

ABSTRACT
Of the various genetic subtypes of human immunodeficiency virus types 1 and 2 (HIV-1 and HIV-2) and simian immunodeficiency virus (SIV), only in subtype C of HIV-1 is a genetically variant NF-κB binding site found at the core of the viral promoter in association with a subtype-specific Sp1III motif. How the subtype-associated variations in the core transcription factor binding sites (TFBS) influence gene expression from the viral promoter has not been examined previously. Using panels of infectious viral molecular clones, we demonstrate that subtype-specific NF-κB and Sp1III motifs have evolved for optimal gene expression, and neither of the motifs can be replaced by a corresponding TFBS variant. The variant NF-κB motif binds NF-κB with an affinity 2-fold higher than that of the generic NF-κB site. Importantly, in the context of an infectious virus, the subtype-specific Sp1III motif demonstrates a profound loss of function in association with the generic NF-κB motif. An additional substitution of the Sp1III motif fully restores viral replication, suggesting that the subtype C-specific Sp1III has evolved to function with the variant, but not generic, NF-κB motif. A change of only two base pairs in the central NF-κB motif completely suppresses viral transcription from the provirus and converts the promoter into heterochromatin refractory to tumor necrosis factor alpha (TNF-α) induction. The present work represents the first demonstration of functional incompatibility between an otherwise functional NF-κB motif and a unique Sp1 site in the context of an HIV-1 promoter. Our work provides important leads as to the evolution of the HIV-1 subtype C viral promoter with relevance for gene expression regulation and viral latency.
IMPORTANCE Subtype-specific genetic variations provide a powerful tool to examine how these variations offer a replication advantage to specific viral subtypes, if any. Only in subtype C of HIV-1 are two genetically distinct transcription factor binding sites positioned at the most critical location of the viral promoter. Since a single promoter regulates viral gene expression, the promoter variations can play a critical role in determining the replication fitness of the viral strains. Our work for the first time provides a scientific explanation for the presence of a unique NF-κB binding motif in subtype C, a major HIV-1 genetic family responsible for half of the global HIV-1 infections. The results offer compelling evidence that the subtype C viral promoter not only is stronger but also is endowed with a qualitative gain-of-function advantage. The genetically variant NF-κB and the Sp1III motifs may be respond differently to specific cell signal pathways, and these mechanisms must be examined.
INTRODUCTION
Based on genetic variation, human immunodeficiency virus type 1 (HIV-1) is classified into four distinct groups (M, N, O, and P), and group M is subclassified into nine molecular subtypes (A, B, C, D, F, G, H, J, and K) and numerous circulating recombinant forms, including A/E (1). The global distribution of HIV-1 subtypes is uneven, with C, A, and B being the most widespread subtypes. Subtype C is predominant in southern and eastern African countries, India, and Nepal and in recombinant forms in China, and it is currently emerging in southern Brazil. Subtype C is responsible for approximately half of the global HIV-1 infections and more than 95% of the infections in India (2). The factors that contribute to the widespread expansion of subtype C are not well understood. Diverse viral subtypes differ from one another up to 30 to 35% in certain gene segments, such as the envelope (3). A genetic variation to this large an extent is expected to have a significant impact on the biological properties of the subtypes influencing their relative fitness properties. Subtype-specific genetic variations in elements such as the viral promoter (enhancer and other regulatory elements), regulatory proteins, and structural proteins may underlie the biological differences, although drawing such a correlation between these factors may not always be possible.
The individual components of the viral promoter, including the modulator region, the enhancer, and the core promoter, are characterized by several subtype-specific molecular variations. Such differences have been mapped to many transcription factor binding sites (TFBS), including USF, c-Myb, NF-AT, Ap-1, NF-κB, and Sp1, and regulatory elements such as the TATA box and the TAR element (4, 5). Of these differences, the genetic sequence variations and copy number differences in the NF-κB and Sp1 binding motifs are of importance especially in the context of subtype C. The Sp1 and NF-κB motifs are the key players in regulating the inducible and basal levels of transcription from the long terminal repeat (LTR), respectively (6–8).
In the HIV-1 LTR, the binding sites of Sp1 and NF-κB are characterized by several unique qualities. First, the binding sites of Sp1 and NF-κB are present in multiple copies, typically arranged in a tandem fashion and in proximity to any two adjacent binding sites separated by very short spacer sequences of only 1 to 3 bases (Fig. 1A). While a large number of HIV-1 and HIV-2 subtypes contain three copies of Sp1 binding sites in the core promoter, a few simian immunodeficiency virus (SIV) strains contain four copies (9). Likewise, while a large number of HIV-1 subtypes (A, B, D, F, G, H, J, and K) and several subtypes of SIV contain two copies of NF-κB binding sites in the enhancer, subtype A/E of HIV-1 (10, 11), all HIV-2 subtypes, and the other strains of SIV contain a single NF-κB binding site. Subtype C of HIV-1 is the only viral family that contains three binding sites for NF-κB (12–15). Second, a great sequence variation characterizes the Sp1 binding sites, especially in Sp1III and Sp1II sites, with only the Sp1I motif being highly conserved among most of the HIV-1 subtypes (16). In particular, the genetic variation of the Sp1III motif located adjacent to the NF-κB sites is so great that no two major subtypes of HIV-1 share the same sequence (Fig. 1B). Of the three motifs, the Sp1III site plays a more deterministic role in gene expression from the LTR, as Sp1 protein binding at this site must physically interact with the p65 protein of NF-κB recruited to the H-κB site located immediately upstream (17). Thus, the Sp1III site critical for the regulation of gene expression for the viral promoter demonstrates subtype-specific sequence variations, and the influence of such variations has not been adequately examined (18, 19). Third, in contrast to the large genetic variation of the Sp1 sites, the sequence context of the NF-κB binding sites is invariable in the viral strains regardless of the copy number difference. Although the NF-κB binding sequence typically is degenerate (5′-GGGPuNNTTCC-3′), a single NF-κB motif is present in all the HIV-1, HIV-2, and SIV strains, and several canonical and noncanonical binding motifs have been identified in various cellular and viral promoters. The unique NF-κB motif that we refer to as the H-κB site (5′-GGGACTTTCC-3′) is present in all of the enhancers of HIV-1, HIV-2, and SIV subtypes (12, 20), alluding to the biological significance of this element in these promoters. In the backdrop of the universal presence of the H-κB site in all of these viral strains, subtype C of HIV-1 alone stands out as an exception in containing a variable, but canonical, NF-κB element, which we refer to as the C-κB site (5′-GGGGCGTTCC-3′; differences from the H-κB motif are underlined), in addition to the presence of two copies of the generic H-κB site (HHC). Lastly, the two TFBS, the C-κB and Sp1III motifs that contain subtype-specific variations in the subtype C LTR (C-LTR), are invariably positioned at the central location where these two binding sites, in the context of subtype B, have been shown to permit physical association between the host factors binding to the respective sites (17, 21). Of note, in almost all of the sequences of subtype C viral strains available in the extant databases, the C-κB motif is present, invariably positioned at the central location and never swapped with the H-κB sequence, alluding to the critical role the variant κB motif may be playing in this genetic subtype (Fig. 2A).
FIG 1.

(A) Schematic diagram comparing the TFBS of the enhancer and core promoter elements of B- and C-LTRs. An outline of the prototype B LTR with some of the basic transcription factor binding sites is shown at the top (not to scale). The enhancer region of the B-LTR contains two κB motifs (HH), in contrast to the C-LTR, which contains three κB elements (HHC). The additional NF-κB motif is genetically distinct and is referred to here as the C-κB site (filled square box). Note that the enhancer-proximal Sp1III site is also characterized by subtype-specific genetic variations, as shown at the bottom. The central TFBS sequences are shaded, and the genetic variations between the subtypes are highlighted by the darker shading. Additionally, the spacer length between the two TFBS is comprised of only two bases in subtype C, unlike the three bases in subtype B. (B) Multiple-sequence comparison of the Sp1 motifs. The nucleotide sequences of Sp1III, Sp1II, and Sp1I motifs representing the consensus sequences of the major HIV-1 genetic subtypes are aligned. Dots in the alignment represent sequence identity. The sequence alignment of the Sp1III motif is highlighted in gray. The Sp1II sequence of subtype C is also included in the alignment of Sp1III sequences for comparison.
FIG 2.

Genetic variation within the C-κB and Sp1III motifs of subtype C LTR. (A) Multiple-sequence alignment of the enhancer and core promoter sequences of the subtype C LTR. The LTR sequences comprising the three NF-κB sites and three Sp1 motifs of a few representative subtype C strains deposited during the years 1989 to 2010 were downloaded from http://www.hiv.lanl.gov/. The sequence of subtype C prototype Indie C1 (GenBank accession number AB023804) was used as the reference sequence. Dots and dashes represent the sequence identity and deletions, respectively. The C-κB and Sp1III motifs are highlighted by shading and the other TFBS by square boxes. (B) Position weight matrix of the C-κB motif. A total of 1,091 subtype C U3 sequences available at the HIV sequence database (http://www.hiv.lanl.gov/) were downloaded. The sequence logo was generated using the WebLogo tool (http://weblogo.berkeley.edu/logo.cgi). (C) Position weight matrix of the Sp1III motif. The sequence logo was generated as described above.
Some studies previously examined the regulation of gene expression from the HIV-1 LTR primarily in the context of subtype B (B-LTR). In previous studies, an active functional interaction in the B-LTR was demonstrated between the centrally positioned NF-κB and Sp1III sites in such a way that not only the spacing between the sites but also the relative orientation of each site with respect to the other cannot be altered, suggesting that the host factors must bind the specific elements in a specific orientation (17, 21). Furthermore, the centrally positioned NF-κB and Sp1III motifs play a role, more critical than that of the flanking sites (one upstream H-κB site and two downstream Sp1 motifs), in controlling viral gene expression (18, 19). Given the special significance attached to the central NF-κB and the Sp1III motifs in the HIV-1 LTR, it is enigmatic that a genetically different NF-κB site (the C-κB motif) is inserted in the subtype C-LTR, disrupting the original association between the H-κB and Sp1III motifs. Additionally, in the C-LTR, the Sp1III motif is subjected to subtype-specific genetic variation (4), alluding to a newly formed association between the newly inserted C-κB site and the subtype-specific Sp1 motif.
Several publications previously demonstrated that an HIV-1 LTR containing three NF-κB binding sites is a stronger viral promoter than the one that contains only two such sites (7, 16, 22, 23). It was further proposed that the stronger viral promoter contributed significantly to the widespread global expansion of subtype C (5, 24). These publications, however, evaluated only the quantitative gain-of-function properties of the C-LTR that contained an additional NF-κB binding site but ignored the significance of genetic variation of this motif that may confer an additional qualitative gain of function on the promoter. If acquiring a stronger promoter is the only objective, the subtype C-LTR could have duplicated an existing H-κB motif, thus attaining a promoter configuration of three H-κB sites, HHH. However, the C-LTR sequences are characterized exclusively by the NF-κB configuration of HHC, where the presence of the C-κB site at the central location appears to be critical, and no exceptions to this condition appear to be permissible.
Here, we attempted to evaluate the quantitative as well as the qualitative differences that may contribute to transcription differences in the C-LTR. Although genetic differences in the centrally located NF-κB and Sp1 elements have been reported previously (5, 12, 25), the present study represents the first analysis to examine the functional implications of these genetic variations to subtype C. The primary objective of the present work was to examine if there exists a newly established functional association between the HIV-1 subtype C unique NF-κB binding site (the C-κB site) and the proximal and subtype-specific Sp1III motif in the C-LTR. A functional association between the two adjacent motifs is expected and remains to be demonstrated. We used a range of experimental strategies to study the functional association between the two regulatory elements in the C-LTR. Importantly, we demonstrate that the creation of a variant NF-κB motif (the C-κB motif) was necessary for subtype C, as the subtype-specific Sp1III site cannot function in association with the generic H-κB element. To the best of our knowledge, in the context of a mammalian promoter, this is the first demonstration that a unique NF-κB motif and a specific Sp1 site are functionally incompatible with each other, although each TFBS is biologically functional in a different context.
MATERIALS AND METHODS
Cell culture.
T-cell lines (Jurkat and CEM-CCR5 cells) were cultured in RPMI 1640 medium (catalog no. R4130; Sigma, St. Louis, MO) supplemented with 10% fetal bovine serum (catalog no. RM10435; HiMedia Laboratories, Mumbai, India), 2 mM glutamine (catalog no. G8540; Sigma, St. Louis, MO), 100 U/ml penicillin G (catalog no. P3032; Sigma, St. Louis, MO), and 100 μg/ml streptomycin (catalog no. S9137; Sigma, St. Louis, MO). The peripheral blood mononuclear cells (PBMC) were isolated from 10 ml of fresh blood, collected from healthy donors, by density gradient centrifugation using HiSep LSM-1077 (catalog no. LS001; HiMedia Laboratories, Mumbai, India). The CD8 cells were depleted from the PBMC using the StemSep human CD8+ depletion kit (catalog no. 14662; Stem Cell Technologies). PBMC were cultured in complete RPMI medium supplemented with 20 U/ml of interleukin-2 (IL-2; catalog no. H7041; Sigma, St. Louis, MO) and 5 μg/ml of PHA-P (catalog no. L1668; Sigma) for 72 h. PBMC were subsequently used either for nucleofection or viral infection in a medium supplemented with 20 U/ml IL-2 (catalog no. H7041; Sigma, St. Louis, MO). The human embryonic kidney 293T and 293 cells were grown in Dulbecco's modified Eagle's medium (DMEM; catalog no. D1152; Sigma, St. Louis, MO).
Construction of the reporter vectors and viral molecular clones.
