

ABSTRACT
A hallmark of Ebola virus (EBOV) infection is the formation of viral inclusions in the cytoplasm of infected cells. These viral inclusions contain the EBOV nucleocapsids and are sites of viral replication and nucleocapsid maturation. Although there is growing evidence that viral inclusions create a protected environment that fosters EBOV replication, little is known about their role in the host response to infection. The cellular stress response is an effective antiviral strategy that leads to stress granule (SG) formation and translational arrest mediated by the phosphorylation of a translation initiation factor, the α subunit of eukaryotic initiation factor 2 (eIF2α). Here, we show that selected SG proteins are sequestered within EBOV inclusions, where they form distinct granules that colocalize with viral RNA. These inclusion-bound (IB) granules are functionally and structurally different from canonical SGs. Formation of IB granules does not indicate translational arrest in the infected cells. We further show that EBOV does not induce formation of canonical SGs or eIF2α phosphorylation at any time postinfection but is unable to fully inhibit SG formation induced by different exogenous stressors, including sodium arsenite, heat, and hippuristanol. Despite the sequestration of SG marker proteins into IB granules, canonical SGs are unable to form within inclusions, which we propose might be mediated by a novel function of VP35, which disrupts SG formation. This function is independent of VP35's RNA binding activity. Further studies aim to reveal the mechanism for SG protein sequestration and precise function within inclusions.
IMPORTANCE Although progress has been made developing antiviral therapeutics and vaccines against the highly pathogenic Ebola virus (EBOV), the cellular mechanisms involved in EBOV infection are still largely unknown. To better understand these intracellular events, we investigated the cellular stress response, an antiviral pathway manipulated by many viruses. We show that EBOV does not induce formation of stress granules (SGs) in infected cells and is therefore unrestricted by their concomitant translational arrest. We identified SG proteins sequestered within viral inclusions, which did not impair protein translation. We further show that EBOV is unable to block SG formation triggered by exogenous stress early in infection. These findings provide insight into potential targets of therapeutic intervention. Additionally, we identified a novel function of the interferon antagonist VP35, which is able to disrupt SG formation.
INTRODUCTION
Ebola virus (EBOV) causes a severe disease in humans characterized by significant immune dysfunction and high levels of viremia, leading to extraordinarily high case fatality rates (1, 2). As a member of the filovirus family, EBOV belongs to the order Mononegavirales and possesses a nonsegmented negative-sense (NNS) RNA genome that is roughly 19 kb and contains seven genes. EBOV genome replication and transcription take place in the cytoplasm of infected cells, where both the viral genome and the replication intermediate, the antigenome, associate with a number of viral proteins to form ribonucleoprotein (RNP) complexes, or nucleocapsids (3, 4). These complexes include the nucleoprotein NP, which packages the viral RNA, the polymerase L, the polymerase cofactor VP35, the transcription enhancer VP30, and the minor matrix protein VP24 (4–9). Within the cytoplasm, the nucleocapsids aggregate into highly ordered structures, termed viral inclusions, which are the sites of viral genome replication, nucleocapsid assembly, and maturation (10–13). The first morphological sign of EBOV replication is the formation of granular material in close proximity to the endoplasmic reticulum (ER) at about 9 h postinfection (p.i.), as observed by electron microscopy (14). Eventually, tubular structures appear in this granular material, representing the newly synthesized nucleocapsids that assemble into small inclusions (10, 14–16). At later stages of infection, the inclusions fuse together to become larger and more irregularly shaped, but they remain dynamic structures (10).
While type I interferons (IFNs) and cytoplasmic pattern recognition receptors are thought of as prototypical components of the host innate antiviral response, the cellular stress response is becoming increasingly appreciated as an important antiviral strategy. Central to this response is the rapid repression of cellular translation in order to prioritize the production of proteins important for cell survival. This translational arrest is mediated by the phosphorylation of the α subunit of eukaryotic translation initiation factor 2 (eIF2α) by one of four cytoplasmic kinases that sense distinct types of environmental stress (17). Most important for the response to viral infection is protein kinase R (PKR), which senses double-stranded RNA (dsRNA) and also serves as a critical component of type I IFN production (18–20). Additionally, heme-regulated inhibitor kinase (HRI) senses oxidative stress, PKR-like endoplasmic reticulum kinase (PERK) monitors ER stress, and general control nonderepressible 2 (GCN2) responds to nutrient deprivation. Phosphorylation of eIF2α prevents the assembly of the ternary preinitiation complex, which is required to bring tRNAmet to the 40S ribosomal subunit. The nontranslating mRNAs and associated RNA-binding proteins remain bound to the stalled preinitiation complexes and further assemble into cytoplasmic stress granules (SGs) (21–23). SGs are highly dynamic mRNA-protein aggregates and are comprised of a variety of components that can vary depending on the environmental conditions (24, 25). The continuous cycling of components into and out of SGs limits their examination to predominantly microscopy or immunofluorescence (IF)-based techniques. However, in addition to mRNA and the small ribosomal subunit, a number of SG proteins are thought to be consistent across all SG types and are therefore considered canonical SG components. These include the SG proteins eukaryotic translation initiation factor 4 gamma (eIF4G), Ras GTPase-activating protein-binding protein 1 (G3BP1), and T-cell-restricted intracellular antigen 1 (TIA-1) (21, 26).
Because viral protein synthesis relies on the host translation machinery, translational arrest mediated by SG formation is an efficient strategy for inhibiting viral replication, and consequently, many viruses have evolved mechanisms to overcome this block (27–29). Common strategies employed by viruses to disrupt SG formation include the degradation or sequestration of key SG nucleation proteins such as G3BP1 or TIA-1 (30–33) or the suppression of PKR activation (34, 35).
During EBOV infection, the virus employs a number of immune evasion strategies, many of which are mediated by the multifunctional protein VP35. In addition to its role as a nucleocapsid component and polymerase cofactor, VP35 counteracts type I interferon induction (36–44), blocks RNA interference pathways (45, 46), and antagonizes the activation of the dsRNA sensor PKR (47, 48). The inhibitory effects of VP35 on PKR, and thus on eIF2α phosphorylation, were observed late in infection (1 day p.i.), when viral protein expression in the infected cells was presumably high (49). Therefore, it is conceivable that PKR might be activated early in infection by incoming EBOV particles or viral transcripts, when the amount of VP35 is still low, and consequently lead to eIF2α phosphorylation and SG formation. This has been observed for poliovirus, which induces SG formation early in infection, before the viral protease 3C is expressed at sufficient levels to cleave G3BP, but then blocks it as infection progresses (32, 50). While many viruses have been shown to disrupt SG formation at various points during infection via diverse mechanisms, little is known about the dynamics of the stress response in EBOV infection.
Here, we show that EBOV infection does not trigger eIF2α phosphorylation or SG formation at any time postinfection. However, SG proteins are sequestered within viral inclusions, where they colocalize with viral mRNA and form distinct granules. Despite this sequestration of SG proteins, EBOV infection only partially blocks SG formation induced by the activation of other eIF2α kinases. Importantly, we identified a novel function for VP35, showing that it not only prevents SG formation by blocking PKR activation but also disrupts SG formation independently of eIF2α phosphorylation and dsRNA binding. We propose that this might contribute to the inhibition of SG formation during late stages of infection.
MATERIALS AND METHODS
Cells and viruses.
Cell lines used in this study include African green monkey kidney cells (Vero and Vero E6; ATCC CRL-1586), human epithelial osteosarcoma cells (U2OS; ATCC HTB-96), human hepatocellular carcinoma cells (Huh7, kindly provided by J. Alonso, Texas Biomedical Research Institute, San Antonio, TX), and HeLa cells (ATCC CCL-2). We also used U2OS cells expressing G3BP1 fused to green fluorescent protein (GFP) (51) and a tetracycline- and doxycycline-inducible U2OS cell line expressing USP10 fused to GFP (52). Cells were maintained in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS), penicillin (50 U/ml), and streptomycin (50 mg/ml). Twenty-four hours prior to infection, U2OS-GFP-USP10 cells were treated with 200 ng/ml of doxycycline to induce USP10 expression.
Zaire ebolavirus (EBOV) isolates Kikwit 1995 (GenBank accession number KR867676.1) and Mayinga (GenBank accession number AF086833.2) were grown in Vero or Vero E6 cells, and virus titers were determined by 50% tissue culture infective dose (TCID50) or plaque assay. All work with EBOV was done under biosafety level 4 conditions at the Integrated Research Facility in the Rocky Mountain Laboratories, Division of Intramural Research, NIAID, NIH, Hamilton, MT, or the State Research Center of Virology and Biotechnology VECTOR, Koltsovo, Novosibirsk, Russian Federation. Sample inactivation and removal were performed according to standard operation protocols approved by the local institutional biosafety committee.
Plasmids and transfections.
EBOV genes encoding NP, VP35, VP30, or VP24 (Mayinga isolate; GenBank accession number AF086833.2) were cloned into either of the pcDNA3.1 and pCAGGS expression vectors (53) as described previously (5, 48, 54). VP35 was also fused to an N-terminal hemagglutinin (HA) epitope tag. The previously described VP35-3A mutant contains the following mutations: R305A, K309A, and R312A (48). Transfections were performed using Lipofectamine LTX transfection reagent (Invitrogen).
IF analysis.
A total of 2.5 × 104 U2OS or U2OS-GFP-G3BP1 cells were seeded per well of an 8-well chamber slide and 1 day later infected with EBOV at a multiplicity of infection (MOI) of 1 or 1.5 PFU or TCID50 units per cell. At the desired time points p.i., cells were inactivated and fixed by treatment with 4% paraformaldehyde (PFA) for 48 h. Cells were permeabilized with 0.1% Triton X-100 for 5 to 8 min at room temperature, treated with 0.1 M glycine for 5 min at room temperature, and incubated in blocking solution (2% bovine serum albumin [BSA], 0.2% Tween 20, 3% glycerol, and 0.05% sodium azide in phosphate-buffered saline [PBS]) for 10 min at room temperature. All antibodies were diluted in blocking solution. Primary antibodies were incubated for a minimum of 1 h at room temperature or overnight at 4°C. Antibodies used for the detection of EBOV proteins include a mouse monoclonal anti-NP antibody (kindly provided by G. G. Olinger, USAMRIID, Frederick, MD), a rabbit anti-VP30 antibody (kindly provided by V. E. Volchkov, Université de Lyon, Lyon, France), a mouse monoclonal anti-VP35 antibody (kindly provided by C. F. Basler, Icahn School of Medicine at Mount Sinai, New York, NY), a mouse anti-VP24 antibody (kindly provided by G. G. Olinger, USAMRIID), and a goat anti-EBOV serum (kindly provided by S. Becker, University of Marburg, Marburg, Germany). HA-tagged VP35 was detected using a mouse anti-HA antibody (Covance). Antibodies used to detect SG proteins include goat anti-eIF3 (Santa Cruz; SC16377), rabbit anti-eIF4G (Santa Cruz; SC11373), goat anti-TIA-1 (Santa Cruz; C-20), mouse anti-PABP (Santa Cruz; SC-32318), mouse anti-G3BP (BD Biosciences; 611126), mouse anti-HuR (Santa Cruz; SC-5261), and rabbit anti-TTP (kindly provided by W. F. C. Rigby, Geisel School of Medicine at Dartmouth College, Lebanon, NH) antibodies. Secondary antibodies were conjugated with Alexa Fluor 350, 488, 594, or 647 (Invitrogen). Each immunofluorescence (IF) analysis was performed at least three times in independent experiments.
