

ABSTRACT
Despite significant progress in reducing peripartum mother-to-child transmission (MTCT) of human immunodeficiency virus (HIV) with antiretroviral therapy (ART), continued access to ART throughout the breastfeeding period is still a limiting factor, and breast milk exposure to HIV accounts for up to 44% of MTCT. As abstinence from breastfeeding is not recommended, alternative means are needed to prevent MTCT of HIV. We have previously shown that oral vaccination at birth with live attenuated Mycobacterium tuberculosis strains expressing simian immunodeficiency virus (SIV) genes safely induces persistent SIV-specific cellular and humoral immune responses both systemically and at the oral and intestinal mucosa. Here, we tested the ability of oral M. tuberculosis vaccine strains expressing SIV Env and Gag proteins, followed by systemic heterologous (MVA-SIV Env/Gag/Pol) boosting, to protect neonatal macaques against oral SIV challenge. While vaccination did not protect infant macaques against oral SIV acquisition, a subset of immunized animals had significantly lower peak viremia which inversely correlated with prechallenge SIV Env-specific salivary and intestinal IgA responses and higher-avidity SIV Env-specific IgG in plasma. These controller animals also maintained CD4+ T cell populations better and showed reduced tissue pathology compared to noncontroller animals. We show that infants vaccinated at birth can develop vaccine-induced SIV-specific IgA and IgG antibodies and cellular immune responses within weeks of life. Our data further suggest that affinity maturation of vaccine-induced plasma antibodies and induction of mucosal IgA responses at potential SIV entry sites are associated with better control of viral replication, thereby likely reducing SIV morbidity.
IMPORTANCE Despite significant progress in reducing peripartum MTCT of HIV with ART, continued access to ART throughout the breastfeeding period is still a limiting factor. Breast milk exposure to HIV accounts for up to 44% of MTCT. Alternative measures, in addition to ART, are needed to achieve the goal of an AIDS-free generation. Pediatric HIV vaccines constitute a core component of such efforts. The results of our pediatric vaccine study highlight the potential importance of vaccine-elicited mucosal Env-specific IgA responses in combination with high-avidity systemic Env-specific IgG in protection against oral SIV transmission and control of viral replication in infant macaques. The induction of potent mucosal IgA antibodies by our vaccine is remarkable considering the age-dependent development of mucosal IgA responses postbirth. A deeper understanding of postnatal immune development may inform the design of improved vaccine strategies to enhance systemic and mucosal SIV/HIV antibody responses.
INTRODUCTION
The WHO and UNAIDS have reported that 200,000 infants worldwide became newly infected with human immunodeficiency virus type 1 (HIV-1) in 2014. Adolescent girls and women are twice as likely to acquire HIV as men and represent about 55 to 60% of HIV-infected adults. Despite improved access to antiretroviral therapy (ART), especially during the peripartum period, the rate of mother-to-child-transmission (MTCT) of HIV is estimated at 16% (1). Main obstacles in the prevention of MTCT of HIV are loss of follow-up for mothers throughout the breast-feeding period and failure to continually test both mother and child for HIV infection during this period. Breast milk transmission of HIV accounts for about half of new pediatric HIV infections. Thus, alternative measures, in addition to ART, are needed to achieve the goal of an AIDS-free generation. Pediatric HIV vaccine development should constitute a core component of such efforts.
Sub-Saharan Africa carries the greatest burden of the pediatric HIV epidemic, which is exacerbated by the limited resources available to prevent new and treat existing HIV infections. Similarly, numbers of pediatric infections with Mycobacterium tuberculosis are highest in sub-Saharan Africa. Building on the positive attributes and wide use of the current bacillus Calmette Guérin (BCG) vaccine for preventing severe complications of tuberculosis (TB) in infants, we aimed to develop a pediatric combination vaccine, consisting of a recombinant attenuated M. tuberculosis vaccine strain expressing immunodeficiency virus antigens that could protect against both HIV and TB infections in human infants.
We previously reported that the auxotroph M. tuberculosis strain mc26435, expressing simian immunodeficiency virus (SIV) Gag, is safe in healthy and SIV-infected neonatal macaques, and we demonstrated that a peroral (p.o.) M. tuberculosis-SIV prime/intramuscular (i.m.) modified vaccinia Ankara (MVA)-SIV boost regimen was effective for inducing cellular and humoral responses against SIV and M. tuberculosis antigens in infant macaques (2, 3). The oral prime was included to specifically elicit SIV-specific immune responses at potential sites of viral entry in the oro-gastrointestinal tract (GI) after oral SIV exposure. The oral mucosa contains lymphoid tissues that are readily accessed by p.o. vaccines. The lymphatic network of the Waldeyer's Ring provides a noninvasive portal for oral vaccine uptake and, as part of the systemic lymphatic network, allows the induction of mucosal and systemic immune responses. The current studies were designed to test the efficacy of a p.o. M. tuberculosis-SIV prime/i.m. MVA-SIV boost regimen for preventing oral SIV acquisition in infant macaques. To simulate breast milk exposure to HIV in human infants, we developed a repeated low-dose oral SIV challenge regimen that permitted determination of the number of exposures required for SIV infection. Compared to vaccine challenge studies with adult macaques, both our vaccination regimen and the time to challenge were accelerated, because the goal of our studies is to identify a vaccination strategy that will prevent HIV breast milk transmission early after birth.
So far, with the exception of passive immunization strategies, only a single pediatric SIV study reported protection against oral SIV challenge in MVA-SIV- or ALVAC-SIV-vaccinated neonatal macaques (4), with the caveat that the potential contribution of genetic host factors, e.g., major histocompatibility complex (MHC) genotype or SIV restriction factors, to protection could not be evaluated at that time. As found for most pediatric SIV vaccines (5–8), vaccination of infant macaques with the M. tuberculosis-SIV prime/MVA-SIV boost regimen was not able to prevent oral SIV acquisition in infant macaques. However, a subset of vaccinated infants was able to partially control viremia throughout the study period. Reduced viremia was associated with higher-avidity SIV Env-specific IgG antibodies in plasma and greater salivary and intestinal SIV Env-specific IgA responses at the time of oral SIV challenge initiation. Despite the inability to provide sterilizing protection, and despite the age-restricted development of IgG and IgA antibodies postnatally, these results suggest that the induction of potent and persistent antibody responses by early pediatric vaccination is feasible.
MATERIALS AND METHODS
Animals.
Infant rhesus macaques (Macaca mulatta) were enrolled from the SIV- and type D retrovirus-negative colony at the California National Primate Research Center (CNPRC; Davis, CA). Animals were housed in nursery facilities and reared according to the Guide for the Care and Use of Laboratory Animals used by the American Association for Accreditation of Laboratory Animal Care (9). To enhance social interactions and normal development, all infants were housed in pairs. All animal protocols were approved by the UC Davis Institutional Animal Care and Use Committee prior to study initiation. Trained animal technicians monitored the animals daily for any clinical symptoms of TB infection. Animals were randomly assigned to groups and were between 3 and 7 days of age at the first immunization (week 0 in Table 1). All procedures were done following intramuscular (i.m.) administration of 10 mg ketamine-HCl (Parke-Davis, Morris Plains, NC) per kg of body weight. Complete blood counts (CBC) were performed on each peripheral blood sample by the CNPRC Clinical Laboratory.
TABLE 1.
Animal groups
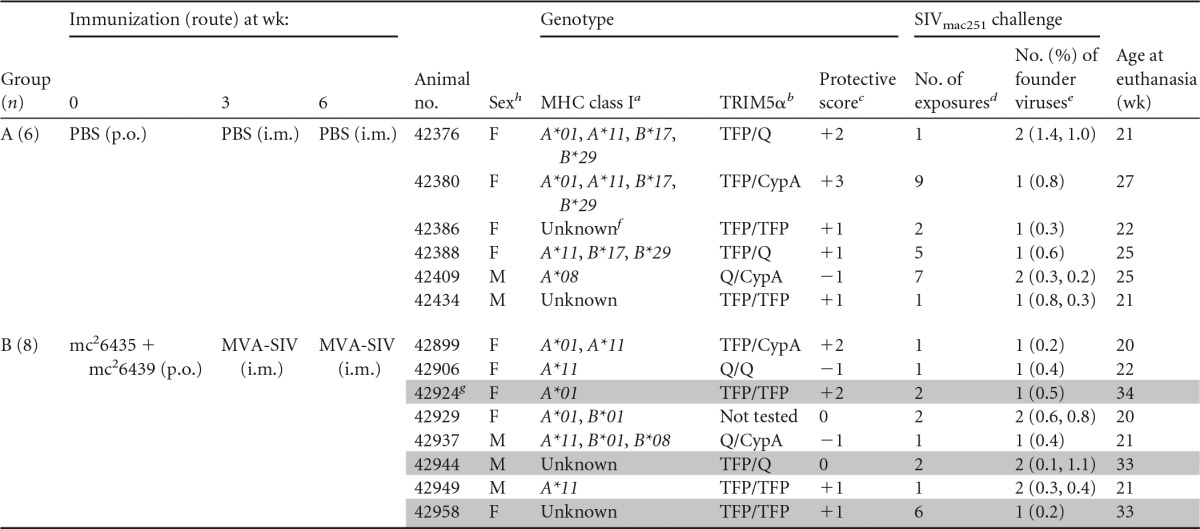
a The alleles Mamu-A*01, -B*08, and -B*17 have been associated with better disease outcome, whereas allele Mamu-B*01 is associated with higher viremia.
b The TRIM5α genotypes TFP/TFP and TFP/Cyp confer resistance to SIV infection, while the genotypes Q/Q and Q/CypA are associated with increased susceptibility to infection.
c Protective class I alleles and resistant TRIM5α alleles were assigned a value of 1. Genotypes associated with increased susceptibility or disease outcome received a score of −1. All other alleles were assigned a score of 0. The protective genotype score represents the sum of all scores for an individual animal (e.g., animal 42376 has MHC of 1 + 0 + 1 + 0 and TRIM5α of 0, for a score of +2).
d The number of exposures necessary to establish systemic infection.
e The number of founder viruses identified by SGA and gp160 sequencing of plasma virus isolated at weeks 1 to 2 postinfection. The numbers in parentheses indicate the percent difference from the inoculum consensus sequence for each viral clone analyzed.
f Unknown indicates not positive for any of the Mamu class I alleles tested (A*01, A*02, A*08, A*11, B*01, B*02, B808, B*04, B*17, and B*26).
g Vaccinated animals with better control of SIV replication are shaded in gray.
h F, female; m, male.
To assess IgG and IgA antibody development in infant macaques, we used genetically outbred healthy non-specific-pathogen-free macaques that were raised in outdoor corrals. Blood and saliva samples were collected without anesthesia during normal monthly health checks.
Vaccines.
