

ABSTRACT
Respiratory syncytial virus (RSV) infection is a common cause of lower respiratory tract illness in infants and children. RSV is a negative-sense, single-strand RNA (ssRNA) virus that mainly infects airway epithelial cells. Accumulating evidence indicates that reactive oxygen species (ROS) production is a major factor for pulmonary inflammation and tissue damage of RSV disease. We investigated immune-responsive gene-1 (IRG1) expression during RSV infection, since IRG1 has been shown to mediate innate immune response to intracellular bacterial pathogens by modulating ROS and itaconic acid production. We found that RSV infection induced IRG1 expression in human A549 cells and in the lung tissues of RSV-infected mice. RSV infection or IRG1 overexpression promoted ROS production. Accordingly, knockdown of IRG1 induction blocked RSV-induced ROS production and proinflammatory cytokine gene expression. Finally, we showed that suppression of IRG1 induction reduced immune cell infiltration and prevented lung injury in RSV-infected mice. These results therefore link IRG1 induction to ROS production and immune lung injury after RSV infection.
IMPORTANCE RSV infection is among the most common causes of childhood diseases. Recent studies identify ROS production as a factor contributing to RSV disease. We investigated the cause of ROS production and identified IRG1 as a critical factor linking ROS production to immune lung injury after RSV infection. We found that IRG1 was induced in A549 alveolar epithelial cells and in mouse lungs after RSV infection. Importantly, suppression of IRG1 induction reduced inflammatory cell infiltration and lung injury in mice. This study links IRG1 induction to oxidative damage and RSV disease. It also uncovers a potential therapeutic target in reducing RSV-caused lung injury.
INTRODUCTION
Human infection with respiratory syncytial virus (RSV) is a major cause of childhood disease of lower respiratory tract infection with common cold-like symptoms. The infection is frequently associated with the development of bronchospasm and bronchiolitis, particularly in children less than 1 year old. Globally, RSV is responsible for over 33 million new episodes of acute lower respiratory tract infection in children, with at least 3.4 million severe cases that require hospital admissions each year (1, 2). A growing body of evidence shows that RSV infection has become a significant burden in the elderly in industrialized countries (3–6). In addition, RSV infection may contribute to the onset of development of type 2 diabetes (7). Proinflammatory response in the lung is thought to play a fundamental role in RSV disease, but the mechanisms regulating RSV disease and long-term consequences are incompletely defined (8–13).
RSV is a negative-sense, single-strand RNA virus of the family Paramyxoviridae, which also includes common respiratory viruses such as those causing measles and mumps. Human RSV infects polarized epithelial cells in the airway and, to lesser degrees, macrophages and dendritic cells (14, 15). RSV stimulates Toll-like receptor 4 (TLR4), TLR2, and the intracellular sensors RIGI and TLR3, and it orchestrates an antiviral milieu involving interferon β and interferon-inducible genes (ISGs) (16, 17). The innate immune response to the fusion protein of RSV is mediated by TLR4 and CD14, leading to the induction of proinflammatory genes (18, 19). TLR4 expression also sensitizes airway epithelial cells to endotoxin and mediates RSV bronchiolitis (20, 21). Although proinflammatory cytokine production and dysregulation of IFN production have been identified as a primary cause of RSV disease (2, 9, 10, 14, 22), accumulating evidence suggests that reactive oxygen species (ROS) have a critical role in RSV disease (23–25). RSV infection induces rapid ROS production that leads to chemokine induction and IRF pathway activation (26–29). Under physiological conditions, ROS are produced as a normal process of cellular metabolism. However, uncontrolled activation of ROS production causes tissue damage, vascular barrier dysfunction, and acute and chronic inflammation by the induction of proinflammatory cytokines and chemokines (30, 31). This, in turn, promotes proinflammatory cell infiltration that exacerbates lung inflammation and induces tissue damage (32, 33). Accordingly, antioxidant treatment ameliorates RSV-induced pulmonary inflammation and disease (23, 29). Reduced expression of antioxidant enzymes has been detected at late stages of RSV infection (34). Lack of antioxidant enzyme expression may contribute to increased production of ROS. The factors contributing to ROS production during RSV infection remain uninvestigated.
Viruses exploit cellular metabolism to provide the energy and biosynthetic building blocks required for their replication. The innate immune system activation and cell metabolic state are intricately linked (35, 36). A recent study identified immune response gene 1 (IRG1) as a critical player linking metabolism to innate immunity in oxidative stress response (37). IRG1 was originally identified as a highly inducible gene in murine macrophages following lipopolysaccharide stimulation (38) and is upregulated by proinflammatory cytokines and by viral and bacterial infections (39–45). The IRG1 gene is highly conserved among different species. Both human and mouse IRG1 have demonstrated activity in catalyzing the conversion of cis-aconitate of the tricarboxylic acid (TCA) cycle to itaconic acid, which was attributed to antibacterial immunity (46, 47). In addition, IRG1 functions within the mitochondria of activated macrophage-lineage cells to enhance the utilization of energetically efficient fatty acid β-oxidation. IRG1-depleted macrophage-lineage cells are impaired in their ability to utilize fatty acids as an energy substrate for oxidative phosphorylation-derived mitochondrial ROS production, resulting in defective antibacterial response (37). The infection by some viruses, including RSV, also promotes IRG1 expression (39, 42, 48, 49). RSV infection increases cellular bioenergetics, characterized by increased mitochondrial O2 consumption and ATP synthesis that is necessary for viral replication (50). What role IRG1 plays during virus infection has not been reported. In this study, we investigated IRG1 induction during RSV infection in mice and of airway epithelial cells and found that IRG1 expression was responsible for ROS production.
MATERIALS AND METHODS
Cells, chemicals, and virus.
Human A549 cells (ATCC CCL-185) and HeLa cells (ATCC CCL-2) were purchased from the ATCC (Manassas, VA, USA), and the Vero cell line (ATCC CCL-81; African green monkey kidney epithelial cells) was purchased from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). Dulbecco's modified Eagle's medium (DMEM), fetal bovine serum, nonessential amino acids, GlutaMAX, and antibiotics (penicillin, streptomycin, and amphotericin B) were purchased from Life Technologies (Carlsbad, CA, USA) and were used to culture the cells. The cells were routinely screened for Mycoplasma contamination. RSV was a type A strain and was propagated in A549 cells as we previously described (51). It is known that ultracentrifugation tends to result in significant loss of virus infectivity. We also used unpurified virus for cell culture studies. The virus stocks were stored at −70°C.
Chemicals were purchased from Sigma-Aldrich (Shanghai, China) except as otherwise stated. 2-(N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl) amino)-2-deoxyglucose (2-NBDG) (N13195) was purchased from Life Technologies. 2′,7′-Dichlorofluorescein diacetate (DCFH-DA) was purchased from Beyotime Biotechnology (Haimen, China). pCMV6-FLAG-IRG1 for mammalian expression of murine IRG1 was purchased from Sino Biological Inc. (Beijing). Lipofectamine 2000 (Life Technologies) was used for transfection studies.
