotinylation and immunodetection of HA after Western blotting demonstrated that plasma membrane steady-state levels of all HA mutants were equivalent to that of standard HA. However, HA proteins carrying either the N154S or T156A substitution showed altered electrophoretic mobility compared to the other HA variants examined (Fig. 6A). Enzymatic removal of complex carbohydrates from precipitated surface proteins by use of PNGase F restored a comparable mobility of the HA mutants in the gel (Fig. 6B), confirming that HA proteins harboring the N154S or T156A substitution indeed lack the membrane-proximal stalk glycan.
FIG 6.

Cooperative resistance to entry inhibition. (A) Surface biotinylation of BSR T7/5 cells transiently transfected with expression plasmids encoding IAV-WSN HA with the specified mutations, carried out as described in the legend to Fig. 4A. Bars show average results ± SD for densitometric quantitation of signal intensities of three independent repeats. One-way ANOVA with Sidak's multiple-comparison posttest was used for statistical analysis (ns, not significant [P ≥ 0.05]; standard HA, unchanged IAV-WSN HA). (B) Immunoblotting of samples prepared as for panel A after deglycosylation with PNGase F. (C) Activity testing of recombinant IAV-WSN strains harboring reintroduced specific HA mutations against all three hit compound classes, as described in the legend to Fig. 5B to E. Values represent averages for three independent experiments ± SD. (D) Single-step growth curves for IAV-WSN-nanoLuc harboring the specified mutations in the HA protein on BEAS-2B cells. Luciferase activities were determined at the specified time points postinfection and are expressed relative to the maximal signal observed for the standard IAV-WSN-nanoLuc strain (standardmax). Values represent averages for three independent experiments ± SD. The dotted line at 0.01% of standardmax marks the signal cutoff (baseline observed at 0 h postinfection).
The N154S and T156A substitutions emerged in context with cooperative mutations in distinct HA subunits, i.e., D85N in HA2 and V323I in HA1, respectively. Only the combinations resulted in viral escape, raising the question of whether the resistance mechanisms are distinct and tied to the individual substitution pairs or whether the N154S and T156A mutations contribute to cooperative resistance solely through elimination of the N-glycan. To differentiate between these alternatives, we generated HA mutants with the artificial alternative combinations HA1 V323I/HA2 N154S and HA2 D85N/HA2 T156A in the recombinant IAV-WSN background. Dose-response testing against the three hit compounds revealed that both double mutants mediated robust escape from inhibition (Fig. 6C) that was comparable to the level of resistance observed previously for the native combinations (Table 3).
While the membrane-proximal stalk glycan is not essential for HA bioactivity, we asked whether the combination of glycan deletion with either the HA1 V323I or HA2 D85N mutation alters the trigger pH for HA refolding. We exposed cells transiently expressing the different HA mutants to different pH conditions, ranging from 5.1 to 5.7, and examined HA-mediated giant cells (syncytia) in a cell-to-cell fusion assay. Standard HA induced appreciable syncytia when cells were exposed to pH 5.3 to 5.4, while the HA K58Q and F110L substitutions slightly lowered the trigger pH, to 5.2 (Fig. 7). In contrast, all HA double mutants lacking the stalk glycan induced syncytia after incubation of cells at pH 5.6, indicating a reduced conformational stability of the metastable prefusion trimer.
FIG 7.

Cell-to-cell fusion assay to assess the trigger pHs of HA proteins carrying resistance mutations. BSR T7/5 cells were transfected with expression plasmids encoding IAV-WSN HA or EGFP at a relative ratio of 10:1, followed by trypsin treatment and exposure to the specified pH range for 5 min at 37°C. Syncytium formation was assessed microscopically after 30 min of incubation at 37°C. Representative fields of view are shown. Magnification, ×100. Standard HA, unchanged IAV-WSN HA.
To assess the relative fitness of the resistant strains in cell culture, we assessed the replication dynamics of the panel of recombinants, including the IAV-WSN HA2-M59I mutant. Of the four resistant strains with different combinations of cooperative double mutations, those harboring the HA2 N154S mutation displayed slight reductions in growth rates and replication success, whereas replication profiles of the pairs containing the T156A exchange matched that of standard IAV-WSN (Fig. 6D). For the strains with individual resistance mutations, the IAV-WSN F110L growth curve was likewise equivalent to that of standard IAV-WSN, while the strains with mutations at HA2 residues (K58Q or M59I) were the most replication impaired.
Taken together, these findings demonstrate that cooperative HA resistance is based on elimination of the membrane-proximal stalk glycan combined with a mutation located near the base of the HA head domain. Lowering the conformational stability of prefusion HA by this combination emerges as a common denominator for cooperative resistance. The precise nature of the second mutation destroying the glycosylation motif is irrelevant for resistance but can influence viral fitness through effects unrelated to N-glycan elimination.
Two distinct mechanisms of IAV resistance to entry inhibitors.
The different trigger pH ranges of HA proteins harboring cooperative and single escape mutations suggest that there are different underlying mechanisms of resistance. With the aim of extracting a candidate docking pose of the compounds, we expressed and purified two soluble HAs with signature mutations of the different escape classes: to facilitate comparison with previous studies (45–47), the HA2 M59I substitution was chosen to represent the cluster of single escape mutations located in proximity to the membrane-distal stalk section, and the HA2 D85N/HA2 T154S combination was selected as a signature pair for cooperative resistance.