Induction of stress granules.
For the induction of stress granules, cells were treated with 0.5 to 2 mM sodium arsenite (Ars) for 30 min at 37°C and fixed in 4% PFA. For infection experiments, cell supernatants were replaced with fresh medium containing the desired amount of Ars, or medium without Ars was added for mock-treated cells. For transfection experiments, the desired amount of Ars or the same volume of PBS (mock treated) was added dropwise onto the cells. Treatment with 1 μM to 1.5 μM hippuristanol (Hipp; kind gift of J. Pelletier, McGill University, Montreal, Canada) was performed as described for Ars. Heat stress was applied by incubating the cells at 44°C for 1 h.
CHX treatment.
U2OS cells were infected with EBOV at an MOI of 1.5 TCID50 units per cell. At 24 h p.i., cell supernatants were removed and replaced with DMEM supplemented with 2% FBS and or DMEM supplemented with 2% FBS and 0.5 mM Ars. After a 30-min incubation period at 37°C, supernatants were removed and replaced with medium containing either 100 μg/ml of cycloheximide (CHX) or dimethyl sulfoxide (DMSO). Cells were incubated for 1 h at 37°C, washed once with PBS, and fixed with DMEM containing 4% PFA.
Western blot analysis.
A total of 2 × 105 HeLa cells were seeded in 10-cm dishes and infected either 24 h (harvested at 24 h p.i.) or 48 h (harvested at 1 and 6 h p.i.) after seeding at an MOI of approximately 5 to 10 or left uninfected. At the desired time points p.i., whole-cell extracts were prepared in 25 μl of cell extraction buffer (Thermo Fisher Scientific) complemented with 1× protease inhibitor mix (25× Complete; Roche) and 1× phosphatase inhibitor (100× HALT phosphatase inhibitor cocktail; Thermo Scientific). Cellular proteins were detected using a rabbit anti-eIF2α phosphospecific antibody (Life Technologies; 44-728G), a mouse total eIF2α antibody (Biosource; AHO0802), or a rabbit lamin B antibody (Abcam; ab16048). Secondary antibodies were either donkey anti-mouse or donkey anti-rabbit IRD-680 or IRD-800 (LI-COR). Protein bands were quantified by using the Odyssey imaging system and software (LI-COR). Attempts to use U2OS cells for the Western blot analysis failed, as the low cell density leading to low protein amounts in combination with harsh inactivation procedures did not allow for reliable Western blot results using phosphospecific antibodies.
FISH.
Fluorescence in situ hybridization (FISH) probes were designed using Stellaris FISH Probe Designer software from Biosearch Technologies. A total of 48 probes, each 20 nucleotides in length and labeled with Alexa Fluor 594 or Cy3, were used to target the EBOV NP mRNA sequence. U2OS cells were infected at an MOI of 1 as described in the section on IF above. Cells were fixed with 4% PFA for 48 h and stored in 70% ethanol at −20°C until usage. Cells were rehydrated in PBS and subsequently subjected to FISH alone or IF followed by FISH. Detection of RNA by FISH analysis was not affected by the additional IF step in this analysis. For IF, all reagents were supplied with 2 mM vanadyl ribonucleoside complex (VRC). For permeabilization and blocking, the IF protocol described above was used. Primary antibodies were incubated for 3 h and secondary antibodies for 2 h at room temperature. After antibody incubation, cells were treated with 3% PFA for 10 min at room temperature for postfixation and subjected to FISH. For FISH, cells were washed and rehydrated in 2× SSC (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate) containing 10% formamide for 5 min. Hybridization was performed at 37°C overnight in 100 μl of hybridization buffer (10% dextran sulfate, 2 mM VRC, 0.02% RNase-free BSA, 50 μg of Escherichia coli tRNA, 2× SSC, 10% formamide) containing labeled FISH probes targeting EBOV NP mRNA.
Click-iT metabolic labeling assay.
For fluorescence-based detection of nascent proteins, the Click-iT l-homopropargylglycine (l-HPG) metabolic labeling system (Invitrogen) was used. U2OS cells were infected as described above. At 25 h p.i., cells were washed with D2-Met/Cys depletion medium (Sigma Gibco) and incubated in D2-Met/Cys depletion medium for 1 h. Cell supernatants were removed and replaced with D2-Met/Cys depletion medium supplemented with 100 μM Click-iT l-HPG or D2-Met/Cys depletion medium alone. After a 30-min incubation period at 37°C, cells were fixed with 4% PFA as described above, permeabilized with methanol for 15 min at −20°C, and blocked with 5% donkey serum for 1 h. The Click-iT reaction buffer containing Alexa Fluor 488 detection reagent was added for 30 min at room temperature, protected from light. The cells were washed with 5% donkey serum and then placed in PBS at 4°C until ready for further IF processing and imaging, which were performed as described above.
Transmission electron microscopy (TEM).
Vero cells were infected with the Mayinga strain of EBOV at an MOI of 1. At 1 day p.i. cells were trypsinized and pelleted by a low-speed centrifugation step. The cell pellets were fixed in 4% paraformaldehyde in Hanks' solution for 24 h, washed three times in Hanks' solution, postfixed in 1% osmium tetraoxide, dehydrated in ethanol and acetone, and embedded in Epon-araldite. To perform ultrathin sectioning a diamond knife was used, with uranyl acetate and lead citrate used to contrast ultrathin sections. The samples were then analyzed with a JEM 1400 transmission electron microscope (JEOL) at 80 kV. All images were collected using a side-mounted Veleta digital camera (SIS). All reagents were obtained from SPI Supplies.
Imaging and processing.
Confocal imaging was performed using a Zeiss 710 confocal microscope with a Plan-Apochromat primary objective (63×; numerical aperture [NA], 1.4). All images were taken with a pinhole diameter set to an Airy unit of 1 and using multitrack scanning for each fluorophore to prevent bleed-through. Images were also acquired with a Zeiss Axiovert 200 M inverted microscope and a Plan-Apochromat primary objective (63×; NA, 1.4). Image acquisition and processing software used includes the Zeiss software Zen (confocal microscope), Zeiss AxioVert (inverted microscope), and ImageJ.
Statistical analysis.
For quantitative analysis of SG-containing cells, an unpaired two-sample t test was performed using GraphPad Prism version 5.04 for Windows (GraphPad Software, La Jolla, CA).
RESULTS
Stress granules do not form during EBOV infection.
To determine if SG formation is induced during EBOV infection, particularly at earlier time points, we analyzed the distribution of the SG marker protein eIF4G in EBOV-infected U2OS cells at different time points p.i. by immunofluorescence (IF) analysis. In uninfected cells, eIF4G was homogenously distributed in the cytoplasm at all time points examined, but it was redistributed into typical punctate SGs when sodium arsenite (Ars) was used to induce oxidative stress (Fig. 1). In EBOV-infected cells at early time points p.i., eIF4G showed homogenous distribution in the cytoplasm and SGs were not observed. EBOV infection, visualized by staining for the nucleocapsid protein VP35, was detected only at 17 and 24 h p.i., when viral protein expression was high. At these late time points, VP35 was observed in viral inclusions, while eIF4G was homogenously distributed throughout the cytoplasm of infected cells. Examining the distribution of two other canonical SG proteins, TIA-1 and eIF3, confirmed that SGs were not present in EBOV-infected cells at any time point examined (data not shown).
FIG 1.
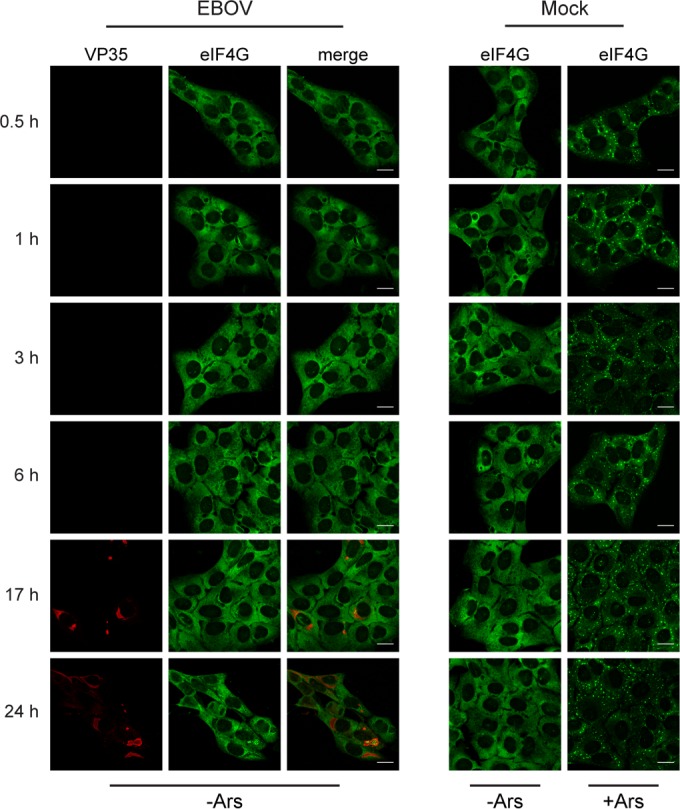
EBOV infection does not induce SG formation. U2OS cells were infected with EBOV at an MOI of 1 or mock infected. At the indicated time points p.i., cells were treated with 0.5 mM Ars (+Ars) for 30 min to induce SG formation or left untreated (−Ars). Immunofluorescence analysis was performed using antibodies against VP35 (red) and eIF4G (green). Scale bars = 20 μm.
SG proteins are sequestered within viral inclusions.