Mouse anti-RSV F0 and F1 proteins (clone 2F7; sc-101362) was purchased from Santa Cruz Biotechnology, and rabbit anti-IRG1 (ab122624) was from Abcam Shanghai. The antibody, generated using an immunogen of 100 amino acid residues of human origin, recognizes both human and mouse IRG1. Mouse anti-FLAG was purchased from ABmart (Shanghai, China), and anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (MB001) was from Bioworld Technology (Minneapolis, MN). Horseradish peroxidase (HRP)-conjugated and Alexa-labeled secondary antibodies were purchased from Sigma-Aldrich and Life Technologies, respectively.
RT-PCR and real-time PCR study.
A549 cells in 6-well plates were uninfected (mock treated) or infected with RSV as indicated. Total RNA was extracted from those samples using TRIzol reagent (Life Technologies). RNA (1 μg) was reverse transcribed into cDNA using reverse transcriptase Moloney murine leukemia virus (RNase H-free; 2641A; TaKaRa) and used for PCR amplification or real-time PCR quantitation (qPCR). The qPCR was carried out using SYBR green master mix (Q141-02/03; Vazyme, Nanjing, China) on a 7300 real-time PCR system (Applied Biosystems). The data were analyzed using the 2−ΔΔCT method (52). We first normalized IRG1 values to GAPDH (housekeeping) values and then normalized those in infected cells to mock-treated cells to determine fold changes. The ratios in mock-treated samples were assigned arbitrarily as 1. The primers for human genes were the following (forward and reverse): IRG1, 5′-CTACCCACTGGGTGGCA and 5′-TCCTCCTGGCTCAGTGG; interleukin-6 (IL-6), 5′-AGACAGCCACTCACCTCTTC and 5′-CTGGCATTTGTGGTTGGGT; IL-1β, 5′-CCAGCTACGAATCTCCGACC and 5′-TATCCTGTCCCTGGAGGTGG; tumor necrosis factor alpha (TNF-α), 5′-TCTCCTTCCTGATCGTGGCA and 5′-GGGTTTGCTACAACATGGGC; GAPDH, 5′-ACAGTCAGCCGCATCTTCTT and 5′-ACGACCAAATCCGTTGACTC.
Western blotting.
Total cell lysates were prepared using a lysis buffer containing 150 mM NaCl, 50 mM Tris-HCl (pH 7.4), 1% NP-40, and a cocktail of protease inhibitors (Roche). The proteins were separated by SDS-PAGE and transferred to a polyvinylidene difluoride (PVDF) membrane (Millipore) for immunoblot analysis using an enhanced chemiluminescence (ECL) reagent kit. The images were captured using a ChemiScope imaging system (Shanghai, China).
siRNA knockdown.
The short interfering RNA (siRNA) or scrambled control siRNA (scRNA) was transfected into A549 cells using Lipofectamine 2000. The cells were harvested for analysis of knockdown effect by Western blotting. The siRNAs against human IRG1 were synthesized by GenePharma (Shanghai, China), and the sequences were the following (sense and antisense strands): IRG1 #1, 5′-GAGGAUGAUUCUAGACACUTT-3′ and 5′-AGUGUCUAGAAUCAUCCUCTT-3′; IRG1 #2, 5′-CCAACUGACUACAUUAAGATT-3′ and 5′-UCUUAAUGUAGUCAGUUGGTT-3′; IRG1 #3, 5′-CUGGCUUUCAAUGUUGGUATT-3′ and 5′-UACCAACAUUGAAAGCCAGTT-3′; scRNA, 5′-UUCUCCGAACGUGUCACGUTT-3′ and 5′-ACGUGACACGUUCGGAGAATT-3′.
Glucose uptake assay.
We used a fluorescent probe to measure glucose uptake (53). Briefly, mock-treated or RSV-infected A549 cells (multiplicity of infection [MOI] of 1 for 24 h) were switched to DMEM (no glucose; Life Technologies) for 2 h, and then 2-NBDG at a final concentration of 30 μM was added. The cells were collected after another 45 min, and 2-NBDG uptake was analyzed by flow cytometry (FACS Aria; BD Biosciences) using 488 nm for excitation and 540 nm for detection. FlowJo software (version 7.6.1; TreeStar, Ashland, OR) was used for data analysis and measurement of fluorescence-positive cells (M2) and the geometric mean of fluorescence intensity (GMFI).
Metabolite extraction, derivatization, and conditions for GC-MS analysis.
For gas chromatography-mass spectrometry (GC-MS) analysis of organic compounds in mock-treated or RSV-infected samples (MOI of 1 for 36 h), the samples were prepared essentially as reported previously (47). Briefly, A549 cells grown in 10-cm dishes were rinsed with ice-cold phosphate-buffered saline (PBS). The cells were collected into 0.7 ml ice-cold water with a cell scraper. After transferring to 1.5-ml Eppendorf tubes, the lipids were removed by extraction with 0.6 ml methanol-chloroform (1:1). The aqueous phase was collected into specific GC glass vials and evaporated under vacuum using a refrigerated SpeedVac concentrator. The dried metabolites were dissolved in pyrimidine containing 2% methoxyamine hydrochloride and trimethylsilylized by reacting with trimethylsilyl chloride.
GC-MS studies were performed using an Agilent 6890 GC equipped with a 30-m DB-35 MS capillary column and MS operated under electron impact ionization at 70 eV. The MS source was held at 230°C and the quadrupole at 150°C. The detector was operated in scan mode, and 1 μl of derivatized sample was injected in splitless mode. Helium was used as a carrier gas at a flow rate of 1 ml/min. The GC oven temperature was held at 80°C for 6 min and then increased at 6°C/min to 300°C and maintained at that temperature for another 10 min, followed by an increase at 10°C/min to 325°C and holding for 4 min. The run time of one sample was 59 min.
ROS production assay.
ROS production was assayed at 24 h after RSV infection (MOI of 1) using DCFH-DA by following a protocol provided by the manufacturer (Beyotime). The mock-treated or RSV-infected cells were rinsed with serum-free medium (SFM) 3 times and then were fed with DCFH-DA fluorescent probe (at 5 μM) in fresh SFM for another hour. At the end of incubation, the cells were washed 3 times with PBS and collected by trypsin-EDTA treatment. ROS production was measured by flow cytometry with an excitation wavelength of 488 nm and emission wavelength of 525 nm. FlowJo software was used to determine fluorescence-positive cells (M2) and the GMFI.
Pulmonary infection in mouse model.