When HA was subjected to BLI-based monitoring of compound docking, KD values for the two hit compounds used in this assay (GRP-71271 and GRP-115249) were approximately 2 orders of magnitude higher for the HA target with the M59I mutation (Fig. 8A) than for standard HA (Fig. 4C), indicating impaired compound binding. In contrast, KD values for these compounds against the HA D85N/T154S double mutant (Fig. 8B) were within a <2.5-fold range of those obtained earlier for standard HA, demonstrating essentially unaffected compound docking for the double mutant. These data suggest that residues located near the membrane-distal stalk section comprising HA2 residues M59 and K58 may be part of the primary target site for all entry inhibitor classes, while cooperative resistance appears to be based on a secondary mechanism and the residues at resistance sites do not directly participate in compound binding.
FIG 8.

Assessment of the compound docking pose. (A and B) BLI analysis of GRP-781271 and GRP-115249 binding to mutated, purified soluble IAV-WSN HA proteins immobilized on high-density streptavidin-coated sensors. Association and dissociation curves for different concentrations are shown. Lines represent regression modeling, and numbers represent dissociation values (KD) and the goodness of fit of the underlying regression model (R2). (C) Structural model of a candidate docking pose for GRP-71271. Coloring of the ribbon model and resistance mutations (shown as solid spheres) is the same as that in Fig. 5A. The inset shows a closeup view of the predicted target site, engaging residues of both the B loop and the central α-helices. (D) 2D transformations of the predicted docking poses of the three hit compounds. All three classes dock preferentially into the microdomain highlighted in panel C, but the nature of predicted interactions and the individual residues engaged are distinct for each pose.
To test this hypothesis, we subjected the compounds to in silico docking simulations, using an induced-fit model with prefusion influenza A/Puerto Rico/8/1934 (H1N1) virus HA (PDB entry 1RU7) as a target for all bioactive scaffolds identified. When we ranked hypothetical docking poses by affinity score, a single target pocket was predicted consistently and scored highest for all three compound classes (Fig. 8C). The lateral sides of this pocket are formed by two short sequences of HA1 (residues 277 to 280 and 314 to 319), a section of the α-helical prefusion stalk, and residues of the HA2 B-loop domain, whereas the top section is covered by HA1 residues 121 to 125 and residues 74 to 79 of the B loop. The prominent candidate mediators of primary resistance, HA2 residues 58 and 59, are positioned in close proximity to this pocket and predicted to affect its spatial geometry. Due to the pocket location between two HA monomers, compounds populating this microdomain may prevent membrane fusion by stabilizing the HA prefusion conformation.
All reported group 1 IAV entry inhibitors are proposed to bind in or near this microdomain (45–47), but predicted docking poses and the nature of key points of contact are unique for each scaffold, as illustrated by two-dimensional (2D) transformations that we generated for the docking poses of the three GRP hit scaffolds (Fig. 8D). The predicted pocket is lined by several hydrophobic surfaces, but some exposed side chains are available for H-bonding and electrostatic interactions. Very favorable docking with minimal surface exposure and a tight and uninterrupted contour plot was obtained for GRP-71271. In addition, HA1 K308 is predicted to act as a side chain donor to anchor the central ring system of GRP-71271 in the site. In contrast, GRP-103594 and GRP-115249 are left slightly more exposed, but docking is stabilized through additional hydrogen bonding and π-interactions, respectively.
To explore the predictive capacity of the model and refine the docking pose, we docked the three hit compounds into a structural model of influenza A/chicken/Potsdam/4705/1984 (H2N2) virus HA and synthesized (see Supplemental Information S1 in the supplemental material) a focused library of GRP-71271 analogs (Table 4) that were subjected in parallel to both biotesting and in silico docking. When the compounds were tested against the H2N2-derived HA, only compound GRP-71271 returned a favorable docking pose, while compounds GRP-103594 and GRP-115249 were sterically unable to populate the candidate target microdomain (Fig. 9). All of the bioactive GRP-71271 analogs identified could be docked in the predicted target site—albeit less favorably than GRP-71271, consistent with the measured actual decrease in inhibitory activity (Fig. 10)—whereas inactive analogs either failed to populate this microdomain (Table 4) or were predicted to violate docking constraints established by the GRP-71271 pose. Taken together, the predicted effects of both target and ligand variations on the docking poses emerged as remarkably consistent with our experimental results obtained for hit compound testing against avian IAV H2N2 and screening of GRP-71271 analogs against IAV-WSN, recommending the docking model as a tangible starting platform for structure-aided development of the GRP-71271 class.
TABLE 4.