Although eIF4G was homogenously distributed throughout the cytoplasm of infected cells, indicating that SGs do not form, it was found aggregated within the viral inclusions starting at 17 h p.i. (Fig. 1). This aggregation was more pronounced at 24 h p.i., when viral inclusions were larger and more prominent (Fig. 1 and 2A, panel i). Sequestration of SG proteins in EBOV inclusions at 24 h p.i. was also observed for other SG marker proteins, including eIF3, PABP (Fig. 2A, panels ii and iii), and G3BP1 (data not shown). While a number of SG proteins were found aggregated within viral inclusions, the homogenous distribution of TIA-1 in both the nucleus and cytoplasm of EBOV-infected cells demonstrated that not every canonical SG protein was sequestered in viral inclusions. TIA-1 distribution looked similar to that observed in uninfected cells and showed little accumulation within viral inclusions (Fig. 2A, panel iv). This indicates that the SG-like granules in EBOV inclusions are different from fully formed canonical SGs and might represent a distinct species of protein granules.
FIG 2.

SG proteins are sequestered within viral inclusions. (A to D) U2OS cells were infected with EBOV at an MOI of 1.5, fixed at 1 day p.i., and analyzed by co-IF analysis. (A) Co-IF analysis using antibodies against eIF4G (i, green), eIF3 (ii, green), PABP (iii, green) or TIA-1 (iv, green). EBOV inclusions (red) were detected using an antibody against VP35 (i, ii, and iv) or an anti-EBOV serum that recognizes both GP and NP (iii). Boxed areas are shown zoomed 2× below the larger images. (B) Co-IF analysis using antibodies against eIF4G (red) and eIF3 (green) or against eIF4G (red) and PABP (green). Boxed areas are shown zoomed 3.4× in insets. (C) Co-IF analysis using antibodies against the viral proteins NP, VP30, and VP24 (all red) and eIF3 (green). Boxed areas are shown zoomed 2× below the larger images. (D) Z-stack analysis of EBOV-infected U2OS cells. The viral proteins NP, VP35, VP30, and VP24 were examined by using antibodies specific for each protein (red). The SG protein eIF3 (green) was detected using an anti-eIF3 antibody. Images on the top and right side of each larger image depict the Z-axis. (E) U2OS cells were transfected with the indicated combinations of plasmids expressing EBOV NP, VP30, VP35, or VP24 and examined 48 h posttransfection by co-IF analysis using antibodies against NP (red) and eIF4G (green). Boxed areas are shown zoomed 2.5× in insets. Scale bars = 20 μm.
We next determined if the detected SG marker proteins colocalize in discrete granules within the viral inclusions. IF analysis of infected cells revealed colocalization between eIF4G and eIF3 and between eIF4G and PABP, suggesting that these proteins are components of the same granules within the viral inclusions (Fig. 2B). Using a U2OS cell line constitutively expressing G3BP1 fused to GFP (51), we also examined the colocalization of GFP-G3BP1 with eIF3 and showed that GFP-G3BP1 followed the same pattern of distribution as eIF3, PABP, and eIF4G (data not shown), confirming that the SG marker proteins observed within EBOV inclusions colocalized within the same granules.
SG proteins colocalize with viral RNA in the inclusions.
We next examined whether individual nucleocapsid proteins were found within the inclusion-bound SG protein granules (IB granules) and did not observe colocalization between any of the nucleocapsid proteins examined (NP, VP35, VP30, and VP24) and the SG marker protein eIF3 (Fig. 2A and C). These data were confirmed by confocal optical sectioning analysis visualizing the distribution of eIF3 within the viral inclusions. The Z-stack analysis showed that eIF3 formed discrete granules that were clearly separated from the cytoplasm and were entirely encompassed by viral inclusion proteins (Fig. 2D). Similar results were obtained using the U2OS-GFP-G3BP1 cell line (data not shown). Together, these results indicate that the nucleocapsid proteins fully encompass, but are excluded from, the SG protein aggregates within viral inclusions.
The driving force for inclusion formation is NP and when expressed in cells in the absence of other viral proteins, NP self-assembles, leading to the formation of inclusion-like structures. Interaction of NP with the other nucleocapsid proteins, either directly or via a linker protein, then redirects them into the inclusion-like structures (6, 8, 55). To analyze if inclusion formation itself was sufficient to trigger the relocation of SG marker proteins into viral inclusions, U2OS cells were transfected with different combinations of plasmids expressing the EBOV nucleocapsid proteins NP, VP35, VP30, and VP24 (Fig. 2E). Although inclusion formation was observed in cells expressing NP alone or NP in combination with the other nucleocapsid proteins, eIF4G was homogenously distributed in the cytoplasm and did not form granules within the inclusion-like structures (Fig. 2E). These results indicate that inclusion formation itself does not induce the aggregation of SG proteins within inclusions.
Many proteins involved in SG formation are RNA-binding proteins important for mRNA stability and translation (56). Because EBOV inclusions are the sites of viral RNA synthesis and contain viral RNA (10, 11), we next examined if viral mRNAs colocalized with the IB granules. To visualize the intracellular distribution of viral mRNAs during infection, we performed fluorescence in situ hybridization (FISH) using probes targeting the positive-sense NP mRNA, which is the most abundant viral mRNA (57). While the probes employed also recognize the positive-sense antigenome RNA, this RNA species is much less abundant in infected cells than the viral mRNA (57, 58). We therefore infer that the FISH probes predominantly bind to mRNA. At 6 h p.i., the NP mRNA was diffusely distributed throughout the cytoplasm of infected cells. At 24 h p.i., however, the NP mRNA accumulated within the inclusions, forming aggregates that resembled those observed for the sequestered SG marker proteins (Fig. 3A, white arrows). When infected cells were analyzed by IF followed by FISH, costaining of NP mRNA and eIF3 confirmed that the accumulated RNA colocalized with IB granules (Fig. 3B). As a control, cells were stained with an antibody directed against tristetraprolin (TTP), an RNA-binding protein that is found in SGs induced by energy deprivation but is not a universal SG protein (59). TTP did not accumulate in EBOV inclusions, nor did it colocalize with NP mRNA (Fig. 3C). Together, these data suggest that viral RNA colocalizes with IB granules and may be involved in their formation.
FIG 3.
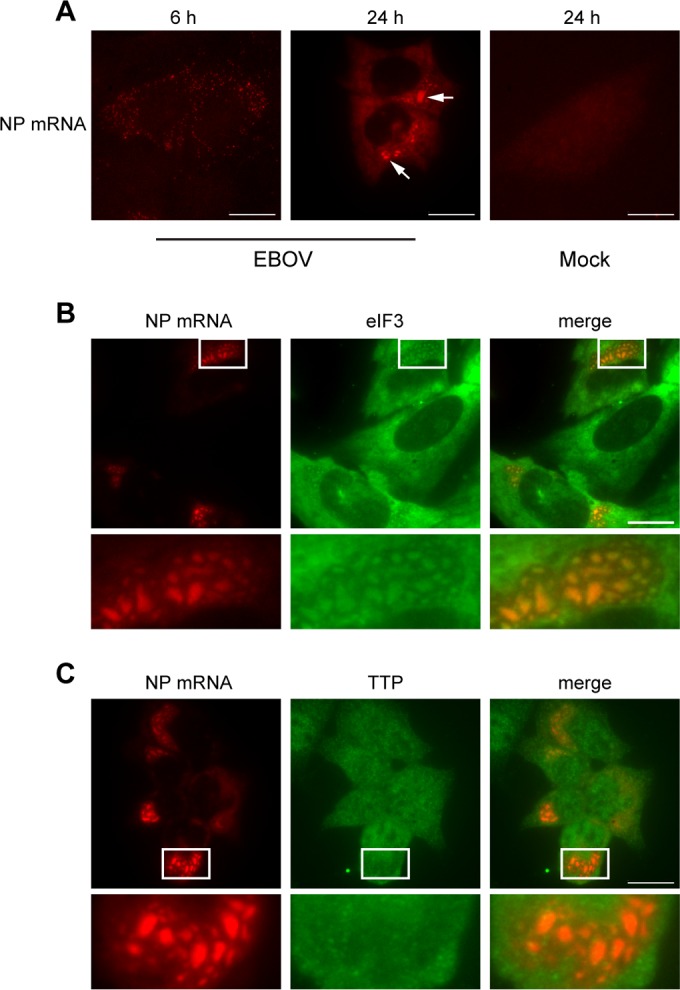
SG proteins colocalize with viral RNA within EBOV inclusions. (A) U2OS cells were infected with EBOV at an MOI of 1, fixed at 6 h or 24 h p.i., and analyzed by FISH using RNA probes directed against the NP mRNA (red). White arrows indicate viral mRNA aggregates within viral inclusions. (B and C) EBOV-infected U2OS cells were fixed at 24 h p.i. and examined by IF assay for the SG proteins eIF3 or TTP (green), followed by FISH using RNA probes against NP mRNA (red). Boxed areas are shown zoomed 3.7× below the larger images.
Inclusion-bound granules are distinct from canonical SGs.
The IB granules were decisively larger and less symmetrical in shape than typical SGs and differed in their protein composition from canonical SGs, as shown by the lack of TIA-1 (Fig. 2A) and TTP (Fig. 3C), suggesting that these structures are distinct from canonical SGs. Other viruses, including chikungunya virus, Semliki Forest virus, and vaccinia virus, induce the formation of granules containing various SG components that are morphologically, structurally, or functionally different from canonical SGs (31, 33, 60, 61). To further characterize the IB granules in EBOV-infected cells and determine potential differences from canonical SGs, we used cycloheximide (CHX), an inhibitor of protein synthesis known to block polysome disassembly and dissolve SGs. Canonical SGs disperse upon exposure to CHX, while some other virally induced SG-like aggregates do not (25, 61, 62). EBOV- or mock-infected U2OS cells were treated with Ars to induce SG formation and then subsequently treated with CHX. In mock-infected cells treated with Ars, SGs completely disassembled upon CHX treatment (Fig. 4A). When EBOV-infected cells were treated with CHX alone, the IB granules persisted and did not disassemble. Ars treatment of EBOV-infected cells led to the formation of cytoplasmic SGs, and IB granules disappeared from the inclusions (Fig. 4A, +Ars +DMSO). While the cytoplasmic SGs in Ars-treated infected cells dissolved upon CHX treatment, the IB granules did not (Fig. 4A, +Ars +CHX), indicating that these granules are, indeed, different from canonical SGs and represent a unique accumulation of SG marker proteins within viral inclusions.
FIG 4.

Inclusion-bound granules are not canonical SGs. (A) U2OS cells were infected with EBOV at an MOI of 1.5, incubated for 24 h, and then treated with 0.5 mM Ars for 30 min (+Ars) or left untreated (−Ars). Cells were subsequently treated with either DMSO or CHX for 1 h and analyzed by co-IF analysis using antibodies against VP35 (red) and eIF4G (green). Boxed areas are shown zoomed 2× in insets. Scale bars = 20 μm. (B) U2OS-eGFP-USP10 cells were treated with doxycycline (+ Doxy) for 24 h to induce eGFP-USP10 expression or left untreated (− Doxy). Cells were then treated with 0.5 mM Ars for 30 min or left untreated. After fixation, SGs were visualized using an antibody against HuR (red). White arrows indicate cells expressing eGFP-USP10 that do not form SGs. Yellow arrowheads indicate cells that do not express eGFP-USP10 and form SGs. (C) U2OS-eGFP-USP10 cells were treated with doxycycline for 24 h to induce eGFP-USP10 expression and then infected with EBOV at an MOI of 1.5 in the presence of doxycycline, fixed at 24 h p.i., and stained using an antibody against G3BP (red). Boxed areas are shown zoomed 3.75× in insets.