Animal studies were approved by the Medical School of Nanjing University for Animal Use and Care Committee in accordance with the guidelines of the U.S. NIH. Female BALB/c mice, 4 to 6 weeks old, were obtained from the Model Animal Research Center of Nanjing University. The animals were kept in a specific-pathogen-free environment and fed food and water ad libitum. For intranasal instillation, the animals were sedated with ketamine at 100 mg/kg of body weight intraperitoneally.
To conduct RNA interference (RNAi) studies in mice, we first performed in vitro screening studies to identify an siRNA that blocks lipopolysaccharide (LPS)-induced IRG1 expression using Raw264.7 cells. The siRNA that significantly blocked LPS-induced IRG1 expression (sequence 1) was then used in animal studies. Lightly sedated mice (5 per group) were given 5 nmol siRNA or a scrambled control siRNA in Opti-MEM-Lipofectamine 2000 (7:1; total volume, 100 μl) via nasal instillation as reported previously (54). Forty-eight h post-siRNA administration, RSV (5 × 106 PFU/100 μl/mouse) was delivered via nasal instillation. The animals were monitored daily and were killed at 48 h postinfection (pi). The lungs were removed and weighed, and the right lung was used for titration of infectious virus by plaque-forming assay and for RNA extraction. The left lung was used for flow cytometry analysis of T lymphocyte and macrophage infiltration or for hematoxylin and eosin (H&E) staining by first flushing with ice-cold sterile PBS and then with 10% formalin. The samples were then submerged in formalin for fixation and subsequent histological sections.
Sequences of siRNA used to knock down mouse IRG1 (mIRG1) expression are the following (siRNA #1 was selected and used for the in vivo studies with the above-mentioned scrambled control oligonucleotides as a control): mIRG1 #1, 5′-CAAGCUUCGACACGCUAUATT-3′ and 5′-UAUAGCGUGUCGAAGCUUGTT-3′; mIRG1 #2, 5′-CACCUUGUGACACCAGAAATT-3′ and 5′-UUUCUGGUGUCACAAGGUGTT-3′; mIRG1 #3, 5′-GGACGAUUAAUGCACUUCUTT-3′ and 5′-AGAAGUGCAUUAAUCGUCCTT-3′. The following primer pairs were used for murine gene detection by PCR (forward and reverse): IL-6, 5′-CCACTTCACAAGTCGGAGGC and 5′-TGCAAGTGCATCATCGTTGTTC; IL-8, 5′-CGGCAATGAAGCTTCTGTAT and 5′-CCTTGAAACTCTTTGCCTCA; monocyte chemoattractant protein 1 (MCP-1), 5′-ACAGCACCAAGGTCCATTCC and 5′-TCCTGCCTGAGAATGCTTGG; TNF-α, 5′-ATCCGCGACGTGGAACTGGC and 5′-CCATGCCGTTGGCCAGGAGG; GAPDH, 5′-ATCTCCGCCCCTTCTGCCGA and 5′-CCACAGCCTTGGCAGCACCA.
Flow cytometry analysis for macrophage and T lymphocyte infiltration.
Single-cell suspensions were prepared from lungs by being cut into small fragments and were digested at 37°C with type I collagenase (3 mg/ml) and DNase I (30 μg/ml) in RPMI 1640 medium for 45 min. Digested lungs were mechanically disrupted by passage through a sterile 100-μm strainer (Falcon; BD Biosciences) using the flat portion of a plunger from a 3-ml syringe followed by passage through an additional 40-μm strainer (Falcon; BD Biosciences). Red blood cells were lysed with 150 mM NH4Cl, 10 mM KHCO3, and 0.1 mM EDTA. After washing with ice-cold PBS, the cells were collected, resuspended in PBS, and stained with allophycocyanin (APC)-labeled anti-F4/80 and fluorescein isothiocyanate (FITC)-labeled anti-CD11b, APC-labeled anti-CD3 and phycoerythrin (PE)-labeled anti-CD8, or APC-labeled anti-CD3 and FITC-labeled anti-CD4 (eBioscience, San Diego, CA). The samples were analyzed on a FACSCalibur. FlowJo software was used for data analysis.
RESULTS
RSV infection upregulates IRG1 expression in A549 cells.
IRG1 is a highly inducible gene by viral and bacterial infections. To investigate whether IRG1 played a role in RSV infection, we first examined IRG1 expression during RSV infection of A549 cells, a type II alveolar epithelial cell of human origin. IRG1 expression was relatively low in A549 cells. RSV infection induced increased expression of IRG1, which was first detected by RT-PCR and quantified by real-time PCR (qPCR). As shown in Fig. 1A and B, RSV infection dose- and time-dependently induced IRG1 expression as early as 30 min pi. The induction was verified by immunoblotting studies (Fig. 1C). IRG1 induction was noticeable at approximately 2 h pi and became more significant at approximately 9 to 12 h pi. The induction was dependent on RSV infection, since inclusion of ribavirin during infection or deactivation of RSV by UV irradiation or heat treatment significantly suppressed or ablated IRG1 expression (Fig. 1D).
FIG 1.

Induction of IRG1 in RSV-infected A549 cells. (A and B) Dose response and time course of IRG1 induction. A549 cells were mock treated or infected with RSV at the MOIs indicated for 24 h or at an MOI of 3 for the times indicated. IRG1 induction was determined by RT-PCR (upper) and qPCR (lower). Data in the bar graphs are means ± SE from duplicate samples. Ctrl, control; rel. exp, relative expression. (C) Immunoblot detection of IRG1 induction. A549 cells were infected with RSV under the conditions indicated. IRG1 expression was detected by Western blotting. RSV F0 protein expression was blotted as a control for infection, and GAPDH was used as a loading control. The numbers under the blots indicate relative levels of IRG1 expression. (D) IRG1 induction depends on RSV infection. A549 cells were infected with RSV in the absence or presence of ribavirin (Rib) at various concentrations or treated in parallel with UV-irradiated (UV) or heat-inactivated (HK) RSV. IRG1 expression was detected by Western blotting. GAPDH and F0 expression were determined as controls for sample loading and infection, respectively. All experiments were performed at least 2 times independently.
Transient expression of IRG1 has no effect on RSV infection.
IRG1 was identified as one of three interferon-inducible genes determining the susceptibility to viral infections in the brain (48). We therefore tested whether IRG1 expression inhibited RSV infection. To this end, we chose to transfect HEK293T cells with a plasmid for mouse IRG1 expression, since the cells are more readily transfected than A549 cells, and tested whether overexpression of IRG1 had an effect on RSV infection. IRG1 expression showed little effect on RSV production or RSV F0 protein expression (Fig. 2A), indicating that preexisting IRG1 had no effect on RSV infection. IRG1 was reported to function as a suppressor of proinflammatory cytokine expression (55). We found that transient expression of IRG1 in A549 cells did not promote proinflammatory cytokine expression, as was determined by RT-PCR. The protein seemed to potentiate IL-1β and IL-6 expression in RSV-infected samples (Fig. 2B, right lanes).
FIG 2.
