

ABSTRACT
The hepatitis C virus NS5A protein is tethered to cellular membranes via an amphipathic amino-terminal helix that is inserted in-plane into the outer endoplasmic reticulum (ER)-derived membrane leaflet. The charged face of the helix faces the cytoplasm and may contribute to interactions involved in replicase assembly and function. Using an aggressive charge flip mutagenesis strategy, we identified a number of essential residues for replication on the charged face of the NS5A anchor and identified a double charge face mutant that is lethal for RNA replication but generates suppressor mutations in the carboxy-terminal helix of the NS4B protein. This suppressor restores RNA replication of the NS5A helix double flip mutant (D1979K/D1982K) and, interestingly, seems to function by restoring the proper localization of NS5A to the viral replicase. These data add to our understanding of the complex organization and assembly of the viral replicase via NS4B-NS5A interactions.
IMPORTANCE Information about the functional role of the cytosolic face of the NS5A anchoring helix remains obscure. In this study, we show that while the hydrophobic face of the NS5A anchor helix mediates membrane association, the polar cytosolic face of the helix plays a key role during hepatitis C virus (HCV) replication by mediating the interaction of NS5A with other HCV nonstructural proteins via NS4B. Such an interaction determines the subcellular localization of NS5A by engaging NS5A in the HCV replication process during the formation of a functional HCV replication complex. Thus, collectively, it can be stated that the findings in the present study provide further information about the interactions between the HCV nonstructural proteins during HCV RNA replication and provide a platform to gain more insights about the molecular architecture of HCV replication complexes.
INTRODUCTION
Hepatitis C virus (HCV) is an enveloped, positive-sense, single-stranded RNA virus belonging to Hepacivirus genus within the Flaviviridae family. The HCV open reading frame (ORF) encodes a polyprotein of ∼3,011 amino acids (aa), which is cleaved co- and posttranslationally by viral and host proteases into 10 viral proteins: the core; the envelope glycoproteins E1 and E2; the viroporin p7; and the nonstructural (NS) proteins NS2, NS3, NS4A, NS4B, NS5A, and NS5B. Among the NS proteins, NS2 with the protease activity is required for virion assembly; NS3 is a multifunctional protein with serine protease activity at the N-terminal region, while the C-terminal portion of the protein holds helicase/nucleotide triphosphatase (NTPase) activity; NS4A acts as a cofactor for the NS3 serine protease; NS4B serves the scaffold for the HCV replication complex (RC) by inducing the formation of the membranous web (MW); NS5A is a phosphoprotein that plays a key role in HCV RNA replication and viral assembly processes; and NS5B is the RNA-dependent RNA polymerase (1–5).
Like all other positive-strand RNA viruses, HCV RNA replicates in close association with cellular membranes. HCV alters the endoplasmic reticulum (ER) to create a large number of predominantly double-membrane vesicles for replication (6–8). This HCV-specific membrane alteration is referred to as the MW (5). Several previous studies demonstrated that the rearrangement of the ER into the MW is induced by the viral NS4B and NS5A proteins (6–9).
Among the HCV nonstructural proteins, NS5A plays a central role in RNA replication and viral assembly (10–15). NS5A is a homodimeric multifunctional phosphoprotein composed of three domains separated by two low-complexity sequences (LCSs) (16, 17). During RNA replication, NS5A domains I and II play an essential role, while domain III is important for the viral assembly process (10–13). The LCS-I region between NS5A domain I and domain II holds phosphorylation sites (18, 19) giving rise to NS5A in two phosphorylation states, namely, the basal (p56) and hyperphosphorylated (p58) NS5A forms. The basal phosphorylated form of NS5A favors the replication of HCV, while the hyperphosphorylated form supports virus production (13, 20–22). NS5A binds viral RNA, is an integral part of the HCV RC, and also likely connects the RC with the site of virion assembly (15, 23). Thus, collectively, these observations indicate that NS5A is a multifunctional protein that has distinct roles in both HCV replication and virus production.
NS5A is associated posttranslationally with the cytoplasmic face of the ER, and most likely the MW, via an amphipathic amino-terminal α-helix, and this association is critical for the proper formation of a functional HCV RC (24–28). Disruption of the NS5A anchoring helix results in the complete loss of HCV replication, indicating the importance of NS5A membrane insertion for HCV RNA replication (29, 30). During the membrane association of the NS5A anchor helix, the tryptophan-rich hydrophobic face of the helix is inserted into one leaflet of the ER membrane, while the charged, polar face of the helix is oriented toward the cytosol, resulting in a topological conformation parallel to the lipid bilayer in the cytoplasmic leaflet of the ER membrane (25, 26). Driven by structural and functional analyses and the conserved nature of the polar residues of the NS5A anchor helix, it is hypothesized that while the hydrophobic side of the anchor helix mediates membrane associations, the polar residues exposed to the cytosol play an important role in the formation of a functional RC (5, 25, 26). Among the multiple polar residues of the NS5A anchor helix, Penin et al. (25) studied the effect of three residues on HCV replication by an alanine substitution approach. Despite these efforts, there is still a dearth of information on the role of these charged, polar residues of the NS5A anchor helix in the establishment of a functional HCV RC.
In this study, we used an HCV subgenomic replicon containing the minimum portion of the HCV nonstructural protein-coding region (NS3 to NS5B) sufficient to support HCV RNA replication in vitro and a selectable marker gene (20, 31). A panel of 12 mutants was generated by introducing individual changes to the 12 polar residues comprising the polar face of the helix, and the mutants were analyzed for their impact on HCV RNA replication. The mutations introduced were designed to be dramatic, with charge residues being changed to amino acids of the opposite charge, polar residues being altered to hydrophobic residues, and other similar alterations of amino acid character, with the goal of forcing potential second-site reversions in other HCV nonstructural proteins. This effort identified a patch of residues in the center portion of the NS5A anchor that were intolerant of the introduced mutations but failed to generate significant second-site reversions. We therefore generated a more dramatic double mutant in which two adjacent aspartic acid residues on the face of the helix were altered to lysine residues. This mutation produced a near-lethal phenotype in replication and generated a revertant in the HCV NS4B protein that was able to rescue replication in the context of the double aspartic-acid-to-lysine mutant. Surprisingly, further characterization of the effects of the double mutant suggested that the localization of NS5A to the MW was disrupted, despite the mutant having normal membrane insertion. Proper localization of NS5A and functional RNA replication were restored with the introduction of the second-site reversion in NS4B. These data provide a glimpse into how NS5A might be recruited to the RC and provide additional evidence that NS4B serves as a scaffold on which the RC assembles, both of which provide new insights into HCV RNA replication and the enigmatic organization of the viral RC.
MATERIALS AND METHODS
Cell culture.
Huh-7.5 and Lunet-T7 cells were used for this study. Briefly, cells were maintained in Dulbecco's modified Eagle's medium (DMEM; Gibco, Life Technologies) supplemented with 10% fetal bovine serum (FBS), nonessential amino acids (1× final concentration), 100 U/ml penicillin, and 100 μg/ml streptomycin (complete DMEM) at 37°C with 5% CO2.
In vitro mutagenesis and plasmid constructs.
All mutagenesis was performed by using the QuikChange II in vitro mutagenesis kit (Stratagene) according to the manufacturer's instructions. For the generation of mutations, shuttle vector pSL1180GIT containing the BsrG1-MfeI fragment of the Con1/SG-neo GIT replicon encompassing the NS5A coding sequence and surrounding regions was used as described previously (11, 12). The Con1/SG-neo GIT replicon is identical to a subgenomic replicon reported previously (20), with the exception of two adaptive mutations in NS3 (E1202G and T1280I) and one in NS4B (K1846T) identified previously by Lohmann et al. (32). All the generated mutations were confirmed by DNA sequencing with the pSL1180GIT construct and were subcloned back into the parental Con1/SG-neo GIT replicon plasmid. As a negative control, the parental Con1/SG-neo GIT construct bearing substitutions that inactivate the NS5B RNA-dependent RNA polymerase, generating Con1/SG Neo GIT (pol−), was used.
Fused pmVenus(L68V)-mTurquoise2 (33), a gift from Dorus Gadella (vector backbone of enhanced green fluorescent protein [EGFP]-C1) (Addgene plasmid 60493) (with pmVenus as yellow fluorescent protein [YFP] and mTurquoise2 as cyan fluorescent protein [CFP]), was used as a positive control as well as to generate all other constructs for fluorescence resonance energy transfer (FRET) analyses. Briefly, by using a PCR-based restriction-free cloning approach (34) using the FRET positive-control plasmid, various experimental constructs were generated. By using PCR-based deletion approaches, YFP-only and CFP-only constructs were generated to serve as negative controls (34). In the basic positive-control YFP-CFP plasmid, there is a 12-amino-acid linker between CFP and YFP. Full-length NS4B and NS5A sequences (from Con1/SG-neo GIT) were inserted in place of YFP and CFP, respectively, to obtain the YFP-NS5A and NS4B-CFP fusion proteins, keeping the native linker intact.
In vitro RNA transcription, electroporation of viral RNAs, and G418 selection.
The preparation of in vitro transcripts, their electroporation into Huh-7.5 cells, and the G418 transduction assay were performed as described previously (12). G418-resistant colonies were subsequently counted and used to calculate the transduction efficiency as CFU per microgram of input RNA. All experimental results represented are the means of data from three independent experiments.
Selection of revertants, amplification of HCV RNA by RT-PCR, and sequencing.
After transient transfection of Huh-7.5 cells with in vitro RNA transcripts and G418 selection for 2 weeks (as discussed above), the mutant replicons that produced lower number of colonies were subjected to revertant analysis. Briefly, the few colonies that survived and grew under G418 selection were isolated with cloning cylinders and expanded further as single clones. Total RNA from these clones was extracted with a Qiagen RNeasy kit according to the manufacturer's instructions. The extracted RNA was used for reverse transcription-PCR (RT-PCR), and subsequent direct sequencing was done as described previously (35). Initial sequence analysis was performed for the region of the viral RNA encoding the NS5A protein, and clones that featured same-site reversions or same-site pseudoreversions were not analyzed further. Clones that retained the input mutations in the NS5A anchor helix were subjected to sequence analysis of the entire nonstructural protein-coding regions.