To further characterize the IB granules, we used a doxycycline-inducible U2OS cell line that allows overexpression of the SG inhibitory protein ubiquitin carboxyl-terminal hydrolase 10 (USP10), a deubiquitinating enzyme that binds to G3BP and can prevent SG formation by blocking G3BP aggregation (52, 63). In the absence of doxycycline, the cells did not express enhanced GFP (eGFP)-USP10 and formed SGs when treated with Ars (Fig. 4B). When these cells were treated with doxycycline to induce eGFP-USP10 overexpression and subsequently treated with Ars, SG formation did not occur, as expected (Fig. 4B, compare cells marked by white arrows to cells marked by yellow arrowheads). Overexpression of eGFP-USP10 had no discernible effect on EBOV replication or inclusion formation (data not shown). Interestingly, although SGs were unable to form in cells overexpressing eGFP-USP10, G3BP was still sequestered in viral inclusions in EBOV-infected, doxycycline-treated eGFP-USP10 cells (Fig. 4C). This corroborates our findings that IB granules are distinct from canonical SGs.
Inclusion-bound granules do not block protein translation.
Next we addressed the question of whether IB granules exert antiviral activity in EBOV-infected cells. SG-like granules that are distinct from SGs but have antiviral activity have been observed for various viruses, including poxviruses and influenza virus (19, 61, 62). Formation of antiviral granules during vaccinia virus infection interferes with protein translation, leading to decreased viral titers (61, 62). To determine if the IB granules blocked protein translation during EBOV infection, we examined nascent protein synthesis in EBOV-infected cells using Click-iT labeling technology to label newly synthesized proteins in EBOV-infected cells at 24 h p.i. Viral inclusions and SG marker proteins were visualized by co-IF analysis. While nascent proteins (green) were distributed throughout the cytoplasm in mock-infected cells, newly synthesized proteins accumulated in the viral inclusions in EBOV-infected cells despite the presence of SG proteins, suggesting that the IB granules are not detrimental to protein translation (Fig. 5A). Transmission electron microscopy (TEM) analyses of EBOV-infected Vero cells showed that ribosomes were not present within viral inclusions but were found in close proximity (Fig. 5B, red arrowheads). Regardless of the size or density of viral inclusions, ribosomes were never found interspersed with nucleocapsids (yellow arrows) or viral granular material (blue arrowheads), suggesting that the inclusions are not the sites of viral protein translation. Rather, translation of viral proteins likely occurs in close proximity to inclusions with the newly synthesized proteins then transported into viral inclusions.
FIG 5.

Inclusion-bound granules are not antiviral. (A) U2OS cells were mock infected or infected with EBOV at an MOI of 1.5. At 24 h p.i., cells were pulse-labeled with the amino acid analog l-homopropargylglycine (l-HPG) for 30 min. Nascent proteins were visualized with an Alexa Fluor 488 detection reagent specific for l-HPG (green), while virus infection was detected using an anti-VP35 antibody (blue) and inclusion-bound granules using an eIF4G specific antibody (red). The rightmost images show EBOV-infected cells labeled with l-HPG 488 in the absence of VP35 and eIF4G antibodies. Boxed areas are shown zoomed 2.6× in insets. Scale bars = 20 μm. (B) Vero cells were infected with EBOV at an MOI of 1, fixed at 24 h p.i. and examined by transmission electron microscopy. Cytoplasmic areas containing viral inclusions are shown. Boxed areas are shown zoomed 1.6× in insets. Blue arrowheads indicate viral granular material, yellow arrows nucleocapsids, and red arrowheads ribosomes.
EBOV does not efficiently block SG formation induced by various forms of exogenous stress.
Because the IB granules did not negatively impact protein translation, we next investigated if their sequestration might play a more proviral role and prevent canonical SG formation in EBOV-infected cells. A number of viruses have been shown to not only avoid the induction of the cellular stress response but also actively prevent SG formation during infection (28, 64). To examine if EBOV was able to block SG formation induced by oxidative stress, EBOV-infected U2OS cells were treated with Ars at different time points p.i. and analyzed by co-IF analysis using antibodies against VP35 and a panel of SG marker proteins. SG formation was observed in the cytoplasm of EBOV-infected, Ars-treated cells at all time points p.i. but was reduced at 24 h p.i. compared to that in uninfected cells treated with Ars and in EBOV-infected cells at earlier stages of infection (Fig. 6A and B). While the majority of EBOV-infected, Ars-treated cells formed cytoplasmic SGs at early time points p.i., eIF4G was homogenously distributed in a significant proportion of infected, Ars-treated cells at 24 h p.i. (Fig. 6B and C). A quantitative analysis of cells containing canonical SGs showed that nearly 100% of the mock-infected, Ars-treated cells displayed typical punctate SGs, while there was a significant reduction in the number of SG-containing cells in EBOV-infected cells treated with Ars at 24 h p.i. (Fig. 6B). In addition, the SGs in many infected, Ars-treated cells appeared more diffuse and less punctate than in Ars-treated, mock-infected cells (Fig. 6C).
FIG 6.

SGs are reduced late during EBOV infection. (A) U2OS cells were mock infected or infected with EBOV at an MOI of 1.5. At the indicated time points p.i., cells were treated with 0.5 mM Ars for 30 min, fixed, and examined by IF analysis using antibodies against eIF4G (green) and VP35 (red). (B) Quantification of SG-containing cells at 24 h p.i. Counting was performed for three separate experiments. For each experiment, 24 random fields were counted containing total numbers of 290, 187, and 262 cells, respectively. Counting was performed twice by different lab members. Statistical analysis was performed by GraphPad Prism software, using an unpaired two sample t test. (C) Representative fields showing the three outcomes of SG induction observed in EBOV-infected U2OS cells at 24 h p.i., shown zoomed 1.4× below with separated color channels. (D) U2OS cells were infected with EBOV as described for panel A. At 24 h p.i., cells were treated with 0.5 mM Ars for 30 min. Accumulation of SG proteins within viral inclusions was examined by staining for VP35 (red) and eIF4G (green). Boxed areas are shown zoomed 2× in insets. (E) U2OS-GFP-G3BP1 cells were infected as for panel D but treated with 1 mM Ars for 30 min. Cells were stained for VP30 (red), and G3BP1 was visualized by its GFP tag. Boxed areas are shown zoomed 1.5× in insets. Scale bars = 20 μm.
To determine if the sequestration of SG proteins within inclusions impaired cytoplasmic SG formation and accounted for the reduction in SGs observed late in infection, EBOV-infected U2OS cells that were left untreated or treated with Ars at 24 h p.i. were closely examined to determine if there was a correlation between the presence of IB granules and the absence of cytoplasmic SGs. As expected, eIF4G was sequestered in a majority of viral inclusions in EBOV-infected, untreated cells (Fig. 6D). Surprisingly, EBOV-infected cells treated with Ars showed little to no aggregation of eIF4G within inclusions, and SGs could still form in the cytoplasm of most cells (Fig. 6D). This shows that the sequestration of SG proteins within viral inclusions is not sufficient to prevent SG formation induced by cellular stress. It also indicates that inclusion-bound SG proteins are not rigidly restrained by viral components and may be released into the cytoplasm upon the induction of stress. Interestingly, when U2OS-GFP-G3BP1 cells were infected with EBOV and treated with Ars, GFP-G3BP1 remained clustered within viral inclusions even in cells that showed cytoplasmic SG formation (Fig. 6E). The overexpression of GFP-G3BP1 in this cell line suggests that the availability of excess G3BP1 in the cytoplasm reduces the need for inclusion-bound G3BP1 when cellular stress is induced.
Similar to the case with Ars treatment, SG formation was induced in EBOV-infected U2OS-GFP-G3BP1 cells exposed to heat stress or hippuristanol (Hipp), and GFP-G3BP1 remained sequestered in viral inclusions (data not shown). Hipp induces SG formation independently of eIF2α phosphorylation by inhibiting the RNA helicase eIF4A and preventing 60S ribosomal subunit joining (65). These data confirm that the sequestration of SG proteins within viral inclusions is not sufficient to fully prevent SG formation induced by multiple forms of cellular stress.
SG formation does not occur within viral inclusions.
Despite the ability of the inclusion-bound SG proteins to respond to cytoplasmic stress, canonical SGs were not present within viral inclusions in EBOV-infected, Ars-treated cells (Fig. 7A). Rather, we observed assorted distributions of SG marker proteins and viral inclusions. In a majority of cells, the viral inclusions were completely devoid of SG proteins but were surrounded by cytoplasmic SGs (Fig. 7A, panel i). A significant number of EBOV-infected cells contained no SGs within either the cytoplasm or viral inclusions (Fig. 7A, panel ii). SG-like aggregates were rarely found within large viral inclusions (Fig. 7A, panel iii) but were occasionally seen in very small viral inclusions (Fig. 7B, left set of images). Although canonical SGs were predominately absent in large inclusions, they were often seen in close proximity to, or surrounded by, viral inclusions (Fig. 7B, middle set of images). Late stages of EBOV infection are characterized by large, more diffuse inclusions that eventually disperse (11). In these instances, SGs were often scattered throughout dispersing inclusions but did not colocalize with viral proteins (Fig. 7B, right set of images). Based on these observations, we propose that while viral inclusions do not entirely insulate SG proteins from cytoplasmic stress signals, SG formation itself is prevented within inclusions.
FIG 7.

Stress granules do not form in viral inclusions. U2OS cells were infected with EBOV at an MOI of 1.5. At 24 h p.i., cells were treated with 0.5 mM Ars for 30 min, fixed, and analyzed by IF analysis using antibodies against eIF4G (green) and VP35 (red). Representative fields of infected cells are shown. (A) Arrows indicate three examples of SG formation around viral inclusions, shown zoomed 1.4× to the right with separated color channels. (B) Boxed areas are shown zoomed 2.6× below the larger images. Scale bars = 20 μm.
VP35 is able to disrupt SG formation.