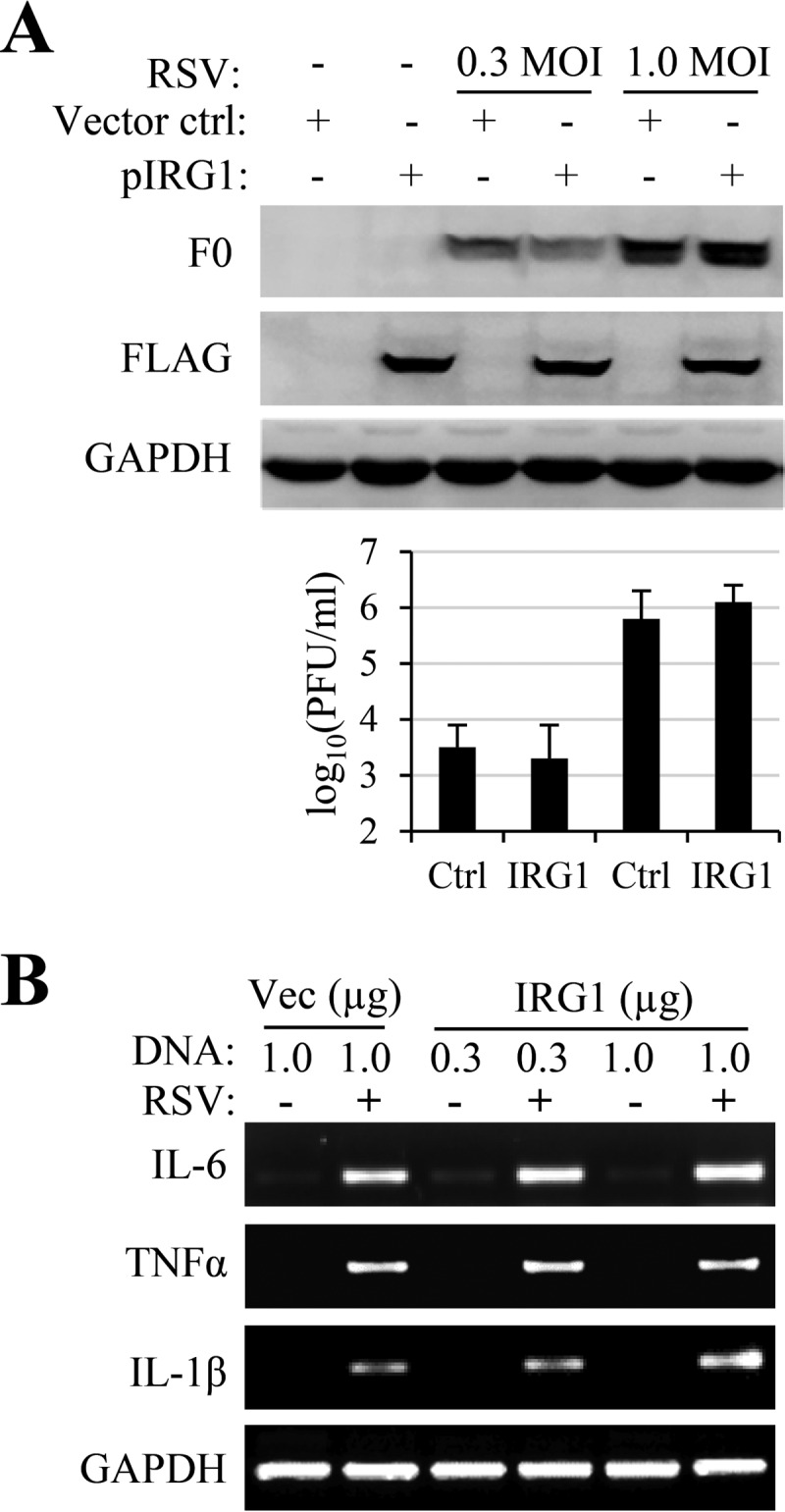
Overexpression of IRG1 on RSV infection and cytokine gene expression. (A) HEK 293T cells were transfected with pCMV-FLAG-IRG1 (pIRG1) or p3x-FLAG-CMV (vector control). The cells were then infected with RSV at an MOI of 0.3 or 1.0, respectively. Virus production and F0 production were determined, and viral titers are reported as means ± standard deviations (SD) from triplicate samples. The experiments were performed 3 times independently. (B) A549 cells were transfected with p3x-FLAG-CMV (Vec; at 1 μg/well) or with pCMV-IRG1 (IGR1) at 0.3 and 1.0 μg/well. The cells were left uninfected or were infected with RSV (at an MOI of 3) for 24 h. Proinflammatory cytokine gene expression was determined by RT-PCR. The experiment was performed 2 times independently.
RSV infection promotes bioenergetics but not increased production of itaconic acid.
We next measured whether there was increased itaconic acid production during RSV infection, since both mouse and, to a lesser degree, human IRG1 have recently been shown to catalyze cis-aconitate conversion to itaconic acid (46, 47). The metabolites from control and RSV-infected A549 cells were extracted and analyzed as trimethylsilyl derivatives by GC-MS. There was significant accumulation of citrate, isocitrate, and cis-aconitate, intermediates of the tricarboxylic acid (TCA) cycle, from RSV-infected samples (Fig. 3A). This suggests that there was increased bioenergetics during RSV infection, consistent with results from glucose uptake assays (Fig. 3B). However, no significant production of itaconic acid was detected from RSV-infected samples, suggesting that IRG1 did not interpose the TCA cycle during RSV infection.
FIG 3.

RSV infection on glucose uptake and itaconic production. (A) RSV infection promotes production of intermediates of the TCA cycle but not itaconic acid production. Water-soluble metabolites from mock or RSV infection (MOI of 1 for 36 h) were separated by organic aqueous extraction. The metabolites were analyzed by GC-MS as trimethylsilyl derivatives. The identities of citric acid, cis-aconitic acid, isocitric acid, and itaconic acid were determined by comparison to the database and by an itaconic acid standard purchased from Sigma. The signal strength (area) of corresponding peaks in the control (S ctrl) and infected samples (S RSV) was used for comparison of abundance of those compounds. RT, retention time in minutes; ND, not detected; CoA, coenzyme A. (B) RSV infection promotes glucose uptake. A549 cells were left uninfected or were infected with various amounts of RSV (MOI of 1, 3, and 10) for 24 h and tested for glucose uptake by feeding with 2-NBDG, a fluorescent probe for glucose uptake. The percentage of fluorescence-positive cells (M2) and the geometric mean fluorescence intensity (GMFI) for each sample are presented. The experiments were performed 3 times independently.
RSV infection promotes ROS production through IRG1 induction.