Transient-replication assay by real-time reverse transcriptase PCR.
For transient-replication assays, cells were electroporated with 1 μg of HCV RNA as described above, and the cells were plated at a density of 5 × 105 cells per well on a six-well dish. At various time points postelectroporation, cells were washed twice in phosphate-buffered saline (PBS), and RNAs were then extracted and purified by using the Qiagen RNeasy kit according to the manufacturer's instructions. After extraction, for each reaction mixture, 100 ng of total RNA was used for real-time reverse transcriptase PCR. All real-time PCR experiments were performed by using a Roche LightCycler 480 instrument, using HCV-specific primers and probe described previously (4). Known in vitro-transcribed HCV RNA standards with 1-log dilution series starting from 107 copies per reaction to 101 copies per reaction were used as standards. During quantification, the copy number for each sample was determined in triplicate, and the average was considered to be the mean viral RNA concentration.
Transient expression of HCV proteins.
Transient expression of the HCV polyprotein was accomplished by using two different approaches. In the first approach, following infection with a recombinant vaccinia virus expressing T7 RNA polymerase (vTF7-3 expression system) (36), Huh-7.5 cells were transiently transfected by using replicon plasmid DNA. Briefly, 5 × 105 Huh-7.5 cells were infected with vTF7-3 at a multiplicity of infection of 10 for 30 min at room temperature and then transfected with 1 μg of replicon plasmid DNA by using Fugene 6 transfection reagent. At 24 h postinfection, cells were lysed, and lysates were used for Western blot analysis of NS5A. In the second approach, 5 × 105 Lunet-T7 cells were cotransfected with 750 ng of replicon plasmid DNA and 750 ng of plasmid pT7-T7 as described previously (37). At 24 h posttransfection, transfected cells were enriched by G418 selection (500 μg/ml) for two cell passages over a course of 6 days. After selection, transfected cells were used for immunofluorescence assays and membrane flotation assays. All experiments were done within two to three passages following selection to minimize the chances of potential artifacts.
Immunofluorescence assay for NS5A.
Transiently transfected Lunet-T7 cells after selection (as described above) were plated at a density of 5 × 104 cells per well in a four-well chamber slide. After 48 h, cells were washed with PBS twice and fixed with 4% paraformaldehyde at room temperature for 20 min. Cells were washed twice with PBS, and blocking was performed at room temperature for 20 min with blocking buffer (PBS containing 3% [vol/vol] FBS and 0.1% [wt/vol] saponin). Cells were then incubated with monoclonal anti-NS5A antibody 9E10 (13) at 1:500 dilutions in blocking buffer for 1 h at room temperature. After incubation with the primary antibody, the cells were washed three times with wash buffer (PBS containing 0.1% [wt/vol] saponin) and subsequently incubated with an Alexa 488 (Molecular Probes)-conjugated secondary antibody at 1:500 dilutions in PBS containing 3% FBS and 0.1% saponin for 1 h at room temperature. Finally, cells were washed three times with PBS containing 0.1% saponin and mounted with Prolong Diamond Antifade Mountant with 4′,6-diamidino-2-phenylindole (DAPI) (Life Technologies, Thermo Fisher Scientific). After 24 h of curing, cells were observed, and images were captured under a fluorescence microscope.
Additionally, to confirm the findings of the immunofluorescence assay, substitutions that inactivate the NS5B RNA-dependent RNA polymerase, from the Con1/SG Neo GIT (pol−) replicon negative control, were introduced into all the experimental constructs, and following transfection of Huh-7.5 cells with these pol(−) negative constructs, the vTF7-3 expression system was used to transiently express the viral proteins as described above for Western blotting for NS5A. At 6 h postinfection, cells were fixed, and immunofluorescence assays were performed as described above.
Membrane flotation assay.
Transiently transfected Lunet-T7 cells after selection (as described above) were lysed with a Dounce homogenizer in 1.2 ml of low-salt TNE buffer (25 mM Tris, 25 mM NaCl, and 5 mM EDTA [pH 7.4]) containing a complete protease inhibitor cocktail (Roche). The cell lysate was spun at 5,000 × g for 10 min at 4°C, and the supernatant was collected. Five hundred microliters of the supernatant was mixed with 3.5 ml of a 72% sucrose solution to obtain a final sucrose concentration of 63%, placed at the bottom of an 11-ml ultracentrifugation tube (Beckman Coulter), and overlaid with 6 ml of a 55% sucrose solution and 1 ml of a 10% sucrose solution in PBS. Ultracentrifugation was carried out at 38,000 × g overnight at 4°C in a Beckman Coulter Optima L-100 XP ultracentrifuge. After centrifugation, 11 total fractions were collected (1 ml each) from top to bottom and subsequently analyzed by Western blotting for the NS5A protein. Note that for every sample, the same amount of the supernatant was mixed with 1% NP-40 (Sigma), and a similar flotation assay was performed as a control for non-membrane-associated protein.
Fluorescence resonance energy transfer.
NS4B and NS5A interactions were examined by an in-cell FRET assay. Huh-7.5 cells were transfected with the CFP- and YFP-tagged constructs, and at 24 h posttransfection, cells were analyzed for FRET by using a fluorescence activated cell sorter (FACS)-based method (38). FACS-FRET measurements were accomplished by using a BD LSRII instrument (BD Bioscience) equipped with 405-nm, 488-nm, and 633-nm lasers. To measure CFP levels and FRET, cells were excited with the 405-nm laser, and fluorescence was collected with a standard 480/30-nm-band-pass filter, while the FRET signal was measured with a 530/30-nm-band-pass filter. To measure YFP levels, cells were excited with the 488-nm laser, while emission was also detected with a 530/30-nm-band-pass filter. For each sample, we evaluated a minimum of 2,500 CFP/YFP-positive cells that fell within the background-adjusted gate. By performing forward-scatter area by forward-scatter width, doublet exclusion was achieved, and dead cells were excluded by using SYTOX red dead-cell stain (Thermo Fisher Scientific).
The FRET experiment was performed without fixing of the cells. We used a gating strategy described previously (38). Briefly, we started with gating of live cells and further selected cells that were double positive for the CFP/CFP-fused protein and the YFP/YFP-fused protein by additional gating. Finally, we gated out the CFP- and YFP-cotransfected cells (FRET negative) to obtain FRET-positive cells due to the interaction of CFP and YFP, such as cells transfected with the CFP-YFP-fused construct (see Fig. 8B). In all experimental replicates, <2% of cells cotransfected with separate CFP and YFP constructs fell within the FRET-positive gate, while >95% of cells transfected with CFP-YFP-fused proteins were found to be FRET positive.
FIG 8.

FRET analysis of the interaction between NS4B and NS5A. (A) Subcellular localization of the CFP, YFP, CFP-YFP, NS4B-CFP, and YFP-NS5A proteins 24 h following transfection of Huh-7.5 cells. Images were captured at a ×40 magnification. Note that no differences are observed at the level of expression for NS4B-CFP and NS4B-CFP(K1958R) or for YFP-NS5A and NS5A(D1979K/D1982K) (data not shown). (B) Representative FACS plots showing the efficiency of the interaction between NS4B and NS5A in terms of percentages of FRET-positive cells. The experiment was repeated three times, and the average numbers of FRET-positive cells are shown in the triangular gates for controls as well as for NS4B-CFP, YFP-NS5A, and the respective mutant versions of the fused proteins. Due to space limitations, the NS5A D1979K/D1982K mutant is abbreviated NS5A(DD/KK). (C) For all three independent FRET experiments, after correcting the percentage of FRET-positive cells of the CFP- and YFP-dual-positive (i.e., cotransfected) cell population to zero, the percentages of FRET-positive cells for NS4B-CFP and YFP-NS5A were adjusted to 100%, and subsequently, the percentages of FRET-positive cells were normalized accordingly for experimental groups such as NS4B-CFP plus YFP-NS5A(D1979K/D1982K), NS4B-CFP(K1958R) plus YFP-NS5A(D1979K/D1982K), and NS4B-CFP(K1958R) plus YFP-NS5A.
Western blotting.
Cells were lysed with cold NETN buffer (50 mM Tris-HCl [pH 8.0], 120 mM NaCl, 1 mM EDTA, 0.5% NP-40) supplemented with a complete protease inhibitor cocktail (Roche). Soluble fractions of the lysates were isolated following centrifugation at 16,000 × g for 20 min at 4°C. Equal amounts of protein from each sample were mixed with 2× Laemmli sample buffer (Bio-Rad) containing 5% 2-mercaptoethanol (Sigma), boiled for 5 min, and separated on 10% SDS-PAGE gels prior to electroblotting onto nitrocellulose membranes. Following blocking, the membrane was probed with diluted primary anti-NS5A monoclonal antibody 9E10 in blocking buffer at 4°C overnight with gentle shaking. After incubation with horseradish peroxidase (HRP)-conjugated secondary antibody, ECL detection reagent (Thermo Scientific) was then added to the membrane. The membrane was then exposed to a chemiluminescence film (GE Healthcare Life Sciences, Piscataway, NJ) and developed.
Statistical analysis.
For all experiments, the numerical data (such as CFU or HCV titer) obtained were analyzed with GraphPad Prism software version 5.01 (GraphPad Software, La Jolla, CA). All the bar graphs and line diagrams were generated with the same software. All results were expressed as means ± SD (standard deviations). Comparison between two groups was performed with Student's t test. To compare means within multiple groups, one-factor analysis of variance (ANOVA) was performed. Data from FRET analysis was analyzed by using a single-tailed paired t test with a confidence interval of 90%. A P value of <0.05 was considered to be statistically significant.
RESULTS
Residues on the charged face of the NS5A anchor helix are critical for HCV RNA replication.