To dissect the underlying mechanism(s) for the observed reduction in the number of SGs in EBOV-infected, Ars-treated cells at 24 h p.i. (Fig. 6), we first analyzed if EBOV was able to block eIF2α phosphorylation triggered by oxidative stress. We performed Western blot analysis to examine the phosphorylation state of eIF2α in EBOV-infected cells that were treated with Ars or left untreated at both early and late time points p.i. Since high cell density can trigger eIF2α phosphorylation (66), we used subconfluent HeLa cells for this experiment. Ars treatment led to eIF2α phosphorylation in mock-infected as well as EBOV-infected HeLa cells at all time points examined, indicating that although EBOV is able to block PKR signaling and antagonize eIF2α phosphorylation (47, 49), it is unable to prevent Ars-induced activation of the signaling pathways leading to SG formation (Fig. 8A). eIF2α was not phosphorylated in EBOV-infected, untreated cells at any time examined, which corroborates our results showing that EBOV infection per se does not induce eIF2α-triggered SG formation (Fig. 1). Importantly, robust eIF2α phosphorylation was observed in EBOV-infected, Ars-treated cells at 24 h p.i., indicating that the observed impairment of SG formation cannot be attributed to the inhibition of eIF2α phosphorylation induced by oxidative stress. We therefore explored if EBOV was able to disrupt SG formation downstream of eIF2α phosphorylation.
FIG 8.

VP35 disrupts SG formation. (A) HeLa cells were infected with EBOV at an MOI of 1.5 or mock infected and harvested for Western blot analysis at the indicated time points p.i. Prior to lysis, cells were treated with 0.5 mM Ars for 30 min to induce SG formation or left untreated. Western blot analysis was performed using a phosphospecific eIF2α antibody and antibodies against total eIF2α and lamin B (loading control). (B) U2OS-GFP-G3BP1 cells were transfected with 0.75 μg of HA-VP35 pcDNA3.1 expression plasmid. At 1 day (1 d) or 2 days (2 d) posttransfection, cells were treated with 1 mM Ars for 30 min or left untreated and examined by IF analysis. VP35 (red) was visualized using an HA-specific antibody. SGs (green) were visualized by GFP-G3BP1 expression. Boxed areas are shown zoomed 4× in insets. (C and D) Huh7 cells were transfected with 0.5 μg or 1.5 μg of HA-VP35 pCAGGS expression plasmid. At 2 days posttransfection, cells were left untreated (C) or treated with 0.5 mM Ars for 30 min (D) and fixed. Co-IF analysis was performed using an HA-specific antibody to detect VP35 (red) and an anti-eIF4G antibody (green). (E) Huh7 cells were transfected as described for panel D. At 2 days posttransfection, cells were left untreated or treated with 1.5 μM Hipp for 30 min and fixed. Co-IF analysis was performed as for panel D. White arrows in panels D and E indicate cells expressing VP35, with little to no SG formation. Yellow arrowheads indicate cells that do not express VP35 and show canonical SG formation. Boxed areas are shown zoomed 4× in insets. Scale bars = 20 μm.
Because SG formation was prevented within viral inclusions (Fig. 6 and 7), we analyzed whether any of the EBOV nucleocapsid proteins were able to interfere with the formation of SGs. EBOV NP, VP35, VP30, and VP24 were individually expressed in Huh7 cells or U2OS-GFP-G3BP1 cells. At 2 days posttransfection, the cells were treated with Ars or left untreated and examined by IF analysis for the impact of individual viral proteins on SG formation. Of the proteins tested, VP35 was found to disrupt the intracellular aggregation of SG proteins under conditions of oxidative stress. When U2OS-GFP-G3BP1 cells were transfected with a VP35 expression plasmid in the absence of Ars and examined by IF at 1 day posttransfection, VP35 showed a granular distribution in the cytoplasm, while GFP-G3BP1 was homogenously distributed (Fig. 8B, 1 d, left set of images). At 2 days posttransfection, when VP35 expression levels were higher, VP35 accumulated in larger cytoplasmic aggregates, while GFP-G3BP1 remained homogenous but showed some areas of minor redistribution to VP35-dense areas (Fig. 8B, 2 d, left set of images). VP35 expressed in smaller amounts at 1 day posttransfection was directed to SGs formed in U2OS-GFP-G3BP1 cells after Ars treatment. Higher magnification revealed that these SGs were surrounded by globular VP35-positive aggregates (Fig. 8B, 1 d, right set of images). At 2 days posttransfection, VP35 was observed in irregularly shaped, large aggregates that colocalized with GFP-G3BP1 in Ars-treated cells. Canonical GFP-G3BP1-containing SGs were almost entirely absent in these VP35-positive cells (Fig. 8B, 2 d, right set of images). To examine if, indeed, the expression level of VP35 correlated with its ability to disrupt SG formation, Huh7 cells were transfected with either 0.5 μg or 1.5 μg of a VP35 expression plasmid and analyzed at 2 days posttransfection. When expressed at lower levels, VP35 colocalized with or was in close proximity to SGs in Ars-treated cells. While SGs could still form in these cells, they started to appear more diffuse and less punctate than those in Ars-treated cells lacking VP35 (Fig. 8D, VP35 [0.5 μg], compare cells indicated by white arrow and yellow arrowhead). As also observed with GFP-G3BP1-expressing U2OS cells, SGs were frequently surrounded by small, globular VP35 aggregates. At higher expression levels, VP35 was found in patch-like structures in the cytoplasm of both untreated and Ars-treated cells (Fig. 8C and D) and SG formation was strongly impaired in the Ars-treated cells (Fig. 8D, VP35 [1.5 μg], compare cells indicated by white arrows and yellow arrowheads). Together, these data indicate that VP35 interacts with SGs and is able to disrupt SG formation when expressed at sufficiently high levels. To determine if VP35 was able to disrupt SG formation induced by other forms of stress, Huh7 cells were transfected with 0.5 or 1.5 μg of a VP35 expression plasmid, and treated with 1.5 μM Hipp at 2 days posttransfection. Similar to Ars-treated cells, in Hipp-treated cells expressing lower levels of VP35, VP35 formed clusters around eIF4G and showed some areas of colocalization (Fig. 8E, VP35 [0.5 μg], insets). SGs were almost entirely absent from Hipp-treated cells transfected with 1.5 μg of a VP35 expression plasmid (Fig. 8E, VP35 [1.5 μg], insets). Collectively, these data indicate that VP35 is able to disrupt SG formation induced by diverse forms of stress.
VP35 is able to disrupt SG formation in the presence of NP and prevents NP-driven inclusion formation.
VP35 is not diffusely distributed in the cytoplasm of EBOV-infected cells but is directed into the viral inclusions via binding to NP-RNA complexes (67). It has been reported previously that the ratio of NP to VP35 is crucial for NP-induced inclusion formation and that excess expression of VP35 prevents archetypal inclusion formation in cells coexpressing NP and VP35 (68). To examine if VP35 was still able to disrupt SG formation in the presence of NP, Huh7 cells were transfected with NP and VP35 plasmid DNA in a 1:1 ratio and 2 days after transfection were treated with Ars or left untreated. As expected, VP35 was no longer diffusely distributed in the cytoplasm of untreated cells but was directed to NP-induced inclusions (Fig. 9A, left set of images, top row, compare to VP35 distribution in Fig. 8). Upon Ars treatment, VP35 remained bound to NP-induced inclusions and did not colocalize with eIF4G-containing SGs that formed in the cytoplasm (Fig. 9A, right set of images, top row). When NP concentrations were kept low but VP35 was expressed at 3-fold-larger amounts in excess of NP, inclusions were dispersed, confirming the previous observations by Noda and colleagues (68) (Fig. 9B, bottom rows). In addition, SG formation was significantly reduced under these conditions (Fig. 9A and B, bottom rows). This might recapitulate the disruption in SG formation seen late in EBOV infection, when VP35 expression is high. In summary, our data provide strong evidence that VP35 interacts with SGs and plays a role in preventing SG formation late in infection when concentrations of VP35 are high.
FIG 9.

Excess VP35 disrupts both SGs and viral inclusions. Huh7 cells were transfected with pCAGGS expression plasmids expressing EBOV NP or HA-tagged VP35. Top rows show cells transfected in a 1:1 plasmid ratio at 0.5 μg each per well in a 6-well plate. Cells shown in bottom rows were transfected in a NP-to-VP35 plasmid ratio of 1:3 (0.5 μg of NP DNA and 1.5 μg of VP35 DNA). At 2 days posttransfection, cells were treated with 0.5 mM Ars for 30 min or left untreated and then imaged by IF analysis. Staining was performed using antibodies targeting HA to visualize VP35 (A; red), NP (B; red), and eIF4G (A and B; green). Scale bars = 20 μm.
Double-stranded RNA binding is not required for VP35's ability to disrupt SG formation.
Previous extensive mutational analyses of VP35 have shown that residues R305, K309, and R312 within the interferon inhibitory domain (IID) are essential for its ability to bind dsRNA and block antiviral host responses but have minor effects on its polymerase cofactor functions (38, 41, 45, 48). To determine if dsRNA binding is also important for VP35's capacity to disrupt SG formation, we used the previously described VP35-3A mutant (48), in which the three basic amino acids at positions 305, 309, and 312 are replaced with alanine. U2OS cells were transfected with 0.5 μg or 1 μg of a VP35-3A expression plasmid and at 2 days posttransfection treated with Ars or left untreated. In the absence of Ars, VP35-3A was homogenously distributed in the cytoplasm when transfected at a low level (0.5 μg) and more aggregated at a higher expression level (1 μg), similar to the wild type (Fig. 10A, left set of images). When cells transfected with 0.5 μg of VP35-3A plasmid were subsequently treated with Ars, although SGs could still form in these cells, VP35-3A often colocalized with or formed aggregates around SGs (Fig. 10A, compare white arrows to yellow arrowheads). Furthermore, SGs appeared more diffuse and less punctate than those in Ars-treated cells lacking VP35. In cells transfected with a larger amount (1 μg) of VP35-3A plasmid, VP35-3A protein did not colocalize with eIF4G upon Ars treatment, and there was a clear reduction in SG formation (Fig. 10A, middle rows, compare yellow arrowheads to blue arrows). When coexpressed in a 1:2 ratio with NP, VP35-3A colocalized with NP inclusions, indicating that NP binding was not disrupted by the mutation (Fig. 10B). Furthermore, when NP and VP35-3A expression plasmids were cotransfected at a 1:5 ratio, NP inclusions began to disperse and resembled those disrupted by wild-type VP35. Taken together, these data suggest that VP35-3A behaved similarly to wild-type VP35, indicating that dsRNA binding is not required for VP35's function to disrupt SGs or NP-based viral inclusions.
FIG 10.