IRG1 expression has been linked to ROS production. We therefore questioned whether IRG1 induction correlated with ROS production, since RSV infection promotes ROS production. To determine ROS production, mock-treated or RSV-infected A549 cells were fed with DCFH-DA, a nonfluorescent probe that can be converted to the fluorescent DCF after ROS oxidation. DCF intensity was quantitatively measured by fluorescence-activated cell sorter (FACS) analysis. Consistent with previous reports, RSV infection promoted ROS production dose dependently (Fig. 4A). ROS production was detected at early stages and persisted during RSV infection (Fig. 4B). The effect of IRG1 on ROS production was confirmed by transient expression. As shown in Fig. 4C, expression of IRG1 by transient transfection dose-dependently increased ROS production. Accordingly, knockdown of IRG1 expression by RNAi markedly reduced ROS production (Fig. 4D), indicating that RSV promotes ROS production through IRG1 expression. We found that suppression of IRG1 induction also blocked RSV-induced proinflammatory gene induction (Fig. 4E).
FIG 4.

IRG1 promotes ROS production. ROS production in mock-treated or RSV-infected A549 cells was detected by FACS analysis using DCFH-DA as a probe. (A) Dose response of ROS production to RSV infection. ROS production was detected at 24 h pi. The percentage of fluorescence-positive cells (M2) and the geometric mean fluorescence intensity (GMFI) for each sample are presented. (B) Time course of ROS production during RSV infection. The cells were infected with RSV at an MOI of 3 for the times indicated. M2 values relative to the mock-treated control were plotted as an indicator of increased ROS production. The data are presented as means ± SE from duplicate samples. (C) Transient expression of IRG1 promotes ROS production. A549 cells in 12-well plates were transfected with various amounts of pCMV-FLAG-IRG1 (IRG1). p3x-FLAG-CMV was included as a vector control. ROS production was detected at 24 h posttransfection. (D) Suppression of IRG1 by siRNA reduces ROS production. A549 cells were untreated or were transfected with scRNA or with siRNA #1 for human IRG1. The cells were mock treated or RSV infected (MOIs of 1 and 3) at 24 h after transfection. ROS production was determined 24 h later. The M2 values relative to the noninfected controls were plotted. The inset shows the knockdown effect of 3 different siRNAs targeting IRG1. The relative levels of IRG1 expression are presented as numbers beneath the blots. (E) Knockdown of IRG1 expression suppresses cytokine gene induction. A549 cells were transfected with scRNA or siIRG1#1 for 24 h. The cells then were infected with RSV (MOI of 3) for another 24 h. Gene expression was detected by RT-PCR. All experiments were performed at least 2 times independently.
Suppression of IRG1 expression in mice prevents lung injury by RSV infection.
ROS production has recently been identified as a factor contributing to RSV disease. We performed animal studies to determine whether suppression of IRG1 induction had an impact on RSV disease. To this end, we first selected an siRNA (siRNA #1) that showed significant effect in knocking down IRG1 induction in LPS-treated Raw264.7 cells (by approximately 70%). siRNA #1 or a scrambled control siRNA (scRNA) was then administered to BALB/c mice via intranasal instillation. The animals were then infected with RSV at 48 h after siRNA treatment. The animals were examined 5 days later for signs of inflammation and viral production. RSV infection caused body weight drop initially that started to recover on the 2nd day postinfection (Fig. 5A). When examined for IRG1 expression in mouse lungs, we found that RSV infection caused profound induction of IRG1, which was significantly suppressed by siIRG1 treatment (Fig. 5B). We next examined histological changes of H&E-stained samples to address whether siIRG1 treatment had an impact on the diseases. As shown in Fig. 5C, RSV infection recapitulated previously reported abnormal histology of lung sections. H&E staining showed alveolar walls and alveolar spaces filled with moderate to severe inflammatory infiltrates of neutrophils, macrophages, and lymphocytes in the mock-treated or scRNA-treated group (Fig. 5C, RSV, scRNA+RSV). Samples from the IRG1 knockdown group had significant reduction of interstitial mononuclear cell infiltration and improvement in the overall structure of the alveolar spaces (Fig. 5C, siRNA), indicating suppression of IRG1 induction ameliorated inflammatory conditions. In agreement with results from the HE staining study, we noticed that tissue samples from the siIRG1-treated group also had reduced expression of IL-6, IL-8, and MCP-1 (Fig. 5D). In addition, the viral titers were lowered by approximately 0.8 log in siIRG1-treated samples compared to that in the mock-treated control group (Fig. 5E).
FIG 5.

Effect of IRG1 knockdown on lung injury by RSV infection. Mice were left untreated or were treated with a control siRNA (scRNA) or with siRNA targeting mouse IRG1 (siIRG1) for 48 h. The animals were left uninfected or were infected with RSV (5 × 106 PFU in 100 μl PBS). (A) Body weight changes. Body weight prior to siRNA treatment (day −2) was used as the baseline (100%) for comparison. Data are presented as means ± SD (for clarity in plotting). (B) IRG1 expression in untreated or RSV-challenged animals. Lung tissues were homogenized and the lysates were analyzed for IRG1 expression. Three mice from each group were used. (C) Lung sections of RSV-challenged mice. Images show representative H&E-stained sections from uninfected (control), infected (RSV), and treated and infected animals (scRNA+RSV and siRNA+RSV). The bar represents 100 μm. (D) Gene expression of proinflammatory cytokines and chemokines in lung tissues determined by qPCR. Cytokine expression in lung tissues of three individual mice was determined by qPCR. Data are normalized to uninfected controls and are represented as means ± SD. (E) Viral load determination. Lung tissues of 3 mice were homogenized and viral titers were determined by plaque-forming assay. Data are presented as means ± SD for 3 mice from duplicate measurements. A Student t test was performed for statistical analysis. (F) FACS analysis of T lymphocytes and macrophages in lung tissues. Representative results are shown. The panel of bar graphs summarizes quantitative measurement of CD3+ CD4+, CD3+ CD8+, or F4/80+ CD11b+ cell populations in those samples. The data are means ± SD from 3 to 5 mice. All experiments were performed 2 times independently, except the FACS study, which was performed once using 2 mice per group independently.
Finally, we performed FACS analysis to determine T lymphocyte and macrophage infiltration. There were significantly increased populations of CD8+ and CD4+ T lymphocytes and CD11b- and F4/80-positive cells in the lung tissues of RSV-infected mice (Fig. 5F). Suppression of IRG1 induction resulted in significant reduction of those cells.
Together, these results demonstrated that suppression of IRG1 induction ameliorates the adverse effect of RSV infection on lung injury.