To elucidate the functional role of the cytoplasmically exposed polar residues of the NS5A anchor helix, a panel of site-directed mutants was generated (Fig. 1). Overall, we targeted 12 polar residues with individual amino acid mutations, and a double mutant with a substitution of two spatially adjacent amino acids was also generated. During the generation of the mutants, instead of an alanine-scanning-substitution approach (except for S1997A), we decided to introduce very aggressive amino acid changes to both test the plasticity of the various amino acids of the helix to dramatic substitutions and, hopefully, drive the formation of second-site suppression mutants in other HCV proteins, and perhaps other parts of NS5A, that might make contacts with the charged face of the helix. For this approach, basic residues were replaced with acidic residues (such as R1978E, K1992D, and K1998D) and vice versa (such as D1979K, D1982K, and D1990K). In the case of polar uncharged residues such as C, T, Q, and S, substitutions with nonpolar hydrophobic residues such as L and I (such as T1986L, T1989L, T1993L, and Q1996I) or with polar acidic residues such as E (C1985E) were performed.
FIG 1.

Schematic representation of the domain organization of NS5A indicating the locations of introduced mutations. The location of the membrane-anchoring helix is also shown by the dark gray box at the start of domain I (labeled helix). The three domains predicted for NS5A are shown separated by predicted low-complexity sequence regions (LCS-I and -II). Numbers indicate amino acid positions on the Con1 genotype 1b polyprotein sequence. Sequences of the N-terminal membrane anchor helix of NS5A, indicated by a dark gray box, are represented according to data for HCV 1b Con1 (GenBank accession number AJ238799). The polar amino acid residues of the NS5A anchor helix that are replaced are depicted in boldface type, and arrows indicate the corresponding amino acid residues introduced during the generation of the mutants. Single mutation indicates the panel of mutants generated with a single amino acid substitution (12 in total), and double mutation indicates the generation of a mutant bearing two amino acid substitutions.
The replication capacities of the mutant replicons were determined by transient transfections with in vitro transcripts followed by G418 selection, with parental GIT and GIT-pol(−) serving as positive and negative controls, respectively. The replication efficiencies of the single-polar-residue mutant replicons are depicted in Fig. 2A. Clearly, the parental GIT replicon (1.84 × 106 CFU/μg of RNA) replicated efficiently, and a complete lack of replication was observed for the GIT-pol(−) replicon (0 CFU/μg of RNA). Among the 12 single-polar-residue mutant replicons, 4 mutants were near lethal or severely impaired in terms of replication, as evident from the low or very low transduction efficiencies (0.5 × 101 CFU/μg of RNA for the C1985E mutant, 6.0 × 101 CFU/μg of RNA for T1989L, 2.05 × 102 CFU/μg of RNA for D1990K, and 3.0 × 102 CFU/μg of RNA for K1992D) (Fig. 2A). For the rest of the eight single-polar-residue mutant replicons, the efficiency of replication was comparable to that of the parental GIT replicon (1.13 × 106 CFU/μg of RNA for the R1978E mutant, 8.6 × 105 CFU/μg of RNA for D1979K, 2.01 × 105 CFU/μg of RNA for D1982K, 2.26 × 106 CFU/μg of RNA for T1986L, 1.69 × 106 CFU/μg of RNA for T1993L, 1.29 × 105 CFU/μg of RNA for Q1996I, 1.93 ×106 CFU/μg of RNA for S1997A, and 4.44 ×105 CFU/μg of RNA for K1998D). These data suggest that the central portion of the NS5A membrane anchor charged face is less plastic than the ends of the helix.
FIG 2.

Characterization of NS5A anchor helix single mutants. (A) RNA replication efficiencies of the replicons bearing mutations in the NS5A anchoring helix region. The bar diagram represents the transduction efficiencies of replicon RNAs in CFU per microgram of HCV RNA. pol(−) indicates a Con1/SG-neo replicon RNA bearing a nonfunctional RNA-dependent RNA polymerase sequence that served as a negative control, while the transduction efficiency of the parental Con1/SG-neo GIT replicon (labeled GIT) served as a positive control for replication experiments. The assay was performed in triplicates, and each bar from the graph represents the average number of colonies (with standard deviations) obtained from three experimental replicates for each experimental mutant and control replicons. (B) Assessment of transient RNA replication of selected mutants (mutants identified as lethal or near lethal by a replication assay [C1985E, T1989L, D1990K, and K1992D]) of HCV RNA by real-time RT-PCR. The numbers of HCV RNA copies present in the case of each experimental mutant per 100 ng of total RNA extracted at 4 h, 24 h, 48 h, and 72 h postelectroporation are plotted in the graph. Each sample was assayed by RT-PCR in triplicates. (C) Following transient expression, NS5A processing for the lethal or near-lethal mutants was analyzed by Western blotting with an anti-NS5A monoclonal antibody (9E10).
It can be considered that the replicons generating lower levels of G418 transduction may actually represent revertants that evolved during the extended course of the selection process (G418 selection), ultimately leading to the restoration of some replicative fitness (12). To obtain a more realistic picture of the RNA replication phenotype of the mutant replicons with a decreased replication capacity in the G418 transduction assay, we tested the replication fitness of the four single-polar-residue mutants (C1985E, T1989L, D1990K, and K1992D) by using a transient-RNA-replication assay using quantitative real-time reverse transcriptase PCR. This PCR assay directly measures HCV RNA replication over a short time frame, thereby eliminating the limitations of the G418 transduction assay. For all four single-polar-residue mutants (C1985E, T1989L, D1990K, and K1992D) tested, a clear lack of replication over a period of 72 h was observed (Fig. 2B), indicating that these changes were lethal for RNA replication and that colonies generated by G418 transduction were likely revertants (Fig. 2B). Serving as a positive control during the experiments, only the parental GIT replicon demonstrated efficient RNA replication, while that of GIT-pol(−), used as a negative control, was very similar to those of all of the mutants assayed.
The lethal phenotype for NS5A N-terminal anchor helix mutants (C1985E, T1989L, D1990K, and K1992D) is not due to disruption of NS5A processing.
All the mutations that were generated in the amino-terminal anchoring helix of NS5A are very close to the NS4B-NS5A polyprotein junction; hence, there is a possibility for potential alteration of these sites, affecting the proper cleavage of NS5A. To test if there are NS5A processing defects among the lethal mutants (C1985E, T1989L, D1990K, and K1992D), we used a recombinant vaccinia virus expressing T7 RNA polymerase (vTF7-3 expression system) followed by transient transfection using mutant replicon plasmid DNA to generate the HCV polyprotein and cleaved mature proteins. Cell lysates were then collected after 24 h and subjected to Western blotting to detect NS5A. No gross defects in terms of NS5A expression or processing were observed for any of the lethal mutants, and the appearance and electrophoretic migration of the NS5A protein bands of the mutant replicons were similar to those of the NS5A protein band from the control parental GIT replicon (Fig. 2C), indicating that the mutations in the anchoring helix do not grossly alter HCV polyprotein processing. No evidence of any NS4B-NS5A processing intermediate was observed, although we did not perform a more sensitive pulse-chase-type analysis to investigate the presence of differences in rates of NS4B-NS5A processing and to allow the accumulation of small amounts of uncleaved precursors. We also observed no obvious defects in NS5A phosphorylation (data not shown).
Identification of putative second-site reversions that rescue replication for lethal helix mutants.
Second-site reversions that compensate for the replication defects observed for our impaired helix mutants were investigated as a potential means to provide information on the surfaces of HCV proteins that might contact the NS5A anchor charged face. Hence, all four single-polar-residue mutants (C1985E, T1989L, D1990K, and K1992D) that failed to support robust HCV RNA replication were included for reversion analysis. RNA from each mutant was electroporated into Huh-7.5 cells, and G418 selection was performed, resulting in the selection of a number of colonies, indicating the chance of evolution of revertants in HCV RNA that restored replicative fitness. As no HCV RNA replication was detected for these mutants by quantitative real-time reverse transcriptase PCR (Fig. 2B), the evolution of revertants during prolonged G418 selection seemed likely. For each mutant, multiple colonies were picked and expanded (range of 5 to 24 colonies per mutant), and RNA was extracted for RT-PCR and DNA sequence analyses. Our analysis initially focused on the NS5A protein-coding region, particularly the site of the original introduced mutations, to ensure that no reversion occurred at these sites (same-site reversion). Mutant colonies that acquired same-site reversions (same amino acid) or pseudoreversions (reversion at the same site to a non-wild-type amino acid) were not included for further analysis, as the goal of this analysis was to potentially define interaction partners for the NS5A anchor. The mutant colonies that retained the input mutations, suggesting that their restoration of replication resided with a compensatory mutation elsewhere in the polyprotein, were subjected to sequence analysis of the entire HCV polyprotein-coding region to identify any second-site reversions. Details of the reversion-scanning analysis of the four lethal mutants that were assayed are described in Table 1. For the C1985E mutant, none of the colonies picked for expansion survived, and hence, no further study of this mutant was possible. For the D1990K and K1992D mutants, all of the expanded colonies were same-site pseudoreversions (for D1990K, the pseudoreversions are K1990N and K1990E, and for K1992D, the pseudoreversions are D1992V, D1992G, D1992A, and D1992H). The nature of residues associated with pseudoreversion at these positions was rather broad, with position 1992 typically reverting to small nonpolar amino acids (V, G, and A) and position 1990 reverting back to a negatively charged residue (E) or a polar residue (N) with a size similar to that of the original residue present at this position (D).
TABLE 1.