The construction and immunological characterization of the recombinant Mycobacteria tuberculosis-SIV strain mc26435 has been previously described in detail (3). Briefly, strain mc26435, derived from human H37Rv Mycobacterium tuberculosis, had deletions in genes panCD and leuCD to produce a double auxotroph strain with severe attenuations for replication. The nonessential secretory gene secA2 was also eliminated to limit immune evasion. In addition, full-length SIVmac239 cassettes, codon optimized for expression by mycobacteria, were combined to create the recombinant mc26435 (Gag) (3) and mc26439 (Env) (U. D. Ranganathan, unpublished data) vaccine strains used in this study. Recombinant MVA SIVmac239 Gag/Pol/Env (MVA-SIV; kindly provided by Bernie Moss and Patricia Earl, National Cancer Institute) was used to boost SIV-specific responses. We previously demonstrated that vaccination with strain mc26435, despite its very low replicative capacity, could safely induce persistent Mycobacterium tuberculosis- and SIV-specific T cell and antibody responses in neonatal macaques following oral vaccination at birth (2).
Immunizations.
The vaccination regimen is outlined in Table 1. Naive control animals (group A; n = 6) received sterile phosphate-buffered saline (PBS) at birth by the p.o. route and at weeks 3 and 6 by the i.m. route. Group B (n = 8) animals were p.o. primed with 109 CFU each of M. tuberculosis-SIV strains mc26435 (expressing Gag) and mc26439 (expressing Env) at week 0. Group B mice then were i.m. boosted with MVA-SIV (108 infectious units, divided over 4 injection sites) at weeks 3 and 6 of age.
SIV challenge.
Beginning at week 9, all animals were challenged by the p.o. route once per week with 5 × 103 50% tissue culture infective doses (TCID50) of SIVmac251 (stock 6/04 grown in rhesus macaque peripheral blood mononuclear cells [PBMC]) in a volume of 1 ml until plasma viremia was confirmed at 3 consecutive time points. Once systemic infection was confirmed, the first PCR-positive time point was considered week 1 postinfection. Animals were monitored for a minimum of 9 weeks postinfection; animals with controlled viremia were monitored longer. All animals were euthanized prior to meeting established criteria for AIDS (10).
SIV RNA testing.
Longitudinal plasma samples were used for virological analysis by reverse transcription-PCR (RT-PCR) for SIV RNA as described previously (11). Note that limits of detection depended on input plasma volume and ranged from 3 to 65 copies/ml, with a mean of 26 and median of 30. Samples showing transient low viremia followed by SIV RNA-negative time points were retested to confirm SIV RT PCR results. Data are reported as SIV RNA copy equivalents per ml of plasma.
Tissue samples were stabilized in RNAlater (Ambion), rinsed in cation-free phosphate-buffered saline, and then processed and analyzed for levels of SIV gag RNA and SIV gag DNA, normalized to diploid genome equivalents of DNA based on simultaneous determination of copy numbers for a single-copy sequence in the rhesus macaque CCR5 gene, essentially as described previously (12, 13).
SIV sequence analysis.
To characterize founder virus envelope variants, SIV gp160 was amplified using single-genome amplification (SGA), sequenced, and compared among vaccinated and control animals. TRIzol reagent (Life Technologies) was used to purify viral RNA from ultracentrifuged plasma samples collected 1 to 2 weeks postinfection. RNA was reverse transcribed using Superscript III (Invitrogen) with oligo(dT) primers, the resulting cDNA was serially diluted, and the SIV gp160 was amplified in a nested PCR assay. The first round of PCR utilized forward primer GAACTCCGAAGAAGGCTAAGGCTAAT and reverse primer CAGGGCTCAATCTGCCAGCCT with Q5 polymerase (New England BioLabs) in a 25-μl reaction mixture with the following cycling conditions: 98°C for 30 s, followed by 25 cycles of 98°C for 8 s, 70°C for 20 s, and 72°C for 2 min, and a final hold at 72°C for 2 min. Second-round PCR utilized 1 μl of the first-round mixture in a 25-μl reaction mixture with forward primer ATGGGATGTCTTGGGAATCAGCTGCTT and reverse primer CCCCTTGTGGAAAGTCCCTGC and similar cycling conditions to yield an amplicon of approximately 3,295 bp that was confirmed by agarose gel electrophoresis. The dilution of cDNA that produced <30% positive reactions was used to perform multiple SGA PCRs to yield approximately 10 envelope amplicons for sequence analysis from each animal as well as the inoculum stock. SGA envelope amplicons were directly sequenced using a set of 8 SIV-specific primers that produced overlapping reads (performed by the University of Chicago DNA Sequencing Facility). Primers included ATGGGATGTCTTGGGAATCAGCTGCTT, CAGTCACAGAACAGGCAATAGA, AAGCAAAGCATAACCTGGAGGT, CTTCATGCACAAGGATGAT, CACTTGGCATAAAGTAGGCAAAAATG, ACACTACTGTACCATGGCCAAATG, CACTGTAATAAATCCCTTCCAGTCC, and CCCCTTGTGGAAAGTCCCTGC. Geneious v8.1.3 (14) was used to assemble reads and generate contigs, which were manually screened and edited for quality. Full-length sequences obtained for each animal were aligned using the Geneious alignment tool, and overall diversity was assessed. Envelope sequences found in each animal with <0.1% differences were identified as a single founder virus genotype, while sequences with ≥0.1% differences were identified as distinct founder virus genotypes. A representative sequence for each genotype was used for comparisons among animals. The predominant envelope genotype contained in the SIVmac251 inoculation stock was used for comparisons. Phylogenetic comparisons of SGA variants identified in each animal were done with a Jukes-Cantor genetic distance model and Neighbor-joining trees with 10,000 bootstrap resampling replicates. Trees were rooted to the predominant inoculum stock envelope genotype.
Sample collection and processing.
Peripheral blood, saliva, and stool samples were collected prior to all interventions and throughout the study period at weeks 0, 3, 6, and 9 and then every 2 weeks until necropsy. Peripheral blood was collected in EDTA, and plasma was separated by centrifugation and stored in aliquots at −80°C for antibody and viremia measurements. PBMC were purified using density gradient centrifugation with lymphocyte separation medium (MP Biomedicals, Santa Ana, CA) as described previously (2, 5). Mononuclear cells in tissues collected at the terminal time point were isolated as described previously (2, 5). Salivary secretions were collected using Weck-Cel sponges (Beaver Visitec, Waltham, MA), immediately snap-frozen, and later eluted as described previously (2). When possible, 2 to 3 g of fresh stool was added to 5 ml sterile PBS supplemented with 1/100 protease inhibitor cocktail (Sigma) and snap-frozen. Fecal extracts were prepared as described previously (2) by homogenization, removal of debris by centrifugation, 0.45-μm filtration, and concentration to roughly 0.5 ml using a Millipore-Amicon Ultra-4 50K centrifugal filter unit.
Antiviral binding antibodies and avidity.
Antibodies and total IgA or IgG were measured by enzyme-linked immunosorbent assay (ELISA) as previously described (15). Viral antigens were recombinant SIVmac251 gp140, gp120 (both from Immune Technology, New York, NY), gp70-V1V2 (kindly provided by David Montefiori, Duke Medical School), HIV-2 gp36 (Prospec, East Brunswick, NJ), which has 85% homology to the SIV gp41 ectodomain (16), and SIVmac251 viral lysate (Advanced Biotechnologies, Columbia, MD), which was used to measure anti-Gag and anti-Pol antibodies, as it lacks detectable Env antigen at the dilution used. All assays for IgA were performed using specimens depleted of IgG as described previously (17). Antigen-specific IgA or IgG antibody concentrations measured in secretions were normalized in accordance with the total IgA or IgG content. The specific activity (nanograms of IgA or IgG antibody per microgram of total IgA or IgG) in secretions was deemed significant if it was greater than the mean + 3 standard deviations (SD) for negative controls. The avidity of gp140-specific IgG in plasma was measured using microtiter plates coated with SIV gp140 in an NaSCN displacement ELISA as described previously (18). The avidity index was calculated by dividing the concentration of anti-gp140 IgG measured in 1.5 M NaSCN-treated wells by that in untreated wells on the same plate.
Neutralizing antibodies.
Neutralizing activity was measured as described previously (19) using heat-inactivated plasma, TZM-bl cells (AIDS Reagent Program), and a clone of SIVmac251 (clone C) derived in the laboratory (GenBank accession number KT447167). This clone was deemed a tier 1 virus based on the finding that clone C neutralizing antibody titers in plasma of SIV-infected adult macaques were virtually identical to those previously measured in assays with an established tier 1 SIVmac251 (20).
ADCVI.
Antibody-dependent cellular viral inhibition (ADCVI) assays were performed as described previously (21) using 1/100 dilutions of heat-inactivated week 9 plasma, human PBMC as effector cells, and SIVmac251-infected CCR5+ CEM-NKr T cells as target cells. The extent of infection was determined by measuring SIV p27 in culture medium using the p27 ELISA described below. Because a neutralization-resistant SIVmac251 was used, results of this assay primarily reflected antibody-dependent cytotoxicity (ADCC) and phagocytic activity in plasma.
SIVp27 ELISA.
An in-house ELISA, developed as described previously (22), was used to measure p27 concentration in culture medium. Briefly, plates were coated overnight with 125 ng/ml goat anti-mouse IgG2a (SouthernBiotech). The following day, plates were washed, blocked, and treated for 1 h with 1/100 culture medium harvested from the 2F12 anti-SIV p27 hybridoma (AIDS Reagent Program). The plates were washed, loaded with samples and a recombinant p27 standard (ImmuneDiagnostics, Woburn, MA), and then stored overnight at 4°C. Plates were then washed and treated for 1 h at 37°C with biotinylated anti-SIV IgG that had been purified from pooled SIV-positive macaque serum in the laboratory using protein G Sepharose (GE Healthcare) and subsequently biotinylated using EZ-link NHS-Sulfo (Pierce). The plates were developed using 1/2,000 neutralite avidin-peroxidase, TMB (both from SouthernBiotech), and 2N H2SO4 stop solution.
Antibody-dependent phagocytosis (ADP).
Phagocytosis assays were performed as described previously (23), with modifications noted below. Briefly, 1 × 109 1-μm NeutrAvidin fluorospheres (Invitrogen) were labeled with 7 μg biotinylated recombinant SIV gp140mac251 (Immune Technology). After washing, 1 × 107 beads per well were added to V-bottom plates containing triplicate serial dilutions of heat-inactivated serum. THP-1 cells (2 × 104 per well) were then added. After 6 h at 37°C in 5% CO2, the cells were washed with Ca2+/Mg2+-free Dulbecco's PBS (DPBS) and incubated for 10 min with 50 μl of 0.05% Trypsin-EDTA (Gibco) to remove beads that nonspecifically adhere to cell surfaces, allowing specific assessment of internalized beads. The cells were then washed in DPBS, resuspended in 1% paraformaldehyde, and analyzed for fluorescence. The phagocytic score was calculated as described previously (23) by multiplying the number of bead-positive cells by their median fluorescent intensity. The average score for naive monkey serum at the same dilution was subtracted from test samples to obtain the final score.
Histopathology and immunohistochemistry.