DISCUSSION
RSV infection is the primary cause of childhood disease of infants and young children. The infection is exquisitely restricted to the mucosa of the respiratory tract, since the virus does not normally replicate outside the bronchopulmonary tree. Although inflammatory mediators play a major role in the pathogenesis of RSV disease, glucocorticoids, anti-inflammatory in nature, have been shown to be ineffective in the treatment of RSV-induced bronchiolitis and wheezing (56–58). Several groups have recently shown ROS may be important regulators of RSV-induced cellular signaling and may play a fundamental role in the pathogenesis of RSV-associated lung injury due to oxidative stress and the production of inflammation mediators (23). We studied IRG1 induction during RSV infection of human airway epithelial cells and in a mouse model. We found that IRG1 induction regulates ROS production. Casola and colleagues showed that antioxidants ameliorate pulmonary inflammation in RSV disease (23, 29). Consistent with their findings, our data show that suppression of IRG1 induction improves RSV-caused lung injury, underlining the importance of ROS production to RSV disease.
Virus infection promotes bioenergetics for energy metabolism and production of building blocks for viral assembly. Michelucci and colleagues recently identified IRG1 as a cis-aconitate decarboxylase that catalyzes the conversion of cis-aconitate to itaconic acid, an inhibitor of isocitrate lyase. Some bacteria, when grown under a carbon source limitation, synthesize isocitrate lyase for energy metabolism through the glyoxylate shunt, effectively linking metabolism to antibacterial immunity (47). Cho et al. reported that IRG1 was among three upregulated ISGs with antiviral effects against different neurotropic viruses (48). Although RSV infection promotes IRG1 expression, our data show that overexpression of IRG1 had no effect on RSV infection in 293T or A549 cells. Whether an unknown host factor(s) contributes to the observations is unknown, since epigenetic state and microRNAs have been implicated in ISG antiviral activity specific to granule cell neurons (48). Different from those reports, our work shows that IRG1 regulates ROS production during RSV infection. Importantly, suppression of IRG1 induction resulted in reduction in lung injury after RSV infection, uncovering a novel target for therapeutically reducing lung injury by RSV infection.
ACKNOWLEDGMENT
We thank Wei Zhang for proofreading the manuscript.
Funding Statement
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
REFERENCES
- 1.Nair H, Nokes DJ, Gessner BD, Dherani M, Madhi SA, Singleton RJ, O'Brien KL, Roca A, Wright PF, Bruce N, Chandran A, Theodoratou E, Sutanto A, Sedyaningsih ER, Ngama M, Munywoki PK, Kartasasmita C, Simoes EA, Rudan I, Weber MW, Campbell H. 2010. Global burden of acute lower respiratory infections due to respiratory syncytial virus in young children: a systematic review and meta-analysis. Lancet 375:1545–1555. doi: 10.1016/S0140-6736(10)60206-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Collins PL, Graham BS. 2008. Viral and host factors in human respiratory syncytial virus pathogenesis. J Virol 82:2040–2055. doi: 10.1128/JVI.01625-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Matias G, Taylor R, Haguinet F, Schuck-Paim C, Lustig R, Shinde V. 2014. Estimates of mortality attributable to influenza and RSV in the United States during 1997-2009 by influenza type or subtype, age, cause of death, and risk status. Influenza Other Respir Viruses 8:507–515. doi: 10.1111/irv.12258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fleming DM, Taylor RJ, Lustig RL, Schuck-Paim C, Haguinet F, Webb DJ, Logie J, Matias G, Taylor S. 2015. Modelling estimates of the burden of respiratory syncytial virus infection in adults and the elderly in the United Kingdom. BMC Infect Dis 15:443. doi: 10.1186/s12879-015-1218-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang Y, Yuan L, Zhang Y, Zhang X, Zheng M, Kyaw MH. 2015. Burden of respiratory syncytial virus infections in China: systematic review and meta-analysis. J Glob Health 5:020417. doi: 10.7189/jogh.05.020417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Falsey AR, Hennessey PA, Formica MA, Cox C, Walsh EE. 2005. Respiratory syncytial virus infection in elderly and high-risk adults. N Engl J Med 352:1749–1759. doi: 10.1056/NEJMoa043951. [DOI] [PubMed] [Google Scholar]
- 7.Chen R, Mias GI, Li-Pook-Than J, Jiang L, Lam HY, Chen R, Miriami E, Karczewski KJ, Hariharan M, Dewey FE, Cheng Y, Clark MJ, Im H, Habegger L, Balasubramanian S, O'Huallachain M, Dudley JT, Hillenmeyer S, Haraksingh R, Sharon D, Euskirchen G, Lacroute P, Bettinger K, Boyle AP, Kasowski M, Grubert F, Seki S, Garcia M, Whirl-Carrillo M, Gallardo M, Blasco MA, Greenberg PL, Snyder P, Klein TE, Altman RB, Butte AJ, Ashley EA, Gerstein M, Nadeau KC, Tang H, Snyder M. 2012. Personal omics profiling reveals dynamic molecular and medical phenotypes. Cell 148:1293–1307. doi: 10.1016/j.cell.2012.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Collins PL, Crowe JEJ. 2007. Respiratory syncytial virus and metapneumovirus. In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE (ed), Fields virology, 5th ed Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 9.Bueno SM, Gonzalez PA, Riedel CA, Carreno LJ, Vasquez AE, Kalergis AM. 2011. Local cytokine response upon respiratory syncytial virus infection. Immunol Lett 136:122–129. doi: 10.1016/j.imlet.2010.12.003. [DOI] [PubMed] [Google Scholar]
- 10.Goritzka M, Makris S, Kausar F, Durant LR, Pereira C, Kumagai Y, Culley FJ, Mack M, Akira S, Johansson C. 2015. Alveolar macrophage-derived type I interferons orchestrate innate immunity to RSV through recruitment of antiviral monocytes. J Exp Med 212:699–714. doi: 10.1084/jem.20140825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ogra PL. 2004. Respiratory syncytial virus: the virus, the disease and the immune response. Paediatr Respir Rev 5(Suppl A):S119–S126. doi: 10.1016/S1526-0542(04)90023-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lambert L, Sagfors AM, Openshaw PJ, Culley FJ. 2014. Immunity to RSV in early-life. Front Immunol 5:466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gonzalez PA, Bueno SM, Carreno LJ, Riedel CA, Kalergis AM. 2012. Respiratory syncytial virus infection and immunity. Rev Med Virol 22:230–244. doi: 10.1002/rmv.1704. [DOI] [PubMed] [Google Scholar]