Potential-reversion-scanning analysis of the five lethal mutantsa
| Mutant | No. of clones picked (no. of viable picks) | Expanded clone(s) | Potential reversion(s) harbored by HCV gene |
||||
|---|---|---|---|---|---|---|---|
| NS5A | NS3 | NS4A | NS4B | NS5B | |||
| C1985E | 5 (0) | None | ND | ND | ND | ND | ND |
| T1989L | 5 (4) | 1, 2 | L1989T | NA | NA | NA | NA |
| 3 | T1989L | I1278V | E2437K, N2529S | ||||
| 4 | T1989L | E2437K, N2529S, E2590K | |||||
| D1990K | 10 (4) | 1 | K1990N | NA | NA | NA | NA |
| 2, 3, 4 | K1990E | NA | NA | NA | NA | ||
| K1992D | 24 (16) | 1, 14, 15 | D1992V | NA | NA | NA | NA |
| 2, 5, 8, 9, 10 | D1992G | NA | NA | NA | NA | ||
| 3, 6, 11, 12, 13, 16 | D1992A | NA | NA | NA | NA | ||
| 4, 7 | D1992H | NA | NA | NA | NA | ||
| D1979K/D1982K | 18 (7) | 2, 6, 8, 17 | K1982E | NA | NA | NA | NA |
| 5, 9 | K1979E | NA | NA | NA | NA | ||
| 13 | D1979K/D1982K | K1958R | K2469Q, N2529S | ||||
Clones that showed potential reversions are indicated in boldface type. ND, not done; NA, not analyzed.
For the T1989L mutant, two expanded colonies acquired a same-site reversion back to threonine (L1989T), while the other two expanded colonies that retained the input mutation showed potential second-site reversions. For the one clone that retained the input mutation (clone 3 of the T1989L mutant) (Table 1), the viral RNA acquired an I1278V mutation in NS3 and also E2437K and N2529S mutations in NS5B. For the other clone that retained the input mutation (clone 4 of the T1989L mutant) (Table 1), a total of three mutations were found in NS5B, namely, E2437K, N2529S, and E2590K. We chose mutations common to both expanded clones (E2437K and N2529S), reengineered these reversions either individually or in combination into the primary mutant replicon (T1989L), and tested them in a transient-transfection-based G418 transduction assay for replication restoration (Fig. 3A). None of the mutations, alone or in combination, led the primary lethal mutant T1989L to propagate in the G418 transduction assay (Fig. 3B). It is possible that our selection of the common mutations shared by both independent clones for analysis was in error, with the other mutations seen in only one clone (I1278V and E2590K) being responsible for the suppression of the T1989L replication defect, or perhaps a combination of some of these common and unique mutants was required; however, this remains to be assessed. Nonetheless, the data generated highlighted that the interior portions of the NS5A membrane anchor charged face were less tolerant of mutations than the ends of the helix, even if we were not successful at identifying mutations that suppressed the defect of our introduced mutations.
FIG 3.
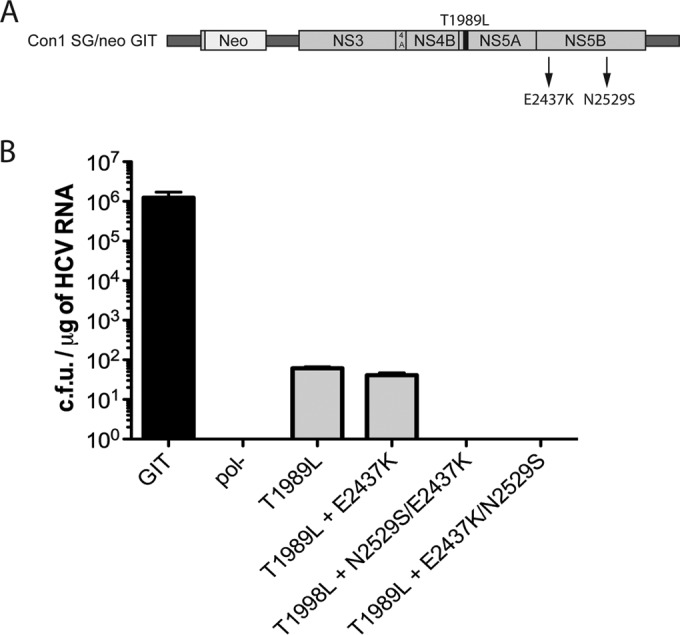
Analysis of replication efficiency of the T1989L anchor helix mutant bearing reversions. (A) Location of the input helix mutation (T1989L) (black bar) shown on a schematic representation of a subgenomic HCV replicon. Potential revertants identified from replicons harboring the input mutation T17L are designated by arrows and amino acid positions relative to the replicon model. (B) Transduction efficiencies for the NS5A T1989L anchor mutant bearing the identified reversions (E2437K, N2529S, and E2437K plus N2529S) were assayed following RNA electroporation into Huh-7.5 cells and selection with G418 for a period of 2 weeks. Values are presented as CFU per microgram of HCV RNA. Results shown are the averages of data from three independent experiments. Cells electroporated with replication-efficient GIT and replication-defective pol(−) RNAs served as positive controls and negative controls, respectively.
As our initial efforts aimed at identifying functional second-site suppressors for our mutants were not successful, we decided to make a more aggressive mutation in the NS5A membrane anchor's charged face. We selected two spatially adjacent aspartic acid residues at positions 1979 and 1982 of NS5A as a location for our first double mutant in the helix. Although both of these positions tolerated substitution individually to residues of the opposite charge, we hypothesized that changing both residues, thereby eliminating a large negatively charged patch on the helix, might be more deleterious to replication than the single mutations at each position and that the mutation of two residues might reduce the potential generation of same-site reversions. We therefore generated a double mutation of the two aspartic acid residues to lysines (D1979K/D1982K) (Fig. 1). When the replication efficiency of the double flip (D1979K/D1982K) mutant-containing replicon was tested, a very low transduction efficiency was observed (1.42 × 106 CFU/μg of RNA for the GIT replicon versus 7.0 × 101 CFU/μg of RNA for the D1979K/D1982K mutant) (Fig. 4A), demonstrating that although single changes of each residue individually did not have any major impact on replication, the combination of the two mutations clearly did. As shown for the single mutant panel, we assessed the double flip mutant (D1979K/D1982K) replicon by using real-time RT-PCR analysis of transient RNA replication and observed that the mutation was lethal for RNA replication (Fig. 4B), and the small number of colonies observed under G418 selection might be revertants. As was the case for our above-described single mutants, it was possible that the impairment of RNA replication observed for the double flip mutant (D1979K/D1982K) was due to a loss of correct NS5A processing from the viral polyprotein, so a similar analysis of the transient expression of the HCV polyprotein, followed by NS5A Western blotting, was performed to eliminate this possibility (Fig. 4C).
FIG 4.

Characterization of an NS5A anchoring helix double flip mutant (D1979K/D1982K). (A) RNA replication efficiency of the double flip mutant replicon. As described in the legend of Fig. 2A, the bar diagram represents the transduction efficiency of replicon RNAs in CFU per microgram of RNA, where pol(−) and GIT served as negative controls and positive controls, respectively. The assay was performed in triplicates, and each bar in the graph represents the average number of colonies (with standard deviations) obtained from three experimental replicates. (B) Assessment of transient RNA replication of the double flip mutant by a real-time reverse transcription-PCR assay of HCV RNA. The numbers of HCV RNA copies present in the case of each experimental mutant per 100 ng of total RNA extracted at 4 h, 24 h, 48 h, and 72 h postelectroporation are plotted. Each sample was assayed in triplicate. Replication-competent GIT RNA and replication deficient pol(−) RNA were used as the respective controls. (C) Following transient expression, NS5A processing for the double flip mutant was analyzed by Western blotting with an anti-NS5A monoclonal antibody (9E10).
We performed a similar expansion of colonies from the double flip mutant (D1979K/D1982K) with G418 selection to investigate if these colonies represented interesting reversions to the dramatic input mutation. We were able to clone and expand seven colonies from this mutant. Six expanded colonies acquired same-site pseudoreversions affecting only one residue of the double flip mutant (either a K1979E or a K1982E mutation), with preferences for the acidic residue glutamic acid, which is not surprising considering that the wild-type residue that was preferred at these positions was aspartic acid. Interestingly, it seems that only a single negatively charged residue is needed in this region of the helix, and it does not matter at which of the two positions tested (Table 1). As the single mutations introduced at these positions had a minimal effect on RNA replication efficiency (Fig. 2A), and the reversions were all substitutions at one site or the other, we opted not to explore these clones further. One clone isolated from the double flip mutant (D1979K/D1982K) that retained the input mutations (clone 13) (Table 1) contained a K1958R mutation in NS4B and K2469Q and N2529S mutations in NS5B (Fig. 5A). When all three reversions were individually reengineered back into the primary mutant replicon (D1979K/D1982K) and tested by a transient-transfection-based G418 transduction assay, the two NS5B mutants (K2469Q and N2529S) failed to exert any replication-rescuing effect. Interestingly, the NS4B reversion (K1958R) alone rescued replication of the lethal double flip mutant (D1979K/D1982K) to within 1 log of the parental replicon (7.8 × 105 CFU/μg of RNA for the parental GIT replicon versus 4.0 × 101 CFU/μg of RNA for the D1979K/D1982K mutant and 3.4 × 104 CFU/μg of RNA for the D1979K/D1982K mutant with the K1958R reversion) (Fig. 5B), indicating a clear suppression of the double flip mutant replication defect and genetic evidence of a possible interaction between NS4B and NS5A.
FIG 5.

Analysis of replication efficiency for the anchor helix double flip mutant (D1979K/D1982K) bearing reversions. (A) Location of the double flip input helix mutation (D1979K/D1982K) (black bar) shown on a schematic representation of a subgenomic HCV replicon. Potential revertants identified from replicons harboring the double flip input mutation are indicated by arrows and amino acid positions relative to the replicon model. (B) Transduction efficiencies for the NSS5A D1979K/D1982K anchor helix double flip mutant bearing the identified potential reversions (K1958R, K2469Q, and N2529S) assayed following RNA electroporation into Huh-7.5 cells and selection with G418 for a period of 2 weeks in triplicate experiments. Cells electroporated with replication-efficient GIT RNA and replication-defective pol(−) RNA served as positive controls and negative controls, respectively.