Formalin-fixed, paraffin-embedded tissues were cut into 5-μm serial sections and hematoxylin and eosin stained to evaluate SIV-induced pathology by a pathologist blinded with respect to treatment groups. Criteria included, among others, tissue structure, hyperplasia, hypertrophy, cellular influx indicative of inflammation, hemorrhages, pneumonitis, colitis, bronchitis, spirochetosis, and evidence of coinfections. A tissue disease score scale was used to measure disease severity: 0, no indications of SIV-associated pathology; 1, mild disease, multifocal; 2, mild disease, diffuse; 3, moderate disease, multifocal; 4, moderate disease, diffuse; 5, severe disease with mild T cell depletion; 6, severe disease with massive T cell depletion. Similarly, tissues were evaluated in a blinded manner for TB-indicative pathologies and for the presence of cryptosporidiosis, an indicator of opportunistic infections.
Intracellular cytokine staining.
Flow cytometry was performed as described previously (2, 3, 5) using peripheral blood mononuclear cells or single-cell suspensions prepared from fresh tissues. Cytokine production following a 6-h incubation with SIVmac239 Gag and Env peptide pools (Gag, number 6204; Env, number 6883; NIH AIDS Reagent Program) was used to assay SIV-specific T cells. Phorbol myristate acetate (PMA) and ionomycin (Sigma) stimulation served as a positive control. Background responses by unstimulated cell suspensions were subtracted to account for nonspecific immune responses. Cells were stained for flow cytometry using standard protocols (2, 3, 5) and included antibodies for CD3 (clone SP34-2), CD4 (clone L200), CD8 (clone RPA-T8), interleukin-2 (IL-2; clone MQ1-17H12), tumor necrosis factor alpha (TNF-α; clone MAb11), gamma interferon (IFN-γ; clone B27) (all from BD Biosciences), and IL-17 (clone eBio64CAP17; eBioscience) (2, 5). Intracellular staining using fixation/permeabilization solution and perm/wash (BD Biosciences) was performed according to the manufacturer's instructions. Samples were resuspended in PBS following fixation and acquired within 24 h on an LSRII instrument. Samples were analyzed in FlowJo (TreeStar, Ashland, OR) using the following gating procedure prior to panel-specific analysis: (i) lymphocyte gate, (ii) forward scatter area versus forward scatter height and side scatter height versus side scatter width to eliminate doublets, and (iii) fixable live/dead discriminator (Invitrogen) to discard dead cells and debris. Boolean gating was applied when appropriate.
Animal genotyping.
Snap-frozen splenic cell pellets were typed for the common MHC class I variants Mamu-A*01, -A*02, -A*08, -A*11, -B*01, -B*03, -B*04, -B*08, and -B*17, chosen for their possible roles in restricting SIV infection and/or control of viremia. Genotyping was completed by the MHC Genotyping Service at the University of Miami Miller School of Medicine, and the established methods have been reported previously (24–29). Genotypes of the animals are reported in Table 1.
TRIM5α genotyping.
The TRIM5α genotype of macaques was determined by sequence analysis of the C-terminal B30.2/SPRY domain of the TRIM5 gene (Table 1). Briefly, the region was amplified by PCR utilizing purified genomic DNA and amplification protocols and primers as described previously (30). Resulting PCR fragments were cloned using the TOPO-TA vector system (Invitrogen), and TRIM5 sequences for each animal were determined from 5 to 7 clones. The genotypes of TRIM5TFP, TRIM5Q, and TRIM5CypA were identified and animals were classified as resistant, intermediate, or sensitive as described previously (30–32).
Statistical analysis.
Data were analyzed using GraphPad Prism software, version 6.0f (GraphPad Software, Inc., La Jolla, CA), and R, version 3.1.2. The numbers of infections per SIV exposure for unvaccinated or vaccinated animals were used to estimate the risk of infection per exposure, and results were plotted using the Kaplan Meier method and analyzed by an exact log-rank test (33–35). For comparisons of three or more groups, the nonparametric Kruskal-Wallis test was performed. The nonparametric Mann-Whitney exact test was used for comparison of two groups. Correlations were evaluated using the Spearman rank test. Two-tailed statistical tests were conducted and P values of ≤0.05 were considered statistically significant. No adjustments were made for multiple hypothesis testing.
Nucleotide sequence accession number.
The sequence determined in the course of this work was deposited in GenBank under accession number KT447167.
RESULTS
Establishment of a repeated low-dose oral SIVmac251 challenge model.
To mimic the repeated and continuous exposure of human infants to HIV-1 in breast milk in a relevant animal model while addressing the practical constraints of nonhuman primate studies, we previously developed a repeated oral SIVmac251 challenge model in which infant macaques were bottle fed 3× daily for 5 consecutive days with 104 TCID50 of SIVmac251 in 1 ml of RPMI (4, 5). This oral SIVmac251 exposure model resulted in an 80% infection rate within 2 weeks of the first viral exposure (4, 5). A limitation of this model, however, was that the frequent exposures in rapid succession did not permit quantification of the number of exposures required for SIV acquisition. For the present studies, we used a repeated once-weekly low-dose challenge model to more precisely evaluate vaccine efficacy by measuring (i) protection against infection (sterilizing immunity) and/or (ii) partial efficacy by quantifying the number of challenges required for infection (4). To achieve 100% infection of control animals, our regimen aimed for 20 to 50% probability of infection per SIV exposure.
We began once-weekly oral SIVmac251 exposures in naive infant macaques at 9 weeks of age for a maximum of 10 weeks (Table 1, group A). All group A animals became persistently infected, although the number of exposures leading to SIV infection varied greatly within the group (Fig. 1A). Two naive animals became infected following a single exposure and one animal each became infected after 2, 5, 7, and 9 exposures (Fig. 1A). The animal that required 7 exposures (number 42409) experienced low transient viremia at 2 consecutive time points (550 and 380 copies/ml, respectively), but viral RNA was undetectable 1 week later. A second plasma aliquot from the negative time point was tested and confirmed to be negative for SIV RNA. Based on our definition of persistent viral infection as three consecutive plasma SIV RNA-positive time points, this animal was defined as being uninfected during the period of low transient viremia. Peak viremia (defined as the highest viral load during the first 3 weeks of detectable persistent viremia) in nonvaccinated group A animals ranged from 7.3 × 106 to 3.4 × 108 viral RNA copies/ml plasma (mean, 7.7 × 107; median, 2.4 × 107), and viral replication was maintained at high levels in all animals (Fig. 1A). The estimated probability of infection per oral SIV exposure in nonvaccinated group A infant macaques was 0.24. Although this per-exposure risk of oral SIV infection is higher than the estimated risk of breast milk HIV transmission in human infants, we deemed the per-exposure infection rate for this repeated low-dose oral SIVmac251 challenge model to be within a relevant range for vaccine efficacy studies aimed at recapitulating frequent, low-dose breast milk exposure with balancing efforts to mimic clinical circumstances and the practical logistics associated with infant macaque studies.
FIG 1.

Challenge outcome after repeated low-dose oral SIVmac251 exposure. (A and B) Plasma viremia in naive controls (A) and vaccinated animals (B). Starting at week 9 of age (black arrow), animals were exposed once weekly with 5,000 TCID50 of SIVmac251 by the oral route. Naive animals are shown as open circles, vaccinated animals with high and uncontrolled viremia (noncontrollers) are represented by black diamonds, and vaccinated animals that could partially control viremia (controllers) are shown as gray diamonds. The dashed line at 106 copies of SIV RNA/ml plasma indicates the threshold to define partial control of viremia in the current study, because most of the nonvaccinated animals had higher peak and chronic viremia throughout the study period. (C) Representing the per-exposure risk of SIV infection in naive control and vaccinated infant macaques, the Kaplan-Meier survival plot shows the percentage of naive and vaccinated animals that remain uninfected after each oral SIV exposure. (D) Median acute viremia (horizontal line), defined as area under the curve (AUC) of plasma viremia from weeks 1 to 3 post-SIV infection (PI) in controller and noncontroller animals of group B. (E) Analogous to panel D, but showing differences in early chronic viremia, defined as the AUC of plasma viremia between weeks 6 and 8 post-SIV infection in controller and noncontroller group B animals. Each symbol represents an individual animal.
Challenge outcome in vaccinated infant macaques.
The current study was designed to test the protective efficacy of the p.o. mc26435 prime/i.m. MVA-SIV boost vaccine regimen, which we previously showed to be safe and immunogenic (2, 3) against oral SIVmac251 transmission in infant macaques (Table 1). As in our prior study (2), animals were orally primed at birth to induce SIV-specific immune responses proximal to the presumed sites of virus entry after oral SIV exposure. To enhance the immunogenicity and the potential efficacy of the vaccine regimen, we used a cocktail of the newly developed M. tuberculosis strain mc26439 expressing SIV Env and the previously described mc26435 strain as an oral prime at birth (week 0). Infant macaques were then boosted i.m. with MVA-SIV at 3 and 6 weeks of age (group B). Naive controls (group A) were age matched and received sterile PBS at the same time points and by the same routes as vaccinated animals (Table 1). Beginning at week 9, all infant macaques were exposed to SIVmac251 using the weekly oral challenge regimen described above for the nonvaccinated animals. Our results showed that this M. tuberculosis-SIV/MVA-SIV regimen was unable to prevent oral SIV acquisition (Fig. 1B). In fact, 87.5% of vaccinated animals became infected after only 1 or 2 exposures compared to 50% of naive controls. The per-exposure risk of SIV infection in vaccinated animals was 0.5 (Fig. 1C), which was 2-fold higher than that of naive animals. However, due to the small group sizes, the difference in the per-exposure risk of SIV infection was not statistically significant (P = 0.17). Despite acquisition of SIV, the viral trajectories in the vaccine group showed that 3 of 8 vaccinated infants exhibited reduced acute viremia (Fig. 1D) and continued to exhibit reduced viremia during the early chronic phase of infection (Fig. 1E). These 3 animals were subsequently referred to as “controllers” and were monitored for 6 months to evaluate long-term challenge outcome. One of the controller animals (number 42958) eventually showed an increase in viremia (after week 21) (Fig. 1B), whereas the other 2 infant controller animals maintained lower viremia than the noncontrollers until the study end. We deemed it unlikely that MHC genotype and/or TRIM5α genotype influenced oral SIV challenge outcomes in this study, because there was no consistent genotype that distinguished controllers from noncontrollers (Table 1). In addition, TRIM5α genotypes have not been associated with resistance to SIVmac239 or SIVmac251 infection (36).
There was also no difference in the number of founder viruses identified between group A and B animals or between vaccinated noncontroller and controller animals. One or two envelope genotypes were identified in each of the animals (Table 1), and phylogenetic comparisons of founder viruses did not identify significant clustering between naive and vaccinated animals or between vaccinated noncontroller and controller animals (Fig. 2). Differences in founder viruses compared to the predominant inoculum genotype ranged from 0.1 to 1.4% and did not differ among groups.
FIG 2.

SIV Env sequence diversity in naive and vaccinated animals. Phylogenetic tree depicting the 19 founder viral variants found in SIV-infected group A and B animals. The predominant gp160 variant found in the SIVmac251 inoculum (stock 6/04) was used to root the tree. The horizontal bar shows the scale of genetic distance. Sequence labels from group B vaccinated animals are boxed, and those from vaccinated controller animals are double boxed.
Anamnestic SIV-specific T cell responses may have contributed to control of viremia.