VP35's dsRNA binding ability is not required for SG disruption. (A) U2OS cells were transfected with 0.5 μg or 1 μg of an HA-tagged VP35-3A expression plasmid and 2 days posttransfection treated with 0.5 mM Ars for 30 min or left untreated. VP35-3A distribution was visualized using an HA-specific antibody (red). SG formation was shown using an antibody directed against eIF4G (green). White arrows indicate cells expressing VP35-3A showing VP35-3A colocalizing with or surrounding SGs. Blue arrows indicate cells that express VP35-3A and show significantly disrupted SG formation. Yellow arrowheads indicate cells that do not express VP35-3A and show canonical SG formation. (B) U2OS cells were transfected with an expression plasmid encoding EBOV NP (0.5 μg) alone or in combination with an HA-tagged VP35-3A expression plasmid to a 1:2 or 1:5 ratio with the NP plasmid. Antibodies directed against NP (green) or HA (red) were used for co-IF analysis. Insets show boxed regions zoomed 4×. Scale bars = 20 μm.
DISCUSSION
The increasing number of viruses shown to disrupt or evade SG formation during infection has led to the recognition of the cellular stress response as an important antiviral mechanism. In this study, we showed that EBOV infection does not induce SG formation at any time point p.i. In line with this observation, we also showed that phosphorylation of eIF2α, a common trigger of SG formation (21), does not occur in EBOV-infected cells during the course of infection. Despite the ability to inhibit PKR-induced eIF2α phosphorylation (47–49), EBOV did not antagonize phosphorylation of eIF2α or block the induction of SGs when exposed to Ars-induced oxidative stress, which leads to the activation of HRI (69, 70). Furthermore, EBOV was unable to prevent SG formation after exposure to heat stress, which induces SGs via HRI activation, or Hipp treatment, which acts downstream of eIF2α phosphorylation (65, 71). This suggests that EBOV is unable to antagonize an antiviral stress response mediated by stress sensors apart from PKR. In line with this result is a previous study showing that EBOV replication was significantly reduced after treatment with the stress inducer thapsigargin (72).
A majority of EBOV-infected cells exposed to exogenous stress formed canonical SGs early, but SG formation was significantly decreased late in infection. Although it cannot be ruled out that homeostatic cellular processes required for proper SG formation are compromised at late stages of infection, we propose that the observed impairment of SG formation occurs downstream of eIF2α phosphorylation and is mediated by a newly identified function of VP35. Blocking SG formation downstream of eIF2α has been shown for influenza A virus (IAV), mediated by the viral proteins NP and PA-X (73). Similar to VP35, IAV NP was found to disrupt SG formation when expressed in the absence of other viral proteins (73). Our data suggest that VP35 is able to disrupt SG formation by physically interfering with SG protein aggregation. This function is independent of the ability of VP35 to interact with dsRNA. It is conceivable that VP35 interacts with an unidentified SG component that then recruits VP35 to SGs upon stress. Subsequently, when VP35 is present at high enough levels, it might prevent the oligomerization of its binding partner or inhibit binding of other SG components. A number of host proteins with known localization to SGs have been shown to bind VP35, including DRBP76 and various members of the heterogeneous nuclear ribonucleoprotein (hnRNP) protein family (74, 75).
In line with previous reports, our data show that VP35 is also able to disrupt NP-mediated inclusion formation when expressed at large amounts (68). Similar to the SG nucleating proteins, NP is the assembly factor of RNA-protein aggregates, the viral inclusions, and serves as a hub for the recruitment of other viral proteins, including VP35 (6, 76). The disruption in inclusion formation by VP35 might be related to a recently identified intrinsically disordered region within VP35, which mediates binding to monomeric NP and interferes with NP self-aggregation (77, 78). It is conceivable that through this type of interaction, high levels of VP35 interfere with the nucleation of SGs, possibly through competitive binding, in a manner similar to its disruption of NP-based inclusion formation.
Although SGs were not induced by EBOV infection, numerous SG proteins were sequestered exclusively within viral inclusions late in infection. However, these IB granules were not canonical SGs. Other viruses, including chikungunya virus, vaccinia virus, Semliki Forest virus, and vesicular stomatitis virus, have been shown to redistribute SG-associated proteins into distinct cytoplasmic aggregates with altered protein composition or function compared to those of canonical SGs (31, 33, 60−62, 79). The IB granules we observed during EBOV infection were devoid of the canonical SG marker protein TIA-1 and, similar to the antiviral granules observed in vaccinia virus infection and the proviral G3BP aggregates induced by chikungunya virus, did not dissolve upon CHX treatment (33, 61, 62, 79). CHX-mediated dissolution of SGs is driven by the stabilization of polysomes, indicating that SG formation requires a pool of free messenger ribonucleoprotein (mRNP) complexes (25). The resistance of IB granules to CHX treatment suggests that these aggregates are composed of translationally silent (viral) mRNAs in complex with mRNA-binding proteins. Furthermore, the inhibition of SG formation by overexpression of USP10 had no effect on the aggregation of G3BP within inclusions, indicating that SG formation itself is not required for the sequestration of SG proteins within inclusions.
The IB granules did not impair the accumulation of newly synthesized proteins within inclusions, indicating that they are not detrimental to viral protein translation. This observation, in conjunction with the ability of EBOV to grow to very high titers despite the presence SG proteins within inclusions, indicates that the IB granules are not antiviral and might even be proviral.
While EBOV nucleocapsid proteins in viral inclusions did not colocalize with any of the SG proteins examined, the accumulated SG proteins, specifically eIF3, colocalized with viral NP mRNA (and viral antigenomes) within the inclusions. TEM analysis showed that ribosomes were not found within viral inclusions, indicating that they are not sites of viral protein translation, which most likely occurs in very close proximity to viral inclusions. This led us to speculate that a potential function of these SG proteins within the inclusions, many of which are translation initiation factors, may be to prepare viral mRNAs for translation. It is also conceivable that binding of the SG proteins to the accumulated viral RNA within EBOV inclusions at late stages of infection prevents dsRNA formation. Since the viral inclusions are the sites for replication and transcription, they contain both positive- and negative-sense RNAs, enabling dsRNA formation, especially at late stages of infection, when the amount of replicated and transcribed viral RNA is high. However, we did not detect any signs of dsRNA within the viral inclusions using an antibody directed against dsRNA (data not shown). This strengthens the hypothesis that covering the viral mRNA with cellular RNA-binding proteins might help to prevent dsRNA formation with nonencapsidated negative-sense RNA, thereby evading the cellular dsRNA sensing machinery.
The presence of cellular proteins within viral inclusions may be a common proviral mechanism among those viruses that form cytoplasmic inclusion bodies. Marburg virus (MARV), another member of the filovirus family, recruits components of the endosomal complex required for transport (ESCRT) machinery to viral inclusions to enhance budding (80). The sequestration of cellular proteins within viral inclusions, including HuR and proteins involved in antiviral signaling, was also observed in respiratory syncytial virus (RSV) infection and dampened the antiviral host response (81–83). This supports the hypothesis that the sequestration of certain cellular proteins within viral inclusions promotes viral infection by disrupting antiviral signaling pathways.
Even though the sequestered SG proteins still responded to stress signals despite being subverted into viral inclusions, SGs did not form within inclusions themselves, suggesting that viral inclusions provide an environment that is resistant to SG formation. We hypothesize that this may in part be mediated by the disruption in SG formation observed with high expression levels of VP35. Inclusions are rich in VP35 and therefore might prevent canonical SG formation despite the abundance of many canonical SG components.
In summary, our data show that EBOV does not induce SG formation during the course of infection but does sequester many SG proteins within viral inclusions, which we propose contributes to the proviral environment created by viral inclusions. Furthermore, the absence of canonical SGs in inclusions may in part be mediated by a newly identified function of VP35, which we showed was able to disrupt cytoplasmic SG formation independently of eIF2α phosphorylation. The sequestration of SG proteins in EBOV inclusions leaves many open questions: Which mechanisms are used to selectively transport SG proteins into inclusions? Are cellular mRNAs bound to SG proteins sequestered in viral inclusions? Do IB granules affect EBOV infection? We feel that this is an exciting and novel research area worth pursuing in the future.
ACKNOWLEDGMENTS
We are indebted to H. Feldmann (LV, NIAID, NIH), F. Feldmann (RMVB, NIAID, NIH), and members of the Laboratory of Virology for training of K.M.S. in the high-containment laboratory and support with conducting biosafety level 4 experiments at the Rocky Mountain Laboratories, NIH, NIAID, Hamilton, MT. We thank T. Taylor (University of Toledo) and S. Best (NIH, NIAID, Rocky Mountain Laboratories) for help with the confocal imaging, D. K. Rozelle (BU) and J. Connor (BU) for technical advice, and J. Pacheco (BU) for excellent technical assistance. We are grateful to J. Alonso, C. F. Basler, S. Becker, G. G. Olinger, J. Pelletier, W. F. C. Rigby, and V. E. Volchkov for providing material.
This work was supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under award numbers U01-AI082954 (E.M.), R03-AI114293 (E.M.), UC6AI058618, AI0655858 (N.K.), CA168872 (N.K.), and U19-AI083025 (T.H.) and by the Division of Intramural Research, NIAID, NIH (H.E.).