With good evidence of a genetic interaction between the charged face of the NS5A helix and the NS4B protein, we proceeded to assess the replication capacity of replicons harboring this collection of mutants via a quantitative real-time reverse transcriptase PCR assay. As described above, the double flip helix mutant (D1979K/D1982K) was lethal and showed no accumulation of RNA over a 72-h time course, similar to the phenotype of the GIT-pol(−) negative-control construct (Fig. 6A). The addition of the K1958R mutation to the double flip helix mutant (D1979K/D1982K) replicon led to a restoration of replication to levels only slightly lower than those of the parental GIT replicon (a 2-fold reduction relative to that of GIT, which was not statistically significant). The K1958R mutation alone also produced RNA replication levels and kinetics similar to those of the combination of K1958R and D1979K/D1982K, with values not being statistically different from those for the parental replicon, indicating that the K1958R mutation does not enhance RNA replication or otherwise function as an adaptive mutation in this assay. Interestingly, the RNA replication phenotype of the combination of the K1958R and D1979K/D1982K mutations in the real-time PCR assay showed a very small reduction, ∼2-fold, in replication relative to that of the parental GIT replicon, but this same combination produced nearly a 20-fold decrease in replicative fitness in the G418 transduction assay relative to that of the GIT parent. The reason for this discrepancy is not clear, other than that the assays are different, but it is also possible that the K1958R/D1979K/D1982K triple mutant replicon produces less stable replication complexes and that this is apparent only in the longer G418 selection process and not in the 72-h transient real-time PCR assay.
FIG 6.

Replication efficiencies for the NS5A anchor helix double flip mutant (D1979K/D1982K) and the rescue revertant on NS4B (K1958R). (A) Comparison of transient RNA replication of the double flip mutant (D1979K/D1982K), the double flip mutant bearing an NS4B reversion (D1979K/D1982K + K1958R), and the NS4B revertant only (K1958R) by a real-time reverse transcription-PCR assay of HCV RNA. The numbers of HCV RNA copies present in the case of each experimental mutant per 100 ng of total RNA extracted 4 h, 24 h, 48 h, and 72 h postelectroporation are plotted. Each sample was assayed by RT-PCR in triplicates. Replication-competent GIT RNA and replication-deficient pol(−) RNA were used as controls. (B) RNA replication efficiency of the NS4B revertant (K1958R). The bar diagram represents the transduction efficiencies of replicon RNAs in CFU per microgram of RNA, where pol(−) and GIT served as a negative controls and positive controls, respectively, for the replication assay. The assay was performed in triplicates, and each bar in the graph represents the average number of colonies (with standard deviations) obtained from three experimental replicates for each experimental mutant and control.
Finally, we assessed the K1958R mutation alone by a G418 transduction assay to demonstrate that this mutation was not merely an adaptive mutation that generally increased RNA replication in replicon transduction (Fig. 6B). The G418 transduction efficiency of the K1958R-harboring replicon demonstrated no statistically significant difference in G418 transduction relative to that of the control parental GIT replicon (2.04 × 106 CFU/μg of RNA for the parental GIT replicon versus 1.07 × 106 CFU/μg of RNA for the K1958R mutant). Collectively, these data suggest that the K1958R mutation is almost phenotypically null unless presented in the context of the double flip helix mutant (D1979K/D1982K), where it suppresses the defect seen with this mutation and restores replication capacity.
NS4B K1958R mutation restores correct subcellular localization of the NS5A double flip helix mutant.
Functional HCV RCs are observed by fluorescence imagining as punctate, perinuclear foci on the ER-like membranes where HCV nonstructural proteins accumulate, including NS5A (30, 39–42). Hence, once we observed that the lethal replication phenotype of the NS5A helix double mutant is restored by an NS4B carboxyl-terminal suppressor mutation, we were curious to examine the subcellular localization of NS5A in light of the proposed role of NS4B as a scaffold upon which the RC assembles. We therefore performed an immunofluorescence analysis to determine the subcellular localization of the NS5A protein for the lethal double flip helix mutant (D1979K/D1982K) alone, the double flip helix mutant containing the NS4B rescue reversion (D1979K/D1982K plus K1958R), the NS4B mutation alone (K1958R), and the parental GIT replicon as a positive control. To avoid issues with protein expression from the lethal double flip helix mutant, these experiments were conducted by using Lunet-T7 cells that express T7 RNA polymerase and transfection of a replicon plasmid under the control of a T7 promoter. We observed similar levels of protein expression and mRNA from these cells regardless of which replicon construct was transfected, suggesting that the effects that we observed were independent of RNA replication. As expected for the wild-type control GIT replicon, distinct perinuclear punctate staining was observed (Fig. 7A). Strikingly, the double flip mutant (D1979K/D1982K) lacked the normal punctate staining of NS5A and presented a diffuse ER-like staining pattern. Interestingly, perinuclear punctate staining was restored for the double-helix mutant in the presence of the rescue reversion mutant in NS4B (D1979K/D1982K plus K1958R), generating an appearance similar to that of the parental GIT control. The K1958R mutation alone produced perinuclear punctate NS5A staining similar to that observed for the GIT replicon. Although we did not observe any evidence of T7-launched RNA replication in these experiments, it remained a possibility that GIT, D1979K/D1982K plus K1958R, and K1958R alone were capable of launching RNA replication, resulting in a punctate NS5A localization characteristic of the HCV RC, whereas D1979K/D1982K was replication impaired and produced a diffuse pattern of NS5A, as no RCs formed. To address this possibility, we cloned our various replicons into a backbone harboring a lethal lesion in the RNA-dependent RNA polymerase (Fig. 2) and expressed polyproteins from these T7 promoter-based constructs using a vaccinia virus expressing T7 RNA polymerase. The short time frame of vaccinia virus expression (6 to 8 h), combined with the replication-defective backbone, provides additional evidence that our observed localization differences are not dependent on HCV RNA replication or a lack thereof. Figure 7B presents data from this analysis, with GIT, D1979K/D1982K plus K1958R, and K1958R producing perinuclear punctate staining and D1979K/D1982K generating overall diffuse membrane staining (with very few possible punctate foci), results that were identical to that that we observed with the Lunet-T7 assay system (Fig. 7A). No difference was observed at the level of NS5A expression or processing among the experimental replicon constructs (D1979K/D1982K, D1979K/D1982K plus K1958R, and K1958R) and the control (parental GIT) (Fig. 7C), indicating that the differences in NS5A subcellular localization were not due to staining artifacts or due to variations at the level of expression.
FIG 7.

Characterization of NS5A subcellular localization, membrane association, and phosphorylation. (A) Following cotransfection of Lunet-T7 cells (with plasmid pT7-T7 and an experimental plasmid) and one round of G418 selection, cells were plated into four-well chamber slides, and after 48 h, cells were fixed and stained for NS5A expression (9E10, anti-NS5A, and Alexa-488 secondary antibody) in an indirect-immunofluorescence assay. Nuclei were stained with DAPI. The images were colored and merged by using ImageJ software. For parental GIT, the anchor helix double flip mutant (D1979K/D1982K) bearing the NS4B reversion (K1958R), and the NS4B revertant only (K1958R), perinuclear punctate staining consistent with the membranous web was observed, while the anchor helix double flip mutant (D1979K/D1982K) alone presented disrupted perinuclear staining consistent with diffuse ER localization. (B) vTF7-3 (expressing T7 RNA polymerase)-infected Huh-7.5 cells were transfected with HCV replicon constructs as described above for panel A but with each construct harboring a lethal mutation in the HCV polymerase sequence, thereby eliminating any chance of RNA replication. Parental GIT, the anchor helix double flip mutant bearing the NS4B reversion (D1979K/D1982K + K1958R), and the construct harboring the NS4B revertant alone (K1958R) produced punctate perinuclear staining consistent with the membranous web. The double helix flip mutant (D1979K/D1982K) alone produced a diffuse ER staining pattern with relatively few punctate structures. Cells were fixed, stained, and photographed at 6 h posttransfection to avoid vaccinia virus-induced cytopathic effects. (C) NS5A expression analyzed by Western blotting with an anti-NS5A (9E10) monoclonal antibody. Beta actin served as a loading control. The location of the NS5A hyperphosphorylated species is indicated with an asterisk. (D) Assessment of membrane association of NS5A by a membrane flotation assay. Homogenized lysates of Lunet-T7 cells transiently transfected with parental GIT, the anchor helix double flip mutant (D1979K/D1982K), the double flip mutant bearing the NS4B reversion (D1979K/D1982K + K1958R), and the construct bearing the NS4B revertant only (K1958R) were separated through a sucrose gradient, followed by Western blotting of the collected fractions. For every experimental sample, a similar flotation assay was performed with 1% NP-40-treated homogenized lysates as a control.
Of note, none of the mutants presented any difference in NS5A phosphoform distribution (Fig. 7C, asterisk), suggesting that alteration of NS5A phosphorylation does not impact the observed phenotypes. As we observed a striking difference in the subcellular localization of the double flip helix mutant in our imaging experiments, we wanted to know if the differences seen might be due to the incorrect insertion of NS5A into target membranes. We therefore harvested cell lysates from cells transfected with our various constructs (as shown in Fig. 7A) and subjected them to a membrane flotation assay to see if NS5A was associated with cellular membranes (Fig. 7D). The lethal NS5A helix double flip mutant (D1979K/D1982K), the helix mutant with the NS4B reversion (D1979K/D1982K plus K1958R), and the NS4B mutant only (K1958R) were all inserted into the low-density-membrane-containing gradient fractions in a manner similar to that for the parental GIT construct, indicating that no defect in membrane association was present.