A dual-reporter viral vector, LTR-sLuc-IRES-GFP, that simultaneously expresses two different reporter genes, secreted Gaussia Luciferase (sLuc) and green fluorescent protein (GFP), was constructed using a parental vector, pCMV-sLuc-IRES-GFP, reported previously (12, 26). The cytomegalovirus (CMV) promoter was replaced by the full-length HIV-1 subtype C LTR of 634 bp, amplified from the Indie C1 subtype C molecular clone (GenBank accession number AB023804) using primers N1515 (5′-GGATCCACGCGTTGGAAGGGTTAATTTACTG-3′) and N1359 (5′-CCGCGGGAATTCCTGCTAGAGATTTTCC-3′). The PCR amplicon was cloned directionally using the restriction sites MluI and EcoRI. Subsequently, a series of isogenic variant LTR reporter vectors was generated from the parental vector, LTR-sLuc-IRES-GFP, using the overlap PCR strategy. Transcription factor binding sites were inactivated by mutating critical residues using site-directed mutagenesis, including the H-κB binding site (GGGACTTTCC to TCTACTTTCC), the C-κB binding site (GGGGCGTTCC to GTCTCATTCA), Sp1III (GAGGTGTGGT to GATTCGTGGT), Sp1II (TGGGCGGGAC to TGTTCGGGAC), and Sp1I (TGGGAGTGGT to TGTTAGTGGT).
Truncated LTR vectors.
A panel of minimal LTR reporter vectors expressing luciferase was constructed by deleting the 313-bp upstream modulator region from the viral promoter using the BspEI and MluI restriction sites in the pLTR-sLuc-IRES-GFP vector. The vector was self-ligated to generate pmLTR-sLuc-IRES-GFP vector. The panel of vectors consisted of three different LTRs, HHC:321 (wild type), HHC:231, and HHC:213, with the numerals representing the Sp1 motifs.
The pLGITC series of vectors.
A minivirus reporter plasmid, pLGIT (a kind gift from David Schaffer, University of California), expressed enhanced GFP (EGFP) and Tat under the HIV-1 LTR (18). This vector was modified further in two successive steps to replace Tat and the LTR of subtype B origin with the subtype C counterparts. First, the subtype B Tat expression segment was replaced with that of subtype C (GenBank accession number FJ765005.1). C-Tat was amplified using pcTat.BL43.CS as a template and primers N1140 (5′-TCCAGTCCACAACCATGGATGGAGCCAGTAGATCCTAAC-3′) and N1141 (5′-GGGCCCCTCGAGCTAGTCGAAGGGGTCTGTCTC-3′), and the amplified fragment was cloned between BstXI and XhoI sites on pLGIT to give rise to the vector pLGITC. Second, the subtype B LTR in pLGITC vector was replaced with the subtype C LTR (Indie C1; GenBank accession number AB023804). The subtype C LTR was amplified using primers N1145 (5′-GGCGCCCTCGAGACGCGTTGGAAGGGTTAATTTACTCC-3′) and N1146 (5′-TCTAGAGTTTAAACGCGGCCGCTGCTAGAGATTTTTCCCACACTAC-3′) and cloned between XhoI and PmeI restriction sites to generate the pcLGIT vector. Further, the 3′-LTR of the pcLGIT vector was subjected to additional mutations in the NF-κB and Sp1 binding sites to generate a panel of reporter viral vectors with isogenic LTRs analogous to the panel of reporter plasmid vectors described above.
Infectious viral strains.
The viral molecular clones Indie HHC:C, HHC:B, HHH:C, and HHH:B were constructed using the pIndie FHHC molecular clone reported previously from our laboratory (12). Using overlap PCR, variant LTRs were generated and substituted for the original FHHC LTR at the 3′ end of the virus between the MluI and SacII sites.
Reporter gene expression analysis.
Jurkat cells were transiently transfected with the reporter expression vectors using the Lipofectamine 2000 transfection reagent (catalog no. 11668-019; Invitrogen BioServices). The cells were seeded into 48-well plates at a density of 0.2 × 106 cells/well in 250 μl of RPMI 1640 medium supplemented with 10% fetal calf serum. A plasmid DNA pool of 500 ng, containing 400 ng of one of the reporter plasmids and 100 ng of pGL3 vector expressing Firefly luciferase (catalog no. E1741; Promega Corporation), included as a control for the transfection efficiency, was prepared in 50 μl of serum-free RPMI medium. One microliter of Lipofectamine was diluted with 49 μl of serum-free RPMI medium to prepare the lipid transfection reagent. The plasmid pool was mixed with the lipid reagent, and the plasmid-lipid mix was incubated for 20 min at room temperature and then added to appropriate wells. Four hours following the transfection, 350 μl of 10% RPMI with or without tumor necrosis factor alpha (TNF-α; 200 ng/ml; catalog no. T0157; Sigma, St. Louis, MO) was added to the wells. The plates were incubated, and the reporter gene expression was monitored periodically. A BioLux Gaussia Luciferase assay kit (catalog no. E3300L; New England BioLabs, Ipswich, MA) was used for monitoring the levels of Gaussia Luciferase secreted into the culture supernatant. The luciferase assay was performed using a SpectraMax L luminescence 96-well microplate reader (model no. s/n Lu 03094; MDS, Inc.) by mixing 20 μl of the culture supernatant and an equal volume of the 1× BioLuxGLuc substrate reagent. The assays were performed in triplicate wells, and every experiment was repeated at least two times. Expression of Firefly luciferase in the cell extracts was measured using a Bright-Glo luciferase assay kit (catalog no. E2620; Promega Corporation, Madison, WI) per the manufacturer's instructions. The primary data for each well were normalized for transfection efficiency.
Preparation of viral stocks and estimation of viral titer.
Viral stocks of different molecular clones were generated in HEK 293T cells using the standard calcium-phosphate protocol. Briefly, cells were seeded in a 100-mm dish at a density of 3 × 106 cells and transfected with 20 μg of the viral vector DNA along with 0.2 μg of the CMV-red fluorescent protein (RFP) expression vector as an internal control for transfection efficiency. Six hours posttransfection, medium was replaced with complete DMEM. Culture supernatants were harvested at 48 h, filtered through a 0.22-μm filter, and stored in a deep freezer in multiple aliquots of 1 ml. The p24 levels of the viral stocks were measured using a p24 enzyme-linked immunosorbent assay (ELISA) kit (4th generation p24 ELISA kit; J. Mitra and Co. Pvt. Ltd., New Delhi, India) per the manufacturer's instructions. The 50% tissue culture infectious dose (TCID50) titer of the viral stocks was determined using a β-galactosidase assay in TZM-bl cells. Briefly, 104 TZM-bl cells were seeded in 100 μl of complete DMEM in a flat-bottom 96-well culture plate. After 12 h, 50 μl of serially diluted viral stocks (a serial 4-fold dilution) was added to appropriate wells in complete DMEM supplemented with 25 μg/ml of DEAE-dextran (catalog no. D9885; Sigma, St. Louis, MO) (27). Six hours postinfection, medium in the wells was replaced with 100 μl of complete DMEM. The plates were incubated at 37°C in the presence of 5% CO2. To examine β-galactosidase expression, 48 h postinfection, cells in each well were washed using 1× phosphate-buffered saline (PBS; 137 mM NaCl, 2.7 mM KCl, 4.3 mM Na2HPO4, 1.47 mM KH2PO4, pH 7.4). After washing, cells were fixed with 100 μl of a fixing solution (1.0% formaldehyde and 0.2% glutaraldehyde in 1× PBS), and the plates were incubated for 5 min at room temperature. The cells were washed with 1× PBS and stained with 100 μl of the freshly prepared β-galactosidase staining solution (4 mM potassium ferrocyanide, 4 mM potassium ferricyanide, 2 mM MgCl2, and 1 mM 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside [X-Gal] in PBS), and the plates were incubated for 4 h at 37°C. The wells were washed with 1× PBS, and the stained cells were counted manually under a low-resolution microscope. The infectious titer of each viral stock was determined by multiplying the cell count by the dilution factor. Pseudotyped lentiviral vectors used for the chromatin immunoprecipitation (ChIP) analysis were packaged in HEK 293T cells (18). Briefly, cells were seeded in 100-mm culture dishes and transfected using the calcium phosphate transfection protocol with a total of 20 μg of plasmid DNA pool. The plasmid DNA pool consisted of four different vectors: 10 μg of pcLGIT reporter virus containing one of the 3′-LTR variant constructs, 5 μg psPax2, 3.5 μg pVSV-G, and 1.5 μg pCMV-Rev. Culture supernatant was harvested 48 h following the transfection and stored in a deep freezer in 1-ml aliquots. The p24 levels and the TCID50 titers of the viral stocks were determined as described above.
Protein expression and purification.
The expression plasmid pGEX CD-p50 was a kind gift from Neil D. Perkins (University of Dundee, Scotland, United Kingdom). The p50-glutathione S-transferase (GST) fusion protein was expressed in Escherichia coli BL21(DE3) cells. The p50-GST fusion protein was purified as described previously (12), with a small modification. Briefly, the cells were grown in 250 ml of the LB broth supplemented with 100 μg/ml of ampicillin (catalog no. A9518; Sigma, St. Louis, MO) for 4 h at 28°C, until the optical density (OD) at 600 nm reached 0.4. Protein expression was induced for 2 h by supplementing the medium with 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG). The bacterial cells were pelleted and resuspended in 10 ml of homogenization buffer (20 mM HEPES, pH 7.9, 10 mM MgCl2, 20% glycerol, 0.1% ND-40, 1 mM dithiothreitol [DTT], 0.5 mM phenylmethylsulfonyl fluoride [PMSF], and 1× protease inhibitor cocktail [catalog no. P8454; Sigma, St. Louis, MO]). The resuspended cells were sonicated using a Vibra cell sonicator (Sonics and Materials Inc., Newtown, CT) at an amplitude of 35%, with 2 s on and 4 s off for 2 min. The sonicated bacterial cell lysate was centrifuged at 45,000 × g for 30 min, and to the supernatant 400 μl of prewashed GST-bind resin (catalog no. 70541; Novagen, Madison, WI) was added. The samples were incubated for 2 h at 4°C with constant mixing. The supernatant-resin suspension was centrifuged at 100 × g for 5 min, and the supernatant was carefully aspirated. The GST-resin pellet was washed three times with 10 ml of homogenization buffer. The washed GST-resin was transferred to a fresh 1.5-ml vial and incubated for 10 min with 200 μl of the elution buffer (50 mM Tris-Cl, pH 7.9, containing 10 mM reduced glutathione). The sample was centrifuged for 5 min at 100 × g, and the clear supernatant was transferred to a fresh vial. The supernatant was dialyzed each time against 200 ml of dialysis buffer (5 mM HEPES, pH 7.6, 1 mM DTT, 1 mM EDTA, 10% glycerol, 1× protease inhibitor cocktail, and 0.2 mM PMSF) for 4 h at 4°C for three rounds and stored in a −20°C deep freezer.
Electrophoretic mobility gel-shift assay (EMSA) and supershift assay.
Jurkat cells (10 × 106 cells), harvested and resuspend in 1 ml of ice-cold 1× PBS, were transferred to a 1.5-ml plastic vial and centrifuged at 500 × g for 5 min at 4°C. The buffer was aspirated and the nuclear extract was prepared using a commercial kit (cytoplasm and nuclear protein extraction kit; catalog no. K0311; Fermentas Life Sciences, Waltham, MA). The protein concentration of the nuclear extracts was determined using a commercial bicinchoninic acid (BCA) protein assay kit (catalog no. 23227; Pierce Biotech, Waltham, MA). Aliquots of 50 μl of the nuclear extract were snap-frozen in liquid nitrogen and stored in a −80°C deep freezer until use.
A single-stranded DNA probe was radiolabeled using 5 μg of DNA with 1 U of T4 polynucleotide kinase (catalog no. M0201; New England BioLabs) in a solution containing 2 μCi of [γ-32P]ATP. The reaction mixture was incubated for 1 h at 37°C, and the enzyme was heat inactivated at 65°C for 20 min. The radiolabeled oligonucleotide was then hybridized to 10 μg of the complementary strand in the annealing buffer (50 mM NaCl, 20 mM Tris, pH 7.5, 10 mM MgCl2, 20 μM EDTA) by incubating the reaction mixture for 5 min in boiling water, followed by gradual cooling at room temperature for 2 h. The radiolabeled annealed double-stranded probe was gel purified using a 6% (29:1) polyacrylamide gel. The gel was exposed to an X-ray film for 1 h to develop the autoradiogram. The developed autoradiogram was superimposed over the gel containing radiolabeled probed, and the corresponding gel piece was excised out using a scalpel. The radiolabeled probe was extracted from the excised gel piece using the crush-and-soak method with minor modifications (28). Briefly, the excised gel piece was placed in a fresh 1.5-ml plastic vial and crushed into fine pieces using a fresh needle. The weight of gel pieces was determined and the gel pieces were soaked in 2 volumes of acrylamide gel extraction buffer (0.5 M ammonium acetate, 10 mM magnesium acetate, 1 mM EDTA), and the vial was incubated at 37°C for 6 to 8 h. The sample was vortexed briefly and centrifuged at 10,000 rpm for 5 min, and the supernatant was transferred to a fresh vial. The gel pieces were treated once again with a 0.5 volume of acrylamide extraction buffer, and the supernatant was pooled with the extraction solution from the previous step. Two volumes of 100% ethanol were added to the supernatant, the sample was incubated at −20°C for 1 h, and the vial was centrifuged at 10,000 rpm for 5 min at 4°C. The supernatant was carefully aspirated and the pellet was dissolved in 200 μl of 10 mM Tris-EDTA (pH 8.0). The probe was reprecipitated with 2 volumes of 100% ethanol, and the pellet was washed with 1 ml of 70% ethanol, air-dried, and resuspended in 50 μl of 10 mM Tris-EDTA buffer. The radioactivity counts of each probe were determined using a liquid scintillation counter (Wallac 1409; PerkinElmer, Waltham, MA). Probes of the following sequences, sense and antisense in that order, were used in the assay. The uppercase letters in the sequences denote the respective transcription factor binding sites and the lowercase letters the native flanking sequences for the C-κB probe (5′-ccactGGGGCGTTCCagga-3′ and 5′-tcctGGAACGCCCCagtgg-3′), the H-κB probe (5′-ccactGGGACTTTCCagga-3′ and 5′-tcctGGAAAGTCCCagtgg-3′), the NFAT binding sequence from the IL-2 promoter (5′-tcgagcccaaagaGGAAAatttgtttcatg-3′ and 5′-catgaaacaaatTTTCCtctttgggctcga-3′), and a nonspecific oligonucleotide sequence (5′-ctactgtctcattaagaa-3′ and 5′-ttcttaatgagacagtag-3′). For the EMSA, 25 μg of nuclear extract was mixed with 30,000 cpm of the labeled probe in the binding buffer (10 mM Tris, pH 7.9, 150 mM KCl, 1 mM DTT, 0.5 mM EDTA, 0.1% Triton X-100, and 10% glycerol) and supplemented with 1 μg poly(dI-dC) per reaction and 1 μg/ml of bovine serum albumin (BSA) in a reaction volume of 30 μl. The binding reaction mixture was incubated for 20 min at room temperature and resolved on a 6% polyacrylamide gel (16 by 16 cm) at 250 V for 2.5 h at 4°C. The competition experiments were performed with a 5-, 10-, or 50-fold excess of unlabeled oligonucleotides added to the reaction mixture. For the supershift assay, the nuclear extracts were preincubated with 2 μg of affinity-purified rabbit polyclonal antibodies raised against p50, p52, p65, RelB, or cRel proteins. For the supershift of NFAT complexes, antibodies specific for NFAT1 (catalog no. ab2722; Abcam, Cambridge, United Kingdom), NFAT2 (catalog no. ab2796; Abcam, Cambridge, United Kingdom), and NFAT5 (catalog no. ab3446; Abcam, Cambridge, United Kingdom) were used. A nonspecific antibody raised against HIV-1 p24 (generated in-house) was used as a negative control. Thirty minutes following incubation, radiolabeled probes were added to the mix and EMSA was performed as described above. Gels were dried and exposed to a Kodak Biomax film at −80°C. Quantitative EMSA was performed using recombinant p50 protein to estimate the affinity of the recombinant p50 protein for the C-κB or H-κB binding site (29).