Efficacy testing of 14 chemical analogs of GRP-71271 against IAV-WSN-nanoLuca
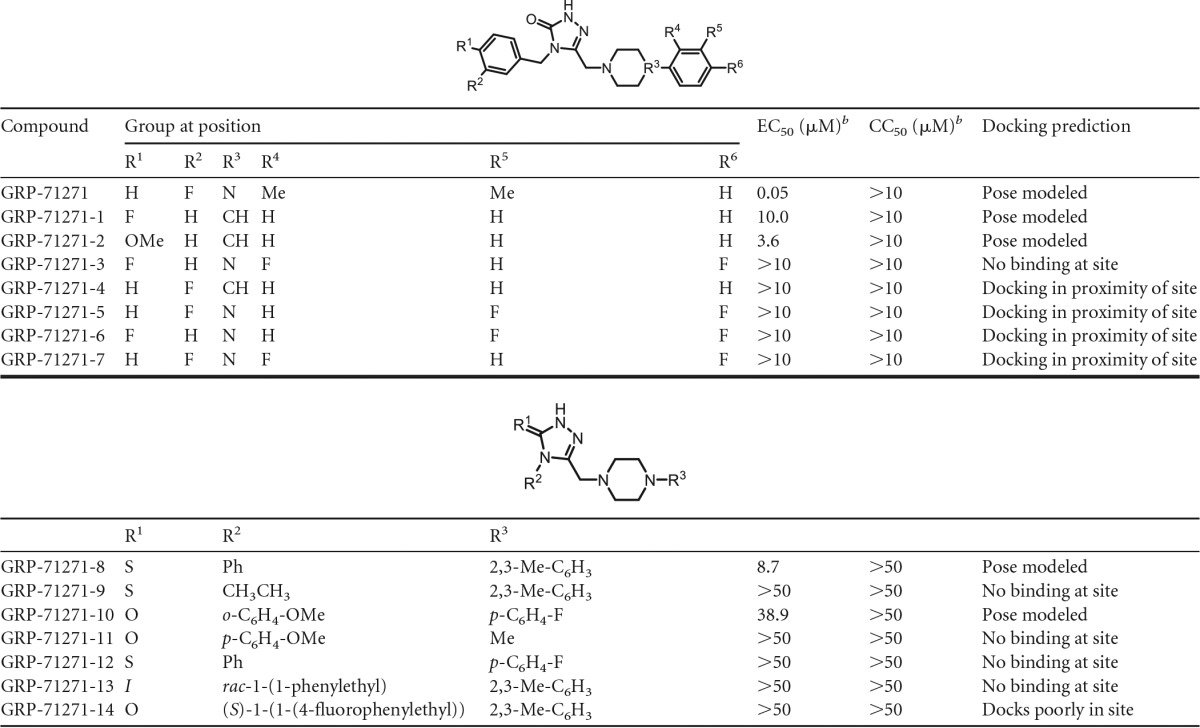
Values represent three independent repeats and were calculated through four-parameter variable-slope regression modeling.
b For the values in the top half of the table, the highest concentration assessed was 10 μM; for those in the bottom half of the table, the highest concentration assessed was 50 μM.
FIG 9.

2D transformations of predicted docking poses of the three hit classes for the influenza A/chicken/Potsdam/4705/1984 (H2N2) virus (PDB entry 2WRF)-derived HA protein. Blue markings highlight solvent-exposed areas of GRP-71271. GRP-103594 and GRP-115249 were force docked in silico into the candidate binding domain, but moieties highlighted in red stand in steric violation to the side chains lining the pocket.
FIG 10.

2D transformations of predicted docking poses of GRP-71271 analogs to A/Puerto Rico/8/1934 (H1N1) HA (PDB entry 1RU7) with EC50 values of ≤10 μM in biotesting. Analogs are predicted to populate the candidate target site, but interrupted contour plots and large solvent-exposed substructures indicate less favorable docking than that predicted for GRP-71271.
DISCUSSION
Two innovations characterize the drug discovery approach implemented in this study; they are the application of a fully replication-competent IAV reporter strain to a large-scale HTS campaign and the combination of two clinically relevant representatives of the myxovirus families for the simultaneous identification of hit candidates with distinct inhibitory profiles. This first application of the myxovirus coinfection protocol (35) to a large open discovery library revealed the following distinct advantages of the strategy: robust assay parameters are maintained for each target strain under real screening conditions; the approach is highly resource and time effective; use of replication-competent reporter strains provides compatibility with a variety of host cell lines, including the human respiratory cells used in our study; and the simultaneous assessment of two distinct viral targets reliably eliminates undesirable cytotoxic compounds from the virus-specific hit pool. While the frequency of confirmed IAV inhibitors identified in this screen was relatively low, we have no indication to assume that this outcome may be linked to the coinfection protocol itself. Rather, the conservative hit rate may reflect a feature of our rigorously curated compound collection.
In comparison, the approach was less effective at identifying host-directed antiviral hits with a broadened indication spectrum, since approximately 40% of the hit candidates with activity against both myxovirus targets failed subsequent toxicity tests and most of the remaining candidates underperformed against one or the other viral target in direct counterscreens. This experience underscores that a majority of host antiviral targets may be compromised permanently by unacceptable cytotoxicity, at least for use in pediatric indications, such as RSV infection or the treatment of seasonal influenza.
We therefore focused this study on three chemically distinct hit compounds that specifically inhibited influenza virus, with active concentrations in the nanomolar range against the attenuated IAV-WSN screening strain and a recombinant based on an isolate of the 2009 pandemic. Mechanistic characterization demonstrated that each of these hits blocks viral membrane fusion, suggesting binding to the viral HA protein. We confirmed this notion through both biochemical and virological identification of the HA protein as the molecular target, using label-free BLI and resistance profiling.
Small-molecule viral entry inhibitors have proven efficacious against different viral targets in clinical and preclinical settings (50–52). In the past few decades, a number of influenza virus fusion blockers were described that originated from structure-aided drug discovery or screening campaigns (53–58). Recently, in three screening campaigns, six different compounds were identified that all block entry of group 1 IAVs (45–47), supposedly through interaction with the same target pocket as that suggested by our docking exercises. One of these studies employed a lentivirus vector system pseudotyped with IAV HA, limiting the hit pool to inhibitors of IAV entry (47), but the other two campaigns were open by design, assessing either IAV-induced cytopathicity as a readout (46) or using a second-generation IAV reporter system (45). Considering the two open screens only combined with our study employing a third-generation reporter strain, over 1,100,000 drug-like compounds have been used to interrogate potential druggable targets of IAVs.