Although vaccine-induced SIV-specific immune responses were not sufficient to prevent oral SIV acquisition, we examined the possibility that the immune responses at the time of oral SIV challenge initiation (week 9) were associated with reduced viremia in the controller animals. At week 9, SIV Gag-specific CD4+ and CD8+ T cells producing IL-2, IFN-γ, IL-17, and/or TNF-α were detectable in all group B animals except in one controller, animal number 42944 (Fig. 3A). However, this animal had SIV-specific T cell responses at week 6 (data not shown). We also measured SIV Env-specific T cell responses, but due to the small blood volumes available from infant macaques, these responses could not be tested at all time points for all animals (Fig. 3B). Therefore, we only sought correlations between SIV Gag-specific T cell responses and acute viremia. Although neither SIV Gag-specific CD4+ nor CD8+ T cell responses of group B animals at the time of oral challenge correlated with acute peak viremia (Fig. 3C), SIV Gag-specific CD4+ T cell responses at week 2 postinfection inversely correlated with acute viremia (Fig. 3D). A similar trend was observed for CD8+ T cell responses but did not reach statistical significance. Thus, anamnestic SIV Gag-specific T cell responses may have contributed to lower viremia of group B controller animals.
FIG 3.

Prechallenge (week 9) SIV-specific T cell responses in group B vaccinated infant macaques. (A) SIV Gag-specific T cell responses in peripheral blood CD4+ (left) and CD8+ T cells (right). Each bar represents the sum of the percentage of single-cytokine-positive CD4+ or CD8+ T cells for a specific animal. Cytokine-positive T cells were measured by flow-cytometric analysis that included TNF-α (black), IL-17 (striped), IFN-γ (gray), and IL-2 (white). (B) Analogous data for SIV Env-specific T cell responses. N.T. (not tested) indicates that no cells were available for measuring SIV Env-specific responses in these animals. (C) Prechallenge (week 9) SIV Gag-specific CD4+ (left) and CD8+ T cell (right) responses were log transformed and plotted against acute peak viremia, defined as area under the curve of plasma viremia in weeks 1 to 3 post-SIV infection, to test for a potential correlation. Note that a value of 0.001 was assigned to animal number 42944, which did not have detectable T cell responses. (D) SIV Gag-specific CD4+ (left) and CD8+ (right) T cell responses at week 2 post-SIV infection were tested for their correlation with acute plasma viremia.
Vaccine-induced systemic IgG antibody responses.
Only 1 vaccinated animal (number 42949) developed SIV Env-specific plasma IgG antibodies before MVA-SIV boosting, and these were only at low levels. This was not surprising, because we have previously found that even 2 immunizations 3 weeks apart with M. tuberculosis-SIV did not elicit SIV Env-specific IgG, suggesting that Env expression by mc26439 is insufficient for induction of these antibodies (data not shown). In all group B animals, plasma IgG antibodies to gp140 and gp120 were detected after receiving the first MVA-SIV boost at week 3, and these were further increased by the second MVA-SIV boost on week 6 (Fig. 4A). In addition, group B animals also developed IgG antibodies to gp36 and to gp70-V1V2 by week 9 (Fig. 4B), but the magnitude of these responses at the time of challenge did not differ between controller and noncontroller animals (Fig. 4B). Plasma from vaccinated animals at week 9 was able to neutralize a tier 1 SIVmac251 (Fig. 4C) and to mediate ADP of gp140-coated fluorescent beads (Fig. 4D). However, these activities were not associated with control of viremia and did not differ between controller and noncontroller animals, and none of the plasma samples had antibodies capable of mediating ADCVI against a neutralization-resistant SIVmac251 (data not shown). However, at week 9, the avidity of gp140-specific IgG antibodies was slightly higher, albeit not significantly, in controller than noncontroller animals (Fig. 4E), and acute peak viremia, defined as the area under the curve (AUC) for viremia measured at weeks 1 to 3 postinfection, was inversely correlated with the avidity index (Fig. 4F). In both controller and noncontroller vaccinated animals, much greater avidity indices were recorded at necropsy (Fig. 4G), indicating that the quality of Env-specific IgG antibodies increased throughout the postinfection period, although statistically avidity indices did not differ between controller and noncontroller animals. As functional activities of plasma antibodies also likely improved during this time (but were not measured), SIV Env-specific IgG may have contributed to control of viral replication. No correlations were observed between plasma anti-SIV Gag, Pol IgG levels, and viremia (data not shown).
FIG 4.

Vaccine-induced SIV Env-specific plasma IgG responses at week 9. (A) Plasma SIV gp140-specific IgG concentrations at weeks 0, 3, 6, and 9 in naive controls, noncontroller animals, and controller animals. The times of immunization and the vaccine constructs are indicated by arrows. (B) Plasma IgG concentrations for gp140, gp120, gp36, and gp70-V1V2 in controller and noncontroller animals at week 9, just prior to oral SIV challenge initiation. (C to E) Week 9 SIVmac251 neutralization titer (50%), ADP score, and avidity index for controller and noncontroller animals, respectively. (F) The correlation between anti-gp140 plasma IgG avidity at week 9 and acute viremia. (G) The gp140 plasma IgG avidity index for group B controller and noncontroller animals at the time of euthanasia. Nx, necropsy.
Vaccine-induced systemic and mucosal IgA antibody responses.
As found for plasma IgG, SIVgp140 Env-specific plasma IgA developed in all group B animals after the first MVA-SIV boost and was increased further by the second boost (Fig. 5A). Vaccine-induced gp140-specific IgA was also detected in saliva and in feces at week 9, the time of the first oral SIV exposure (Fig. 5B and C). It should be pointed out that 2 of the controller animals had specific SIV Env IgA activities of 97.8 and 117.8 ng/μg total IgA in saliva, a magnitude much higher than was observed in previous pediatric SIV vaccine studies (5). Plasma IgA antibodies to gp140 were not associated with acute viremia (Fig. 5D). However, both SIV gp140-specific salivary and fecal IgA responses at week 9 correlated positively with each other and were inversely correlated with acute viremia (Fig. 5E to G), whereas there was little or no correlation between mucosal and systemic IgA (Fig. 5H and I), indicating that the vaccine-induced SIV Env IgA antibodies in saliva and intestine were produced locally. In contrast to IgA, SIV gp140-specific IgG in fecal samples was undetectable at week 9. IgG antibodies were not measured in saliva due to limited sample volume. Table 2 summarizes the correlation between vaccine-induced plasma and mucosal antibody responses at the time of oral SIV challenge initiation and acute viremia.
FIG 5.

Vaccine-induced SIV Env-specific IgA responses prior to SIV challenge. (A) SIV gp140-specific plasma IgA levels at weeks 6 and 9 in naive animals, noncontroller animals, and controller animals. (B and C) gp140-specific IgA activity in saliva and fecal samples of controller and noncontroller animals at week 9. (D to F) Correlation between plasma, salivary, and fecal gp140-specific IgA at week 9 and acute viremia, respectively. (G) Linear positive correlation between salivary and fecal IgA at week 9. (H and I) Lack of correlation between plasma IgA and saliva IgA (H) or fecal IgA (I).
TABLE 2.
Correlations between vaccine-induced antibody responses at week 9 and acute viremia
| Parametera | Spearman correlationb |
||
|---|---|---|---|
| Correlation | r value | P value | |
| IgG | |||
| Plasma anti-SIV Env IgG | |||
| gp140 IgG BAb | NS | 0.2381 | 0.5821 |
| gp120 IgG BAb | NS | −0.0476 | 0.9349 |
| V1V2 IgG BAb | NS | −0.2143 | 0.6191 |
| gp36 IgG BAb | NS | 0.0952 | 0.8401 |
| Fecal anti-SIV Env IgG | |||
| gp140 IgG BAb | Not detectable | NA | NA |
| Plasma Gag, Pol IgG | NS | −0.2143 | 0.6191 |
| Fecal Gag, Pol IgG | Not detectable | NA | NA |
| IgA | |||
| Plasma gp140 IgA BAb | NS | −0.4286 | 0.2992 |
| Salivary gp140 IgA BAb | Inverse | −0.8571 | 0.0107 |
| Fecal gp140 IgA BAb | Inverse | −0.9550 | 0.0028 |
| Plasma Gag, Pol IgA | NS | −0.4048 | 0.3268 |
| Fecal Gag, Pol IgA | Not detectable | NA | NA |
| Function | |||
| Plasma gp140 IgG avidity | Inverse | −0.7381 | 0.0458 |
| Plasma ADP | None | 0.0000 | >0.9999 |
| Plasma ADCVI | <5% (negative) | NA | NA |
| Plasma tier 1 neutralization | None | 0.3995 | 0.3894 |
BAb, binding antibody.
NS, not significant; NA, not applicable.
After the vaccinated animals were infected with SIV, we observed no differences in salivary gp140-specific IgA responses between controller and noncontroller animals (Fig. 6A). In contrast, the fecal IgA responses to gp140 continued to increase in controllers and were significantly greater than those of noncontroller animals at week 6 postinfection and the terminal time point (Fig. 6B). However, there was no correlation between gp140-specific fecal IgA and plasma SIV RNA levels at the time of necropsy (data not shown).
FIG 6.

Postinfection salivary and fecal IgA antibodies in vaccinated animals. (A) Salivary SIV gp140-specific IgA activities at weeks 4 and 8 and at the time of euthanasia in controller and noncontroller animals. (B) Fecal gp140-specific IgA activities at weeks 2 and 6 and at the time of euthanasia in controller and noncontroller animals. The different time points in panels A and B are a result of sample availability. Nx, necropsy.
Total IgA and IgG in plasma and saliva during the first year of life.
The significance of the magnitude of vaccine-induced mucosal Env-specific IgA antibodies and their contribution to control of virus replication, even if observed in only a subset of animals, becomes apparent when the above-described IgA results are viewed in the context of normal postnatal antibody development in infant macaques. In plasma, transplacentally transferred maternal IgG is present at birth and is lower at 1 month of age (Fig. 7A). Once infants begin producing their own IgG, total plasma IgG levels rapidly reach adult levels (Fig. 7A). A similar decline followed by an increase in total IgG was also observed in saliva (Fig. 7B). Interestingly, levels of total salivary IgG in 6- to 7-month-old infants were significantly higher than those in adult macaques. Together, these results suggest that more plasma IgG in infant than in adult macaques is passively transudated into the oral cavity.
FIG 7.

Postnatal antibody development in rhesus macaques. The analysis of plasma and salivary antibodies was performed using cross-sectional samples from infant macaques (black symbols) between the ages of 0 (birth) to 7 months (Mo). Adult antibody levels (open diamonds) are shown for comparison. The number of animals in each group is listed in parentheses below the relevant age group. Plasma and salivary total IgG levels (A and B) and plasma and salivary IgA levels (C and D) in infant compared to adult macaques are shown. Each symbol represents an individual animal, with horizontal bars indicating the median value. The nonparametric Mann-Whitney test was performed to determine differences between two specific infant age groups and between 6- to 7-month-old infants and adults. Note that a monomeric serum IgA standard was used to measure the polymeric IgA. Therefore, the total IgA is likely to be underestimated by a factor of 2.5.