Generation of the cell pools with stable viral integration.
In a volume of 700 μl of 10% RPMI supplemented with 25 μg/ml DEAE-dextran in a 12-well tissue culture plate, 0.3 × 106 Jurkat cells were infected at a multiplicity of infection (MOI) of 0.02 to 0.05 with pseudotyped viruses harboring one of the LTR viral variants. The cells were washed after 6 h of incubation and resuspended in 1 ml of complete RPMI medium. Fifteen days following the viral infection, cells positive for GFP fluorescence were selected using a flow sorter (Aria III sorter; BD Biosciences, Franklin Lakes, NJ). Sorted cell pools were allowed to proliferate, and stable GFP expression was confirmed. To confirm comparable levels of integration with different viral variants, a quantitative real-time PCR was used to measure the mean number of integrated proviral copies in a fixed quantity of the genomic DNA extracted from the cells. A 129-bp fragment spanning the R region to the U5 region (+18 to +147) was amplified using the primers N2208 [5′-GATCTGAGCC(T/C)GGGAGCTCTCTG-3′] and N2209 (5′-TCTGAGGGATCTCTAGTTACCAGAGTC-3′) in a real-time PCR that has the sensitivity to detect as few copies as 101 in the assay. Genomic DNA extracted from TZM-bl cells that harbor a single copy of the integrated virus per cell was used to construct the standard curve for the quantification of the number of integrated viral copies using regression analysis. For normalization of the viral real-time PCR data, the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) housekeeping gene was amplified using primers N2232 (5′-GAGCTGAACGGGAAGCTCACTG-3′) and N2233 (5′-GCTTCACCACCTTCTTGATGTCATC-3′).
ChIP assay.
The ChIP assay was performed using genomic DNA extracted from Jurkat cell pools harboring integrated proviruses containing variations in the NF-κB and/or Sp1 sites. Cells (20 × 106 cells per assay) were incubated in the absence or presence of TNF-α (100 ng/ml) for 12 h. The cells were washed once with 5 ml of 1× PBS, resuspended in 1 ml of plain RPMI supplemented with 1% formaldehyde, and agitated in a shaker incubator for 10 min at room temperature. After 10 min, the cross-linking reaction was stopped by adding glycine to a final concentration of 0.125 M. The cells were centrifuged at 3,000 rpm for 5 min at 4°C, supernatant was aspirated, and the cells were washed once with 1 ml of ice-cold 1× PBS. The cells were resuspended in 0.6 ml of the chilled lysis buffer [5 mM piperazine-N,N′-bis(2-ethanesulfonic acid) (PIPES), pH 8.0, 85 mM KCl, 0.5% NP-40] supplemented with the protease inhibitor cocktail (catalog no. 11836170001; Roche Applied Science) and incubated for 10 min on ice. The nuclear suspension was divided into 100-μl fractions, and each fraction was subjected to 22 cycles of sonication at the high-level setting, using 30-s-on and 30-s-off pulses, in ice-chilled water using Bioruptor plus equipment (Diagenode, Liege, Belgium). The magnitude of DNA shearing was monitored in gel electrophoresis, and an average shearing size of 200 to 600 bp was targeted. The sonicated lysate was centrifuged at 14,000 rpm for 10 min at 4°C and the supernatant was collected. Immunoprecipitation was performed using 2 μg of one of the antibodies against p50 (catalog no. ab7971; Abcam, Cambridge, United Kingdom), p65 (catalog no. ab 7970; Abcam, Cambridge, United Kingdom), Sp1 (catalog no. CS200631; Upstate, Syracuse, NY), RNA polymerase II (Pol II; catalog no. C15200004; Diagenode, Seraing, Belgium), RNA Pol II CTD phospho-S2 (catalog no. ab5095; Abcam, Cambridge, United Kingdom), RNA Pol II phospho-S5 (catalog no. C15200007; Diagenode, Seraing, Belgium), HDAC1 (catalog no. C15410053; Diagenode, Seraing, Belgium), HDAC3 (catalog no. ab 7030; Abcam, Cambridge, United Kingdom), H3K9Ac (catalog no. ab441; Abcam, Cambridge, United Kingdom), or HIV-1 p24 (as a negative control; generated in-house). The immunoprecipitated DNA was subjected to PCR using the primer pair N1054 (5′-GAAGTATTAAAGTGGAAGTTTGACATTC-3′) and N1056 (5′-AGAGACCCAGTACAGGCGAAAAGC-3′), which amplified a 240-bp region flanking the κB and Sp1 elements in the LTR. A 239-bp fragment was amplified in the gag region to observe the occupancy of RNA Pol II S5 (associated with transcription initiation) using primer pair N2467 (5′-GTAATAGAGGAGAAGGCTTTTAGCC-3′) and N2468 (5′-AACCAAGGGGAAGTGACATAGC-3′). Occupancy of RNA Pol II S2 was observed by amplifying a 218-bp region in the tat gene using the primer pair N2469 (5′-GATCCTAACCTAGAGCCCTGGA-3′) and N2470 (5′-GTTCGGGGTAAGGGTTGCTTTG-3′). The amplified fragments were subjected to agarose gel electrophoresis to confirm the presence or absence of transcription factors over the transcription factor binding sites.
Viral proliferation and replication kinetics.
CEM-CCR5 cells or PBMC (0.3 × 106) were infected with the virus Indie HHC3C21, Indie HHH3C21, Indie HHC3B21, or Indie HHH3B21 at approximately 500 IU. The infectious titers of the viral stocks were determined as described above. Prior to viral infection, the PBMC were CD8 cell depleted and activated with 5 μg/ml phytohemagglutinin (PHA) in complete RPMI medium supplemented with 20 U/ml of IL-2 for 72 h. Following this, the cells were incubated with the viruses in complete RPMI medium supplemented with 10 μg/ml of DEAE-dextran (30) for 6 h at 37°C in the presence of 5% CO2. Six hours following the infection, the cells were washed three times in PBS, suspended in complete RPMI 1640 medium, and incubated. Supernatants from the third wash were saved as the day zero time point for p24 ELISA. The cells were monitored for p24 production at weekly intervals for a period of a month. The p24 ELISA was performed for each time point, and the viral growth curve was constructed for each monoinfection.
Postentry events of viral infection.
Full-length Indie C1 viruses with variant LTRs (HHC3C21, HHH3C21, HHC3B21, and HHH3B21) were allowed to infect the CEM-CCR5 cells (1 × 106) at an infectious titer of 1,000 TCID50 in a 6-well tissue culture plate. To analyze the late reverse transcription (RT) products, the cellular DNA was isolated using the GenElute blood genomic DNA kit (catalog no. NA2020; Sigma, St. Louis, MO). Primer pair N1734 (5′-TGTGTGCCCGTCTGTTGT-3′) and N1735 (5′-GAGTCCTGCGTCTAGAGGATC-3′) was used to amplify a 145-bp amplicon from the U5-ψ region. A standard curve was constructed using a 10-fold serial dilution of the plasmid Indie-C1 (copy number ranging from 101 to 105) using salmon sperm DNA (5 ng/μl). The real-time PCR was performed using a commercial kit (catalog no. BIO-94002; SensiFAST SYBR Mastermix kit; Bioline, London, United Kingdom) and with 250 ng of the cellular DNA derived from the infection of each virus for the template.
To determine the rate of transcription of the viruses, real-time PCR analysis was performed for the proximal (TAR) and distal (Tat) transcripts. Forty-eight hours postinfection, cells were treated with 100 ng/ml of TNF-α for 2 h or were left untreated. The total cellular RNA was extracted from 1 × 106 cells using TRIzol (catalog no. T9424; Sigma, St. Louis, MO). The RNAs were subjected to cDNA synthesis using a commercial kit (catalog no. BIO-65042; Tetro cDNA synthesis kit; Bioline, London, United Kingdom). The cDNA was diluted 10-fold using salmon sperm DNA and subjected to real-time PCR analysis. Eighty-nine bp of TAR transcript was amplified using the primer pair N2367 (5′-GGTAGACCAGATCTGAGCC-3′) and N2368 (5′-CTCAGAGCACTCAAGGCAAG-3′). The distal transcript of a 152-bp fragment was amplified in Tat using the primer pair N2270 (5′-TGGAGCCAGTAGATCCTAACCTAGAGCC-3′) and N1784 (5′-CTTCGTCGCTGTCTCCGCTTCTTCCTG-3′). For normalization, a transcript from the GAPDH gene was amplified using primers N2232 (5′-GAGCTGAACGGGAAGCTCACTG-3′) and N2233 (5′-GCTTCACCACCTTCTTGATGTCATC-3′). All of the real-time PCR analyses were performed using the Bio-Rad CFX96 Touch real-time PCR machine.
RESULTS
Physical proximity between the C-κB and Sp1III motifs is critical for optimal gene expression from subtype C LTR.
Compared with the HIV-1 subtype B LTR (B-LTR), three important molecular differences could be seen in the subtype C LTR (C-LTR) within the NF-κB and Sp1 binding sites. First, the total number of NF-κB binding sites in the viral enhancer of C-LTR is three, as opposed to only two in B-LTR. Second, the additional NF-κB binding site is invariably located immediately upstream of the Sp1 motifs and is genetically different (5′-GGGGCGTTCC-3′; referred to as the C-κB site) from the two upstream generic motifs (5′-GGGACTTTCC-3′; referred to as the H-κB sites). Finally, the Sp1III binding site located immediately downstream of the C-κB site contains subtype-specific variations. Thus, in C-LTR, the variant NF-κB site and the subtype-specific Sp1III motif are invariably juxtaposed with each other in the core of the viral promoter (Fig. 1A). Of note, while the subtype-specific C-κB site and Sp1III motifs are highly conserved among subtype C strains (Fig. 2B and C), a functional association between these two adjacent motifs is expected and remains to be demonstrated. To delineate the nature of the association between the C-κB and proximal Sp1 sites, we constructed two different panels of expression vectors under the control of C-LTR, using a parental bicistronic vector, pLTR-sLuc-IRES-EGFP, reported previously (12). The initial rounds of standardization did not show a significant difference in the profile of reporter gene expression whether a complete C-LTR or a truncated one that lacked the upstream modulator region (−150 to −463) was used in the assay (data not shown). In all subsequent analyses in the present study, we used only the full-length (634-bp) promoter (Indie C1; GenBank accession no. AB023804).
In one of the two panels of the reporter vectors (Fig. 3A), consisting of three different promoters (HHC, HCH, and CHH) and keeping the Sp1III site constant, the C-κB motif was moved progressively upstream, thus increasing the distance between the Sp1III site and the C-κB element. In the other panel of three vectors (Fig. 3B), doing the contrary, i.e., keeping the C-κB site constant, the subtype C-specific Sp1III site was moved progressively downstream, again increasing the distance between the elements. Of note, in all of the expression vectors, all 6 TFBS (three each NF-κB and Sp1 sites) remained intact except for the positional shifts described.
FIG 3.

C-κB site is functionally associated with the Sp1III motif in HIV-1 subtype C LTR. Jurkat cells were transiently transfected with one of the reporter vectors, and 8 h following transfection the cells were exposed to TNF-α activation (100 ng/ml) or left without activation (filled black and gray bars, respectively). The luciferase secretion was evaluated from the culture supernatant at 24 h following the activation or at later times. Gene expression was significantly attenuated when the subtype C-specific C-κB site (A) or subtype C-specific Sp1 motif (B) was shifted away from their respective natural positions, when the orientation of the two motifs (middle construct) was changed, or when a 5-bp linker was inserted between the two elements (lower construct) (C). (D) The native Sp1III motif of subtype C was replaced by the heterologous Sp1 sequences from the other subtypes of HIV-1. Sequences used at motif Sp1III are depicted in Fig. 1B. Two-way analysis of variance (ANOVA) was used for statistical analysis.
Jurkat cells were transiently transfected with the vectors of panel 1 containing the positional shift of the C-κB site. The cells were activated with TNF-α (100 ng/ml) or left without activation, and the expression pattern of secreted luciferase or GFP (data not shown) was measured 24 h after activation and at later time points. The wild-type LTR (HHC) demonstrated 588,286 ± 428 relative light units (RLU) of luciferase activity that was upregulated 2.3-fold (1,359,497 ± 42,491 RLU) following TNF-α activation. The basal and induced promoter activities were significantly reduced when the C-kB motif was progressively shifted upstream to one (HCH) or the other (CHH) position away from the original location. Although the loss of the promoter activity was statistically significant (P < 0.05), gene expression was not completely abrogated. These results suggested that although the H-κB motif can function from the central location, this motif is not an ideal substitute for the natural C-κB element located here in the C-LTR. Comparable results were also obtained in HEK293 cells (data not presented). Further, in Jurkat cells, a loss of promoter activity was also manifested from the vectors in panel 2, where the subtype C-specific Sp1III motif was shifted progressively downstream away from the C-κB element (Fig. 3B). The loss of reporter function was quite severe, suggesting that the Sp1II motif is not a functional substitute for Sp1III in the C-LTR. Comparable results were obtained in HEK293 cells (data not presented). Collectively, the strategy of the TFBS shifting suggested that in the C-LTR, the C-κB and Sp1III motifs must be located proximal to each other and the alternate TFBS are not functional substitutes for either of the two original regulatory motifs.