Notably, IAV entry inhibitors have emerged as the predominant inhibitor class in these campaigns; five compounds were reported previously, in addition to the three scaffolds identified in our study. Of the five compounds discovered previously, two (MBX 2546 and FA-617 [46, 47]) are direct analogs of each other, and another (45) is chemically related, resulting in a total of six truly independent chemical scaffolds. Despite their chemical diversity, the antiviral profiles of these six compound classes are remarkably similar: the indication spectrum of each is limited to group 1 IAVs, all are sensitive to resistance mutations at B-loop position 58 or 59, and all are predicted to populate the same pocket near the membrane-distal end of the prefusion HA stalk. These results spotlight an interesting dominance of the HA protein as the target for small-molecule group 1 IAV inhibitors identified in open screening campaigns. The available previous in silico docking exercises are typically backed up by resistance testing (46) and, in some cases, BLI binding studies and/or NMR analysis (45). If the resulting predictions are accurate, then potent small-molecule HA inhibition furthermore emerges as being governed by a single target site for compound binding that can be engaged productively by diverse chemical structures through different docking poses.
Support for this hypothesis of chemodominance comes from two directions. First, independently identified entry blockers of group 2 IAV entry (53) were found in cocrystal structures to dock into a defined pocket in the HA stalk domain that is located in proximity to that targeted by group 1 inhibitors (59). Second, a bias toward interaction of small-molecule screening hits with viral glycoproteins and the presence of a single predominant target site is not entirely unprecedented for small-molecule drug screens against myxovirus family members. A number of anti-RSV HTS campaigns were conducted in recent years that likewise yielded entry inhibitors at a high frequency (60). The molecular target site of these chemically diverse compound classes was originally discussed controversially. However, recent mapping of resistance mutations (61) and cocrystallization of structurally different inhibitor classes with the RSV fusion (F) protein (62) spotlighted a defined microdomain located at the base of the prefusion RSV F protein head domain as the common docking site. Analogous to our prediction for the different HA inhibitor classes, all RSV compounds tested engage the same F microdomain, but different avenues exist to productively populate the target (60, 62, 63).
Viral resistance against current therapeutics constitutes a major driving force for the development of novel influenza virus inhibitors; clinical use of the adamantanes is no longer advised, and preexisting resistance increasingly compromises the neuraminidase inhibitors (11, 22). Resistance profiles should therefore be considered early during development of novel drug candidates. Our findings revealed two distinct mechanisms of viral escape from the entry inhibitor classes identified in this study. A mutation in the HA2 B-loop domain reduced the binding affinity of the compounds for HA by approximately 1 order of magnitude, suggesting a primary resistance mechanism. B-loop residues may be part of the primary compound binding site or may sterically control compound access to the target. While our resistance mapping yielded a substitution at position 58, previous adaptations against structurally different entry inhibitors identified the neighboring residue 59 as a resistance hot spot (45, 46). Since the M59I exchange resulted in robust cross-resistance to our scaffolds, both mutations very likely mediate resistance through equivalent mechanisms. Whereas in silico docking positions HA2 residues 58 and 59 in close proximity to the predicted target site, the second single escape mutation, HA2 F110L, is positioned in the central section of the triple helix prefusion HA stalk, considerably distal from the postulated compound binding pocket. We certainly cannot exclude alternative binding poses not anticipated by in silico docking. However, phenylalanine residues stabilize helix-helix interactions, as demonstrated, for instance, for the conserved HA2 B-loop phenylalanines 63 and 70, which function as a conformational lock at the trimer interface in the postfusion HA structure (64–66). As an alternative to a secondary resistance mechanism, it is therefore conceivable that the F110L exchange induces a spatial distortion of the target pocket through a long-range mechanism.
In contrast to the effect of the B-loop substitutions, elimination of the most membrane-proximal N-linked glycan in the HA stalk combined with either an HA1 V323I or HA2 D85N exchange resulted in a cooperative resistance that is rarely experienced with small-molecule entry inhibitors. We propose that cooperative resistance is mediated by a secondary escape mechanism, based on several observations, as follows: sterically, a small molecule cannot engage both mutation sites simultaneously; the compound binding affinity for an HA protein with a cooperative double mutation was virtually identical to that for standard HA; and the cooperative double mutations raised the HA trigger pH, indicating a reduced conformational stability of the mutant prefusion HA trimer. Interestingly, different escape mechanisms also seem to be responsible for RSV pan-resistance to all entry inhibitor classes that were profiled in regard to resistance (60–62). In the case of RSV, however, secondary resistance does not depend on cooperative mutations; rather, the actual nature of amino acid substitutions at the same position controls the mechanism of escape.
Of the resistance hot spots identified in HA, the prominent B-loop mutations K58Q and M59I severely impaired viral growth in cell culture, suggesting that they may compromise viral fitness in vivo. It was also shown in a previous study that, in contrast to the other two HA stalk glycans, the N-glycan at HA2 N154 can be eliminated without major penalties for the virus in tissue culture (49). However, since the N154 glycan is conserved in circulating strains, we anticipate that loss of this glycosylation site will likely coincide with reduced viral fitness in the field. The HA2 F110L mutation did not result in reduced growth rates in cell culture, and group 2 IAV strains with mutations of HA2 positions 109 and 112 show resistance to amantadine (67). Assessments of the pathogenicity and transmission success of viruses carrying the F110L mutation will clarify whether this route of escape may lead to a reduction in overall viral fitness.