Funding Statement
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
REFERENCES
- 1.Feldmann H, Geisbert TW. 2011. Ebola haemorrhagic fever. Lancet 377:849–862. doi: 10.1016/S0140-6736(10)60667-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Messaoudi I, Amarasinghe GK, Basler CF. 2015. Filovirus pathogenesis and immune evasion: insights from Ebola virus and Marburg virus. Nat Rev Microbiol 13:663–676. doi: 10.1038/nrmicro3524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mühlberger E. 2007. Filovirus replication and transcription. Future Virol 2:205–215. doi: 10.2217/17460794.2.2.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brauburger K, Deflubé LR, Mühlberger E. 2015. Filovirus transcription and replication, p 515–555. In Pattnaik AK, Whitt MA (ed), Biology and pathogenesis of rhabdo- and filoviruses. World Scientific Publishing Co Pte Ltd, Singapore. [Google Scholar]
- 5.Mühlberger E, Weik M, Volchkov VE, Klenk H-D, Becker S. 1999. Comparison of the transcription and replication strategies of Marburg virus and Ebola virus by using artificial replication systems. J Virol 73:2333–2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huang Y, Xu L, Sun Y, Nabel GJ. 2002. The assembly of Ebola virus nucleocapsid requires virion-associated proteins 35 and 24 and posttranslational modification of nucleoprotein. Mol Cell 10:307–316. doi: 10.1016/S1097-2765(02)00588-9. [DOI] [PubMed] [Google Scholar]
- 7.Noda T, Aoyama K, Sagara H, Kida H, Kawaoka Y. 2005. Nucleocapsid-like structures of Ebola virus reconstructed using electron tomography. J Vet Med Sci 67:325–328. doi: 10.1292/jvms.67.325. [DOI] [PubMed] [Google Scholar]
- 8.Bharat TA, Noda T, Riches JD, Kraehling V, Kolesnikova L, Becker S, Kawaoka Y, Briggs JA. 2012. Structural dissection of Ebola virus and its assembly determinants using cryo-electron tomography. Proc Natl Acad Sci U S A 109:4275–4280. doi: 10.1073/pnas.1120453109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Watt A, Moukambi F, Banadyga L, Groseth A, Callison J, Herwig A, Ebihara H, Feldmann H, Hoenen T. 2014. A novel life cycle modeling system for Ebola virus shows a genome length-dependent role of VP24 in virus infectivity. J Virol 88:10511–10524. doi: 10.1128/JVI.01272-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hoenen T, Shabman RS, Groseth A, Herwig A, Weber M, Schudt G, Dolnik O, Basler CF, Becker S, Feldmann H. 2012. Inclusion bodies are a site of ebolavirus replication. J Virol 86:11779–11788. doi: 10.1128/JVI.01525-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nanbo A, Watanabe S, Halfmann P, Kawaoka Y. 2013. The spatio-temporal distribution dynamics of Ebola virus proteins and RNA in infected cells. Sci Rep 3:1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schudt G, Dolnik O, Kolesnikova L, Biedenkopf N, Herwig A, Becker S. 2015. Transport of Ebolavirus nucleocapsids is dependent on actin polymerization: live-cell imaging analysis of Ebolavirus-infected cells. J Infect Dis 212(Suppl 2):S160–S166. [DOI] [PubMed] [Google Scholar]
- 13.Dolnik O, Kolesnikova L, Stevermann L, Becker S. 2010. Tsg101 is recruited by a late domain of the nucleocapsid protein to support budding of Marburg virus-like particles. J Virol 84:7847–7856. doi: 10.1128/JVI.00476-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ryabchikova E, Price BBS. 2004. Ebola and Marburg viruses: a view of infection using electron microscopy. Battelle Press, Columbus, OH. [Google Scholar]
- 15.Noda T, Ebihara H, Muramoto Y, Fujii K, Takada A, Sagara H, Kim JH, Kida H, Feldmann H, Kawaoka Y. 2006. Assembly and budding of Ebolavirus. PLoS Pathog 2:e99. doi: 10.1371/journal.ppat.0020099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Olejnik J, Ryabchikova E, Corley RB, Mühlberger E. 2011. Intracellular events and cell fate in filovirus infection. Viruses 3:1501–1531. doi: 10.3390/v3081501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Anderson P, Kedersha N. 2009. Stress granules. Curr Biol 19:R397–R398. doi: 10.1016/j.cub.2009.03.013. [DOI] [PubMed] [Google Scholar]
- 18.McAllister CS, Taghavi N, Samuel CE. 2012. Protein kinase PKR amplification of interferon beta induction occurs through initiation factor eIF-2alpha-mediated translational control. J Biol Chem 287:36384–36392. doi: 10.1074/jbc.M112.390039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Onomoto K, Jogi M, Yoo JS, Narita R, Morimoto S, Takemura A, Sambhara S, Kawaguchi A, Osari S, Nagata K, Matsumiya T, Namiki H, Yoneyama M, Fujita T. 2012. Critical role of an antiviral stress granule containing RIG-I and PKR in viral detection and innate immunity. PLoS One 7:e43031. doi: 10.1371/journal.pone.0043031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Onomoto K, Yoneyama M, Fung G, Kato H, Fujita T. 2014. Antiviral innate immunity and stress granule responses. Trends Immunol 35:420–428. doi: 10.1016/j.it.2014.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Anderson P, Kedersha N. 2008. Stress granules: the Tao of RNA triage. Trends Biochem Sci 33:141–150. doi: 10.1016/j.tibs.2007.12.003. [DOI] [PubMed] [Google Scholar]
- 22.Anderson P, Kedersha N. 2009. RNA granules: post-transcriptional and epigenetic modulators of gene expression. Nat Rev Mol Cell Biol 10:430–436. doi: 10.1038/nrm2694. [DOI] [PubMed] [Google Scholar]
- 23.Kedersha N, Anderson P. 2009. Regulation of translation by stress granules and processing bodies. Prog Mol Biol Transl Sci 90:155–185. doi: 10.1016/S1877-1173(09)90004-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Buchan JR, Parker R. 2009. Eukaryotic stress granules: the ins and outs of translation. Mol Cell 36:932–941. doi: 10.1016/j.molcel.2009.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kedersha N, Cho MR, Li W, Yacono PW, Chen S, Gilks N, Golan DE, Anderson P. 2000. Dynamic shuttling of TIA-1 accompanies the recruitment of mRNA to mammalian stress granules. J Cell Biol 151:1257–1268. doi: 10.1083/jcb.151.6.1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Anderson P, Kedersha N, Ivanov P. 2015. Stress granules, P-bodies and cancer. Biochim Biophys Acta 1849:861–870. doi: 10.1016/j.bbagrm.2014.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Montero H, Trujillo-Alonso V. 2011. Stress granules in the viral replication cycle. Viruses 3:2328–2338. doi: 10.3390/v3112328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.White JP, Lloyd RE. 2012. Regulation of stress granules in virus systems. Trends Microbiol 20:175–183. doi: 10.1016/j.tim.2012.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Beckham CJ, Parker R. 2008. P bodies, stress granules, and viral life cycles. Cell Host Microbe 3:206–212. doi: 10.1016/j.chom.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Matthews JD, Frey TK. 2012. Analysis of subcellular G3BP redistribution during rubella virus infection. J Gen Virol 93:267–274. doi: 10.1099/vir.0.036780-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Panas MD, Varjak M, Lulla A, Eng Merits KEA, Karlsson Hedestam GB, McInerney GM. 2012. Sequestration of G3BP coupled with efficient translation inhibits stress granules in Semliki Forest virus infection. Mol Biol Cell 23:4701–4712. doi: 10.1091/mbc.E12-08-0619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.White JP, Lloyd RE. 2011. Poliovirus unlinks TIA1 aggregation and mRNA stress granule formation. J Virol 85:12442–12454. doi: 10.1128/JVI.05888-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Scholte FE, Tas A, Albulescu IC, Zusinaite E, Merits A, Snijder EJ, van Hemert MJ. 2015. Stress granule components G3BP1 and G3BP2 play a proviral role early in Chikungunya virus replication. J Virol 89:4457–4469. doi: 10.1128/JVI.03612-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Elde NC, Child SJ, Geballe AP, Malik HS. 2009. Protein kinase R reveals an evolutionary model for defeating viral mimicry. Nature 457:485–489. doi: 10.1038/nature07529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dauber B, Wolff T. 2009. Activation of the antiviral kinase PKR and viral countermeasures. Viruses 1:523–544. doi: 10.3390/v1030523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Basler CF, Mikulasova A, Martinez-Sobrido L, Paragas J, Mühlberger E, Bray M, Klenk HD, Palese P, Garcia-Sastre A. 2003. The Ebola virus VP35 protein inhibits activation of interferon regulatory factor 3. J Virol 77:7945–7956. doi: 10.1128/JVI.77.14.7945-7956.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hartman AL, Towner JS, Nichol ST. 2004. A C-terminal basic amino acid motif of Zaire ebolavirus VP35 is essential for type I interferon antagonism and displays high identity with the RNA-binding domain of another interferon antagonist, the NS1 protein of influenza A virus. Virology 328:177–184. doi: 10.1016/j.virol.2004.07.006. [DOI] [PubMed] [Google Scholar]
- 38.Cárdenas WB, Loo YM, Gale M Jr, Hartman AL, Kimberlin CR, Martinez-Sobrido L, Saphire EO, Basler CF. 2006. Ebola virus VP35 protein binds double-stranded RNA and inhibits alpha/beta interferon production induced by RIG-I signaling. J Virol 80:5168–5178. doi: 10.1128/JVI.02199-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hartman AL, Bird BH, Towner JS, Antoniadou ZA, Zaki SR, Nichol ST. 2008. Inhibition of IRF-3 activation by VP35 is critical for the high level of virulence of ebola virus. J Virol 82:2699–2704. doi: 10.1128/JVI.02344-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Prins KC, Cardenas WB, Basler CF. 2009. Ebola virus protein VP35 impairs the function of interferon regulatory factor-activating kinases IKKepsilon and TBK-1. J Virol 83:3069–3077. doi: 10.1128/JVI.01875-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Leung DW, Prins KC, Borek DM, Farahbakhsh M, Tufariello JM, Ramanan P, Nix JC, Helgeson LA, Otwinowski Z, Honzatko RB, Basler CF, Amarasinghe GK. 2010. Structural basis for dsRNA recognition and interferon antagonism by Ebola VP35. Nat Struct Mol Biol 17:165–172. doi: 10.1038/nsmb.1765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kimberlin CR, Bornholdt ZA, Li S, Woods VL Jr, MacRae IJ, Saphire EO. 2010. Ebolavirus VP35 uses a bimodal strategy to bind dsRNA for innate immune suppression. Proc Natl Acad Sci U S A 107:314–319. doi: 10.1073/pnas.0910547107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chang TH, Kubota T, Matsuoka M, Jones S, Bradfute SB, Bray M, Ozato K. 2009. Ebola Zaire virus blocks type I interferon production by exploiting the host SUMO modification machinery. PLoS Pathog 5:e1000493. doi: 10.1371/journal.ppat.1000493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Leung LW, Park MS, Martinez O, Valmas C, Lopez CB, Basler CF. 2011. Ebolavirus VP35 suppresses IFN production from conventional but not plasmacytoid dendritic cells. Immunol Cell Biol 89:792–802. doi: 10.1038/icb.2010.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Haasnoot J, de Vries W, Geutjes EJ, Prins M, de Haan P, Berkhout B. 2007. The Ebola virus VP35 protein is a suppressor of RNA silencing. PLoS Pathog 3:e86. doi: 10.1371/journal.ppat.0030086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fabozzi G, Nabel CS, Dolan MA, Sullivan NJ. 2011. Ebolavirus proteins suppress the effects of small interfering RNA by direct interaction with the mammalian RNA interference pathway. J Virol 85:2512–2523. doi: 10.1128/JVI.01160-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Feng Z, Cerveny M, Yan Z, He B. 2007. The VP35 protein of Ebola virus inhibits the antiviral effect mediated by double-stranded RNA-dependent protein kinase PKR. J Virol 81:182–192. doi: 10.1128/JVI.01006-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schümann M, Gantke T, Mühlberger E. 2009. Ebola virus VP35 antagonizes PKR activity through its C-terminal interferon inhibitory domain. J Virol 83:8993–8997. doi: 10.1128/JVI.00523-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Olejnik J, Alonso J, Schmidt KM, Yan Z, Wang W, Marzi A, Ebihara H, Yang J, Patterson JL, Ryabchikova E, Mühlberger E. 2013. Ebola virus does not block apoptotic signaling pathways. J Virol 87:5384–5396. doi: 10.1128/JVI.01461-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.White JP, Cardenas AM, Marissen WE, Lloyd RE. 2007. Inhibition of cytoplasmic mRNA stress granule formation by a viral proteinase. Cell Host Microbe 2:295–305. doi: 10.1016/j.chom.2007.08.006. [DOI] [PubMed] [Google Scholar]