We generated an additional panel of reporter vectors by flipping the C-κB and Sp1III sites or inserting a five-residue linker between the two sites (Fig. 3C). The vector in which the positions of the C-κB and Sp1III sites were interchanged did not show any reporter activity in Jurkat cells (Fig. 3C, middle) or HEK293 cells (data not shown). The DNA-binding domain of the p65 subunit can interact with the DNA-binding domain of Sp1 only when the NF-κB motif is present upstream of the Sp1 binding element, thus juxtaposing the two factors in a specific fashion as demonstrated previously (17). Likewise, when the spacer length between the two sites was increased to 7 bp from the natural 2 bp (GGGGCGTTCCtgcagagGAGGTGTGGT; the lowercase letters represent the spacer sequence and the added bases are underlined), a complete loss of reporter activity was observed in Jurkat (Fig. 3C, lower) or HEK293 (data not shown) cells. Collectively, these data ascertained that the association between the C-κB and Sp1III motifs is orientation and distance dependent.
C-κB motif of subtype C can functionally associate with the heterologous Sp1III elements of other subtypes.
It was unexpected that the Sp1III site of subtype C could not be replaced by Sp1II sequence of the same viral promoter (Fig. 3B, vectors II:III:I and II:I:III). Of the three genetically diverse Sp1 sites in the core promoter of HIV-1, the Sp1III site is the most variable element, demonstrating subtype-specific variations (Fig. 1B) (31). Importantly, experimental evidence is suggestive that the Sp1III site is functionally more important for the viral promoter than the other two (18, 19, 32). Given the biological significance of the centrally located Sp1III motif and the subtype-associated variations within this motif among HIV-1 subtypes, we asked if the heterologous Sp1III sequences derived from the other HIV-1 subtypes can substitute for the original Sp1III sequence in C-LTR. To this end, we generated a panel of reporter vectors by grafting subtype-specific Sp1III sequences of subtypes A, B, D, and E at the NF-κB proximal location in the C-LTR. The expression of luciferase reporter was determined in Jurkat (Fig. 3D) or HEK 293 (data not shown) cells. The wild-type subtype C-LTR demonstrated 22,975 ± 7,343 and 1.02 × 108 ± 2.0 × 107 RLU/s of reporter activity in Jurkat cells under control and TNF-α-induced conditions, respectively. The gene expression profile from other chimeric viral promoters containing the heterologous Sp1III motifs was comparable to that of wild-type C-LTR with an important difference. Although the induced reporter activity of all of the different LTRs in the panel is comparable, the basal-level promoter function of the wild-type LTR was the lowest in both cell lines. Consequently, the fold transactivation of the wild-type C-LTR was the highest (6- to 7-fold versus 2- to 3-fold) and significantly different (P < 0.05) among all of the viral promoters compared. The biological significance of the low-basal-level activity of the wild-type C-LTR for the establishment of viral latency needs further evaluation. In summary, regardless of the genetic variation, the heterologous Sp1III motifs of all four viral subtypes (A, B, D, and E) functioned in the context of C-LTR with comparable efficiency, suggesting that each of these motifs can complement efficiently the C-κB site. Thus, these heterologous Sp1III elements can serve as functional substitutes for the subtype C Sp1III motif.
Functional association between the C-κB and Sp1III motifs in the absence of the flanking NF-κB and Sp1 sites.
Although the wild-type subtype C-LTR contains three each of NF-κB and Sp1 binding sites in the promoter, a direct association between the bound TF is possible only between the centrally positioned Sp1III site and the C-κB, but not the upstream H-κB, motif. In the context of the viral promoter, the relative magnitude of gene expression due to the specific interaction between the central C-κB and Sp1III sites is likely to be modulated by interference from the two upstream NF-κB sites and the two downstream Sp1 motifs. To overcome this overlapping activity of the flanking TFBS and examine the direct influence of the C-κB and Sp1III elements on gene expression from the C-LTR, we introduced inactivating mutations into the four flanking TFBS sequences using site-directed mutagenesis, leaving only the centrally positioned C-κB and Sp1III sites intact. By introducing mutations into critical residues of the corresponding motifs, we generated a panel of four reporter viral vectors, using the pcLGIT vector where the C-κB and/or the Sp1III sites were intact or both were inactivated (C:C, C:X, X:C, and X:X, where C represents the subtype-specific NF-κB or Sp1III site and X the inactivated TFBS) (Fig. 4). To maintain the same DNA helical conformation as that in the wild-type LTR, no sequences were deleted from any of the vectors. Therefore, the overall length of the promoter sequences remained constant.
FIG 4.

Gene expression in the absence of the flanking TFBS. (A) GFP expression profile of the promoter variant reporter viruses used in flow cytometry. A panel of four different reporter viruses with TFBS variations engineered in the 3′-LTR was constructed. Following viral infection, Jurkat cell pools harboring stable proviruses for each of the four viruses were generated. Jurkat cells were activated with TNF-α (100 ng/ml) for 24 h or were left without activation, and the mean fluorescence intensity (MFI) of the EGFP was measured. Each assay was performed in duplicate wells. Data are representative of three independent experiments and are presented as MFI ± standard deviations (SD). Two-way ANOVA was used for statistical analysis. (B) Comparable levels of viral integration in the four Jurkat stable pools. Genomic DNA was extracted from the cell pools used in panel A and subjected to quantitative real-time PCR, amplifying a 129-bp region spanning the R-U5 region in the LTR. Data are presented as mean quantification cycle (Cq) ± SD. The data were normalized using the GAPDH housekeeping gene. (C) ChIP analysis to examine the occupancy of transcription factors in the variant LTRs. The Jurkat stable cell pools harboring the C:C- or X:X-LTR reporter virus were activated with TNF-α for 4 h or left without activation. The chromatin complexes were immune precipitated using 2 μg of antibodies specific to different cellular factors as shown. An antibody against HIV-1 p24 was used as a negative control in the experiment. The ChIP analysis was performed as described in Materials and Methods. One-tenth of the input chromatin was loaded as the input control. The data are representative of three independent experiments. (D) The C-κB and Sp1III sites of subtype C are functionally associated when the flanking κB and Sp1 sites are inactivated. A panel of reporter expression vectors was constructed that contained only the C-κB site (C:X), Sp1III site (X:C), both (C:C), or none (X:X). While C represents subtype C-specific κB or Sp1III sequences, X denotes a debilitating mutation in the corresponding motif. Luciferase activity was determined in Jurkat cells (left) or PMA-activated PBMC of a healthy subject (right) at 24 h following the transfection. The assay was performed using PBMC derived from three different subjects, and the data from one representative subject are presented. The assay is representative of three independent experiments.
Viruses pseudotyped with the vesicular stomatitis virus G glycoprotein (VSV-G) envelope were used for the infection of Jurkat T cells at a low MOI of 0.01. Cell pools stably expressing EGFP were sorted 21 days after the viral infection and used in the assay with or without TNF-α activation. The three reporter viruses that lacked one (X:C or C:X) or both (X:X) TFBS demonstrated a minimal basal level of activity that did not increase significantly following TNF-α activation (Fig. 4A). The reporter virus containing both motifs (C:C) demonstrated the highest basal level of fluorescence (mean fluorescence intensity [MFI], 2,128 ± 9) that increased 2.2-fold (MFI, 4,783 ± 115; P < 0.01) following TNF-α activation. To rule out the possibility that different rates of viral infection influenced the assay outcome, we used a real-time PCR targeting the R-U5 region of the LTR to confirm equivalent levels of viral infection (Fig. 4B). The results are highly reproducible when virus-infected cell pools were established independently (data not shown).
To correlate the magnitude of gene expression from the integrated viral promoter with the occupancy of the host factors at the promoter, we performed a chromatin immunoprecipitation (ChIP) analysis using the cellular DNA extracted from Jurkat cells harboring integrated X:X or C:C reporter viral strains. Fragmented chromatin was immunoprecipitated using antibodies specific to p50, p65, Sp1, RNA polymerase II (RNA Pol II), phosphorylated RNA Pol II-Ser-2, or a nonspecific antibody (an antibody against the p24 antigen of HIV-1) as a negative control (Fig. 4C). The X:X viral promoter did not bind any of the tested host factors regardless of TNF-α activation, thus evidencing the lack of viral proliferation. In contrast, the promoter containing intact C-κB and Sp1III sites, in the absence of activation, bound p50 and Sp1 but little p65 or RNA polymerase II, suggesting the occupancy of the enhancer core by the repressive p50 homodimer. Following TNF-α activation, however, the promoter was occupied by p50, p65, Sp1, and significantly higher quantities of RNA Pol II that was phosphorylated at serine 2. Thus, following activation, the suppressive p50:p50 homodimer was replaced by a transcription-supportive p50-p65 heterodimer complex at the viral enhancer. Additionally, the presence of the phosphorylated RNA Pol II is suggestive of an efficient transcription elongation. The relative quantity of Sp1 immobilized on the promoter was not significantly different under the basal and induced conditions. The ChIP data not only are consistent with the proliferation analysis of the reporter viruses but also confirmed that regardless of the subtype-specific genetic variations that characterize the C-κB and Sp1III motifs, these sequences regulate gene expression by recruiting the appropriate host factors. Additionally, comparable results were obtained in Jurkat cells or PBMC isolated from different healthy donors from a panel of plasmid reporter vectors (X:X, C:X, X:C, and C:C) that are analogous to the corresponding reporter viral vectors (Fig. 4D). Collectively, the data evidenced the pivotal role of the association between the C-κB and Sp1III sites in regulating gene expression from the subtype C viral promoter in the absence or presence of the flanking regulatory elements.
The C-κB motif recruits the p50-p65 heterodimer.
Compared with the canonical H-κB site, the C-κB motif in C-LTR is genetically distinct, with variations found at positions 4 (A to G) and 6 (T to G) (Fig. 1A). Using gel shift analysis (cold competition and supershift assays), a couple of publications previously claimed the absence of NF-κB binding to the C-κB sequence with an underlying implication that the motif is not biologically functional (11, 33). A close examination of the probes used in these reports reveals variations in the probe sequences used or an improper experimental design (33). Importantly, in contrast to the above-described reports, two other publications demonstrated that the C-κB motif is biologically functional (5, 16). With the backdrop of this controversy, we set out to examine the ability of the C-κB probe to recruit NF-κB from nuclear extracts. Using double-stranded radiolabeled DNA probes comprised of either the canonical H-κB (5′-ccactGGGACTTTCCagga-3′; the flanking sequences are in lowercase and the differences are underlined), the C-κB (5′-ccactGGGGCGTTCCagga-3′), or a scrambled (5′-CTACTGTCTCATTAAGAA-3′) probe sequence, we compared the binding of cellular factors from Jurkat cell nuclear extract in the presence or absence of TNF-α activation. Distinct cellular complexes were recruited by both H- and C-κB DNA probes under no-activation conditions, and the intensity of the complexes increased severalfold following TNF-α activation (Fig. 5A, compare lanes 2 and 3). These specific complexes were outcompeted with progressively increasing concentrations of the cold probes (10-, 20-, and 50-fold molar excesses) representing H- or C-κB sequences (Fig. 5A, lanes 4 to 6) but not with a nonspecific probe at a 50-fold molar excess concentration (lanes 10). The nature and intensity of the shifted complexes appear to be comparable between the H- and C-κB probes with no significantly visible differences. The binding of the nuclear complexes to the C-κB DNA probe was highly reproducible. To examine the nature of the host factors binding the DNA probes, we performed a supershift assay using nuclear extract isolated from TNF-α-activated Jurkat cells and immunoaffinity-purified rabbit antibodies raised against each of the five Rel family members (12). The analysis revealed the presence of a p50-p65 heterodimer, but not the other members of the Rel family, in the recruited complexes. The slower-moving complexes were evident only in the p50 and p65 lanes (Fig. 5B, lanes 4 and 5). The presence of the p50-p65 heterodimer also was reproducible using the nuclear extract of HeLa cells (data not shown). Furthermore, the nature of the shifted and supershifted complexes was comparable between the H- and C-κB probes, with a small difference. The intensity of the supershifted complexes was clearly higher when the C-κB probe was used in the assay rather than the H-κB probe, suggesting a higher affinity of binding of the nuclear complexes to the C-κB element (see below).
FIG 5.

C-κB probe binds NF-κB from the Jurkat nuclear extract. (A) Electrophoretic mobility shift analysis. Radiolabeled double-stranded probes (40,000 cpm) comprised of the H-κB (left) or the C-κB motif (right) were incubated with 30 μg of the Jurkat nuclear extract prepared from cells not activated or activated with TNF-α for 2 h. Cold competition was performed by incubating the complexes with 0-, 10-, 20-, or 50-fold molar excesses of the cold probes (H-κB or C-κB probe). A nonspecific probe was used only at the highest concentration. (B) Supershift analysis. The nuclear extract prepared from the activated Jurkat cells was incubated with antibodies specific to each of the Rel family members before the addition of radiolabeled H-κB or C-κB probe. Oct-1 was used as a loading control in the experiment. FP, free probe; NS, nonspecific probe; NS Ab, nonspecific antibody (anti-p24 antibody); NT, no TNF-α treatment; SC, shifted complex; SSC, supershifted complex. The data are representative of three independent experiments.
The C-κB element demonstrates higher binding affinity for NF-κB.