Consistent with the fact that IgA antibodies cannot cross the placenta, median plasma total IgA levels (50.2 μg/ml) in infant macaques were more than 2 logs lower than median plasma IgG levels (12,262 μg/ml) in the first month and gradually increased with age (Fig. 7C). Median plasma IgA levels in 6- to 7-month-old infants were still roughly 1 log10 μg/ml lower than adult plasma IgA levels (Fig. 7C). Salivary IgA was almost undetectable at birth and remained low for the first few months, but it reached adult levels by 6 to 7 months (Fig. 7D). Thus, considering the very low levels of total IgA being produced in infant macaques in the first 3 months of life, it is encouraging that the SIV M. tuberculosis/MVA vaccine was able to induce SIV-specific mucosal IgA responses at a magnitude that was associated with partial control of viremia in a subset of infant animals.
Clinical outcome and tissue pathology.
To test if the effects of reduced viremia impacted disease morbidity and progression, we evaluated clinical markers of HIV/SIV progression in the vaccinated controller infant macaques. In lymphoid tissues, SIV-induced pathology ranged from mild hyperplasia to severe disease with massive T cell depletion. Overall, there was a lower median pathology score in vaccinated than naive control animals, but tissue pathology did not differ between animals that showed partial control of viremia and noncontrollers (Fig. 8A). Cryptosporidiosis, an opportunistic infection, was present in 20% of mock-vaccinated animals and 100% of noncontrollers in the ileum, whereas none of the controller animals developed this opportunistic infection. In the colon, 40% of noncontroller compared to 0% of the controller animals showed cryptosporidiosis, but the nonvaccinated animals also had no signs of cryptosporidiosis (Fig. 8B). To test whether reduced tissue pathology was indeed associated with better control of virus replication, we measured colon SIV RNA and DNA levels. The 3 controller animals had significantly lower SIV RNA copies per 1 million cell equivalents than noncontroller animals, whereas median colon RNA levels were indistinguishable between the noncontroller and naive control animals (Fig. 8C). SIV DNA levels, however, were only slightly reduced in the controller animals. A similar pattern of lower SIV RNA and DNA levels was observed in the submandibular lymph nodes and in spleen (data not shown).
FIG 8.

Clinical assessment of SIV-infected animals. (A) The median disease score, determined by histological analysis, for animals in group A (naive controls) and group B animals at the time of euthanasia. Controller and noncontroller animals are represented by gray and black symbols, respectively. (B) The percentage of animals with detectable cryptosporidiosis in the colon and ileum of naive controls and vaccinated animals at the time of euthanasia. (C) SIV RNA (left y axis) and DNA levels (right y axis) were measured in colon tissue samples by PCR and are reported as copy numbers per 106 cell equivalents. (D) The percentage of CD4+ T cells within the CD3 population at the time of oral SIV challenge initiation (week 9), at weeks 1 to 2 (acute phase) and weeks 7 to 8 (chronic phase), and at the time of euthanasia in nonvaccinated animals and in controller and noncontroller vaccinated infant macaques. Median values per group are represented by horizontal bars. (E) Comparative analysis of CD4+ T cell frequencies in selected tissues of mock-vaccinated and vaccinated animals at the time of euthanasia. Differences between animals in 2 different groups were determined using the nonparametric Mann-Whitney test. Mes, mesenteric; Ret, retropharyngeal; Nx, necropsy.
Over time, vaccinated controller animals showed better preservation of peripheral blood CD4+ T cells than nonvaccinated or noncontroller animals (Fig. 8D). Controller animals also better maintained tissue CD4+ T cell populations than noncontroller animals (Fig. 8E). While the differences in CD4+ T cell frequencies were only statistically significant in mesenteric lymph node (LN), retropharyngeal LN, and tonsil, a similar trend was observed in other tissues (representatively shown for colon and bronchoalveolar fluid) (Fig. 8E). Even the animal that eventually lost control of viremia after week 24 (number 42958) benefitted from preserved CD4+ T cell maintenance, which could be further indicative of reduced morbidity from partial viremia control early after infection. We could not detect a targeted loss of memory and/or effector CD4+ T cells, as all memory subset ratios were maintained (data not shown). The improved outcome in vaccinated controller animals with regard to preservation of CD4+ T cells and reduced tissue pathology was noteworthy, especially given the duration of SIV infection in these 3 animals was extended by 10 weeks compared to that of the noncontroller animals.
DISCUSSION
The current study tested the efficacy of a recombinant attenuated M. tuberculosis-SIV vaccine to prevent oral SIV acquisition in infant macaques. To simulate breast milk transmission of HIV in human infants, we applied a repeated oral SIVmac251 challenge model. In unvaccinated animals, the per-exposure risk of oral SIV infection using this model was 0.24. This per-exposure infection rate balances the practical logistics of pediatric nonhuman primate studies while maintaining clinical relevance for efficacy studies testing vaccines for the prevention of breast milk transmission of HIV in human infants.
Applying this weekly oral challenge model, our p.o. M. tuberculosis-SIV prime/i.m. MVA-SIV boost strategy was unable to prevent oral SIV acquisition. In fact, more than 80% of the vaccinated animals became infected after only 1 to 2 oral SIV exposures compared to 50% of mock controls, but this difference was not statistically significant. Despite the lack of protection against oral SIV acquisition, 3 of 8 vaccinated infants were able to partially control virus replication. In these controller animals, SIV Env-specific plasma IgG antibody avidity and the magnitude of SIV Env-specific mucosal IgA responses at the time of challenge inversely correlated with acute peak viremia. SIV-specific T cells may have contributed to control of viremia as well, because acute viremia inversely correlated with SIV-specific peripheral blood CD4+ T cell responses. These results also emphasize that certain immune responses might be induced by vaccination and contribute to control of SIV replication after infection, even though at the time of challenge, these responses are not detected or do not differ between vaccinated noncontroller and controller infants. In the current study, we could not measure SIV-specific T cell responses in tissues prior to SIV challenge, but it is possible that SIV-specific T cell responses, including follicular T helper cell responses, contributed to stronger SIV-specific antibody responses in controller animals and maybe also promoted stronger SIV-specific T cell responses during chronic viremia.
To date, only three human HIV vaccine efficacy trials have been performed, and these were conducted in adults. The RV144 trial was the only HIV vaccine trial that showed some efficacy, but the effect was only transient. Although broadly neutralizing antibodies are considered the gold standard for preventing HIV acquisition, nonneutralizing antibodies were identified as correlates of protection in RV144 (37, 38). Env-specific plasma IgG binding antibodies, especially against the gp120 V1V2 loops, were inversely correlated with infection risk (39). The protective efficacy of these IgG antibodies was not associated with their avidity (39–43) but rather with FcR-mediated functions, such as ADCC (44). Conversely, high levels of gp120-specific plasma IgA were positively correlated with an increased risk of HIV infection in RV144 vaccine recipients (39) expressing a nonfunctional FcγRIIc receptor (45), which has been associated with reduced IgG/NK cell-mediated ADCC (46). Presumably, IgA interfered with IgG-mediated ADCC in these subjects (47).
Our pediatric p.o. M. tuberculosis-SIV/i.m. MVA-SIV vaccine strategy was able to induce high levels of Env-specific IgG in plasma, including V1V2-specific IgG antibodies. In animals with lower peak viremia and reduced viremia throughout the course of infection, gp140-specific plasma IgG antibodies had higher avidity than those of vaccinated animals with persistently high viremia. However, the functional capacity of these antibodies was limited. In particular, we could not detect ADCVI in assays with neutralization-resistant SIV, suggesting that ADCC antibodies were not generated in these animals. The failure to induce these antibodies could be due to inherent properties of the vaccine regimen and/or related to age-dependent immune ontogeny in infants. ADCC is thought to be mediated primarily by NK cells, although monocytes may also mediate this function (48). Regardless of which cell type is more critical for ADCC, both monocyte and NK cell functions in infants are known to be quantitatively and qualitatively different from those of adults (reviewed in reference 49). Thus, additional vaccine optimization may be needed to enhance the functional capacity of innate immune cells and thereby enhance adaptive immunity. Similarly, antigen-specific antibodies in infants have been described as deficient in affinity maturation and somatic hypermutation (50). This is consistent with the relatively low avidity of the SIV gp140-specific plasma IgG antibodies measured before challenge in our study. SIV/HIV vaccine studies in adult macaques have induced anti-Env plasma IgG antibodies with much higher avidity (51–54). The fact that vaccinated infants had maximal prechallenge avidity indices of 14, whereas protective vaccines in adults typically generate Env-specific IgG with avidity indices of 30, clearly suggests that there is a need to induce higher-avidity plasma antibodies in the infants.
Several SIV vaccine studies in adult macaques have highlighted the importance of mucosal IgA in protection against SIV infection (15, 20, 54–57), but the role of IgA in vaccine-mediated protection against HIV acquisition remains unclear. The fact that plasma IgA responses in the RV144 trial were positively correlated with HIV infection risk does not negate the potential role of mucosal IgA in the protection of HIV acquisition. In fact, monomeric plasma IgA and polymeric mucosal IgA exhibit distinct functions. In the studies described here, we observed a significant inverse relationship between SIV Env-specific mucosal IgA antibodies and acute viremia. Both vaccine-induced salivary and fecal SIV Env-specific IgA activities at the time of challenge inversely correlated with reduced acute viremia. In contrast, there was no correlation between plasma SIV Env-specific IgA and viremia. Furthermore, while SIV Env-specific salivary and fecal IgA antibodies strongly correlated with each other, the lack of a correlation between plasma and mucosal IgA demonstrated that mucosal IgA antibodies were produced locally and not due to transudation.
The importance of local IgA responses was highlighted in a recent study demonstrating that SIV DNA/MVA-vaccinated adult macaques protected against rectal challenge with SIV showed viral “blips,” but no persistent systemic viremia, when rechallenged with SIV repeatedly by the rectal route roughly 6 months later. Control of local virus replication was thought to be due to increased SIV Env-specific rectal IgA observed in these animals (58). Our finding that Env-specific fecal IgA levels further increased in our controller animals compared to noncontrollers is reminiscent of this finding. Due to the small volumes of saliva and stool samples, we could not evaluate potential SIV-specific mucosal IgG responses or the functional mechanisms responsible for IgA-mediated control of viremia. However, mucosal IgA antibodies could control replication of neutralization-resistant SIV in several ways. In the intestinal lamina propria or salivary glands, nonneutralizing Env-specific polymeric IgA could mediate clearance of complexed virions by binding to the polymeric immunoglobulin receptor on columnar epithelial cells and transporting virions to the lumen (59). Resident macrophages in the GI tract typically downregulate Fc receptors (60), but locally synthesized anti-Env IgA antibodies could mediate phagocytosis of SIV virions by FcRα+ neutrophils, monocytes, and dendritic cells recruited to infected tissues. Finally, HIV-specific IgA has been shown to be capable of mediating ADCC in the presence of blood monocytes (61), and it is likely that nonneutralizing IgA and neutrophils can also mediate this activity (62).