Group specificity naturally reduces the developmental potential of an IAV hit candidate. However, the GRP-71271 influenza virus entry inhibitor class, for instance, offsets this limitation with an otherwise attractive hit profile that is characterized by a high starting potency, a very low cytotoxicity (and thus high original SI values), and a chemical scaffold that is free of immediate structural liabilities and amenable to synthetic modification, as demonstrated by the nascent structure-activity relationship developed in this study. It is unlikely that the group specificity of this compound or any other of the entry inhibitor classes can be overcome synthetically, but combination with an equally potent entry inhibitor with a different group specificity may outline a path forward toward clinical use for a promising but currently underexplored type of influenza therapeutics.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to D. A. Steinhauer and R. Fouchier for providing the IAV-WSN and IAV-NL rescue systems, respectively, to J. Rose and M. Schnell for the VSV rescue system, to J. J. Yoon for assistance with PBMC preparation, to R. T. Jacob for IT support, and to A. L. Hammond for a critical reading of the manuscript. The MScreen software package was kindly provided by the Center for Chemical Genomics of the University of Michigan under a license agreement by the University of Michigan Office of Technology Transfer, JChem was used for structure database management, search, and prediction (JChem 6.2; ChemAxon), and Marvin was employed for drawing, displaying, and characterizing chemical structures, substructures, and reactions (Marvin 14.9.22.0; ChemAxon).
This work was supported in part by Public Health Service grants AI119196, AI011002, and HD079327 from the NIH/NIAID and NIH/NICHD (to R.K.P.).
R.K.P. is an inventor on a disclosure filing describing the use of small-molecule inhibitors for antimyxovirus therapy and the generation of recombinant reporter viruses. This study could affect his personal financial status.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.00898-16.
REFERENCES
- 1.Fischer WA II, Gong M, Bhagwanjee S, Sevransky J. 2014. Global burden of influenza as a cause of cardiopulmonary morbidity and mortality. Global Heart 9:325–336. doi: 10.1016/j.gheart.2014.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Johnson NP, Mueller J. 2002. Updating the accounts: global mortality of the 1918-1920 “Spanish” influenza pandemic. Bull Hist Med 76:105–115. doi: 10.1353/bhm.2002.0022. [DOI] [PubMed] [Google Scholar]
- 3.Salomon R, Webster RG. 2009. The influenza virus enigma. Cell 136:402–410. doi: 10.1016/j.cell.2009.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Neumann G, Kawaoka Y. 2011. The first influenza pandemic of the new millennium. Influenza Other Respir Viruses 5:157–166. doi: 10.1111/j.1750-2659.2011.00231.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reid AH, Taubenberger JK. 1999. The 1918 flu and other influenza pandemics: “over there” and back again. Lab Invest 79:95–101. [PubMed] [Google Scholar]
- 6.Thompson WW, Moore MR, Weintraub E, Cheng PY, Jin X, Bridges CB, Bresee JS, Shay DK. 2009. Estimating influenza-associated deaths in the United States. Am J Public Health 99(Suppl 2):S225–S230. doi: 10.2105/AJPH.2008.151944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Osterholm MT, Kelley NS, Sommer A, Belongia EA. 2012. Efficacy and effectiveness of influenza vaccines: a systematic review and meta-analysis. Lancet Infect Dis 12:36–44. doi: 10.1016/S1473-3099(11)70295-X. [DOI] [PubMed] [Google Scholar]
- 8.Ebell MH, Call M, Shinholser J. 2013. Effectiveness of oseltamivir in adults: a meta-analysis of published and unpublished clinical trials. Fam Pract 30:125–133. doi: 10.1093/fampra/cms059. [DOI] [PubMed] [Google Scholar]
- 9.Le QM, Wertheim HF, Tran ND, van Doorn HR, Nguyen TH, Horby P, Vietnam H1N1 Investigation Team . 2010. A community cluster of oseltamivir-resistant cases of 2009 H1N1 influenza. N Engl J Med 362:86–87. doi: 10.1056/NEJMc0910732. [DOI] [PubMed] [Google Scholar]
- 10.Hatakeyama S, Sugaya N, Ito M, Yamazaki M, Ichikawa M, Kimura K, Kiso M, Shimizu H, Kawakami C, Koike K, Mitamura K, Kawaoka Y. 2007. Emergence of influenza B viruses with reduced sensitivity to neuraminidase inhibitors. JAMA 297:1435–1442. [DOI] [PubMed] [Google Scholar]
- 11.van der Vries E, Stelma FF, Boucher CA. 2010. Emergence of a multidrug-resistant pandemic influenza A (H1N1) virus. N Engl J Med 363:1381–1382. doi: 10.1056/NEJMc1003749. [DOI] [PubMed] [Google Scholar]
- 12.Belongia EA, Kieke BA, Donahue JG, Greenlee RT, Balish A, Foust A, Lindstrom S, Shay DK, Marshfield Influenza Study Group . 2009. Effectiveness of inactivated influenza vaccines varied substantially with antigenic match from the 2004-2005 season to the 2006-2007 season. J Infect Dis 199:159–167. doi: 10.1086/595861. [DOI] [PubMed] [Google Scholar]
- 13.Chamberlain AT, Wells K, Seib K, Kudis A, Hannan C, Orenstein WA, Whitney EA, Hinman AR, Buehler JW, Omer SB, Berkelman RL. 2012. Lessons learned from the 2007 to 2009 Haemophilus influenzae type B vaccine shortage: implications for future vaccine shortages and public health preparedness. J Public Health Manag Pract 18:E9–E16. doi: 10.1097/PHH.0b013e31821dce27. [DOI] [PubMed] [Google Scholar]