To compare the relative strengths of binding of the cellular complexes to the H- versus C-κB probes, we performed a quantitative cold competition assay (29) using Jurkat nuclear extract. The binding of 30,000 cpm of the labeled H- or C-κB probe to the host factors in 25 μg of nuclear extract was competed with incrementally increasing concentrations (0-, 4-, 16-, 64-, 128-, and 258-fold molar excesses) of cold H- or C-κB probes. The band intensities were measured using densitometry, and the percent binding was determined at each concentration of the competing probe considering the intensity of the probe binding as 100% in the absence of competition. The percent binding of the probe was plotted against the concentration of the competing probe. The amount of the competing probe required to reduce the probe binding to the host factors by half was determined using Scatchard plot analysis (Fig. 6A). When the H-κB probe was used in the assay, 13.0 pmol of the cold H-κB oligonucleotide was required for a 50% reduction in probe binding, whereas approximately only half of this quantity of the C-κB cold probe, 6.6 pmol, was sufficient for a comparable level of inhibition (Fig. 6A, left), confirming higher affinity of the C-κB motif for NF-κB. The results were comparable when the binding of the C-κB probe to the host factors was outcompeted with the H- or C-κB cold probe. For a 50% inhibition of the probe binding to the nuclear complexes, 35.2 and 18.0 pmol of H- and C-κB competitors, respectively, were required (Fig. 6A, right). NF-κB DNA probes can bind recombinant p50 protein in the absence of posttranslation modification and other host factors. The recruitment of p50 homodimers to the NF-κB motifs is critical for the establishment and maintenance of viral latency (34). Therefore, we compared the binding profile of recombinant p50 to those of H- and C-κB probes to determine the values of the affinity constant. We performed a quantitative EMSA using increasing amounts of recombinant p50 protein (0, 0.6, 1.3, 2.6, 5.2, and 10.4 nM) incubated with a constant amount of H- or C-κB radiolabeled probe (60,000 cpm) (35, 36). The quantities of free and complexed probes were quantitated using densitometry and plotted against the probe concentration used in the assay. Using Scatchard plot analysis, the dissociation constant (Kd) values for the H- and C-κB probes for the recombinant p50 protein were found to be 4.6 and 2.8 nM, respectively (Fig. 6B). The data from the gel shift analyses collectively demonstrate that compared to the H-κB element, the C-κB motif contains an approximately 2-fold higher affinity for NF-κB present in the nuclear extract or the recombinant p50 homodimer. A higher binding affinity of the C-κB site for NF-κB could have a significant impact on the functioning of the viral promoter in subtype C.
FIG 6.
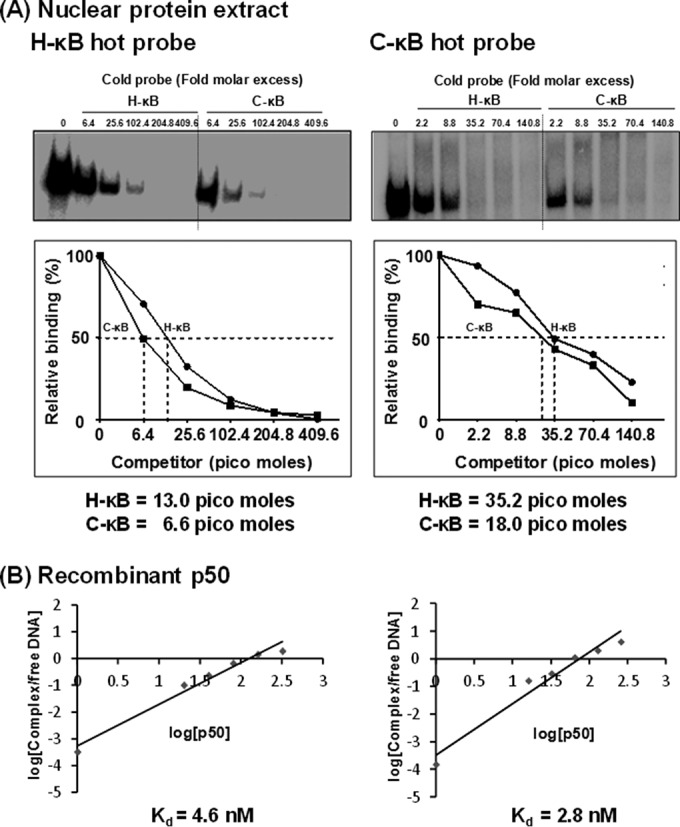
C-κB probe demonstrates a higher binding affinity for NF-κB in the Jurkat nuclear extract as well as recombinant p50 protein. (A) Radiolabeled double-stranded DNA probe (40,000 cpm) comprised of the H-κB or C-κB probe was incubated with 30 μg of the Jurkat nuclear extract. The competition was performed with progressively increasing quantities (fold excess) of H- or C-κB cold probe as shown. (Lower) The intensities of the DNA complexes were determined by densitometry. The percentage of the DNA band intensity was plotted against the concentration of competitor probe. The intercept on the x axis is considered the affinity of binding of the probes for the nuclear factors. The data are representative of three independent experiments. (B) Radiolabeled double-stranded DNA probe (60,000 cpm) comprised of the H-κB or C-κB probe was incubated with 0 to 10.4 nmol of the recombinant p50 protein. The competition was performed with progressively increasing quantities (fold excess) of H- or C-κB cold probe as shown. (Lower) The intensities of the DNA complexes were determined by densitometry. The percentage of the DNA band intensity compared to that of the no-competition control was plotted as log(cold probes) versus log(FP/SC). The intercept on the x axis is considered the affinity of binding of the probes for the nuclear factors. The data are representative of three independent experiments. FP, free probe; SC, shifted complex.
The C-κB motif binds NFAT.
The canonical H-κB element contains an overlapping binding site for another important family of transcription factors called the nuclear factor of activated T cells (NFAT). The NFAT family contains four different members (NFAT1-NFAT4) and one calcium-independent transcription factor (NFAT5) (37, 38). The core sequence for NFAT binding is GGAAA (38, 39). Of the NFAT family members, the NFAT1 homodimer can occupy the complementary sequence of 5 bp in the 3′ half of the H-κB motif (GGGACTTTCC; the NFAT binding sequence within the H-κB motif is underlined) and modulate HIV-1 gene expression (40, 41). Due to a partial overlap in the H-κB motif, NFAT and NF-κB could compete, leading to the mutually exclusive binding and alternate transactivation by these two factors (42). While NFAT1 binding to the H-κB site has a negative modulatory effect on NF-κB-mediated transactivation from the HIV-1 LTR, NFAT2 binding positively regulates HIV-1 LTR gene expression (43).
Importantly, it is not known if the T-to-G genetic variation at position 6 of the C-κB site (GGGGCGTTCC) has an impact on NFAT binding, as only four residues at the 3′ end of this element will be available for the binding. To this end, we compared NFAT binding to H- versus C-κB probes in a gel shift assay. Double-stranded radiolabeled DNA probe containing the NFAT binding sequence, derived from the IL-2 promoter, was incubated with the nuclear extract prepared from Jurkat cells induced with phorbol myristate acetate (20 ng/ml) and ionomycin (10 nM) for 1 h. The intensity of the shifted complexes increased following cell induction (Fig. 7A, compare lanes 2 and 3). In self-competition, the NFAT cold probe outcompeted the probe binding to the nuclear complexes at 5- and 25-fold molar excesses (Fig. 7A, compare lane 3 to lanes 4 and 5). Importantly, the C-κB cold oligonucleotide was as efficient as the H-κB cold probe in inhibiting the binding of the nuclear factors to the NFAT probe (Fig. 7A, compare lanes 4 and 5 to lanes 8 and 9). A scrambled cold DNA oligonucleotide, used as a negative control, was not able to compete in the assay, confirming the specificity of the assay (Fig. 7A, lanes 10 and 11). Collectively, the data confirmed that the C-κB sequence is capable of binding NFAT efficiently regardless of the variation at position 6.
FIG 7.

C-κB probe binds NFAT regardless of genetic variation. (A) Electrophoretic mobility shift analysis. A radiolabeled double-stranded DNA probe (40,000 cpm), derived from the IL-2 promoter sequence and containing an NFAT binding site, was incubated with 30 μg of the nuclear extract prepared from Jurkat cells after PMA/ionomycin activation for 1 h or control cells without activation. The cold competition was performed by incubating the complexes with a 5- or 25-fold excess of cold probes (NFAT, H-κB, C-κB, or a nonspecific probe). (B) Supershift analysis. Thirty micrograms of total protein of the nuclear extract prepared from PMA/ionomycin-induced Jurkat cells was preincubated with 2 μg of antibodies specific to NFAT1, NFAT2, or NFAT5 before the addition of radiolabeled NFAT, H-κB, or C-κB probes. Oct-1 was used as a loading control. FP, free probe; NS, nonspecific probe; NS Ab, nonspecific anti-p24 antibody; NT, no PMA/ionomycin treatment; SC, shifted complexes. The data are representative of three independent experiments.
To determine the identity of the NFAT family members present in the nuclear complexes bound to the probes, we performed a supershift analysis using commercial antibodies specific to three of the important members of the family, NFAT1, NFAT2, and NFAT5. The resolved nuclear bands clearly demonstrated the presence of NFAT1 and NFAT2, but not NFAT5, in the nuclear complexes recruited by all three DNA probes, including the C-κB probe (Fig. 7B). Although a supershifted complex was not visible under the experimental conditions, even with the NFAT positive-control probe, the reduced intensity of the bands when specific antibodies were added to the reaction mixture confirmed the presence of the specific host factors (Fig. 7B, compare lanes 3 to lanes 4 and 5). Importantly, an antibody against the p24 antigen of HIV-1, used as a negative control in the assay, did not reduce the intensity of the shifted bands (Fig. 7B, compare lanes 3 and 7). In summary, the C-κB motif of subtype C can efficiently bind NFAT; hence, it should be capable of exploiting the NFAT-mediated signaling to modulate gene expression from the viral promoter of subtype C.
The restricted gene expression from the HHH viral promoter is manifested in the integrated provirus.
In the reporter assays, the replacement of the native C-κB motif with the H-κB site led to a significant loss of subtype C promoter activity (Fig. 3A, HHC versus HCH or CHH LTRs). The expression profile of the viral promoter in reporter plasmid vectors may not be representative of that of an integrated provirus. To examine if the restricted promoter activity of the HCH- or CHH-LTRs could be manifested in the context of an integrated virus, we constructed a pair of infectious reporter viruses (Fig. 8A, HHC and HHH) using the pcLGIT vector reported previously that coexpressed EGFP and Tat under the control of the C-LTR (12, 18). While HHC represents the wild-type LTR configuration of the NF-κB motifs, in HHH LTR, two mutations were introduced in the C-κB motif, at positions 4 (G to A) and 6 (G to T), to convert the motif into H-κB. Of note, the Sp1 motif located proximal to the NF-κB site in both viruses was subtype C specific. The daughter viruses were pseudotyped with the VSV-G envelope, and their replication profile was compared in CEM-CCR5 or Jurkat T cells at a low MOI of 0.01. The cell pools stably expressing EGFP were sorted 18 h following TNF-α activation or without activation, and the MFI of the cell population was measured using flow cytometry at 48 and 96 h. A significant reduction in the mean fluorescence intensity was evident for the HHH-LTR compared to that of the wild-type HHC-LTR with or without TNF-α induction in both cell lines (Fig. 8). In CEM-CCR5 cells at 48 h, the MFI was reduced from 31,488 ± 1,040 to 14,131 ± 18 RLU (P < 0.002) between HHC and HHH LTRs under control conditions (Fig. 8A). When the cells were induced with TNF-α, these values were 54,864 ± 916 and 41,657 ± 1,855 RLU (P < 0.01) for the HHC and HHH LTRs, respectively. Comparable results were obtained in Jurkat cells (Fig. 8B) and at 96 h (data not shown). Collectively, the data confirmed a compromised promoter function when the natural C-κB motif is replaced by the generic H-κB site in the context of C-LTR in an integrated form.
FIG 8.
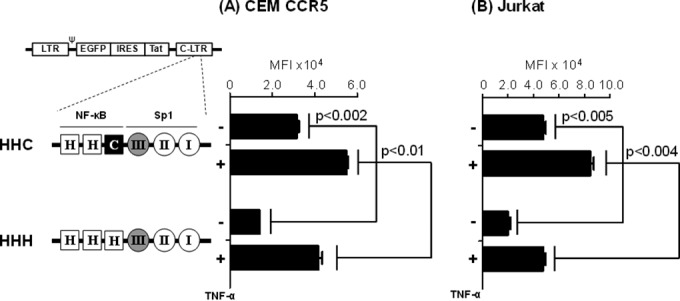
Compromised viral proliferation of the HHH viral strain in a single round of infection. A panel of two different reporter viruses, HHC-LGIT (wild type) and HHH-LGIT, was constructed with the TFBS variations engineered in the 3′ LTR. The reporter viruses were pseudotyped with VSV-G envelope, and each virus coexpresses GFP and Tat from the LTR. CEM-CCR5 (A) or Jurkat (B) cells were infected with equivalent TCID50 units of one of the two viral strains, and 6 h later the unbound virus was removed by washing. The cells were activated with TNF-α (100 ng/ml) after 18 h of incubation or left without activation. The mean fluorescence intensity of GFP was monitored using flow cytometry at 48 h (data presented) and 96 h (data not presented) following TNF-α activation. The data are representative of two independent experiments.
Replication potential of the infectious HHH virus is severely compromised in target T cells.
In the above-described assay, as the pseudotyped viruses lacking their own envelopes cannot proliferate in the target cells, the measured gene expression differences between the promoters would derive from a single round of viral integration and therefore cannot represent the real magnitude of this effect on viral fitness. To this end, to examine the magnitude of compromised LTR function in the context of an infectious virus, we constructed a pair of infectious viruses (Fig. 9A, HHC and HHH) using the subtype C prototype molecular clone Indie C1 (44). Except for the variations in two base pairs in the central κB element of the 3′ LTR (G to A and G to T substitutions at positions 4 and 6, respectively), the two sister molecular clones, HHC and HHH, were genetically identical in the rest of the viral backbone. The Sp1III motif proximal to the NF-κB site in both viruses was subtype C specific. Daughter viruses produced from these vectors would copy the 3′ U3 into the 5′ viral promoter, thus placing the expression of the viral genes under the control of the engineered LTRs. The replication kinetics of the daughter viruses derived from the two viral molecular clones was compared over a period of 4 weeks or longer in different target cells. CEM-CCR5 T cells coexpress the coreceptor CCR5 along with CD4. Hence, the cells can be infected by subtype C viral strains that require these two receptors for a productive infection.
FIG 9.