- 51.Kedersha N, Tisdale S, Hickman T, Anderson P. 2008. Real-time and quantitative imaging of mammalian stress granules and processing bodies. Methods Enzymol 448:521–552. doi: 10.1016/S0076-6879(08)02626-8. [DOI] [PubMed] [Google Scholar]
- 52.Kedersha N, Panas MD, Achorn CA, Lyons S, Tisdale S, Hickman T, Thomas M, Lieberman J, McInerney GM, Ivanov P, Anderson P. 2016. G3BP-Caprin1-USP10 complexes mediate stress granule condensation and associate with 40S subunits. J Cell Biol 212:845–860. doi: 10.1083/jcb.201508028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Niwa H, Yamamura K, Miyazaki J. 1991. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene 108:193–199. doi: 10.1016/0378-1119(91)90434-D. [DOI] [PubMed] [Google Scholar]
- 54.Valmas C, Grosch MN, Schümann M, Olejnik J, Martinez O, Best SM, Krähling V, Basler CF, Mühlberger E. 2010. Marburg virus evades interferon responses by a mechanism distinct from ebola virus. PLoS Pathog 6:e1000721. doi: 10.1371/journal.ppat.1000721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Boehmann Y, Enterlein S, Randolf A, Mühlberger E. 2005. A reconstituted replication and transcription system for Ebola virus Reston and comparison with Ebola virus Zaire. Virology 332:406–417. doi: 10.1016/j.virol.2004.11.018. [DOI] [PubMed] [Google Scholar]
- 56.Kedersha N, Anderson P. 2002. Stress granules: sites of mRNA triage that regulate mRNA stability and translatability. Biochem Soc Trans 30:963–969. doi: 10.1042/bst0300963. [DOI] [PubMed] [Google Scholar]
- 57.Shabman RS, Jabado OJ, Mire CE, Stockwell TB, Edwards M, Mahajan M, Geisbert TW, Basler CF. 2014. Deep sequencing identifies noncanonical editing of Ebola and Marburg virus RNAs in infected cells. mBio 5:e02011-14. doi: 10.1128/mBio.02011-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Weik M, Enterlein S, Schlenz K, Mühlberger E. 2005. The Ebola virus genomic replication promoter is bipartite and follows the rule of six. J Virol 79:10660–10671. doi: 10.1128/JVI.79.16.10660-10671.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Stoecklin G, Stubbs T, Kedersha N, Wax S, Rigby WF, Blackwell TK, Anderson P. 2004. MK2-induced tristetraprolin:14-3-3 complexes prevent stress granule association and ARE-mRNA decay. EMBO J 23:1313–1324. doi: 10.1038/sj.emboj.7600163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dinh PX, Beura LK, Das PB, Panda D, Das A, Pattnaik AK. 2013. Induction of stress granule-like structures in vesicular stomatitis virus-infected cells. J Virol 87:372–383. doi: 10.1128/JVI.02305-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rozelle DK, Filone CM, Kedersha N, Connor JH. 2014. Activation of stress response pathways promotes formation of antiviral granules and restricts virus replication. Mol Cell Biol 34:2003–2016. doi: 10.1128/MCB.01630-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Simpson-Holley M, Kedersha N, Dower K, Rubins KH, Anderson P, Hensley LE, Connor JH. 2011. Formation of antiviral cytoplasmic granules during orthopoxvirus infection. J Virol 85:1581–1593. doi: 10.1128/JVI.02247-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Panas MD, Schulte T, Thaa B, Sandalova T, Kedersha N, Achour A, McInerney GM. 2015. Viral and cellular proteins containing FGDF motifs bind G3BP to block stress granule formation. PLoS Pathog 11:e1004659. doi: 10.1371/journal.ppat.1004659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lloyd RE. 2013. Regulation of stress granules and P-bodies during RNA virus infection. Wiley Interdiscip Rev RNA 4:317–331. doi: 10.1002/wrna.1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bordeleau ME, Mori A, Oberer M, Lindqvist L, Chard LS, Higa T, Belsham GJ, Wagner G, Tanaka J, Pelletier J. 2006. Functional characterization of IRESes by an inhibitor of the RNA helicase eIF4A. Nat Chem Biol 2:213–220. doi: 10.1038/nchembio776. [DOI] [PubMed] [Google Scholar]
- 66.Savinova O, Jagus R. 1997. Use of vertical slab isoelectric focusing and immunoblotting to evaluate steady-state phosphorylation of eIF2 alpha in cultured cells. Methods 11:419–425. doi: 10.1006/meth.1996.0438. [DOI] [PubMed] [Google Scholar]
- 67.Trunschke M, Conrad D, Enterlein S, Olejnik J, Brauburger K, Mühlberger E. 2013. The L-VP35 and L-L interaction domains reside in the amino terminus of the Ebola virus L protein and are potential targets for antivirals. Virology 441:135–145. doi: 10.1016/j.virol.2013.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Noda T, Kolesnikova L, Becker S, Kawaoka Y. 2011. The importance of the NP: VP35 ratio in Ebola virus nucleocapsid formation. J Infect Dis 204(Suppl 3):S878–S883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lu L, Han AP, Chen JJ. 2001. Translation initiation control by heme-regulated eukaryotic initiation factor 2alpha kinase in erythroid cells under cytoplasmic stresses. Mol Cell Biol 21:7971–7980. doi: 10.1128/MCB.21.23.7971-7980.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.McEwen E, Kedersha N, Song B, Scheuner D, Gilks N, Han A, Chen JJ, Anderson P, Kaufman RJ. 2005. Heme-regulated inhibitor kinase-mediated phosphorylation of eukaryotic translation initiation factor 2 inhibits translation, induces stress granule formation, and mediates survival upon arsenite exposure. J Biol Chem 280:16925–16933. doi: 10.1074/jbc.M412882200. [DOI] [PubMed] [Google Scholar]
- 71.Ivanova AA, Velichko AK, Kantidze OL, Razin SV. 2015. Heat stress induces formation of cytoplasmic granules containing HSC70 protein. Dokl Biochem Biophys 463:213–215. doi: 10.1134/S1607672915040043. [DOI] [PubMed] [Google Scholar]
- 72.Shabman RS, Hoenen T, Groseth A, Jabado O, Binning JM, Amarasinghe GK, Feldmann H, Basler CF. 2013. An upstream open reading frame modulates ebola virus polymerase translation and virus replication. PLoS Pathog 9:e1003147. doi: 10.1371/journal.ppat.1003147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Khaperskyy DA, Emara MM, Johnston BP, Anderson P, Hatchette TF, McCormick C. 2014. Influenza a virus host shutoff disables antiviral stress-induced translation arrest. PLoS Pathog 10:e1004217. doi: 10.1371/journal.ppat.1004217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Shabman RS, Leung DW, Johnson J, Glennon N, Gulcicek EE, Stone KL, Leung L, Hensley L, Amarasinghe GK, Basler CF. 2011. DRBP76 associates with Ebola virus VP35 and suppresses viral polymerase function. J Infect Dis 204(Suppl 3):S911–S918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pichlmair A, Kandasamy K, Alvisi G, Mulhern O, Sacco R, Habjan M, Binder M, Stefanovic A, Eberle CA, Goncalves A, Burckstummer T, Muller AC, Fauster A, Holze C, Lindsten K, Goodbourn S, Kochs G, Weber F, Bartenschlager R, Bowie AG, Bennett KL, Colinge J, Superti-Furga G. 2012. Viral immune modulators perturb the human molecular network by common and unique strategies. Nature 487:486–490. doi: 10.1038/nature11289. [DOI] [PubMed] [Google Scholar]
- 76.Shi W, Huang Y, Sutton-Smith M, Tissot B, Panico M, Morris HR, Dell A, Haslam SM, Boyington J, Graham BS, Yang ZY, Nabel GJ. 2008. A filovirus-unique region of Ebola virus nucleoprotein confers aberrant migration and mediates its incorporation into virions. J Virol 82:6190–6199. doi: 10.1128/JVI.02731-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Leung DW, Borek D, Luthra P, Binning JM, Anantpadma M, Liu G, Harvey IB, Su Z, Endlich-Frazier A, Pan J, Shabman RS, Chiu W, Davey RA, Otwinowski Z, Basler CF, Amarasinghe GK. 2015. An intrinsically disordered peptide from Ebola virus VP35 controls viral RNA synthesis by modulating nucleoprotein-RNA interactions. Cell Rep 11:376–389. doi: 10.1016/j.celrep.2015.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kirchdoerfer RN, Abelson DM, Li S, Wood MR, Saphire EO. 2015. Assembly of the Ebola virus nucleoprotein from a chaperoned VP35 complex. Cell Rep 12:140–149. doi: 10.1016/j.celrep.2015.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Fros JJ, Domeradzka NE, Baggen J, Geertsema C, Flipse J, Vlak JM, Pijlman GP. 2012. Chikungunya virus nsP3 blocks stress granule assembly by recruitment of G3BP into cytoplasmic foci. J Virol 86:10873–10879. doi: 10.1128/JVI.01506-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Dolnik O, Stevermann L, Kolesnikova L, Becker S. 2015. Marburg virus inclusions: a virus-induced microcompartment and interface to multivesicular bodies and the late endosomal compartment. Eur J Cell Biol 94:323–331. doi: 10.1016/j.ejcb.2015.05.006. [DOI] [PubMed] [Google Scholar]
- 81.Lindquist ME, Lifland AW, Utley TJ, Santangelo PJ, Crowe JE Jr. 2010. Respiratory syncytial virus induces host RNA stress granules to facilitate viral replication. J Virol 84:12274–12284. doi: 10.1128/JVI.00260-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lifland AW, Jung J, Alonas E, Zurla C, Crowe JE Jr, Santangelo PJ. 2012. Human respiratory syncytial virus nucleoprotein and inclusion bodies antagonize the innate immune response mediated by MDA5 and MAVS. J Virol 86:8245–8258. doi: 10.1128/JVI.00215-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Fricke J, Koo LY, Brown CR, Collins PL. 2013. p38 and OGT sequestration into viral inclusion bodies in cells infected with human respiratory syncytial virus suppresses MK2 activities and stress granule assembly. J Virol 87:1333–1347. doi: 10.1128/JVI.02263-12. [DOI] [PMC free article] [PubMed] [Google Scholar]