- 14.Furuta Y, Gowen BB, Takahashi K, Shiraki K, Smee DF, Barnard DL. 2013. Favipiravir (T-705), a novel viral RNA polymerase inhibitor. Antiviral Res 100:446–454. doi: 10.1016/j.antiviral.2013.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.von Itzstein M, Wu WY, Kok GB, Pegg MS, Dyason JC, Jin B, Van Phan T, Smythe ML, White HF, Oliver SW, Colman PM, Varghese JN, Ryan DM, Woods JM, Bethell RC, Hotham VJ, Cameron JM, Pennet CR. 1993. Rational design of potent sialidase-based inhibitors of influenza virus replication. Nature 363:418–423. doi: 10.1038/363418a0. [DOI] [PubMed] [Google Scholar]
- 16.Kim CU, Lew W, Williams MA, Liu H, Zhang L, Swaminathan S, Bischofberger N, Chen MS, Mendel DB, Tai CY, Laver WG, Stevens RC. 1997. Influenza neuraminidase inhibitors possessing a novel hydrophobic interaction in the enzyme active site: design, synthesis, and structural analysis of carbocyclic sialic acid analogues with potent anti-influenza activity. J Am Chem Soc 119:681–690. doi: 10.1021/ja963036t. [DOI] [PubMed] [Google Scholar]
- 17.Hay AJ, Wolstenholme AJ, Skehel JJ, Smith MH. 1985. The molecular basis of the specific anti-influenza action of amantadine. EMBO J 4:3021–3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hayden FG, Hay AJ. 1992. Emergence and transmission of influenza A viruses resistant to amantadine and rimantadine. Curr Top Microbiol Immunol 176:119–130. [DOI] [PubMed] [Google Scholar]
- 19.Wang J, Wu Y, Ma C, Fiorin G, Wang J, Pinto LH, Lamb RA, Klein ML, Degrado WF. 2013. Structure and inhibition of the drug-resistant S31N mutant of the M2 ion channel of influenza A virus. Proc Natl Acad Sci U S A 110:1315–1320. doi: 10.1073/pnas.1216526110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Carr J, Ives J, Kelly L, Lambkin R, Oxford J, Mendel D, Tai L, Roberts N. 2002. Influenza virus carrying neuraminidase with reduced sensitivity to oseltamivir carboxylate has altered properties in vitro and is compromised for infectivity and replicative ability in vivo. Antiviral Res 54:79–88. doi: 10.1016/S0166-3542(01)00215-7. [DOI] [PubMed] [Google Scholar]
- 21.Herlocher ML, Carr J, Ives J, Elias S, Truscon R, Roberts N, Monto AS. 2002. Influenza virus carrying an R292K mutation in the neuraminidase gene is not transmitted in ferrets. Antiviral Res 54:99–111. doi: 10.1016/S0166-3542(01)00214-5. [DOI] [PubMed] [Google Scholar]
- 22.Le QM, Kiso M, Someya K, Sakai YT, Nguyen TH, Nguyen KH, Pham ND, Ngyen HH, Yamada S, Muramoto Y, Horimoto T, Takada A, Goto H, Suzuki T, Suzuki Y, Kawaoka Y. 2005. Avian flu: isolation of drug-resistant H5N1 virus. Nature 437:1108. doi: 10.1038/4371108a. [DOI] [PubMed] [Google Scholar]
- 23.Kiso M, Mitamura K, Sakai-Tagawa Y, Shiraishi K, Kawakami C, Kimura K, Hayden FG, Sugaya N, Kawaoka Y. 2004. Resistant influenza A viruses in children treated with oseltamivir: descriptive study. Lancet 364:759–765. doi: 10.1016/S0140-6736(04)16934-1. [DOI] [PubMed] [Google Scholar]
- 24.Lowen AC, Palese P. 2007. Influenza virus transmission: basic science and implications for the use of antiviral drugs during a pandemic. Infect Disord Drug Targets 7:318–328. doi: 10.2174/187152607783018736. [DOI] [PubMed] [Google Scholar]
- 25.Bright RA, Shay DK, Shu B, Cox NJ, Klimov AI. 2006. Adamantane resistance among influenza A viruses isolated early during the 2005-2006 influenza season in the United States. JAMA 295:891–894. doi: 10.1001/jama.295.8.joc60020. [DOI] [PubMed] [Google Scholar]
- 26.Swinney DC. 2013. The contribution of mechanistic understanding to phenotypic screening for first-in-class medicines. J Biomol Screen 18:1186–1192. doi: 10.1177/1087057113501199. [DOI] [PubMed] [Google Scholar]
- 27.Swinney DC. 2013. Phenotypic vs. target-based drug discovery for first-in-class medicines. Clin Pharmacol Ther 93:299–301. doi: 10.1038/clpt.2012.236. [DOI] [PubMed] [Google Scholar]
- 28.Karlas A, Machuy N, Shin Y, Pleissner KP, Artarini A, Heuer D, Becker D, Khalil H, Ogilvie LA, Hess S, Maurer AP, Muller E, Wolff T, Rudel T, Meyer TF. 2010. Genome-wide RNAi screen identifies human host factors crucial for influenza virus replication. Nature 463:818–822. doi: 10.1038/nature08760. [DOI] [PubMed] [Google Scholar]
- 29.Beyleveld G, White KM, Ayllon J, Shaw ML. 2013. New-generation screening assays for the detection of anti-influenza compounds targeting viral and host functions. Antiviral Res 100:120–132. doi: 10.1016/j.antiviral.2013.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lutz A, Dyall J, Olivo PD, Pekosz A. 2005. Virus-inducible reporter genes as a tool for detecting and quantifying influenza A virus replication. J Virol Methods 126:13–20. doi: 10.1016/j.jviromet.2005.