Replication profiles of the NF-κB and Sp1III chimera viral strains in target cells. (A) The schematic representation of a panel of NF-κB site variant infectious viral vectors constructed using Indie C1. The centrally positioned NF-κB and Sp1III motifs in the 3′ LTR are highlighted by shading, and the genetic variations engineered into these elements are shown. Of the four viral clones, the top two (HHC and HHH) were constructed for the early rounds of the assays, and two additional clones were constructed subsequently. The initial labeling of the first two vectors is shown on the left side and the revised labeling of all four viral strains on the right side. The letters B and C following the colon in the label represent the subtype identity of the Sp1III motif. In all four variant promoters, the other two Sp1 motifs (II and I) are intact but are not shown. Dots in the sequence alignment represent sequence homology. (B) CEM-CCR5 T cells, CN-2 cells, herpesvirus saimiri strain-transformed CN-2 cells, or CD8+ cell-depleted and mitogen-activated PBMC from a healthy donor were infected with 500 TCID50 of Indie-HHC or Indie-HHH virus. The secretion of p24 into the culture medium was monitored at regular intervals for several weeks. (C) A titer comparison of the viral stocks before and after the assay. The TCID50 titers of HHC and HHH viral strains were determined in TZM-bl cells using β-galactosidase staining before and 4 weeks after the target cell infection to determine the replication profile of the viruses performed as described for panel A. Each bar represents the means from triplicate wells. The data are representative of two independent experiments. (D) Replication profile of all four promoter variant viral strains in CEM-CCR5 cells. The data are presented as means of triplicate wells ± SD and are representative of at least three independent experiments. A two-tailed unpaired t test was used for statistical analysis.
The proliferation of the wild-type HHC virus demonstrated a normal profile in CEM-CCR5 T cells by peaking at around days 7 to 14 and decreasing after that (Fig. 9B, left). In stark contrast, the HHH virus failed to show a significant level of proliferation during the observation period. The difference in the proliferation kinetics was statistically significant between the two viral strains that differ from each other in only two base pairs in the Sp1 proximal NF-κB motif. The replication profile of the two viral strains could be recapitulated in CN-2 cells (Fig. 9B, middle), which are human PBMC transformed by herpesvirus saimiri strain yet retain many biological properties of the primary cells (45, 46). Importantly, restricted replication of the HHH viral strain was manifested in PBMC of several healthy and genetically unrelated donors (Fig. 9B, right; data from one representative donor are shown). The restricted replication of the HHH virus was highly reproducible in all of the above-described target cells. Importantly, the TCID50 titers of the two viral stocks were comparable before and 1 month after the assay, thereby ruling out the possibility of selective viral stock instability as an explanation for the observed difference in the proliferation of the two viral strains (Fig. 9C). Interestingly, a 4-fold difference in the titers of the viral stocks was evident and highly reproducible between HHC and HHH viral stocks, probably recapitulating the transcription differences between the two promoters when the viral stocks were prepared in HEK 293T cells using plasmid molecular clones.
The replacement of the homologous Sp1III motif restores the replication competence of the HHH viral strain.
In the Indie-HHH virus, the H-κB site was juxtaposed with the Sp1III motif of subtype C, a configuration (HHH:C) not naturally seen in the context of the subtype C virus. The Sp1III site of subtype C is characterized by subtype-specific variations seen at positions 5 and 10 compared with the homologous sequence in subtype B (GAGGCGTGGC in subtype B and GAGGTGTGGT in subtype C; differences are underlined). If the experimentally manipulated combination of the H-κB site and the subtype C Sp1III motif are not biologically compatible with each other, the restricted replication of the HHH virus in T cells perhaps could be explained. To test the hypothesis that specific combinations of NF-κB and Sp1 elements may not be functional together, we generated two additional viral variants, increasing the number of viral strains to four in the panel (Fig. 9A, revised label). The two new viral strains where the Sp1III site of subtype C was replaced by the subtype B counterpart were designated HHC:B and HHH:B. We reasoned that if the viral strain HHH:C is devoid of replication fitness due to an incompatibility between the H-κB motif and subtype C Sp1III site, replacing the Sp1III element with that of subtype B (HHH:C to HHH:B) should restore the lost replication competence of the viral strain.
CEM-CCR5 cells were infected with each of the four viral strains independently, and the secretion of the viral antigen p24 into the culture medium was determined on days 0, 3, and 7. The differences in the replication kinetics of the viral strains are typically established by day 7 or earlier (Fig. 9B). Hence, we did not monitor viral proliferation beyond day 7. While the wild-type HHC:C viral strain demonstrated the typical replication profile with a progressively increasing p24 concentration at days 3 and 7, the HHH:C virus failed to show a significant level of viral proliferation, as seen before (Fig. 9D). Importantly, the newly generated HHH:B virus, where the central H-κB site was recombined with the homologous subtype B Sp1III site, demonstrated unrestricted viral proliferation that was as efficient as that of the wild-type HHC:C viral strain (Fig. 9D). Additionally, the fourth virus, HHC:B, also proliferated as efficiently as the wild-type HHC:C strain without demonstrating any signs of replication incompetence. Collectively, the data ascertained that the Sp1III motif of subtype B functioned in combination with either the H- or C-κB elements (HHH:B or HHC:B) with comparable efficiency. In contrast, the Sp1III motif of subtype C can function only in association with the homologous C-κB element (HHC:C) and not with the heterologous H-κB element (HHH:C).
The HHH:C strain is compromised at the level of transactivation.
NF-κB plays an important role in the regulation of basal-level transactivation from the viral promoter in the absence of Tat (30). To understand what stage of the viral life cycle the virus containing variations in the NF-κB and Sp1 motifs might achieve replication advantage, CEM-CCR5 T cells were infected independently with HHC:C, HHH:C, HHC:B, or HHH:B viral strains at equivalent titers (500 TCID50). Using real-time PCR, we examined the proliferation of all four viral strains at two different stages, the generation of the reverse transcription products in the cell extract and the formation of proximal versus distal viral transcripts in the nucleus following integration (Fig. 10A). Twelve hours following viral infection, the reverse transcription products were probed using a real-time DNA PCR that targeted a 142-bp fragment from the U5-ψ region. All four viral strains demonstrated comparable levels of reverse transcription products, suggesting a comparable magnitude of viral infection of the target cells by all viruses, including the HHH:C viral strain that had demonstrated replication deficiency (Fig. 10B).
FIG 10.

Replication impairment of Indie HHH:C virus is at the level of transcription. (A) Schematic representation of the double-stranded viral DNA in the cytoplasm (upper) and the provirus after viral integration (lower). The curved arrow represents the transcription start site at R. The open square box represents the target sequence in DNA PCR for the detection of the late reverse transcription product. The filled square boxes represent the target sequences for the RNA PCR to detect the proximal (TAR) and distal (in Tat exon-1, located approximately 5.4 kb downstream) viral transcripts. (B) Analysis of the late RT products. CEM-CCR5 cells were infected with 500 TCID50 of one of the four viruses as shown. Genomic DNA was extracted from the cells 12 h postinfection and subjected to a quantitative real-time DNA PCR using SYBR green. A 145-bp DNA fragment spanning the U5-ψ region was amplified in the PCR. The threshold cycle (Ct) values were converted to template copy numbers using a standard curve. Each bar is representative of three replicate amplifications. The data are representative of two independent experiments. (C) Analysis of the proximal and distal viral transcripts. CEM-CCR5 cells were infected with 500 TCID50 of one of the four viruses as shown. Two hours before the RNA harvest, the cells were activated with TNF-α (100 ng/ml) or left without activation. The total cellular RNA was extracted 48 h postinfection, the cDNA was prepared, and a quantitative real-time PCR was performed as described in Materials and Methods. The proximal viral transcripts were detected using a primer pair amplifying an 89-bp region in the TAR region, and a 152-bp region was amplified in Tat exon-1 to detect the distal transcripts approximately 5.4 kb downstream of the transcription start site (see the schematic in panel A). The Ct values were converted to template copy numbers using a standard curve. Each bar is representative of three replicate amplifications, and the data are presented as mean copy numbers ± SD. A two-tailed unpaired t test was used for the statistical analysis. The data are representative of three independent experiments. (D) ChIP analysis to examine the occupancy of the viral promoters HHC:C and HHH:C with different transcription factors and associated activation or repressive marks. CEM-CCR5 cells were infected with Indie-HHC:C or Indie-HHH:C virus at 1,000 TCID50, and 48 h postinfection the ChIP assay was performed. Immunoprecipitation of the complexes was performed with 2 μg of the antibodies specific to different host factors as shown. An antibody against HIV-1 p24 was used as a negative control in the experiment. One-tenth of the input chromatin was loaded as the input control. The intensity of each band was normalized to the input. The mean band intensity ± SD of each assay parameter was determined from three replicates and plotted in the inset at the bottom. A two-tailed unpaired t test was used for the statistical analysis (***, P < 0.001; **, P < 0.01). The data are representative of two independent experiments.
A significant difference, however, was found between the restricted HHH:C viral strain and the other three viruses at the level of transcription, both transcription initiation and elongation (Fig. 10C). To evaluate the transcripts expressed from the viral promoters, two different real-time RNA PCRs were performed (Fig. 10A), one for the proximal viral transcripts within the TAR region and the other for distal transcripts in the Tat gene located ∼5.4 kb downstream of the transcription start site (12). Significantly higher levels of viral transcripts, from both proximal and distal transcripts, were generated from three of the four viral strains, HHC:C, HHC:B, and HHH:B, in the absence of TNF-α activation or its presence. The HHH:C virus, in contrast, failed to demonstrate considerable transcription under any of the experimental conditions (Fig. 10C). Under the experimental conditions, 1,227 ± 21 copies of the proximal transcripts were detected from the HHC:C wild-type strain in the absence of TNF-α activation. In contrast, only 164 ± 10 copies of the transcripts were detected from HHH:C, with the difference between the wild-type and variant promoters being statistically significant (P < 0.05). Following TNF-α activation, although the number of the viral transcripts from the restricted HHH:C LTR increased to 1,400 ± 40 copies, this number was significantly different from that of the wild-type HHC:C LTR, 17,503 ± 2,190 copies. Similar differences were observed between the two vectors under the other experimental conditions. Thus, the data of the transcription analysis are consistent with the viral proliferation kinetics and mapped the replication defect of the HHH:C viral strain to the lack of transcriptional activity from this viral promoter.
The epigenetic landscape of the restricted HHH:C promoter is transcription suppressive.
The suppressed viral proliferation and the compromised transcription activation from the restricted HHH:C LTR, compared to those of the other three viral promoters, are intriguing. The data collectively suggested the lack of efficient transcription from the HHH:C LTR. It is unexpected that two different transcription factors, NF-κB and Sp1, which function together in regulating the transcription synergistically from HIV-1 LTR (17), fail to demonstrate gene expression from the HHH:C LTR when the corresponding elements were experimentally positioned together in an orientation conducive to transactivation. Importantly, the two centrally positioned TFBS show an efficient transactivation in other closely related viral LTRs (HHC:C, HHC:B, and HHH:B). To the best of our knowledge, this is the first experimental demonstration of a specific combination of NF-κB and Sp1 binding sites not being biologically compatible with each other in the context of the HIV-1 promoter or in the context of any other viral or cellular promoter.
To gain more insights into the nature of the chromatin at the site of integration, we performed a chromatin immunoprecipitation analysis targeting several host factors (p50, p65, and Sp1), chromatin modulators (HDAC1 and HDAC3), epigenetic marks (H3K9Ac), RNA Pol II, or different phosphorylated forms of RNA Pol II (Ser-2 or Ser-5). CEM-CCR5 cells were infected with the wild-type Indie HHC:C or the Indie HHH:C variant strain at an equivalent TCID50 titer. Two days following the infection, a ChIP assay was performed. The assay results showed a transcriptionally active chromatin configuration in the case of the wild-type HHC:C LTR. In contrast, a predominantly suppressive chromatin landscape was evident in the case of the restricted HHH:C LTR (Fig. 10D). In the wild-type HHC:C LTR, significantly higher levels of p50:p65 heterodimer, acetylated H3K9, Pol II, and Pol II phosphorylated at serine 2 and serine 5 were identified compared to those for the restricted HHH:C promoter. The phosphorylated forms of serine 5 and serine 2 of Pol II are indicative of a higher magnitude of transcription initiation and elongation, respectively (47). In contrast, the presence of the transcription suppression markers HDAC1 and HDAC3 was significantly higher on the restricted HHH:C LTR than on the wild-type HHC:C LTR. In summary, the analysis of the chromatin configuration using the ChIP assay provided conclusive experimental evidence that the lack of replication fitness of this viral strain compared to that of the wild-type HHC:C virus is associated with a compromised recruitment of the transcription machinery to the HHH:C viral promoter. Importantly, the difference between the two viral strains, HHC:C and HHH:C, was in only two nucleotides located in the central NF-κB motif. It is indeed remarkable that a difference as small as this could have such a profound impact on the viral promoter that the promoter is not only completely silenced but fails to respond to TNF-α induction to emerge from viral latency. To the best of our knowledge, a phenomenon of this nature has not been reported previously.
DISCUSSION
Although the basic architecture of the promoter in diverse genetic subtypes of HIV-1 is modeled on a similar design, important subtype-specific differences exist in the profile of TFBS. It is important to understand how and to what extent the subtype-associated TFBS variations influence gene expression from the viral promoter. In the subtype C LTR, in the core of the promoter, a genetically variant NF-κB site and a subtype-specific Sp1III motif are seen (Fig. 1A), and a new-found functional association between the two variant sites is expected. Using different panels of reporter expression vectors that contained all 6 TFBS, intact or only the central κB and Sp1 sites, we confirmed that the two core TFBS function synergistically in an orientation- and position-dependent fashion. Collectively, the data revealed superior transcriptional activity from the subtype C promoter when the native C-κB and Sp1III motifs are present at the central location of the LTR.
Functional incompatibility between the generic H-κB motif and the subtype C unique Sp1III element.