As our data suggested that an interaction of the charged face of the membrane anchor of NS5A with the carboxyl-terminal region of NS4B was required for the localization of NS5A to HCV RC-like structures, we wanted to investigate whether we could demonstrate a direct interaction between NS4B and NS5A that was dependent on our helix mutations. We developed an in-cell FRET assay to monitor interactions between NS5A and NS4B by fusing the proteins to fluorescent proteins (Fig. 8A). In these experiments, we observed that ∼30% of cells transfected with NS4B-CFP and YFP-NS5A produced a FRET interaction signal (Fig. 8B, third plot) relative to cells transfected with unfused CFP and YFP, <2% of which produced a FRET signal, and CFP-YFP fusions, which produced 98% FRET-positive cells (Fig. 8B). We set the NS4B-CFP- and YFP-NS5A-transfected cells as 100% of the maximal FRET signal that we might expect and analyzed our mutant cells. The introductions of the D1979K/D1982K mutations into the YFP-NS5A construct significantly reduced FRET transfer to NS4B-CFP, indicating that an interaction had been disrupted (Fig. 8C). The introduction of the K1958R mutation into the NS4B-CFP plasmid did not fully restore the interaction with the D1979K/D1982K YFP-NS5A construct, but the general trend suggested a relative increase in the FRET signal in this context, and values for FRET restoration were statistically significant. The K1958R NS4B-CFP plasmid with wild-type YFP-NS5A also exhibited a slightly reduced FRET interaction signal but less so than that observed for the helix flip mutation alone. Combined with the striking subcellular localization data, these data suggest that NS5A is inserted into membranes and localizes to HCV RC-like structures through a direct interaction with the carboxyl-terminal region of NS4B in a manner dependent on the residues of the helix double flip mutant (D1979K/D1982K). An alternative possibility is that the helix double flip mutation somehow reduces the stability of RCs, resulting in their loss of NS5A.
DISCUSSION
Polar or charged residues, with their electrostatic surface potential, play key roles during protein-protein and protein-RNA interactions. The cytoplasmically exposed polar amino acids of the anchor helix of NS5A are believed to participate in multiple interactions required for RNA replication (25, 26). Hence, in the present study, the role of polar charged residues of the anchor helix region of NS5A in the context of HCV RNA replication was investigated.
With the aid of an aggressive mutagenesis approach, we identified an interesting scenario with respect to the HCV Con1/SG-neo GIT replicon-derived NS5A anchor helix region, where some polar residues are critical for replication individually and some residues are critical as groups but are dispensable individually. Almost one-third of the individual polar residues (4 out of 12; C1985E, T1989L, D1990K, and K1992D) were identified as being critical for replication, producing a lethal or near-lethal phenotype, and they are all situated around the center of the helix (Fig. 9). As expected, three critical polar residues (C1985, D1990, and K1992) are highly conserved (>99% of isolates), and T1989 is also nearly conserved (67% isolates) (comparison among the 245 HCV subtype 1b reference sequences in the HCV Los Alamos Sequence Database). One interesting aspect of the four critical residues is that, despite being polar, no particular nature of the residues could be classified, as residues such as C and T (for positions 1985 and 1989, respectively) are polar but uncharged, while residues such as D and K (for positions 1990 and 1992, respectively) are polar acidic and polar basic, respectively. A previous study by Penin et al. (25) found no effect of the C1985 mutation at the level of replication. Such differences in findings could be attributed to the differing approaches adopted for mutagenesis (alanine substitution versus aggressive substitution in the present study) or differences at the level of experimental constructs, as functional ability, even for a single residue, is greatly influenced by the overall chemical environment subjective to surrounding residues. Among the residues tested in the present study, despite being nearly conserved, polar residues such as R1978, D1979, D1982, T1986, and S1997 (>97.5% of isolates) were found to be quite tolerant even for aggressive substitutions (Fig. 2A). The conserved nature of these polar residues indicates their possible role in the HCV life cycle. Thus, the substitution tolerance of these conserved polar residues was striking and might be due to the fact that such residues play other roles during the HCV life cycle that are not modeled in our experimental system or that some other surrounding residues compensate for the change of these residues, indicating that they may function as a group of residues. A previous study demonstrated that combined alanine substitutions at amino acid positions 1978 and 1979 (yielding the R1978A/D1979A mutant) had no effect on HCV replication (25). In the present study, since all the critical single residues are situated around the center of the anchor helix of NS5A, a double flip charged mutant was made, which resulted in the generation of the D1979K/D1982K mutant. The basic idea behind the generation of such a double mutant was to disrupt the acidic patch (which forms due to one turn of the helix placing the two Asp residues next to each other) and also to unsettle the charge distribution along the helix originating due to the in-line positioning of the negatively charged residues along one cytosolic border of the helix (25). Probably, a single mutation such as D1979K or D1982K alone had a minimal effect on the charge distribution of the helix to have a substantial effect on replication, as evident with the transient-replicon assay (Fig. 2A), but the combination of these individual mutations (D1979K/D1982K) was lethal (Fig. 4A and B). In line with data from previous studies (25, 26), the findings presented here strongly suggest that the polar residues at the cytosolic face of the NS5A anchor helix provide an interaction site that is absolutely crucial for HCV RNA replication.
FIG 9.
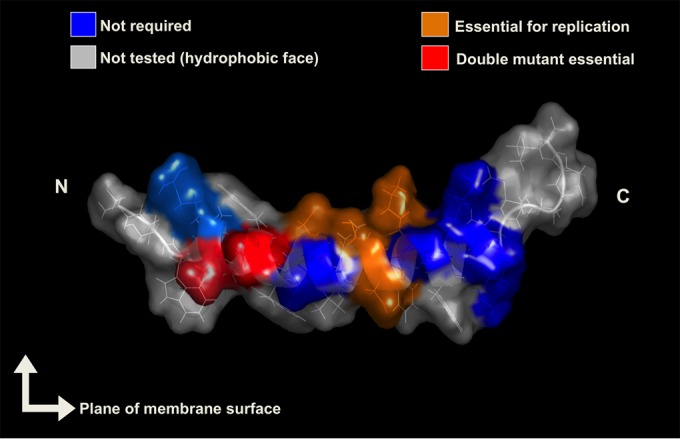
Residues of the NS5A anchor helix that are important for HCV RNA replication. The diagram highlights the residues important for HCV RNA replication either individually or in groups, depicted in yellow and red, respectively. Residues that are dispensable for replication are indicated in blue. “N” and “C” indicate the N-terminal and C-terminal ends, respectively. The two arrows perpendicular to each other depict the positioning of the anchor helix along the membrane surface. The NS5A anchor helix structural illustration was prepared by using PyMOL.
The search for compensatory second-site reversions to suppress our poorly replicating helix mutations revealed an interesting aspect of the probable genetic interaction between the HCV nonstructural proteins required for HCV replication. We analyzed a number of potential second-site revertants (Table 1) from selected colonies that we could grow from our lethal or near-lethal mutant replicons (C1985E, T1989L, D1990K, K1992D, and D1979K/D1982K), but the only second-site reversion that functionally rescued the lethal input mutation phenotype was K1958R in NS4B that rescued D1979K/D1982K (Fig. 5B). The K1958R reversion is somewhat unusual in that it is a very conservative change, with a substitution of one positively charged residue (K) for another (R), and for the fact that although the residue at this position is a K in 94% of HCV genotype 1b isolates, it is an R in isolates of most other HCV genotypes. The overall impression is that this position requires a positive charge, but the absolute identity of the amino acid is not important. Indeed, in our own experiments, the K1958R mutation alone produces replicons that behave very similarly to the parental GIT construct (Fig. 6A and B), erasing the probability of any adaptive mutation type of effect. Of course, something more complex could be happening, as K and R have some different properties, and the function of the substitution as a response to the addition of a more positive charge into the NS5A membrane anchor is rather elusive. Our attempts at modeling the interactions of NS4B carboxy-terminal helix 2 and the membrane anchor of NS5A using the available structures of these regions have not produced an obvious interaction surface on which these residues might interact. Perhaps, the interaction between NS4B and NS5A that we were monitoring involves additional viral or host components and is indirect in nature. Indeed, residues very close to our mutations, such as W1981, are essential for the interaction of NS5A with the required host protein Tip47 (43). A more detailed exploration of the genetics of this NS4B-NS5A interaction is clearly warranted, especially in the context of the diverse HCV genotypes in which K1958 is naturally an R residue, and how these changes might impact host factor associations with NS5A.
The NS4B protein is considered to be the key component of the HCV RCs, as it induces the formation of the membranous web (9, 29, 44). It is believed that with the ability to potentially interact with HCV RNA, as well as with other HCV nonstructural proteins, NS4B acts as a scaffold during the formation of HCV RCs (45–48). The structure of NS4B is relatively complex due to the presence of cytosolic N- and C-terminal domains and a central hydrophobic domain containing four transmembrane segments (49, 50). The relatively conserved carboxyl-terminal domain contains two α-helices, namely, helix 1 and helix 2 (49, 50). Helix 2 is composed of 27 amino acids (aa 1939 to 1965) and has been reported to mediate membrane associations in an in-plane topological manner as well as to play an important role during HCV RC formation (51). So far, several studies demonstrated the interaction between NS4B and NS5A but with limited information about the nature of the interaction. A recent study showed that the interaction between domain I of NS5A and helix 1 of the C-terminal domain of NS4B is involved in HCV RNA replication (52). In another study, revertant analysis showed that the effect of mutations in helix 1 of the C-terminal domain of NS4B is rescued by an intergenic reversion situated around the center of domain I of NS5A (53). It is worth mentioning that a reversion further downstream of NS5A domain I (P2161L) enhances replication for a lethal mutation at the N-terminal domain of NS4B (40). In the present study, the opposite phenomenon was observed, where an NS5A anchor helix lethal mutation was rescued by a reversion situated on the NS4B C-terminal end. The locations of the input mutation and the rescue reversion in the present study were striking. Unlike the previous two studies, where the input mutation resides on helix 1 of the C-terminal domain of NS4B and the rescue reversion or site of interaction resides on domain I of NS5A (52, 53), in the present study, the input mutation resides at the NS5A N-terminal anchor helix region, and the rescue reversion resides on helix 2 of the C-terminal domain of NS4B. Considering that NS4B interacts directly with NS5A during replication, if the findings of the present study are combined with the findings of others (40, 52, 53), and if one assumes that the genetic interactions observed by these efforts represent physical contact surfaces, the arrangement of the input mutation and the rescue reversion suggests that the interaction between NS5A and NS4B occurs at multiple sites in a parallel head-to-tail orientation on the surface of the ER.