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yan D, Krumm SA, Sun A, Steinhauer DA, Luo M, Moore ML, Plemper RK. 2013. Dual myxovirus screen identifies a small-molecule agonist of the host antiviral response. J Virol 87:11076–11087. doi: 10.1128/JVI.01425-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Konig R, Stertz S, Zhou Y, Inoue A, Hoffmann HH, Bhattacharyya S, Alamares JG, Tscherne DM, Ortigoza MB, Liang Y, Gao Q, Andrews SE, Bandyopadhyay S, De Jesus P, Tu BP, Pache L, Shih C, Orth A, Bonamy G, Miraglia L, Ideker T, Garcia-Sastre A, Young JA, Palese P, Shaw ML, Chanda SK. 2010. Human host factors required for influenza virus replication. Nature 463:813–817. doi: 10.1038/nature08699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Heaton NS, Leyva-Grado VH, Tan GS, Eggink D, Hai R, Palese P. 2013. In vivo bioluminescent imaging of influenza A virus infection and characterization of novel cross-protective monoclonal antibodies. J Virol 87:8272–8281. doi: 10.1128/JVI.00969-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Manicassamy B, Manicassamy S, Belicha-Villanueva A, Pisanelli G, Pulendran B, Garcia-Sastre A. 2010. Analysis of in vivo dynamics of influenza virus infection in mice using a GFP reporter virus. Proc Natl Acad Sci U S A 107:11531–11536. doi: 10.1073/pnas.0914994107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yan D, Weisshaar M, Lamb K, Chung HK, Lin MZ, Plemper RK. 2015. Replication-competent influenza virus and respiratory syncytial virus luciferase reporter strains engineered for co-infections identify antiviral compounds in combination screens. Biochemistry 54:5589–5604. doi: 10.1021/acs.biochem.5b00623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hoffmann E, Neumann G, Kawaoka Y, Hobom G, Webster RG. 2000. A DNA transfection system for generation of influenza A virus from eight plasmids. Proc Natl Acad Sci U S A 97:6108–6113. doi: 10.1073/pnas.100133697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jacob RT, Larsen MJ, Larsen SD, Kirchhoff PD, Sherman DH, Neubig RR. 2012. MScreen: an integrated compound management and high-throughput screening data storage and analysis system. J Biomol Screen 17:1080–1087. doi: 10.1177/1087057112450186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hotard AL, Shaikh FY, Lee S, Yan D, Teng MN, Plemper RK, Crowe JE Jr, Moore ML. 2012. A stabilized respiratory syncytial virus reverse genetics system amenable to recombination-mediated mutagenesis. Virology 434:129–136. doi: 10.1016/j.virol.2012.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dahlin JL, Baell J, Walters MA. 18 September 2015. Assay interference by chemical reactivity. In Sittampalam GS, Coussens NP, Nelson H, Arkin M, Auld D, Austin C, Bejcek B, Glicksman M, Inglese J, Iversen PW, Li Z, McGee J, McManus O, Minor L, Napper A, Peltier JM, Riss T, Trask OJ Jr, Weidner J (ed), Assay guidance manual. Eli Lilly & Company and the National Center for Advancing Translational Sciences, Bethesda, MD: http://www.ncbi.nlm.nih.gov/books/NBK326709/. [PubMed] [Google Scholar]
- 40.Brideau C, Gunter B, Pikounis B, Liaw A. 2003. Improved statistical methods for hit selection in high-throughput screening. J Biomol Screen 8:634–647. doi: 10.1177/1087057103258285. [DOI] [PubMed] [Google Scholar]
- 41.Gamblin SJ, Skehel JJ. 2010. Influenza hemagglutinin and neuraminidase membrane glycoproteins. J Biol Chem 285:28403–28409. doi: 10.1074/jbc.R110.129809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sun A, Ndungu JM, Krumm SA, Yoon JJ, Thepchatri P, Natchus M, Plemper RK, Snyder JP. 2011. Host-directed inhibitors of myxoviruses: synthesis and in vitro biochemical evaluation. ACS Med Chem Lett 2:798–803. doi: 10.1021/ml200125r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Krumm SA, Ndungu JM, Yoon JJ, Dochow M, Sun A, Natchus M, Snyder JP, Plemper RK. 2011. Potent host-directed small-molecule inhibitors of myxovirus RNA-dependent RNA-polymerases. PLoS One 6:e20069. doi: 10.1371/journal.pone.0020069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Beckett D, Kovaleva E, Schatz PJ. 1999. A minimal peptide substrate in biotin holoenzyme synthetase-catalyzed biotinylation. Protein Sci 8:921–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.White KM, De Jesus P, Chen Z, Abreu P Jr, Barile E, Mak PA, Anderson P, Nguyen QT, Inoue A, Stertz S, Koenig R, Pellecchia M, Palese P, Kuhen K, Garcia-Sastre A, Chanda SK, Shaw ML. 2015. A potent anti-influenza compound blocks fusion through stabilization of the prefusion conformation of the hemagglutinin protein. ACS Infect Dis 1:98–109. doi: 10.1021/id500022h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lai KK, Cheung NN, Yang F, Dai J, Liu L, Chen Z, Sze KH, Chen H, Yuen KY, Kao RY. 2015. Identification of novel fusion inhibitors of influenza A virus by chemical genetics. J Virol 90:2690–2701. doi: 10.1128/JVI.02326-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Basu A, Antanasijevic A, Wang M, Li B, Mills DM, Ames JA, Nash PJ, Williams JD, Peet NP, Moir DT, Prichard MN, Keith KA, Barnard DL, Caffrey M, Rong L, Bowlin TL. 2014. New small molecule entry inhibitors targeting hemagglutinin-mediated influenza A virus fusion. J Virol 88:1447–1460. doi: 10.1128/JVI.01225-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Foulkes-Murzycki JE, Rosi C, Kurt Yilmaz N, Shafer RW, Schiffer CA. 2013. Cooperative effects of drug-resistance mutations in the flap region of HIV-1 protease. ACS Chem Biol 8:513–518. doi: 10.1021/cb3006193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wagner R, Heuer D, Wolff T, Herwig A, Klenk HD. 2002. N-glycans attached to the stem domain of haemagglutinin efficiently regulate influenza A virus replication. J Gen Virol 83:601–609. doi: 10.1099/0022-1317-83-3-601. [DOI] [PubMed] [Google Scholar]