The most unexpected finding of the present study regarding the association between the core TFBS in C-LTR comes from the use of a panel of four replication-competent viral strains (Fig. 9A). The viral strains are genetically identical except for the differences in the core TFBS representing the four possible combinations of the two NF-κB binding sites (C- and H-κB sites) and the two Sp1III motifs (subtype B- and C-associated sequences). While three of the four viral strains (HHC:C, HHC:B, and HHH:B) proliferated with comparable efficiency, the fourth virus (HHH:C), where the generic H-κB motif and the Sp1III site of subtype C were brought together, demonstrated a severe replication defect (Fig. 9B). The replacement of the subtype C unique Sp1III site in the restricted viral strain with the subtype B equivalent sequence (HHH:C to HHH:B) fully restored the severely compromised replication efficiency of the viral strain (Fig. 9D). Importantly, since the reduction in the promoter activity of the HHH:C-LTR is also manifested in the context of the subgenomic reporter viruses (Fig. 8), in the absence of all viral proteins except Tat, it is reasonable to assume that the compromised transcription is independent of the other viral factors. All four viral strains, including HHH:C, generated the RT products in the cytoplasm at comparable levels, ruling out differences at the level of virus infectivity (Fig. 10B). A direct comparison of the chromatin landscape of the integrated viruses of the wild-type HHC:C and the restricted HHH:C viral strains provided conclusive evidence that the HHH:C viral chromatin is predominantly transcription suppressive in nature (Fig. 10D). It is remarkable that a difference of only two base pairs in the promoter (HHC:C versus HHH:C LTRs) between two full-length viral strains that are otherwise genetically identical could have an impact so profound on the viral transcription that the HHH:C LTR is suppressed.
The impact of functional incompatibility between the C-κB and Sp1III motifs on viral proliferation was profound when evaluated using replication-competent viral molecular clones HHC:C and HHH:C (compare Fig. 8 and 9). The negative impact on transcription of the defective viral promoter may be explained by the possible recruitment of a negative cofactor, a suboptimal recruitment of a positive cofactor, or both. The replication profile of the subgenomic viral strains provides necessary clues that the compromised transcriptional activity manifested by the defective HHH:C viral promoter is due more to the suboptimal recruitment of the positive cofactors than the efficient recruitment of a negative cofactor. In the case of the subgenomic HHH:C viral strain, the compromised transcription from the LTR was only marginal, although it was statistically significant (Fig. 8). In CEM-CCR5 cells, after 48 h of viral infection, the basal-level GFP expression was 31,488 ± 1,041 RLU from the wild-type HHC:C LTR and 14,131 ± 18 RLU from the variant HHH:C LTR, with the difference being significant (P < 0.002). Unlike the replication-competent viral strains that infect the target T-cell populations for several successive rounds, the subgenomic reporter viruses infect the target cells only once. Thus, the cost of the replication compromise per round of viral infection is evident in the case of the subgenomic viral strains. The replication compromise of the defective virus is accumulated over many successive generations, thus leading to a profound defect during the observation period. Although the data presented here do not rule out the involvement of a negative host factor in suppressing the HHH:C LTR, the replication profile of the subgenomic HHH:C virus is strongly indicative of the suboptimal recruitment of a positive transcription activator(s).
We acknowledge a limitation of the present analysis with respect to the specific sequences of the C-κB and Sp1III motifs used. As depicted in Fig. 2, in a subset of randomly selected subtype C LTR sequences, genetic variation was evident in both C-κB and Sp1III sites, raising a question regarding the representative nature of the specific sequences used in the present study. Given that the magnitude of the genetic variation in the C-κB motif is small and the motif GGGGCGTTCC is present in a large majority of subtype C sequences, the sequence motif may be considered representative of subtype C (Fig. 2B). In contrast, the sequences of the Sp1III motif demonstrate a moderate level of genetic diversity. The variations are more pronounced at positions 2 and 5 (Fig. 2C). We considered the specific motif GAGGTGTGGT to be representative of subtype C for the present analysis, as this specific motif is present in nearly half of subtype C sequences. The impact of the genetic variation of the subtype C Sp1III motif remains to be evaluated, especially in the context of the functional incompatibility with the C-κB element.
The probable trajectory of subtype C promoter evolution.
Our data also offer insights into the specific evolutionary path subtype C may have followed to acquire a stronger viral promoter that contained an additional and variant NF-κB motif. A unique subtype C ancestor containing the typical LTR configuration, consisting of two identical H-κB sites and the subtype B-like Sp1III motif, must introduce two different variations to transform the viral LTR into the contemporary subtype C LTR (HH:B to HHC:C). These changes consist of the acquisition of the additional and genetically variant NF-κB binding site (the C-κB motif) and the introduction of the subtype-specific genetic variations into the Sp1 III site. Although it is difficult to ascertain in which specific order these two or more evolutionary changes were prioritized, acquisition of the variant Sp1III element appears to be more critical for subtype C than gaining an additional NF-κB binding site.
While predicting the evolutionary path of the subtype C ancestor promoter, we exploited the new knowledge of the functional incompatibility between the two TFBS and additionally applied two different assumptions. First, to acquire a genetic variation, a viral subtype is likely to follow the shortest evolutionary path involving the least number of evolutionary steps. Second, a viral subtype is more likely to acquire a specific variant TFBS readily available from a cellular source than altering an existing site into a variant motif, especially when this procedure gives the advantage of exploiting fewer evolutionary steps to reach the destination. Using these paradigms, we predicted six or seven different potential pathways for subtype C promoter evolution and weighed the merits of each evolutionary path as depicted in Fig. 11. The pathways differ from one another in the order in which the evolutionary modifications have been introduced into the subtype C promoter.
FIG 11.

Schematic diagram depicting an evolutionary trajectory of a hypothetical HIV-1 subtype C ancestral promoter. We surmise six different pathways by which the ancestral subtype C promoter HH:B may have evolved into the contemporary HHC:C LTR. Two successive steps of an undefined order would be required for promoter evolution, the acquisition of the C-κB motif, and the introduction of the subtype-specific variations into the Sp1III element. Of the six likely pathways of the evolutionary trajectory, pathways 1 and 4 may be disregarded based on the incompatibility observed in the present work between the C-κB and Sp1III elements in subtype C. Of the remaining four pathways, pathway 6 is the most plausible route the viral promoter may have taken considering the smallest number of steps, only two, needed.
Evolutionary path 1 predicts that the Sp1III site was the first element to have been subjected to the modification. The outcome cannot be a viable option, as the resulting LTR, HH:C, is nonfunctional. Therefore, the subtype C ancestor could not have attempted to alter the Sp1III site first. The evolutionary paths 2 and 3 predict that initially, the central H-κB site was changed into the C-κB motif (HH:B to HC:B). The HC:B virus should be viable. By following path 2, the HC:B ancestor would alter the Sp1III site to generate an HC:C intermediate that would acquire an additional H-κB site to make the contemporary C-LTR (HHC:C). Alternatively, the HC:B ancestor could acquire an additional H-κB site first (HHC:B) and alter the Sp1III site to generate the subtype C LTR. Pathways 2 and 3 should be less favored, as these paths not only require the transformation of the existing H-κB site into the C-κB motif but also need three evolutionary steps to generate subtype C LTR, unlike pathway 6 (described below). Pathways 4 and 5 predict that the HH:B ancestor initiates the evolutionary process with the acquisition of an additional H-κB element (HHH:B). Progression through pathway 4 further would have resulted in a replication-incompetent viral intermediate HHH:C that is not a viable option. Alternatively, the ancestor HHH:B could first alter the central H-κB site into C-κB (HHC:B) and then alter the Sp1III site to generate the subtype C LTR. Pathway 5 should be disfavored for the same reasons as pathways 2 and 3, although none of these three pathways can be ruled out logically. Pathway 6 predicts that the HH:B ancestor possibly acquired an additional C-κB element from a cellular promoter, such as the Fas gene, which contains an NF-κB element (GGGCGTTCC) (48) genetically quite similar to the C-κB site (GGGGCGTTCC) and positions the element immediately upstream of the Sp1III site to create the HHC:B LTR. The HHC:B intermediate, though fully functional (Fig. 9D), undergoes additional variations in the Sp1III site to generate the contemporary subtype C LTR HHC:C. Pathway 6 is the most likely evolutionary trajectory the subtype C HH:B ancestor would have taken, as this path needs only two evolutionary steps and does not require a transformation of an existing TFBS.
The acquisition of the variant Sp1III motif appears to be more critical for subtype C.
If pathway 6 is indeed the evolutionary trajectory subtype C used for promoter evolution, it is intriguing that the HHC:B intermediate promoter altered the Sp1III site further despite being fully functional (Fig. 9D). When the intermediate HHC:B promoter was fully functional, what was the need for the additional modification to create the HHC:C contemporary subtype C LTR? What was the nature of the selection forces that have driven the HHC:B promoter into the HHC:C LTR? Presumably, the introduction of the subtype-specific variations into the Sp1III motif is as critical for the subtype C promoter as the acquisition of the C-κB site. If the acquisition of a stronger promoter were the only motive, subtype C could have simply duplicated one of the existing H-κB elements (HH:B to HHH:B) or could have acquired an additional variant NF-κB site (HH:B to HHC:B). Importantly, subtype C acquires the variant Sp1III site even at the cost that the altered site is biologically compatible only with the homologous C-κB site and not the heterologous H-κB motif. No such restrictions are attached to the functioning of the C-κB motif, which can work with the heterologous Sp1III motifs of four different viral subtypes, A, B, D, and E (Fig. 3D). Therefore, it appears that promoter evolution in subtype C was primarily focused on gaining the variant Sp1III motif. A variant NF-κB site was necessary to retain the variant Sp1III motif. It appears that the functional incompatibility between the H-κB motif and Sp1III site of subtype C constitutes the primary selection pressure that forced subtype C into acquiring a genetically variable C-κB element instead of duplicating the existing H-κB site to enhance promoter strength. Thus, the present work provides a convincing answer for the presence of a variant NF-κB binding site at the core of the viral promoter.
Our work raises many additional questions. It remains to be established if the variant C-κB motif is receptive to differential cellular signaling by itself or in combination with the variant Sp1III element. Like the generic H-κB site, the C-κB motif can bind NFAT (Fig. 7) as well as NF-κB (Fig. 5), the latter with higher affinity (Fig. 6). A detailed analysis is required to understand if the variant C-κB motif confers a qualitative gain of advantage on C-LTR in terms of NFAT and NF-κB competition in controlling gene expression from the viral promoter and regulating viral latency. Additionally, despite the sequence variation, the C-κB motif recruited both NFAT1 and NFAT2. Since these two NFAT factors show a contrasting effect on NF-κB binding (39), the biological significance of NFAT binding to the NF-κB sites in the C-LTR needs additional analysis. It is tempting to speculate that the C-κB and subtype C-specific Sp1III combination provides a great replication advantage to subtype C in a major body compartment. Presently we do not know if the proposed pathway for the promoter evolution in subtype C is justified given the lack of ancestor subtype C sequences in the public databases representing the intermediate stages of subtype C. Additionally, it also is not clear what the evolutionary pressures were that forced the transition of the HHC:B promoter to HHC:C despite the fact that the former is as competent as the latter in terms of replication (Fig. 9D). On reflection, the presence of the C-κB motif in the core of HHC:B virus may have catalyzed the subsequent changes in the core Sp1III site to generate the contemporary HHC:C viral strain. The in vitro replication assay we used in the present study is clearly inadequate to represent the complex natural biological conditions necessary to recapitulate the differences in the replication properties of the two viral strains HHC:C and HHH:C.
In summary, our work establishes a new-found functional association between the subtype-specific variant NF-κB motif and subtype-specific variant Sp1III site in the subtype C viral promoter. Further, our data provide fresh leads as to the evolutionary pathway the subtype C promoter may have taken to acquire the contemporary LTR. How the unique genetic elements in the subtype C promoter modulate gene expression and viral latency needs additional investigation.
ACKNOWLEDGMENTS
Several reagents were obtained through the AIDS Research and Reference Reagent Program.
A.V. was a recipient of a research fellowship from The Council of Scientific and Industrial Research, Government of India. T.K.K. is a recipient of the Sir J. C. Bose National Fellowship. This work was supported by grants to T.K.K. and U.R. from The Department of BioTechnology, Government of India (Chromatin and Disease: Program Support; grant no. BT/01CEIB/10/III/01) and to U.R. by the Science and Engineering Research Board, The Department of Science and Technology, Government of India (grant no. SERB/F/3630/2014-15), and by intramural funds from JNCASR.
Author contributions: A.V., conception and design, acquisition of data, analysis and interpretation of data, drafting or revising the article, and providing essential unpublished data; P.R., R.L., R.V., and D.S., acquisition of data, analysis and interpretation of data, drafting or revising the article, and providing essential unpublished data; T.K.K. and U.R., conception and design, analysis and interpretation of data, and drafting or revising the article.
We declare that no competing interests exist.