The punctate foci on the ER at the ultrastructural level represent the membranous web of HCV RNA RCs (39, 41). Like other HCV nonstructural proteins, NS5A localizes within the RC represented by foci (45, 47, 54), and such an NS5A localization appears as perinuclear punctate dots. The localization of NS5A is greatly influenced by NS4B, and the absence of NS4B leads to an increased mobility of NS5A (42, 55). In agreement with this increased mobility of NS5A by NS4B perturbation, we observed a diffuse ER-like NS5A localization for the replication-defective NS5A helix anchor mutant (D1979K/D1982K), and the restoration of the relatively normal punctate focus localization of NS5A by the reversion at helix 2 of the NS4B C-terminal domain (D1979K/D1982K plus K1958R) (Fig. 7A and B) suggests that the localization of NS5A into the punctate RC-like structures that we observed is governed at the level of the NS4B-NS5A interaction. Further experiments on the NS4B-NS5A interaction in the context of in-cell FRET interaction studies indicated that NS5A and NS4B interact and that this interaction is dependent on the NS5A helix, as the double mutant (D1979K/D1982K) reduced the FRET signal in a significant manner. The K1958R mutation partially restored the FRET signal that was lost in the presence of the D1979K/D1982K mutant, but restoration was not complete. The K1958R mutation alone also impaired FRET interactions although not as severely as the NS5A helix mutants (∼15% fewer FRET-positive cells). This is somewhat surprising, as this mutation has a very minor impact on replication. Nonetheless, we observed clear evidence of an NS5A-NS4B interaction in these FRET experiments, and the interaction is altered by the mutations that we have identified in our studies. It is also possible that our NS5A double flip helix mutation somehow prevents RC formation and that this is restored by the K1958R mutation in NS4B; however, it is unclear how this might work. Additionally, during the FRET experiment on the interaction between NS4B and NS5A, the potential function of the other HCV nonstructural proteins was absent, and thus, their obvious complex role as mediators or interactors during the formation of a functional HCV replication complex remains unrevealed and falls under current experimental limitations. A more detailed electron microscopic investigation of the RC structure and the localization of NS4B and NS5A in the context of these mutants is under way.
Besides their potential complex interaction, NS4B and NS5A have far-reaching effects on each other, such as NS5A influencing the structural topology of NS4B (56) or NS4B effecting NS5A phosphorylation (55, 57, 58). In the present study, no differences in the NS5A phosphorylation status (p58 versus p56) were found between the lethal mutant and the revertants (Fig. 7C). Such an observation was not surprising, as other than the K1958R reversion, no real change for NS4B existed to perturb its natural ability to function in terms of affecting interactions between NS5A and cellular kinases.
Thus, collectively, the present study indicates the functional relevance of the conserved polar residues on the charged face of the NS5A membrane anchor and provides evidence for a role of the anchoring helix of NS5A in proper localization to the sites of RNA replication via a complex interaction with NS4B. Mutations of some of these residues disrupt membranous-web focus formation on the ER and hence affect HCV RNA replication. Additionally, background structural and functional information about NS4B and the findings of the present study based on a reverse genetic approach adopted for the replication rescue experiment provide functional evidence for the potential complex interaction between NS4B and NS5A during HCV RNA replication. Nevertheless, further studies are needed to extensively characterize the nature of the NS4B-NS5A interaction to gain comprehensive insights about the molecular architecture of HCV RCs.
ACKNOWLEDGMENTS
We thank the following individuals for supplying reagents used in this research: Bernard Moss (vTF7-3), Ralf Bartenschlager and Volker Lohmann (Lunet-T7 cells), and Charles Rice (Huh-7.5 cells and the 9E10 antibody).
This work was supported by grant 1R01-DK090014-01A1 (NIDDK) and funding from the Burroughs Wellcome Fund Investigators in the Pathogenesis of Infectious Diseases Grant Program.
REFERENCES
- 1.Gouttenoire J, Roingeard P, Penin F, Moradpour D. 2010. Amphipathic alpha-helix AH2 is a major determinant for the oligomerization of hepatitis C virus nonstructural protein 4B. J Virol 84:12529–12537. doi: 10.1128/JVI.01798-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Isken O, Langerwisch U, Jirasko V, Rehders D, Redecke L, Ramanathan H, Lindenbach BD, Bartenschlager R, Tautz N. 2015. A conserved NS3 surface patch orchestrates NS2 protease stimulation, NS5A hyperphosphorylation and HCV genome replication. PLoS Pathog 11:e1004736. doi: 10.1371/journal.ppat.1004736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jirasko V, Montserret R, Appel N, Janvier A, Eustachi L, Brohm C, Steinmann E, Pietschmann T, Penin F, Bartenschlager R. 2008. Structural and functional characterization of nonstructural protein 2 for its role in hepatitis C virus assembly. J Biol Chem 283:28546–28562. doi: 10.1074/jbc.M803981200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jones CT, Murray CL, Eastman DK, Tassello J, Rice CM. 2007. Hepatitis C virus p7 and NS2 proteins are essential for production of infectious virus. J Virol 81:8374–8383. doi: 10.1128/JVI.00690-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moradpour D, Penin F, Rice CM. 2007. Replication of hepatitis C virus. Nat Rev Microbiol 5:453–463. doi: 10.1038/nrmicro1645. [DOI] [PubMed] [Google Scholar]
- 6.Romero-Brey I, Merz A, Chiramel A, Lee J-Y, Chlanda P, Haselman U, Santarella-Mellwig R, Habermann A, Hoppe S, Kallis S, Walther P, Antony C, Krijnse-Locker J, Bartenschlager R. 2012. Three-dimensional architecture and biogenesis of membrane structures associated with hepatitis C virus replication. PLoS Pathog 8:e1003056. doi: 10.1371/journal.ppat.1003056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Romero-Brey I, Berger C, Kallis S, Kolovou A, Paul D, Lohmann V, Bartenschlager R. 2015. NS5A domain 1 and polyprotein cleavage kinetics are critical for induction of double-membrane vesicles associated with hepatitis C virus replication. mBio 6:e00759–15. doi: 10.1128/mBio.00759-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Scheel TK, Rice CM. 2013. Understanding the hepatitis C virus life cycle paves the way for highly effective therapies. Nat Med 19:837–849. doi: 10.1038/nm.3248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Egger D, Wölk B, Gosert R, Bianchi L, Blum HE, Moradpour D, Bienz K. 2002. Expression of hepatitis C virus proteins induces distinct membrane alterations including a candidate viral replication complex. J Virol 76:5974–5984. doi: 10.1128/JVI.76.12.5974-5984.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Appel N, Zayas M, Miller S, Krijnse-Locker J, Schaller T, Friebe P, Kallis S, Engel U, Bartenschlager R. 2008. Essential role of domain III of nonstructural protein 5A for hepatitis C virus infectious particle assembly. PLoS Pathog 4:e1000035. doi: 10.1371/journal.ppat.1000035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tellinghuisen TL, Marcotrigiano J, Gorbalenya AE, Rice CM. 2004. The NS5A protein of hepatitis C virus is a zinc metalloprotein. J Biol Chem 279:48576–48587. doi: 10.1074/jbc.M407787200. [DOI] [PubMed] [Google Scholar]
- 12.Tellinghuisen TL, Foss KL, Treadaway JC, Rice CM. 2008. Identification of residues required for RNA replication in domains II and III of the hepatitis C virus NS5A protein. J Virol 82:1073–1083. doi: 10.1128/JVI.00328-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tellinghuisen TL, Foss KL, Treadaway J. 2008. Regulation of hepatitis C virion production via phosphorylation of the NS5A protein. PLoS Pathog 4:e1000032. doi: 10.1371/journal.ppat.1000032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lindenbach BD, Rice CM. 2013. The ins and outs of hepatitis C virus entry and assembly. Nat Rev Microbiol 11:688–700. doi: 10.1038/nrmicro3098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Paul D, Madan V, Bartenschlager R. 2014. Hepatitis C virus RNA replication and assembly: living on the fat of the land. Cell Host Microbe 16:569–579. doi: 10.1016/j.chom.2014.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tellinghuisen TL, Marcotrigiano J, Rice CM. 2005. Structure of the zinc-binding domain of an essential component of the hepatitis C virus replicase. Nature 435:374–379. doi: 10.1038/nature03580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Love RA, Brodsky O, Hickey MJ, Wells PA, Cronin CN. 2009. Crystal structure of a novel dimeric form of NS5A domain I protein from hepatitis C virus. J Virol 83:4395–4403. doi: 10.1128/JVI.02352-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.LeMay KL, Treadaway J, Angulo I, Tellinghuisen TL. 2013. A hepatitis C virus NS5A phosphorylation site that regulates RNA replication. J Virol 87:1255–1260. doi: 10.1128/JVI.02154-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ross-Thriepland D, Mankouri J, Harris M. 2015. Serine phosphorylation of the hepatitis C virus NS5A protein controls the establishment of replication complexes. J Virol 89:3123–3135. doi: 10.1128/JVI.02995-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Blight KJ, Kolykhalov AA, Rice CM. 2000. Efficient initiation of HCV RNA replication in cell culture. Science 290:1972–1974. doi: 10.1126/science.290.5498.1972. [DOI] [PubMed] [Google Scholar]