- 50.Wang G, Deval J, Hong J, Dyatkina N, Prhavc M, Taylor J, Fung A, Jin Z, Stevens SK, Serebryany V, Liu J, Zhang Q, Tam Y, Chanda SM, Smith DB, Symons JA, Blatt LM, Beigelman L. 2015. Discovery of 4′-chloromethyl-2′-deoxy-3′,5′-di-O-isobutyryl-2′-fluorocytidine (ALS-8176), a first-in-class RSV polymerase inhibitor for treatment of human respiratory syncytial virus infection. J Med Chem 58:1862–1878. doi: 10.1021/jm5017279. [DOI] [PubMed] [Google Scholar]
- 51.Jordan R, Shao M, Mackman RL, Perron M, Cihlar T, Lewis SA, Eisenberg EJ, Carey A, Strickley RG, Chien JW, Anderson ML, McEligot HA, Behrens NE, Gershwin LJ. 2015. Antiviral efficacy of a respiratory syncytial virus (RSV) fusion inhibitor in a bovine model of RSV infection. Antimicrob Agents Chemother 59:4889–4900. doi: 10.1128/AAC.00487-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Henrich TJ, Kuritzkes DR. 2013. HIV-1 entry inhibitors: recent development and clinical use. Curr Opin Virol 3:51–57. doi: 10.1016/j.coviro.2012.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hoffman LR, Kuntz ID, White JM. 1997. Structure-based identification of an inducer of the low-pH conformational change in the influenza virus hemagglutinin: irreversible inhibition of infectivity. J Virol 71:8808–8820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bodian DL, Yamasaki RB, Buswell RL, Stearns JF, White JM, Kuntz ID. 1993. Inhibition of the fusion-inducing conformational change of influenza hemagglutinin by benzoquinones and hydroquinones. Biochemistry 32:2967–2978. doi: 10.1021/bi00063a007. [DOI] [PubMed] [Google Scholar]
- 55.Plotch SJ, O'Hara B, Morin J, Palant O, LaRocque J, Bloom JD, Lang SA Jr, DiGrandi MJ, Bradley M, Nilakantan R, Gluzman Y. 1999. Inhibition of influenza A virus replication by compounds interfering with the fusogenic function of the viral hemagglutinin. J Virol 73:140–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Minagawa K, Kouzuki S, Kamigauchi T. 2002. Stachyflin and acetylstachyflin, novel anti-influenza A virus substances, produced by Stachybotrys sp. RF-7260. II. Synthesis and preliminary structure-activity relationships of stachyflin derivatives. J Antibiot 55:165–171. [DOI] [PubMed] [Google Scholar]
- 57.Luo G, Colonno R, Krystal M. 1996. Characterization of a hemagglutinin-specific inhibitor of influenza A virus. Virology 226:66–76. doi: 10.1006/viro.1996.0628. [DOI] [PubMed] [Google Scholar]
- 58.Staschke KA, Hatch SD, Tang JC, Hornback WJ, Munroe JE, Colacino JM, Muesing MA. 1998. Inhibition of influenza virus hemagglutinin-mediated membrane fusion by a compound related to podocarpic acid. Virology 248:264–274. doi: 10.1006/viro.1998.9273. [DOI] [PubMed] [Google Scholar]
- 59.Russell RJ, Kerry PS, Stevens DJ, Steinhauer DA, Martin SR, Gamblin SJ, Skehel JJ. 2008. Structure of influenza hemagglutinin in complex with an inhibitor of membrane fusion. Proc Natl Acad Sci U S A 105:17736–17741. doi: 10.1073/pnas.0807142105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Weisshaar M, Cox R, Plemper RK. 2015. Blocking respiratory syncytial virus entry: a story with twists. DNA Cell Biol 34:505–510. doi: 10.1089/dna.2015.2896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yan D, Lee S, Thakkar VD, Luo M, Moore ML, Plemper RK. 2014. Cross-resistance mechanism of respiratory syncytial virus against structurally diverse entry inhibitors. Proc Natl Acad Sci U S A 111:E3441–E3449. doi: 10.1073/pnas.1405198111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Battles MB, Langedijk JP, Furmanova-Hollenstein P, Chaiwatpongsakorn S, Costello HM, Kwanten L, Vranckx L, Vink P, Jaensch S, Jonckers TH, Koul A, Arnoult E, Peeples ME, Roymans D, McLellan JS. 2016. Molecular mechanism of respiratory syncytial virus fusion inhibitors. Nat Chem Biol 12:87–93. doi: 10.1038/nchembio.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cox R, Plemper RK. 2016. Structure-guided design of small-molecule therapeutics against RSV disease. Expert Opin Drug Discov 2016:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Xu R, Wilson IA. 2011. Structural characterization of an early fusion intermediate of influenza virus hemagglutinin. J Virol 85:5172–5182. doi: 10.1128/JVI.02430-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bullough PA, Hughson FM, Skehel JJ, Wiley DC. 1994. Structure of influenza haemagglutinin at the pH of membrane fusion. Nature 371:37–43. doi: 10.1038/371037a0. [DOI] [PubMed] [Google Scholar]
- 66.Gruenke JA, Armstrong RT, Newcomb WW, Brown JC, White JM. 2002. New insights into the spring-loaded conformational change of influenza virus hemagglutinin. J Virol 76:4456–4466. doi: 10.1128/JVI.76.9.4456-4466.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Steinhauer DA, Sauter NK, Skehel JJ, Wiley DC. 1992. Receptor binding and cell entry by influenza viruses. Semin Virol 3:91–100. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.