REFERENCES
- 1.Hemelaar J, Gouws E, Ghys PD, Osmanov S. 2011. Global trends in molecular epidemiology of HIV-1 during 2000-2007. AIDS 25:679–689. doi: 10.1097/QAD.0b013e328342ff93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Siddappa NB, Dash PK, Mahadevan A, Desai A, Jayasuryan N, Ravi V, Satishchandra P, Shankar SK, Ranga U. 2005. Identification of unique B/C recombinant strains of HIV-1 in the southern state of Karnataka, India. AIDS 19:1426–1429. doi: 10.1097/01.aids.0000180795.49016.89. [DOI] [PubMed] [Google Scholar]
- 3.Chang SY, Vithayasai V, Vithayasai P, Essex M, Lee TH. 2000. Human immunodeficiency virus type 1 subtype E envelope recombinant peptides containing naturally immunogenic epitopes. J Infect Dis 182:442–450. doi: 10.1086/315730. [DOI] [PubMed] [Google Scholar]
- 4.Jeeninga RE, Hoogenkamp M, Armand-Ugon M, de Baar M, Berkhout B. 2000. Functional differences between the long terminal repeat transcriptional promoters of human immunodeficiency virus type 1 subtypes A through G. J Virol 74:3740–3751. doi: 10.1128/JVI.74.8.3740-3751.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Montano MA, Novitsky VA, Blackard JT, Cho NL, Katzenstein DA, Essex M. 1997. Divergent transcriptional regulation among expanding human immunodeficiency virus type 1 subtypes. J Virol 71:8657–8665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Garcia JA, Wu FK, Mitsuyasu R, Gaynor RB. 1987. Interactions of cellular proteins involved in the transcriptional regulation of the human immunodeficiency virus. EMBO J 6:3761–3770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Montano MA, Nixon CP, Ndung'u T, Bussmann H, Novitsky VA, Dickman D, Essex M. 2000. Elevated tumor necrosis factor-alpha activation of human immunodeficiency virus type 1 subtype C in southern Africa is associated with an NF-kappaB enhancer gain-of-function. J Infect Dis 181:76–81. doi: 10.1086/315185. [DOI] [PubMed] [Google Scholar]
- 8.Pereira LA, Bentley K, Peeters A, Churchill MJ, Deacon NJ. 2000. A compilation of cellular transcription factor interactions with the HIV-1 LTR promoter. Nucleic Acids Res 28:663–668. doi: 10.1093/nar/28.3.663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Regier DA, Desrosiers RC. 1990. The complete nucleotide sequence of a pathogenic molecular clone of simian immunodeficiency virus. AIDS Res Hum Retrovir 6:1221–1231. doi: 10.1089/aid.1990.6.1221. [DOI] [PubMed] [Google Scholar]
- 10.Ranjbar S, Rajsbaum R, Goldfeld AE. 2006. Transactivator of transcription from HIV type 1 subtype E selectively inhibits TNF gene expression via interference with chromatin remodeling of the TNF locus. J Immunol 176:4182–4190. doi: 10.4049/jimmunol.176.7.4182. [DOI] [PubMed] [Google Scholar]
- 11.Roof P, Ricci M, Genin P, Montano MA, Essex M, Wainberg MA, Gatignol A, Hiscott J. 2002. Differential regulation of HIV-1 clade-specific B, C, and E long terminal repeats by NF-kappaB and the Tat transactivator. Virology 296:77–83. doi: 10.1006/viro.2001.1397. [DOI] [PubMed] [Google Scholar]
- 12.Bachu M, Yalla S, Asokan M, Verma A, Neogi U, Sharma S, Murali RV, Mukthey AB, Bhatt R, Chatterjee S, Rajan RE, Cheedarla N, Yadavalli VS, Mahadevan A, Shankar SK, Rajagopalan N, Shet A, Saravanan S, Balakrishnan P, Solomon S, Vajpayee M, Satish KS, Kundu TK, Jeang KT, Ranga U. 2012. Multiple NF-kappaB sites in HIV-1 subtype C long terminal repeat confer superior magnitude of transcription and thereby the enhanced viral predominance. J Biol Chem 287:44714–44735. doi: 10.1074/jbc.M112.397158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bjorndal A, Sonnerborg A, Tscherning C, Albert J, Fenyo EM. 1999. Phenotypic characteristics of human immunodeficiency virus type 1 subtype C isolates of Ethiopian AIDS patients. AIDS Res Hum Retrovir 15:647–653. doi: 10.1089/088922299310944. [DOI] [PubMed] [Google Scholar]
- 14.Munkanta M, Handema R, Kasai H, Gondwe C, Deng X, Yamashita A, Asagi T, Yamamoto N, Ito M, Kasolo F, Terunuma H. 2005. Predominance of three NF-kappaB binding sites in the long terminal repeat region of HIV type 1 subtype C isolates from Zambia. AIDS Res Hum Retrovir 21:901–906. doi: 10.1089/aid.2005.21.901. [DOI] [PubMed] [Google Scholar]
- 15.Novitsky V, Smith UR, Gilbert P, McLane MF, Chigwedere P, Williamson C, Ndung'u T, Klein I, Chang SY, Peter T, Thior I, Foley BT, Gaolekwe S, Rybak N, Gaseitsiwe S, Vannberg F, Marlink R, Lee TH, Essex M. 2002. Human immunodeficiency virus type 1 subtype C molecular phylogeny: consensus sequence for an AIDS vaccine design. J Virol 76:5435–5451. doi: 10.1128/JVI.76.11.5435-5451.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Naghavi MH, Schwartz S, Sonnerborg A, Vahlne A. 1999. Long terminal repeat promoter/enhancer activity of different subtypes of HIV type 1. AIDS Res Hum Retrovir 15:1293–1303. doi: 10.1089/088922299310197. [DOI] [PubMed] [Google Scholar]
- 17.Perkins ND, Edwards NL, Duckett CS, Agranoff AB, Schmid RM, Nabel GJ. 1993. A cooperative interaction between NF-kappa B and Sp1 is required for HIV-1 enhancer activation. EMBO J 12:3551–3558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Burnett JC, Miller-Jensen K, Shah PS, Arkin AP, Schaffer DV. 2009. Control of stochastic gene expression by host factors at the HIV promoter. PLoS Pathog 5:e1000260. doi: 10.1371/journal.ppat.1000260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McAllister JJ, Phillips D, Millhouse S, Conner J, Hogan T, Ross HL, Wigdahl B. 2000. Analysis of the HIV-1 LTR NF-kappaB-proximal Sp site III: evidence for cell type-specific gene regulation and viral replication. Virology 274:262–277. doi: 10.1006/viro.2000.0476. [DOI] [PubMed] [Google Scholar]
- 20.Ilyinskii PO, Desrosiers RC. 1996. Efficient transcription and replication of simian immunodeficiency virus in the absence of NF-kappaB and Sp1 binding elements. J Virol 70:3118–3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Perkins ND, Agranoff AB, Pascal E, Nabel GJ. 1994. An interaction between the DNA-binding domains of RelA(p65) and Sp1 mediates human immunodeficiency virus gene activation. Mol Cell Biol 14:6570–6583. doi: 10.1128/MCB.14.10.6570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Abebe A, Demissie D, Goudsmit J, Brouwer M, Kuiken CL, Pollakis G, Schuitemaker H, Fontanet AL, Rinke de Wit TF. 1999. HIV-1 subtype C syncytium- and non-syncytium-inducing phenotypes and coreceptor usage among Ethiopian patients with AIDS. AIDS 13:1305–1311. doi: 10.1097/00002030-199907300-00006. [DOI] [PubMed] [Google Scholar]
- 23.Kurosu T, Mukai T, Komoto S, Ibrahim MS, Li YG, Kobayashi T, Tsuji S, Ikuta K. 2002. Human immunodeficiency virus type 1 subtype C exhibits higher transactivation activity of Tat than subtypes B and E. Microbiol Immunol 46:787–799. doi: 10.1111/j.1348-0421.2002.tb02766.x. [DOI] [PubMed] [Google Scholar]
- 24.Rodenburg CM, Li Y, Trask SA, Chen Y, Decker J, Robertson DL, Kalish ML, Shaw GM, Allen S, Hahn BH, Gao F. 2001. Near full-length clones and reference sequences for subtype C isolates of HIV type 1 from three different continents. AIDS Res Hum Retrovir 17:161–168. doi: 10.1089/08892220150217247. [DOI] [PubMed] [Google Scholar]
- 25.Van Opijnen T, Kamoschinski J, Jeeninga RE, Berkhout B. 2004. The human immunodeficiency virus type 1 promoter contains a CATA box instead of a TATA box for optimal transcription and replication. J Virol 78:6883–6890. doi: 10.1128/JVI.78.13.6883-6890.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Siddappa NB, Kashi VP, Venkatramanan M, Balasiddaiah A, Jayasuryan N, Mahadevan A, Desai A, Satish KS, Shankar SK, Ravi V, Ranga U. 2007. Gene expression analysis from human immunodeficiency virus type 1 subtype C promoter and construction of bicistronic reporter vectors. AIDS Res Hum Retrovir 23:1268–1278. doi: 10.1089/aid.2006.0305. [DOI] [PubMed] [Google Scholar]
- 27.Konopka K, Stamatatos L, Larsen CE, Davis BR, Duzqunes N. 1991. Enhancement of human immunodeficiency virus type 1 infection by cationic liposomes: the role of CD4, serum and liposome-cell interactions. J Gen Virol 72:2685–2696. doi: 10.1099/0022-1317-72-11-2685. [DOI] [PubMed] [Google Scholar]
- 28.Green MR, Sambrook J. 2012. Working with synthetic oligonucleotide probes, p 10.11–10.16. Molecular cloning: a laboratory manual, 4th ed Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 29.Dahiya S, Liu Y, Nonnemacher MR, Dampier W, Wigdahl B. 2014. CCAAT enhancer binding protein and nuclear factor of activated T cells regulate HIV-1 LTR via a novel conserved downstream site in cells of the monocyte-macrophage lineage. PLoS One 9:e88116. doi: 10.1371/journal.pone.0088116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Barboric M, Nissen RM, Kanazawa S, Jabrane-Ferrat N, Peterlin BM. 2001. NF-kappaB binds P-TEFb to stimulate transcriptional elongation by RNA polymerase II. Mol Cell 8:327–337. doi: 10.1016/S1097-2765(01)00314-8. [DOI] [PubMed] [Google Scholar]
- 31.Naghavi MH, Salminen MO, Sonnerborg A, Vahlne A. 1999. DNA sequence of the long terminal repeat of human immunodeficiency virus type 1 subtype A through G. AIDS Res Hum Retrovir 15:485–488. doi: 10.1089/088922299311240. [DOI] [PubMed] [Google Scholar]
- 32.Yuste E, Moya A, Lopez-Galindez C. 2002. Frequency-dependent selection in human immunodeficiency virus type 1. J Gen Virol 83:103–106. doi: 10.1099/0022-1317-83-1-103. [DOI] [PubMed] [Google Scholar]
- 33.Lemieux AM, Pare ME, Audet B, Lagault E, Sylvain L, Boucher N, Landry S, Opijnen TV, Berkhout B, Naghavi MH, Tremblay MJ, Barbeau B. 2009. T-cell activation leads to poor activation of the HIV-1 clade E long terminal repeat and weak association of nuclear factor-κB and NFAT with its enhancer region. J Biol Chem 279:52949–52960. doi: 10.1074/jbc.M409896200. [DOI] [PubMed] [Google Scholar]
- 34.Colin L, Van LC. 2009. Molecular control of HIV-1 postintegration latency: implications for the development of new therapeutic strategies. Retrovirology 6:111. doi: 10.1186/1742-4690-6-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Beinoraviciute-Kellner R, Lipps G, Krauss G. 2005. In vitro selection of DNA binding sites for ABF1 protein from Saccharomyces cerevisiae. FEBS Lett 579:4535–4540. doi: 10.1016/j.febslet.2005.07.009. [DOI] [PubMed] [Google Scholar]
- 36.Li Y, Jiang Z, Chen H, Ma WJ. 2004. A modified quantitative EMSA and its application in the study of RNA–protein interactions. J Biochem Biophys Methods 60:85–96. doi: 10.1016/j.jbbm.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 37.Hogan PG, Chen L, Nardone J, Rao A. 2003. Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev 17:2205–2232. doi: 10.1101/gad.1102703. [DOI] [PubMed] [Google Scholar]
- 38.Macian F, Lopez-Rodriguez C, Rao A. 2001. Partners in transcription: NFAT and AP-1. Oncogene 20:2476–2489. doi: 10.1038/sj.onc.1204386. [DOI] [PubMed] [Google Scholar]
- 39.Badran BM, Wolinsky SM, Burny A, Willard-Gallo KE. 2002. Identification of three NFAT binding motifs in the 5′-upstream region of the human CD3gamma gene that differentially bind NFATc1, NFATc2, and NF-kappa B p50. J Biol Chem 277:47136–47148. doi: 10.1074/jbc.M206330200. [DOI] [PubMed] [Google Scholar]
- 40.Romanchikova N, Ivanova V, Scheller C, Jankevics E, Jassoy C, Serfling E. 2003. NFAT transcription factors control HIV-1 expression through a binding site downstream of TAR region. Immunobiology 208:361–365. doi: 10.1078/0171-2985-00283. [DOI] [PubMed] [Google Scholar]
- 41.Zhang M, Clausell A, Robinson T, Yin J, Chen E, Johnson L, Weiss G, Sabbaj S, Lowe RM, Wagner FH, Goepfert PA, Kutsch O, Cron RQ. 2012. Host factor transcriptional regulation contributes to preferential expression of HIV type 1 in IL-4-producing CD4 T cells. J Immunol 189:2746–2757. doi: 10.4049/jimmunol.1103129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pessler F, Cron RQ. 2004. Reciprocal regulation of the nuclear factor of activated T cells and HIV-1. Genes Immun 5:158–167. doi: 10.1038/sj.gene.6364047. [DOI] [PubMed] [Google Scholar]
- 43.Kinoshita S, Chen BK, Kaneshima H, Nolan GP. 1998. Host control of HIV-1 parasitism in T cells by the nuclear factor of activated T cells. Cell 95:595–604. doi: 10.1016/S0092-8674(00)81630-X. [DOI] [PubMed] [Google Scholar]
- 44.Mochizuki N, Otsuka N, Matsuo K, Shiino T, Kojima A, Kurata T, Sakai K, Yamamoto N, Isomura S, Dhole TN, Takebe Y, Matsuda M, Tatsumi M. 1999. An infectious DNA clone of HIV type 1 subtype C. AIDS Res Hum Retrovir 15:1321–1324. doi: 10.1089/088922299310223. [DOI] [PubMed] [Google Scholar]
- 45.Vella C, Zheng NN, Easterbrook P, Daniels RS. 2002. Herpesvirus saimiri-immortalized human lymphocytes: novel hosts for analyzing HIV type 1 in vitro neutralization. AIDS Res Hum Retrovir 18:933–946. doi: 10.1089/088922202760265605. [DOI] [PubMed] [Google Scholar]
- 46.Zheng NN, Vella C, Easterbrook PJ, Daniels RS. 2002). Selection following isolation of human immunodeficiency virus type 1 in peripheral blood mononuclear cells and herpesvirus saimiri-transformed T cells is comparable. J Gen Virol 83:1343–1352. doi: 10.1099/0022-1317-83-6-1343. [DOI] [PubMed] [Google Scholar]
- 47.Coiras M, Montes M, Montanuy I, Lopez-Huertas MR, Mateos E, Le Sommer C, Garcia-Blanco MA, Hernández-Munain C, Alcamí J, Suñé C. 2013. Transcription elongation regulator 1 (TCERG1) regulates competent RNA polymerase II-mediated elongation of HIV-1 transcription and facilitates efficient viral replication. Retrovirology 10:124. doi: 10.1186/1742-4690-10-124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chan H, Bartos DP, Owen-Schaub LB. 1999. Activation-dependent transcriptional regulation of the human Fas promoter requires NF-kappaB p50-p65 recruitment. Mol Cell Biol 19:2098–2108. doi: 10.1128/MCB.19.3.2098. [DOI] [PMC free article] [PubMed] [Google Scholar]