Importantly, recent human studies demonstrated that HIV-specific breast milk IgA levels could reduce the risk of HIV acquisition in human infants (63). A subsequent study by the same group showed that strong HIV Env-specific IgA responses could be induced in lactating macaques by vaccination (64). Our results provide evidence that protective mucosal IgA responses can also be induced by vaccination of the infant and thereby have the potential to provide long-term protection against viral acquisition compared to transient protection by maternal antibodies. The induction of potent mucosal IgA antibodies by our vaccine is a remarkable achievement considering the age-dependent development of mucosal IgA responses postbirth. Consistent with human infant antibody development, plasma and salivary IgA antibodies in infant macaques are barely detectable at birth and only slowly increase in the first 3 months. Thus, the fact that our vaccine was able to induce high mucosal IgA levels in some animals confirms the feasibility of developing an effective pediatric HIV vaccine.
The current study did not allow us to distinguish between the relative contribution of systemic IgG and mucosal IgA in the control of viremia. This is an important question to address in future studies. An elegant study by Ruprecht et al. showed that rectal administration of an anti-HIV V3 recombinant dimeric human IgA2 monoclonal antibody (MAb), combined with systemic administration of the parental IgG1 MAb, was able to completely protect adult macaques against rectal SHIV-1157ipEL challenge, whereas passive immunization with only IgG1 or dIgA2 protected only 0% and 17% of macaques, respectively (65).
Similarly, the results of this study highlight the potential importance of vaccine-elicited mucosal Env-specific IgA responses in combination with high-avidity systemic Env-specific IgG in protection against oral SIV transmission and control of viral replication in infant macaques. By gaining a deeper understanding of postnatal immune development, we may be able to design improved vaccine strategies to enhance systemic and mucosal SIV/HIV antibody responses.
ACKNOWLEDGMENTS
We are especially grateful to B. Moss and P. Earl for providing us with the MVA-SIV vaccine vectors, to Jeffrey Americo for growth and quality control of the vaccine stocks (Laboratory of Viral Diseases, National Institute of Allergy and Infectious Diseases, NIH, Bethesda, MD), and to Robert L. Wilson (LSUHSC) for technical assistance in processing secretions and performing ELISAs. In addition, we thank William J. Bosche and Michael Hull of the Quantitative Molecular Diagnostics Core of the AIDS and Cancer Virus Program, Frederick National Laboratory for Cancer Research, Frederick, MD, for their help in measuring SIV RNA and DNA in tissue samples. The SIV Gag and Env peptide pools were provided by the NIH AIDS Research and Reference Reagent Program (Division of AIDS, NIAID, NIH). M. tuberculosis antigens were provided by BEI Resources. In addition, we thank the staff of Colony Services, the Pathology, Veterinary Medicine, and Clinical Laboratory at the CNPRC.
The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, and mention of trade names, commercial products, or organizations does not imply endorsement by the U.S. Government.
Funding Statement
These studies were supported by NIH grants 1R01 DE019064 (to K.D.P., M.H.L., and G.F.) and T32 AI007419: Training in Virology and T32 AI007001-36: Training in Sexually Transmitted Diseases and AIDS (to K.J.), by the Louisiana Vaccine Center funded by the Louisiana Board of Regents (P.A.K.), and in part by federal funds from the National Cancer Institute, NIH, under contract no. HHSN261200800001E. The animal studies at the CNPRC were supported by NIH grant RR00169 from the National Center for Research Resources (NCRR; NIH) and Office of Research Infrastructure Programs grant OD P51 OD011107. Studies at the University of North Carolina (UNC) at Chapel Hill were supported by the Center for AIDS Research (CFAR; NIH grant 2 P30 AI050410), by the UNC Flow Cytometry Core that receives support from the Department of Microbiology and Immunology, by NCI Center Core support grant P30 CA06086 to the UNC Chapel Hill Lineberger Cancer Center, and by UNC OHARA (1 U01 AI068636-NIH/NIDCR). The MHC Genotyping Service is additionally supported by NIH grant 5R24RR016038, awarded to David Watkins at the University of Miami Miller School of Medicine.
REFERENCES
- 1.WHO. 2014. Global update on the health sector response to HIV, 2014. World Health Organization, Geneva, Switzerland. [Google Scholar]
- 2.Jensen K, Pena MG, Wilson RL, Ranganathan UD, Jacobs WR Jr, Fennelly G, Larsen M, Van Rompay KK, Kozlowski PA, Abel K. 2013. A neonatal oral-SIV prime/intramuscular MVA-SIV boost combination vaccine induces both SIV and -specific immune responses in infant macaques. Trials Vaccinol 2:53–63. doi: 10.1016/j.trivac.2013.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jensen K, Ranganathan UD, Van Rompay KK, Canfield DR, Khan I, Ravindran R, Luciw PA, Jacobs WR Jr, Fennelly G, Larsen MH, Abel K. 2012. A recombinant attenuated Mycobacterium tuberculosis vaccine strain is safe in immunosuppressed simian immunodeficiency virus-infected infant macaques. Clin Vaccine Immunol 19:1170–1181. doi: 10.1128/CVI.00184-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Van Rompay KK, Abel K, Lawson JR, Singh RP, Schmidt KA, Evans T, Earl P, Harvey D, Franchini G, Tartaglia J, Montefiori D, Hattangadi S, Moss B, Marthas ML. 2005. Attenuated poxvirus-based simian immunodeficiency virus (SIV) vaccines given in infancy partially protect infant and juvenile macaques against repeated oral challenge with virulent SIV. J Acquir Immune Defic Syndr 38:124–134. doi: 10.1097/00126334-200502010-00002. [DOI] [PubMed] [Google Scholar]
- 5.Marthas ML, Van Rompay KK, Abbott Z, Earl P, Buonocore-Buzzelli L, Moss B, Rose NF, Rose JK, Kozlowski PA, Abel K. 2011. Partial efficacy of a VSV-SIV/MVA-SIV vaccine regimen against oral SIV challenge in infant macaques. Vaccine 29:3124–3137. doi: 10.1016/j.vaccine.2011.02.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Van Rompay KK, Abel K, Earl P, Kozlowski PA, Easlick J, Moore J, Buonocore-Buzzelli L, Schmidt KA, Wilson RL, Simon I, Moss B, Rose N, Rose J, Marthas ML. 2010. Immunogenicity of viral vector, prime-boost SIV vaccine regimens in infant rhesus macaques: attenuated vesicular stomatitis virus (VSV) and modified vaccinia Ankara (MVA) recombinant SIV vaccines compared to live-attenuated SIV. Vaccine 28:1481–1492. doi: 10.1016/j.vaccine.2009.11.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Van Rompay KK, Greenier JL, Cole KS, Earl P, Moss B, Steckbeck JD, Pahar B, Rourke T, Montelaro RC, Canfield DR, Tarara RP, Miller C, McChesney MB, Marthas ML. 2003. Immunization of newborn rhesus macaques with simian immunodeficiency virus (SIV) vaccines prolongs survival after oral challenge with virulent SIVmac251. J Virol 77:179–190. doi: 10.1128/JVI.77.1.179-190.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baba TW, Liska V, Khimani AH, Ray NB, Dailey PJ, Penninck D, Bronson R, Greene MF, McClure HM, Martin LN, Ruprecht RM. 1999. Live attenuated, multiply deleted simian immunodeficiency virus causes AIDS in infant and adult macaques. Nat Med 5:194–203. doi: 10.1038/5557. [DOI] [PubMed] [Google Scholar]
- 9.National Research Council. 2011. Guide for the care and use of laboratory animals, 8th ed National Academies Press, Washington, DC. [Google Scholar]
- 10.Van Rompay KK, Singh RP, Brignolo LL, Lawson JR, Schmidt KA, Pahar B, Canfield DR, Tarara RP, Sodora DL, Bischofberger N, Marthas ML. 2004. The clinical benefits of tenofovir for simian immunodeficiency virus-infected macaques are larger than predicted by its effects on standard viral and immunologic parameters. J Acquir Immune Defic Syndr 36:900–914. doi: 10.1097/00126334-200408010-00003. [DOI] [PubMed] [Google Scholar]
- 11.Cline AN, Bess JW, Piatak M Jr, Lifson JD. 2005. Highly sensitive SIV plasma viral load assay: practical considerations, realistic performance expectations, and application to reverse engineering of vaccines for AIDS. J Med Primatol 34:303–312. doi: 10.1111/j.1600-0684.2005.00128.x. [DOI] [PubMed] [Google Scholar]
- 12.Hansen SG, Ford JC, Lewis MS, Ventura AB, Hughes CM, Coyne-Johnson L, Whizin N, Oswald K, Shoemaker R, Swanson T, Legasse AW, Chiuchiolo MJ, Parks CL, Axthelm MK, Nelson JA, Jarvis MA, Piatak M Jr, Lifson JD, Picker LJ. 2011. Profound early control of highly pathogenic SIV by an effector memory T-cell vaccine. Nature 473:523–527. doi: 10.1038/nature10003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hansen SG, Piatak M Jr, Ventura AB, Hughes CM, Gilbride RM, Ford JC, Oswald K, Shoemaker R, Li Y, Lewis MS, Gilliam AN, Xu G, Whizin N, Burwitz BJ, Planer SL, Turner JM, Legasse AW, Axthelm MK, Nelson JA, Fruh K, Sacha JB, Estes JD, Keele BF, Edlefsen PT, Lifson JD, Picker LJ. 2013. Immune clearance of highly pathogenic SIV infection. Nature 502:100–104. doi: 10.1038/nature12519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, Thierer T, Ashton B, Meintjes P, Drummond A. 2012. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28:1647–1649. doi: 10.1093/bioinformatics/bts199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Manrique M, Kozlowski PA, Cobo-Molinos A, Wang SW, Wilson RL, Montefiori DC, Mansfield KG, Carville A, Aldovini A. 2011. Long-term control of simian immunodeficiency virus mac251 viremia to undetectable levels in half of infected female rhesus macaques nasally vaccinated with simian immunodeficiency virus DNA/recombinant modified vaccinia virus Ankara. J Immunol 186:3581–3593. doi: 10.4049/jimmunol.1002594. [DOI] [PubMed] [Google Scholar]