- 21.Evans MJ, Rice CM, Goff SP. 2004. Phosphorylation of hepatitis C virus nonstructural protein 5A modulates its protein interactions and viral RNA replication. Proc Natl Acad Sci U S A 101:13038–13043. doi: 10.1073/pnas.0405152101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Appel N, Pietschmann T, Bartenschlager R. 2005. Mutational analysis of hepatitis C virus nonstructural protein 5A: potential role of differential phosphorylation in RNA replication and identification of a genetically flexible domain. J Virol 79:3187–3194. doi: 10.1128/JVI.79.5.3187-3194.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miyanari Y, Atsuzawa K, Usuda N, Watashi K, Hishiki T, Zayas M, Bartenschlager R, Wakita T, Hijikata M, Shimotohno K. 2007. The lipid droplet is an important organelle for hepatitis C virus production. Nat Cell Biol 9:1089–1097. doi: 10.1038/ncb1631. [DOI] [PubMed] [Google Scholar]
- 24.Brass V, Bieck E, Montserret R, Wolk B, Hellings JA, Blum HE, Penin F, Moradpour D. 2002. An amino-terminal amphipathic alpha-helix mediates membrane association of the hepatitis C virus nonstructural protein 5A. J Biol Chem 277:8130–8139. doi: 10.1074/jbc.M111289200. [DOI] [PubMed] [Google Scholar]
- 25.Penin F, Brass V, Appel N, Ramboarina S, Montserret R, Ficheux D, Blum HE, Bartenschlager R, Moradpour D. 2004. Structure and function of the membrane anchor domain of hepatitis C virus nonstructural protein 5A. J Biol Chem 279:40835–40843. doi: 10.1074/jbc.M404761200. [DOI] [PubMed] [Google Scholar]
- 26.Penin F, Dubuisson J, Rey FA, Moradpour D, Pawlotsky JM. 2004. Structural biology of hepatitis C virus. Hepatology 39:5–19. doi: 10.1002/hep.20032. [DOI] [PubMed] [Google Scholar]
- 27.Moradpour D, Evans MJ, Gosert R, Yuan Z, Blum HE, Goff SP, Lindenbach BD, Rice CM. 2004. Insertion of green fluorescent protein into nonstructural protein 5A allows direct visualization of functional hepatitis C virus replication complexes. J Virol 78:7400–7409. doi: 10.1128/JVI.78.14.7400-7409.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shirota Y, Luo H, Qin W, Kaneko S, Yamashita T, Kobayashi K, Murakami S. 2002. Hepatitis C virus (HCV) NS5A binds RNA-dependent RNA polymerase (RdRP) NS5B and modulates RNA-dependent RNA polymerase activity. J Biol Chem 277:11149–11155. doi: 10.1074/jbc.M111392200. [DOI] [PubMed] [Google Scholar]
- 29.Elazar M, Liu P, Rice CM, Glenn JS. 2004. An N-terminal amphipathic helix in hepatitis C virus (HCV) NS4B mediates membrane association, correct localization of replication complex proteins, and HCV RNA replication. J Virol 78:11393–11400. doi: 10.1128/JVI.78.20.11393-11400.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gosert R, Jendrsczok W, Berke JM, Brass V, Blum HE, Moradpour D. 2005. Characterization of nonstructural protein membrane anchor deletion mutants expressed in the context of the hepatitis C virus polyprotein. J Virol 79:7911–7917. doi: 10.1128/JVI.79.12.7911-7917.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lohmann V, Korner F, Koch J, Herian U, Theilmann L, Bartenschlager R. 1999. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science 285:110–113. doi: 10.1126/science.285.5424.110. [DOI] [PubMed] [Google Scholar]
- 32.Lohmann V, Hoffmann S, Herian U, Penin F, Bartenschlager R. 2003. Viral and cellular determinants of hepatitis C virus RNA replication in cell culture. J Virol 77:3007–3019. doi: 10.1128/JVI.77.5.3007-3019.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Goedhart J, von Stetten D, Noirclerc-Savoye M, Lelimousin M, Joosen L, Hink MA, van Weeren L, Gadella TWJ Jr, Royant A. 2012. Structure-guided evolution of cyan fluorescent proteins towards a quantum yield of 93%. Nat Commun 3:751. doi: 10.1038/ncomms1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Qi D, Scholthof KBG. 2008. A one-step PCR-based method for rapid and efficient site-directed fragment deletion, insertion, and substitution mutagenesis. J Virol Methods 149:85–90. doi: 10.1016/j.jviromet.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 35.Tellinghuisen TL, Lindenbach BD. 2009. Reverse transcription PCR-based sequence analysis of hepatitis C virus replicon RNA. Methods Mol Biol 510:165–175. doi: 10.1007/978-1-59745-394-3_12. [DOI] [PubMed] [Google Scholar]
- 36.Fuerst TR, Niles EG, Studier FW, Moss B. 1986. Eukaryotic transient-expression system based on recombinant vaccinia virus that synthesizes bacteriophage T7 RNA polymerase. Proc Natl Acad Sci U S A 83:8122–8126. doi: 10.1073/pnas.83.21.8122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen X, Li Y, Xiong K, Wagner TE. 1994. A self-initiating eukaryotic transient gene expression system based on contransfection of bacteriophage T7 RNA polymerase and DNA vectors containing a T7 autogene. Nucleic Acids Res 22:2114–2120. doi: 10.1093/nar/22.11.2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Banning C, Votteler J, Hoffmann D, Koppensteiner H, Warmer M, Reimer R, Kirchhoff F, Schubert U, Hauber J, Schindler M. 2010. A flow cytometry-based FRET assay to identify and analyse protein-protein interactions in living cells. PLoS One 5:e9344. doi: 10.1371/journal.pone.0009344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Aligo J, Jia S, Manna D, Konan KV. 2009. Formation and function of hepatitis C virus replication complexes require residues in the carboxy-terminal domain of NS4B protein. Virology 393:68–83. doi: 10.1016/j.virol.2009.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gouttenoire J, Montserret R, Paul D, Castillo R, Meister S, Bartenschlager R, Penin F, Moradpour D. 2014. Aminoterminal amphipathic α-helix AH1 of hepatitis C virus nonstructural protein 4B possesses a dual role in RNA replication and virus production. PLoS Pathog 10:e1004501. doi: 10.1371/journal.ppat.1004501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gretton SN, Taylor AI, McLauchlan J. 2005. Mobility of the hepatitis C virus NS4B protein on the endoplasmic reticulum membrane and membrane-associated foci. J Gen Virol 86:1415–1421. doi: 10.1099/vir.0.80768-0. [DOI] [PubMed] [Google Scholar]
- 42.Blight KJ. 2011. Charged residues in hepatitis C virus NS4B are critical for multiple NS4B functions in RNA replication. J Virol 85:8158–8171. doi: 10.1128/JVI.00858-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vogt DA, Camus G, Herker E, Webster BR, Tsou C-L, Greene WC, Yen TS, Ott M. 2013. Lipid droplet-binding protein TIP47 regulates hepatitis C virus RNA replication through interaction with the viral NS5A protein. PLoS Pathog 9:e1003302. doi: 10.1371/journal.ppat.1003302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Blight KJ. 2007. Allelic variation in the hepatitis C virus NS4B protein dramatically influences RNA replication. J Virol 81:5724–5736. doi: 10.1128/JVI.02481-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dimitrova M, Imbert I, Kieny MP, Schuster C. 2003. Protein-protein interactions between hepatitis C virus nonstructural proteins. J Virol 77:5401–5414. doi: 10.1128/JVI.77.9.5401-5414.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Einav S, Elazar M, Danieli T, Glenn JS. 2004. A nucleotide binding motif in hepatitis C virus (HCV) NS4B mediates HCV RNA replication. J Virol 78:11288–11295. doi: 10.1128/JVI.78.20.11288-11295.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gao L, Aizaki H, He JW, Lai MM. 2004. Interactions between viral nonstructural proteins and host protein hVAP-33 mediate the formation of hepatitis C virus RNA replication complex on lipid raft. J Virol 78:3480–3488. doi: 10.1128/JVI.78.7.3480-3488.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lin C, Wu JW, Hsiao K, Su MS. 1997. The hepatitis C virus NS4A protein: interactions with the NS4B and NS5A proteins. J Virol 71:6465–6471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gouttenoire J, Penin F, Moradpour D. 2010. Hepatitis C virus nonstructural protein 4B: a journey into unexplored territory. Rev Med Virol 20:117–129. doi: 10.1002/rmv.640. [DOI] [PubMed] [Google Scholar]
- 50.Lundin M, Monne M, Widell A, Von Heijne G, Persson MA. 2003. Topology of the membrane-associated hepatitis C virus protein NS4B. J Virol 77:5428–5438. doi: 10.1128/JVI.77.9.5428-5438.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gouttenoire J, Montserret R, Kennel A, Penin F, Moradpour D. 2009. An amphipathic alpha-helix at the C terminus of hepatitis C virus nonstructural protein 4B mediates membrane association. J Virol 83:11378–11384. doi: 10.1128/JVI.01122-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.David N, Yaffe Y, Hagoel L, Elazar M, Glenn JS, Hirschberg K, Sklan EH. 2015. The interaction between the hepatitis C proteins NS4B and NS5A is involved in viral replication. Virology 475:139–149. doi: 10.1016/j.virol.2014.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Paul D, Romero-Brey I, Gouttenoire J, Stoitsova S, Krijnse-Locker J, Moradpour D, Bartenschlager R. 2011. NS4B self-interaction through conserved C-terminal elements is required for the establishment of functional hepatitis C virus replication complexes. J Virol 85:6963–6976. doi: 10.1128/JVI.00502-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Einav S, Gerber D, Bryson PD, Sklan EH, Elazar M, Maerkl SJ, Glenn JS, Quake SR. 2008. Discovery of a hepatitis C target and its pharmacological inhibitors by microfluidic affinity analysis. Nat Biotechnol 26:1019–1027. doi: 10.1038/nbt.1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jones DM, Patel AH, Targett-Adams P, McLauchlan J. 2009. The hepatitis C virus NS4B protein can trans-complement viral RNA replication and modulates production of infectious virus. J Virol 83:2163–2177. doi: 10.1128/JVI.01885-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lundin M, Lindström H, Grönwall C, Persson MA. 2006. Dual topology of the processed hepatitis C virus protein NS4B is influenced by the NS5A protein. J Gen Virol 87:3263–3272. doi: 10.1099/vir.0.82211-0. [DOI] [PubMed] [Google Scholar]
- 57.Koch JO, Bartenschlager R. 1999. Modulation of hepatitis C virus NS5A hyperphosphorylation by nonstructural proteins NS3, NS4A, and NS4B. J Virol 73:7138–7146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Neddermann P, Clementi A, De Francesco R. 1999. Hyperphosphorylation of the hepatitis C virus NS5A protein requires an active NS3 protease, NS4A, NS4B, and NS5A encoded on the same polyprotein. J Virol 73:9984–9991. [DOI] [PMC free article] [PubMed] [Google Scholar]