- 14.Villenave R, Thavagnanam S, Sarlang S, Parker J, Douglas I, Skibinski G, Heaney LG, McKaigue JP, Coyle PV, Shields MD, Power UF. 2012. In vitro modeling of respiratory syncytial virus infection of pediatric bronchial epithelium, the primary target of infection in vivo. Proc Natl Acad Sci U S A 109:5040–5045. doi: 10.1073/pnas.1110203109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fonceca AM, Flanagan BF, Trinick R, Smyth RL, McNamara PS. 2012. Primary airway epithelial cultures from children are highly permissive to respiratory syncytial virus infection. Thorax 67:42–48. doi: 10.1136/thoraxjnl-2011-200131. [DOI] [PubMed] [Google Scholar]
- 16.Barik S. 2013. Respiratory syncytial virus mechanisms to interfere with type 1 interferons. Curr Top Microbiol Immunol 372:173–191. [DOI] [PubMed] [Google Scholar]
- 17.Borchers AT, Chang C, Gershwin ME, Gershwin LJ. 2013. Respiratory syncytial virus–a comprehensive review. Clin Rev Allergy Immunol 45:331–379. doi: 10.1007/s12016-013-8368-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kurt-Jones EA, Popova L, Kwinn L, Haynes LM, Jones LP, Tripp RA, Walsh EE, Freeman MW, Golenbock DT, Anderson LJ, Finberg RW. 2000. Pattern recognition receptors TLR4 and CD14 mediate response to respiratory syncytial virus. Nat Immunol 1:398–401. doi: 10.1038/80833. [DOI] [PubMed] [Google Scholar]
- 19.Haynes LM, Moore DD, Kurt-Jones EA, Finberg RW, Anderson LJ, Tripp RA. 2001. Involvement of toll-like receptor 4 in innate immunity to respiratory syncytial virus. J Virol 75:10730–10737. doi: 10.1128/JVI.75.22.10730-10737.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Monick MM, Yarovinsky TO, Powers LS, Butler NS, Carter AB, Gudmundsson G, Hunninghake GW. 2003. Respiratory syncytial virus up-regulates TLR4 and sensitizes airway epithelial cells to endotoxin. J Biol Chem 278:53035–53044. doi: 10.1074/jbc.M308093200. [DOI] [PubMed] [Google Scholar]
- 21.Caballero MT, Serra ME, Acosta PL, Marzec J, Gibbons L, Salim M, Rodriguez A, Reynaldi A, Garcia A, Bado D, Buchholz UJ, Hijano DR, Coviello S, Newcomb D, Bellabarba M, Ferolla FM, Libster R, Berenstein A, Siniawaski S, Blumetti V, Echavarria M, Pinto L, Lawrence A, Ossorio MF, Grosman A, Mateu CG, Bayle C, Dericco A, Pellegrini M, Igarza I, Repetto HA, Grimaldi LA, Gudapati P, Polack NR, Althabe F, Shi M, Ferrero F, Bergel E, Stein RT, Peebles RS, Boothby M, Kleeberger SR, Polack FP. 2015. TLR4 genotype and environmental LPS mediate RSV bronchiolitis through Th2 polarization. J Clin Investig 125:571–582. doi: 10.1172/JCI75183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Goritzka M, Durant LR, Pereira C, Salek-Ardakani S, Openshaw PJ, Johansson C. 2014. Alpha/beta interferon receptor signaling amplifies early proinflammatory cytokine production in the lung during respiratory syncytial virus infection. J Virol 88:6128–6136. doi: 10.1128/JVI.00333-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Garofalo RP, Kolli D, Casola A. 2013. Respiratory syncytial virus infection: mechanisms of redox control and novel therapeutic opportunities. Antioxid Redox Signal 18:186–217. doi: 10.1089/ars.2011.4307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hosakote YM, Jantzi PD, Esham DL, Spratt H, Kurosky A, Casola A, Garofalo RP. 2011. Viral-mediated inhibition of antioxidant enzymes contributes to the pathogenesis of severe respiratory syncytial virus bronchiolitis. Am J Respir Crit Care Med 183:1550–1560. doi: 10.1164/rccm.201010-1755OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jamaluddin M, Tian B, Boldogh I, Garofalo RP, Brasier AR. 2009. Respiratory syncytial virus infection induces a reactive oxygen species-MSK1-phospho-Ser-276 RelA pathway required for cytokine expression. J Virol 83:10605–10615. doi: 10.1128/JVI.01090-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu T, Castro S, Brasier AR, Jamaluddin M, Garofalo RP, Casola A. 2004. Reactive oxygen species mediate virus-induced STAT activation: role of tyrosine phosphatases. J Biol Chem 279:2461–2469. doi: 10.1074/jbc.M307251200. [DOI] [PubMed] [Google Scholar]
- 27.Hosakote YM, Liu T, Castro SM, Garofalo RP, Casola A. 2009. Respiratory syncytial virus induces oxidative stress by modulating antioxidant enzymes. Am J Respir Cell Mol Biol 41:348–357. doi: 10.1165/rcmb.2008-0330OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Grandvaux N, Mariani M, Fink K. 2015. Lung epithelial NOX/DUOX and respiratory virus infections. Clin Sci (Lond) 128:337–347. doi: 10.1042/CS20140321. [DOI] [PubMed] [Google Scholar]
- 29.Castro SM, Guerrero-Plata A, Suarez-Real G, Adegboyega PA, Colasurdo GN, Khan AM, Garofalo RP, Casola A. 2006. Antioxidant treatment ameliorates respiratory syncytial virus-induced disease and lung inflammation. Am J Respir Crit Care Med 174:1361–1369. doi: 10.1164/rccm.200603-319OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mittal M, Siddiqui MR, Tran K, Reddy SP, Malik AB. 2014. Reactive oxygen species in inflammation and tissue injury. Antioxid Redox Signal 20:1126–1167. doi: 10.1089/ars.2012.5149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dickinson BC, Chang CJ. 2011. Chemistry and biology of reactive oxygen species in signaling or stress responses. Nat Chem Biol 7:504–511. doi: 10.1038/nchembio.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Whitsett JA, Alenghat T. 2015. Respiratory epithelial cells orchestrate pulmonary innate immunity. Nat Immunol 16:27–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chow CW, Herrera Abreu MT, Suzuki T, Downey GP. 2003. Oxidative stress and acute lung injury. Am J Respir Cell Mol Biol 29:427–431. doi: 10.1165/rcmb.F278. [DOI] [PubMed] [Google Scholar]
- 34.Komaravelli N, Tian B, Ivanciuc T, Mautemps N, Brasier AR, Garofalo RP, Casola A. 2015. Respiratory syncytial virus infection down-regulates antioxidant enzyme expression by triggering deacetylation-proteasomal degradation of Nrf2. Free Radic Biol Med 88:391–403. doi: 10.1016/j.freeradbiomed.2015.05.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pearce EL, Poffenberger MC, Chang CH, Jones RG. 2013. Fueling immunity: insights into metabolism and lymphocyte function. Science 342:1242454. doi: 10.1126/science.1242454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Odegaard JI, Chawla A. 2013. The immune system as a sensor of the metabolic state. Immunity 38:644–654. doi: 10.1016/j.immuni.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hall CJ, Boyle RH, Astin JW, Flores MV, Oehlers SH, Sanderson LE, Ellett F, Lieschke GJ, Crosier KE, Crosier PS. 2013. Immunoresponsive gene 1 augments bactericidal activity of macrophage-lineage cells by regulating beta-oxidation-dependent mitochondrial ROS production. Cell Metab 18:265–278. doi: 10.1016/j.cmet.2013.06.018. [DOI] [PubMed] [Google Scholar]