- 16.Kodama T, Mori K, Kawahara T, Ringler DJ, Desrosiers RC. 1993. Analysis of simian immunodeficiency virus sequence variation in tissues of rhesus macaques with simian AIDS. J Virol 67:6522–6534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kozlowski PA, Lynch RM, Patterson RR, Cu-Uvin S, Flanigan TP, Neutra MR. 2000. Modified wick method using Weck-Cel sponges for collection of human rectal secretions and analysis of mucosal HIV antibody. J Acquir Immune Defic Syndr 24:297–309. [DOI] [PubMed] [Google Scholar]
- 18.Kwa S, Sadagopal S, Shen X, Hong JJ, Gangadhara S, Basu R, Victor B, Iyer SS, LaBranche CC, Montefiori DC, Tomaras GD, Villinger F, Moss B, Kozlowski PA, Amara RR. 2015. CD40L-adjuvanted DNA/modified vaccinia virus Ankara simian immunodeficiency virus (SIV) vaccine enhances protection against neutralization-resistant mucosal SIV infection. J Virol 89:4690–4695. doi: 10.1128/JVI.03527-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Montefiori D. 2004. Evaluating neutralizing antibodies against HIV, SIV and SHIV in luciferase reporter gene assays. Curr Protoc Immunol Chapter 12:Unit 12.11. [DOI] [PubMed] [Google Scholar]
- 20.Manrique M, Kozlowski PA, Cobo-Molinos A, Wang SW, Wilson RL, Martinez-Viedma Mdel P, Montefiori DC, Carville A, Aldovini A. 2014. Resistance to infection, early and persistent suppression of simian immunodeficiency virus SIVmac251 viremia, and significant reduction of tissue viral burden after mucosal vaccination in female rhesus macaques. J Virol 88:212–224. doi: 10.1128/JVI.02523-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Forthal DN, Gilbert PB, Landucci G, Phan T. 2007. Recombinant gp120 vaccine-induced antibodies inhibit clinical strains of HIV-1 in the presence of Fc receptor-bearing effector cells and correlate inversely with HIV infection rate. J Immunol 178:6596–6603. doi: 10.4049/jimmunol.178.10.6596. [DOI] [PubMed] [Google Scholar]
- 22.Wehrly K, Chesebro B. 1997. p24 antigen capture assay for quantification of human immunodeficiency virus using readily available inexpensive reagents. Methods 12:288–293. doi: 10.1006/meth.1997.0481. [DOI] [PubMed] [Google Scholar]
- 23.Ackerman ME, Moldt B, Wyatt RT, Dugast AS, McAndrew E, Tsoukas S, Jost S, Berger CT, Sciaranghella G, Liu Q, Irvine DJ, Burton DR, Alter G. 2011. A robust, high-throughput assay to determine the phagocytic activity of clinical antibody samples. J Immunol Methods 366:8–19. doi: 10.1016/j.jim.2010.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kaizu M, Borchardt GJ, Glidden CE, Fisk DL, Loffredo JT, Watkins DI, Rehrauer WM. 2007. Molecular typing of major histocompatibility complex class I alleles in the Indian rhesus macaque which restrict SIV CD8+ T cell epitopes. Immunogenetics 59:693–703. doi: 10.1007/s00251-007-0233-7. [DOI] [PubMed] [Google Scholar]
- 25.Loffredo JT, Friedrich TC, Leon EJ, Stephany JJ, Rodrigues DS, Spencer SP, Bean AT, Beal DR, Burwitz BJ, Rudersdorf RA, Wallace LT, Piaskowski SM, May GE, Sidney J, Gostick E, Wilson NA, Price DA, Kallas EG, Piontkivska H, Hughes AL, Sette A, Watkins DI. 2007. CD8+ T cells from SIV elite controller macaques recognize Mamu-B*08-bound epitopes and select for widespread viral variation. PLoS One 2:e1152. doi: 10.1371/journal.pone.0001152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Loffredo JT, Maxwell J, Qi Y, Glidden CE, Borchardt GJ, Soma T, Bean AT, Beal DR, Wilson NA, Rehrauer WM, Lifson JD, Carrington M, Watkins DI. 2007. Mamu-B*08-positive macaques control simian immunodeficiency virus replication. J Virol 81:8827–8832. doi: 10.1128/JVI.00895-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yant LJ, Friedrich TC, Johnson RC, May GE, Maness NJ, Enz AM, Lifson JD, O'Connor DH, Carrington M, Watkins DI. 2006. The high-frequency major histocompatibility complex class I allele Mamu-B*17 is associated with control of simian immunodeficiency virus SIVmac239 replication. J Virol 80:5074–5077. doi: 10.1128/JVI.80.10.5074-5077.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Giraldo-Vela JP, Rudersdorf R, Chung C, Qi Y, Wallace LT, Bimber B, Borchardt GJ, Fisk DL, Glidden CE, Loffredo JT, Piaskowski SM, Furlott JR, Morales-Martinez JP, Wilson NA, Rehrauer WM, Lifson JD, Carrington M, Watkins DI. 2008. The major histocompatibility complex class II alleles Mamu-DRB1*1003 and -DRB1*0306 are enriched in a cohort of simian immunodeficiency virus-infected rhesus macaque elite controllers. J Virol 82:859–870. doi: 10.1128/JVI.01816-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Loffredo JT, Sidney J, Piaskowski S, Szymanski A, Furlott J, Rudersdorf R, Reed J, Peters B, Hickman-Miller HD, Bardet W, Rehrauer WM, O'Connor DH, Wilson NA, Hildebrand WH, Sette A, Watkins DI. 2005. The high frequency Indian rhesus macaque MHC class I molecule, Mamu-B*01, does not appear to be involved in CD8+ T lymphocyte responses to SIVmac239. J Immunol 175:5986–5997. doi: 10.4049/jimmunol.175.9.5986. [DOI] [PubMed] [Google Scholar]
- 30.Kirmaier A, Wu F, Newman RM, Hall LR, Morgan JS, O'Connor S, Marx PA, Meythaler M, Goldstein S, Buckler-White A, Kaur A, Hirsch VM, Johnson WE. 2010. TRIM5 suppresses cross-species transmission of a primate immunodeficiency virus and selects for emergence of resistant variants in the new species. PLoS Biol 8:e1000462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lim SY, Chan T, Gelman RS, Whitney JB, O'Brien KL, Barouch DH, Goldstein DB, Haynes BF, Letvin NL. 2010. Contributions of Mamu-A*01 status and TRIM5 allele expression, but not CCL3L copy number variation, to the control of SIVmac251 replication in Indian-origin rhesus monkeys. PLoS Genet 6:e1000997. doi: 10.1371/journal.pgen.1000997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lim SY, Rogers T, Chan T, Whitney JB, Kim J, Sodroski J, Letvin NL. 2010. TRIM5alpha modulates immunodeficiency virus control in rhesus monkeys. PLoS Pathog 6:e1000738. doi: 10.1371/journal.ppat.1000738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hudgens MG, Gilbert PB. 2009. Assessing vaccine effects in repeated low-dose challenge experiments. Biometrics 65:1223–1232. doi: 10.1111/j.1541-0420.2009.01189.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hudgens MG, Gilbert PB, Mascola JR, Wu CD, Barouch DH, Self SG. 2009. Power to detect the effects of HIV vaccination in repeated low-dose challenge experiments. J Infect Dis 200:609–613. doi: 10.1086/600891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nolen TL, Hudgens MG, Senb PK, Koch GG. 2015. Analysis of repeated low-dose challenge studies. Stat Med 34:1981–1992. doi: 10.1002/sim.6462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fenizia C, Keele BF, Nichols D, Cornara S, Binello N, Vaccari M, Pegu P, Robert-Guroff M, Ma ZM, Miller CJ, Venzon D, Hirsch V, Franchini G. 2011. TRIM5alpha does not affect simian immunodeficiency virus SIV(mac251) replication in vaccinated or unvaccinated Indian rhesus macaques following intrarectal challenge exposure. J Virol 85:12399–12409. doi: 10.1128/JVI.05707-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rerks-Ngarm S, Paris RM, Chunsutthiwat S, Premsri N, Namwat C, Bowonwatanuwong C, Li SS, Kaewkungkal J, Trichavaroj R, Churikanont N, de Souza MS, Andrews C, Francis D, Adams E, Flores J, Gurunathan S, Tartaglia J, O'Connell RJ, Eamsila C, Nitayaphan S, Ngauy V, Thongcharoen P, Kunasol P, Michael NL, Robb ML, Gilbert PB, Kim JH. 2013. Extended evaluation of the virologic, immunologic, and clinical course of volunteers who acquired HIV-1 infection in a phase III vaccine trial of ALVAC-HIV and AIDSVAX B/E. J Infect Dis 207:1195–1205. doi: 10.1093/infdis/jis478. [DOI] [PubMed] [Google Scholar]
- 38.Rerks-Ngarm S, Pitisuttithum P, Nitayaphan S, Kaewkungwal J, Chiu J, Paris R, Premsri N, Namwat C, de Souza M, Adams E, Benenson M, Gurunathan S, Tartaglia J, McNeil JG, Francis DP, Stablein D, Birx DL, Chunsuttiwat S, Khamboonruang C, Thongcharoen P, Robb ML, Michael NL, Kunasol P, Kim JH, MOPH-TAVEG Investigators . 2009. Vaccination with ALVAC and AIDSVAX to prevent HIV-1 infection in Thailand. N Engl J Med 361:2209–2220. doi: 10.1056/NEJMoa0908492. [DOI] [PubMed] [Google Scholar]
- 39.Haynes BF, Gilbert PB, McElrath MJ, Zolla-Pazner S, Tomaras GD, Alam SM, Evans DT, Montefiori DC, Karnasuta C, Sutthent R, Liao HX, DeVico AL, Lewis GK, Williams C, Pinter A, Fong Y, Janes H, DeCamp A, Huang Y, Rao M, Billings E, Karasavvas N, Robb ML, Ngauy V, de Souza MS, Paris R, Ferrari G, Bailer RT, Soderberg KA, Andrews C, Berman PW, Frahm N, De Rosa SC, Alpert MD, Yates NL, Shen X, Koup RA, Pitisuttithum P, Kaewkungwal J, Nitayaphan S, Rerks-Ngarm S, Michael NL, Kim JH. 2012. Immune-correlates analysis of an HIV-1 vaccine efficacy trial. N Engl J Med 366:1275–1286. doi: 10.1056/NEJMoa1113425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liao HX, Bonsignori M, Alam SM, McLellan JS, Tomaras GD, Moody MA, Kozink DM, Hwang KK, Chen X, Tsao CY, Liu P, Lu X, Parks RJ, Montefiori DC, Ferrari G, Pollara J, Rao M, Peachman KK, Santra S, Letvin NL, Karasavvas N, Yang ZY, Dai K, Pancera M, Gorman J, Wiehe K, Nicely NI, Rerks-Ngarm S, Nitayaphan S, Kaewkungwal J, Pitisuttithum P, Tartaglia J, Sinangil F, Kim JH, Michael NL, Kepler TB, Kwong PD, Mascola JR, Nabel GJ, Pinter A, Zolla-Pazner S, Haynes BF. 2013. Vaccine induction of antibodies against a structurally heterogeneous site of immune pressure within HIV-1 envelope protein variable regions 1 and 2. Immunity 38:176–186. doi: 10.1016/j.immuni.2012.