- 38.Lee CG, Jenkins NA, Gilbert DJ, Copeland NG, O'Brien WE. 1995. Cloning and analysis of gene regulation of a novel LPS-inducible cDNA. Immunogenetics 41:263–270. [DOI] [PubMed] [Google Scholar]
- 39.Smith J, Sadeyen JR, Paton IR, Hocking PM, Salmon N, Fife M, Nair V, Burt DW, Kaiser P. 2011. Systems analysis of immune responses in Marek's disease virus-infected chickens identifies a gene involved in susceptibility and highlights a possible novel pathogenicity mechanism. J Virol 85:11146–11158. doi: 10.1128/JVI.05499-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Degrandi D, Hoffmann R, Beuter-Gunia C, Pfeffer K. 2009. The proinflammatory cytokine-induced IRG1 protein associates with mitochondria. J Interferon Cytokine Res 29:55–67. doi: 10.1089/jir.2008.0013. [DOI] [PubMed] [Google Scholar]
- 41.Basler T, Jeckstadt S, Valentin-Weigand P, Goethe R. 2006. Mycobacterium paratuberculosis, Mycobacterium smegmatis, and lipopolysaccharide induce different transcriptional and post-transcriptional regulation of the IRG1 gene in murine macrophages. J Leukoc Biol 79:628–638. [DOI] [PubMed] [Google Scholar]
- 42.Janssen R, Pennings J, Hodemaekers H, Buisman A, van Oosten M, de Rond L, Ozturk K, Dormans J, Kimman T, Hoebee B. 2007. Host transcription profiles upon primary respiratory syncytial virus infection. J Virol 81:5958–5967. doi: 10.1128/JVI.02220-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ioannidis I, McNally B, Willette M, Peeples ME, Chaussabel D, Durbin JE, Ramilo O, Mejias A, Flano E. 2012. Plasticity and virus specificity of the airway epithelial cell immune response during respiratory virus infection. J Virol 86:5422–5436. doi: 10.1128/JVI.06757-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rodriguez N, Mages J, Dietrich H, Wantia N, Wagner H, Lang R, Miethke T. 2007. MyD88-dependent changes in the pulmonary transcriptome after infection with Chlamydia pneumoniae. Physiol Genomics 30:134–145. doi: 10.1152/physiolgenomics.00011.2007. [DOI] [PubMed] [Google Scholar]
- 45.Gautam A, Dixit S, Philipp MT, Singh SR, Morici LA, Kaushal D, Dennis VA. 2011. Interleukin-10 alters effector functions of multiple genes induced by Borrelia burgdorferi in macrophages to regulate Lyme disease inflammation. Infect Immun 79:4876–4892. doi: 10.1128/IAI.05451-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Strelko CL, Lu W, Dufort FJ, Seyfried TN, Chiles TC, Rabinowitz JD, Roberts MF. 2011. Itaconic acid is a mammalian metabolite induced during macrophage activation. J Am Chem Soc 133:16386–16389. doi: 10.1021/ja2070889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Michelucci A, Cordes T, Ghelfi J, Pailot A, Reiling N, Goldmann O, Binz T, Wegner A, Tallam A, Rausell A, Buttini M, Linster CL, Medina E, Balling R, Hiller K. 2013. Immune-responsive gene 1 protein links metabolism to immunity by catalyzing itaconic acid production. Proc Natl Acad Sci U S A 110:7820–7825. doi: 10.1073/pnas.1218599110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cho H, Proll SC, Szretter KJ, Katze MG, Gale M Jr, Diamond MS. 2013. Differential innate immune response programs in neuronal subtypes determine susceptibility to infection in the brain by positive-stranded RNA viruses. Nat Med 19:458–464. doi: 10.1038/nm.3108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Preusse M, Tantawy MA, Klawonn F, Schughart K, Pessler F. 2013. Infection- and procedure-dependent effects on pulmonary gene expression in the early phase of influenza A virus infection in mice. BMC Microbiol 13:293. doi: 10.1186/1471-2180-13-293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Alsuwaidi AR, Albawardi A, Almarzooqi S, Benedict S, Othman AR, Hartwig SM, Varga SM, Souid AK. 2014. Respiratory syncytial virus increases lung cellular bioenergetics in neonatal C57BL/6 mice. Virology 454-455:263–269. [DOI] [PubMed] [Google Scholar]
- 51.Guo X, Liu T, Shi H, Wang J, Ji P, Wang H, Hou Y, Tan RX, Li E. 2015. Respiratory syncytial virus infection upregulates NLRC5 and major histocompatibility complex class I expression through RIG-I induction in airway epithelial cells. J Virol 89:7636–7645. doi: 10.1128/JVI.00349-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schmittgen TD, Livak KJ. 2008. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc 3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- 53.Raeisi E, Mir LM. 2012. 2-NBDG, a fluorescent analogue of glucose, as a marker for detecting cell electropermeabilization in vitro. J Membr Biol 245:633–642. doi: 10.1007/s00232-012-9479-6. [DOI] [PubMed] [Google Scholar]
- 54.Stark JM, McDowell SA, Koenigsknecht V, Prows DR, Leikauf JE, Le Vine AM, Leikauf GD. 2002. Genetic susceptibility to respiratory syncytial virus infection in inbred mice. J Med Virol 67:92–100. doi: 10.1002/jmv.2196. [DOI] [PubMed] [Google Scholar]
- 55.Li Y, Zhang P, Wang C, Han C, Meng J, Liu X, Xu S, Li N, Wang Q, Shi X, Cao X. 2013. Immune responsive gene 1 (IRG1) promotes endotoxin tolerance by increasing A20 expression in macrophages through reactive oxygen species. J Biol Chem 288:16225–16234. doi: 10.1074/jbc.M113.454538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Somers CC, Ahmad N, Mejias A, Buckingham SC, Carubelli C, Katz K, Leos N, Gomez AM, DeVincenzo JP, Ramilo O, Jafri HS. 2009. Effect of dexamethasone on respiratory syncytial virus-induced lung inflammation in children: results of a randomized, placebo controlled clinical trial. Pediatr Allergy Immunol 20:477–485. doi: 10.1111/j.1399-3038.2009.00852.x. [DOI] [PubMed] [Google Scholar]
- 57.Lee FE, Walsh EE, Falsey AR. 2011. The effect of steroid use in hospitalized adults with respiratory syncytial virus-related illness. Chest 140:1155–1161. doi: 10.1378/chest.11-0047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hinzey A, Alexander J, Corry J, Adams KM, Claggett AM, Traylor ZP, Davis IC, Webster Marketon JI. 2011. Respiratory syncytial virus represses glucocorticoid receptor-mediated gene activation. Endocrinology 152:483–494. doi: 10.1210/en.2010-0774. [DOI] [PMC free article] [PubMed] [Google Scholar]