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zolla-Pazner S, deCamp AC, Cardozo T, Karasavvas N, Gottardo R, Williams C, Morris DE, Tomaras G, Rao M, Billings E, Berman P, Shen X, Andrews C, O'Connell RJ, Ngauy V, Nitayaphan S, de Souza M, Korber B, Koup R, Bailer RT, Mascola JR, Pinter A, Montefiori D, Haynes BF, Robb ML, Rerks-Ngarm S, Michael NL, Gilbert PB, Kim JH. 2013. Analysis of V2 antibody responses induced in vaccinees in the ALVAC/AIDSVAX HIV-1 vaccine efficacy trial. PLoS One 8:e53629. doi: 10.1371/journal.pone.0053629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zolla-Pazner S, Edlefsen PT, Rolland M, Kong XP, deCamp A, Gottardo R, Williams C, Tovanabutra S, Sharpe-Cohen S, Mullins JI, deSouza MS, Karasavvas N, Nitayaphan S, Rerks-Ngarm S, Pitisuttihum P, Kaewkungwal J, O'Connell RJ, Robb ML, Michael NL, Kim JH, Gilbert P. 2014. Vaccine-induced human antibodies specific for the third variable region of HIV-1 gp120 impose immune pressure on infecting viruses. EBioMedicine 1:37–45. doi: 10.1016/j.ebiom.2014.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gottardo R, Bailer RT, Korber BT, Gnanakaran S, Phillips J, Shen X, Tomaras GD, Turk E, Imholte G, Eckler L, Wenschuh H, Zerweck J, Greene K, Gao H, Berman PW, Francis D, Sinangil F, Lee C, Nitayaphan S, Rerks-Ngarm S, Kaewkungwal J, Pitisuttithum P, Tartaglia J, Robb ML, Michael NL, Kim JH, Zolla-Pazner S, Haynes BF, Mascola JR, Self S, Gilbert P, Montefiori DC. 2013. Plasma IgG to linear epitopes in the V2 and V3 regions of HIV-1 gp120 correlate with a reduced risk of infection in the RV144 vaccine efficacy trial. PLoS One 8:e75665. doi: 10.1371/journal.pone.0075665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu P, Yates NL, Shen X, Bonsignori M, Moody MA, Liao HX, Fong Y, Alam SM, Overman RG, Denny T, Ferrari G, Ochsenbauer C, Kappes JC, Polonis VR, Pitisuttithum P, Kaewkungwal J, Nitayaphan S, Rerks-Ngarm S, Montefiori DC, Gilbert P, Michael NL, Kim JH, Haynes BF, Tomaras GD. 2013. Infectious virion capture by HIV-1 gp120-specific IgG from RV144 vaccinees. J Virol 87:7828–7836. doi: 10.1128/JVI.02737-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li SS, Gilbert PB, Tomaras GD, Kijak G, Ferrari G, Thomas R, Pyo CW, Zolla-Pazner S, Montefiori D, Liao HX, Nabel G, Pinter A, Evans DT, Gottardo R, Dai JY, Janes H, Morris D, Fong Y, Edlefsen PT, Li F, Frahm N, Alpert MD, Prentice H, Rerks-Ngarm S, Pitisuttithum P, Kaewkungwal J, Nitayaphan S, Robb ML, O'Connell RJ, Haynes BF, Michael NL, Kim JH, McElrath MJ, Geraghty DE. 2014. FCGR2C polymorphisms associate with HIV-1 vaccine protection in RV144 trial. J Clin Investig 124:3879–3890. doi: 10.1172/JCI75539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.van der Heijden J, Breunis WB, Geissler J, de Boer M, van den Berg TK, Kuijpers TW. 2012. Phenotypic variation in IgG receptors by nonclassical FCGR2C alleles. J Immunol 188:1318–1324. doi: 10.4049/jimmunol.1003945. [DOI] [PubMed] [Google Scholar]
- 47.Tomaras GD, Ferrari G, Shen X, Alam SM, Liao HX, Pollara J, Bonsignori M, Moody MA, Fong Y, Chen X, Poling B, Nicholson CO, Zhang R, Lu X, Parks R, Kaewkungwal J, Nitayaphan S, Pitisuttithum P, Rerks-Ngarm S, Gilbert PB, Kim JH, Michael NL, Montefiori DC, Haynes BF. 2013. Vaccine-induced plasma IgA specific for the C1 region of the HIV-1 envelope blocks binding and effector function of IgG. Proc Natl Acad Sci U S A 110:9019–9024. doi: 10.1073/pnas.1301456110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Forthal DN, Landucci G, Cole KS, Marthas M, Becerra JC, Van Rompay K. 2006. Rhesus macaque polyclonal and monoclonal antibodies inhibit simian immunodeficiency virus in the presence of human or autologous rhesus effector cells. J Virol 80:9217–9225. doi: 10.1128/JVI.02746-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.PrabhuDas M, Adkins B, Gans H, King C, Levy O, Ramilo O, Siegrist CA. 2011. Challenges in infant immunity: implications for responses to infection and vaccines. Nat Immunol 12:189–194. doi: 10.1038/ni0311-189. [DOI] [PubMed] [Google Scholar]
- 50.Ridings J, Nicholson IC, Goldsworthy W, Haslam R, Roberton DM, Zola H. 1997. Somatic hypermutation of immunoglobulin genes in human neonates. Clin Exp Immunol 108:366–374. doi: 10.1046/j.1365-2249.1997.3631264.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Manrique M, Micewicz E, Kozlowski PA, Wang SW, Aurora D, Wilson RL, Ghebremichael M, Mazzara G, Montefiori D, Carville A, Mansfield KG, Aldovini A. 2008. DNA-MVA vaccine protection after X4 SHIV challenge in macaques correlates with day-of-challenge antiviral CD4+ cell-mediated immunity levels and postchallenge preservation of CD4+ T cell memory. AIDS Res Hum Retrovir 24:505–519. doi: 10.1089/aid.2007.0191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhou Q, Hidajat R, Peng B, Venzon D, Aldrich MK, Richardson E, Lee EM, Kalyanaraman VS, Grimes G, Gomez-Roman VR, Summers LE, Malkevich N, Robert-Guroff M. 2007. Comparative evaluation of oral and intranasal priming with replication-competent adenovirus 5 host range mutant (Ad5hr)-simian immunodeficiency virus (SIV) recombinant vaccines on immunogenicity and protective efficacy against SIV(mac251). Vaccine 25:8021–8035. doi: 10.1016/j.vaccine.2007.09.017. [DOI] [PubMed] [Google Scholar]
- 53.Pegu P, Vaccari M, Gordon S, Keele BF, Doster M, Guan Y, Ferrari G, Pal R, Ferrari MG, Whitney S, Hudacik L, Billings E, Rao M, Montefiori D, Tomaras G, Alam SM, Fenizia C, Lifson JD, Stablein D, Tartaglia J, Michael N, Kim J, Venzon D, Franchini G. 2013. Antibodies with high avidity to the gp120 envelope protein in protection from simian immunodeficiency virus SIV(mac251) acquisition in an immunization regimen that mimics the RV-144 Thai trial. J Virol 87:1708–1719. doi: 10.1128/JVI.02544-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xiao P, Patterson LJ, Kuate S, Brocca-Cofano E, Thomas MA, Venzon D, Zhao J, DiPasquale J, Fenizia C, Lee EM, Kalisz I, Kalyanaraman VS, Pal R, Montefiori D, Keele BF, Robert-Guroff M. 2012. Replicating adenovirus-simian immunodeficiency virus (SIV) recombinant priming and envelope protein boosting elicits localized, mucosal IgA immunity in rhesus macaques correlated with delayed acquisition following a repeated low-dose rectal SIV(mac251) challenge. J Virol 86:4644–4657. doi: 10.1128/JVI.06812-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Manrique M, Kozlowski PA, Cobo-Molinos A, Wang SW, Wilson RL, Montefiori DC, Carville A, Aldovini A. 2013. Immunogenicity of a vaccine regimen composed of simian immunodeficiency virus DNA, rMVA, and viral particles administered to female rhesus macaques via four different mucosal routes. J Virol 87:4738–4750. doi: 10.1128/JVI.03531-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Manrique M, Kozlowski PA, Wang SW, Wilson RL, Micewicz E, Montefiori DC, Mansfield KG, Carville A, Aldovini A. 2009. Nasal DNA-MVA SIV vaccination provides more significant protection from progression to AIDS than a similar intramuscular vaccination. Mucosal Immunol 2:536–550. doi: 10.1038/mi.2009.103. [DOI] [PubMed] [Google Scholar]
- 57.Xiao P, Zhao J, Patterson LJ, Brocca-Cofano E, Venzon D, Kozlowski PA, Hidajat R, Demberg T, Robert-Guroff M. 2010. Multiple vaccine-elicited nonneutralizing antienvelope antibody activities contribute to protective efficacy by reducing both acute and chronic viremia following simian/human immunodeficiency virus SHIV89.6P challenge in rhesus macaques. J Virol 84:7161–7173. doi: 10.1128/JVI.00410-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kannanganant S, Gangadhara S, Lai L, Lawson B, Kozlowski PA, Robinson HL, Amara RR. 2014. Local control of repeated-dose rectal challenges in DNA/MVA-vaccinated macaques protected against a first series of simian immunodeficiency virus challenges. J Virol 88:5864–5869. doi: 10.1128/JVI.00145-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Corthesy B. 2013. Multi-faceted functions of secretory IgA at mucosal surfaces. Front Immunol 4:185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Smith PD, Smythies LE, Shen R, Greenwell-Wild T, Gliozzi M, Wahl SM. 2011. Intestinal macrophages and response to microbial encroachment. Mucosal Immunol 4:31–42. doi: 10.1038/mi.2010.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Black KP, Cummins JE Jr, Jackson S. 1996. Serum and secretory IgA from HIV-infected individuals mediate antibody-dependent cellular cytotoxicity. Clin Immunol Immunopathol 81:182–190. doi: 10.1006/clin.1996.0175. [DOI] [PubMed] [Google Scholar]
- 62.Duval M, Posner MR, Cavacini LA. 2008. A bispecific antibody composed of a nonneutralizing antibody to the gp41 immunodominant region and an anti-CD89 antibody directs broad human immunodeficiency virus destruction by neutrophils. J Virol 82:4671–4674. doi: 10.1128/JVI.02499-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pollara J, McGuire E, Fouda GG, Rountree W, Eudailey J, Overman RG, Seaton KE, Deal A, Edwards RW, Tegha G, Kamwendo D, Kumwenda J, Nelson JA, Liao HX, Brinkley C, Denny TN, Ochsenbauer C, Ellington S, King CC, Jamieson DJ, van der Horst C, Kourtis AP, Tomaras GD, Ferrari G, Permar SR. 2015. Association of HIV-1 envelope-specific breast milk IgA responses with reduced risk of postnatal mother-to-child transmission of HIV-1. J Virol 89:9952–9961. doi: 10.1128/JVI.01560-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nelson CS, Pollara J, Kunz EL, Jeffries TL Jr, Duffy R, Beck C, Stamper L, Wang M, Shen X, Pickup DJ, Staats HF, Hudgens MG, Kepler TB, Montefiori DC, Moody MA, Tomaras GD, Liao HX, Haynes BF, Ferrari G, Fouda GG, Permar SR. 2016. Combined HIV-1 envelope systemic and mucosal immunization of lactating rhesus monkeys induces a robust immunoglobulin A isotype B cell response in breast milk. J Virol 90:4951–4965. doi: 10.1128/JVI.00335-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sholukh AM, Watkins JD, Vyas HK, Gupta S, Lakhashe SK, Thorat S, Zhou M, Hemashettar G, Bachler BC, Forthal DN, Villinger F, Sattentau QJ, Weiss RA, Agatic G, Corti D, Lanzavecchia A, Heeney JL, Ruprecht RM. 2015. Defense-in-depth by mucosally administered anti-HIV dimeric IgA2 and systemic IgG1 mAbs: complete protection of rhesus monkeys from mucosal SHIV challenge. Vaccine 33:2086–2095. doi: 10.1016/j.vaccine.2015.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]