

Abstract
Background Adamantanes resistance in H3N2 viruses has been increasing since 2000, and in 2005–2006 reached nearly 100% in most countries, with the circulation of the N‐lineage. In 2006–2007, however, a significant decrease in resistance was observed in many regions.
Objectives To explore potential links between adamantane resistance and the A(H3N2) viruses that circulated between 2006 and 2008.
Methods A total of 1451 Influenza A (H3N2) viruses collected globally in 2001–2008 were screened for the presence of adamantane resistance markers. A subset of 100 viruses representing the broad genetic and geographic spectrum of these viruses was selected for complete genome sequencing and phylogenetic analyses.
Results Full genome sequence analysis of 2006–2007 viruses revealed co‐circulation of four distinct genotypes, designated A–D. Phylogenetic analyses demonstrated reassortment between viruses from the N‐lineage and other viruses that had circulated in prior seasons, including those bearing an adamantane sensitive marker. Genotype D viruses became dominant in late 2006–2007 and continued to be the main H3N2 genotype in 2007–2008. Viruses of this genotype retained all N‐lineage genome segments except PB2 and NP, which were acquired through reassortment.
Conclusions The decrease in adamantane resistance at that time was due to transient co‐circulation of genotypes that emerged through reassortment. Our findings emphasize the importance of complete genome sequencing in understanding the complex nature of the relationship between influenza virus evolution and antiviral resistance. The recent emergence of the pandemic multi‐reassortant H1N1 virus underscores the importance of whole genome sequence monitoring for rapid detection of such unusual and novel strains.
Keywords: Adamantanes, genome, genotype, phylogenetic analysis, resistance
Introduction
Influenza A viruses are respiratory pathogens that cause annual epidemics and occasional pandemics. The genome of influenza A viruses consists of eight segments of negative sense RNA with each segment encoding one (PB2, PA, HA, NP, and NA) or more (PB1, M, and NS) viral proteins. Influenza A viruses have two major surface antigens, the hemagglutinin (HA) and neuraminidase (NA) glycoproteins. Evolution of influenza viruses is thought to be predominantly driven by a host‐mediated selective pressure that leads to changes at key antigenic sites, a mechanism known as antigenic drift. 1 , 2 A second process, known as antigenic shift, also contributes to the evolution of influenza viruses, though at a much lower frequency. Intra‐subtype segment reassortment is a recurring feature of influenza virus evolution and has contributed to major antigenic changes in both influenza A(H3N2) 3 and A(H1N1). 4
In addition to vaccines, antiviral drugs provide valuable means for the management of influenza infections. Two classes of antiviral drugs, M2 blockers (amantadine and rimantadine) and NA inhibitors (oseltamivir and zanamivir), have been approved for use in the USA by the Food and Drug Administration in the prevention and control of influenza A virus infections. Resistance, intrinsic or acquired through drug selective pressure, can lessen effectiveness of antiviral drugs and thus are a cause for significant public health concern.
The M2 ion channel blockers amantadine and rimantadine, known collectively as adamantanes, were the first antiviral drugs available for prophylaxis and treatment of influenza A infections. Until recently, they have also been the most widely used anti‐influenza drugs due to their low cost and over‐the‐counter availability in some countries. The molecular basis underlying resistance to this class of drugs has been well characterized. Adamantanes inhibit virus replication by blocking the proton channel formed by the M2 protein. 5 , 6 , 7 An amino acid change at any one of five residues in the transmembrane domain of this protein (positions 26, 27, 30, 31, or 34) confers cross‐resistance to amantadine and rimantadine. 5 , 8 , 9 , 10 Despite rapid emergence of drug‐resistant mutants following treatment with adamantanes and the ability of drug‐resistant mutants to transmit, 11 the incidence of adamantane resistance among viruses circulating in communities had been low (<1%) for decades. 12 , 13 , 14 , 15 , 16 At CDC, representative community isolates collected in various geographic regions during a given influenza season are tested for antiviral resistance. A dramatic rise in the frequency of adamantane resistance associated with an S31N mutation in the M2 protein was first reported for influenza A(H3N2) viruses circulating in Asia in 2000 17 which was followed by the spread of resistant A(H3N2) viruses to different parts of the world in subsequent seasons. 18 , 19 , 20 , 21 , 22 , 23 Because of this, CDC recommended that adamantanes not be used for prophylaxis or treatment of influenza infections in the USA. 24
The genesis of adamantane resistance in influenza A(H3N2) viruses and mechanisms underlying their global spread have been the subject of intense scientific investigation. Adamantanes used locally during the SARS and avian flu scare in Asia might have contributed to the initial emergence of drug resistant virus variants in recent years; however, there is no evidence supporting the role of adamantanes usage in maintenance of adamantane resistant A(H3N2) viruses in human populations. Simonsen et al. 25 described possible evolutionary mechanisms, unrelated to drug selective pressure, which could lead to the globally increasing frequency of adamantane resistance in A(H3N2) influenza viruses. Based on a full genome phylogenetic survey, Simonsen et al. 25 concluded that the resistance‐conferring S31N mutation did not come to dominance until it was put into a suitable genetic background via segment reassortment. This event led to the genesis of the adamantane‐resistant ‘N‐lineage’. Viruses from this lineage circulated widely in Asia in 2005, before dominating in the Northern hemisphere during 2005–2006 season in a number of countries, including the USA. During the 2005–2006 influenza season, the frequency of drug resistance reached 100% among viruses circulating in several countries and was estimated at 96·4% in the USA. 21 In the following season, the frequency of resistance was reduced in several but not all countries, for instance, it was estimated at 30·1% in Canada and 86·3% in the USA. 16 , 26 , 27 In addition to the variance in frequency of resistance, we observed co‐circulation of several antigenic variants identified by HA inhibition assay (data not shown). The present study is aimed at investigating whether the decreased frequency in resistance among A(H3N2) viruses that circulated during 2006–2007 season was associated with the appearance of new genetic groups distinct from the N‐lineage. The study also provides further insight into the nature of beneficial mutations responsible for the enhanced fitness of the adamantane‐resistant A(H3N2) viruses and the genomic events underlying the genesis of these viruses.
Materials and methods
Virus isolates
A total of 1451 Influenza A (H3N2) viruses collected from 1 October 2006 through 30 September 2007 in the USA and other countries were submitted to the WHO Collaborating Center for Surveillance, Epidemiology and Control of Influenza at the CDC, Atlanta, GA, USA. The viruses were screened for the presence of adamantane resistance markers using pyrosequencing as described elsewhere by Bright et al. 18
Initially, 54 A(H3N2) were selected from a larger sample of viruses based on their genetic and geographic diversity. Theses viruses that co‐circulated globally in the 2006–2007 season were chosen for HA1 gene sequencing in addition to 10 A(H3N2) reference viruses, representing vaccine strains as well as other historic viruses from previous seasons. Also, 36 A(H3N2) viruses collected in different seasons from various geographic locations, with complete genome sequences available from public domain databases were included for comparison. From the early 2007–2008 season, 55 viruses that were available at the time when the study was undertaken were used for HA1 and NA sequence analysis for comparison with the set of 64 viruses from previous seasons. For further detailed genetic analyses, complete genome sequencing was performed on the 64 viruses from the 2001–2007 season as well as 11 representative A(H3N2) viruses from the 2007–2008 season (details on viruses sequenced during this study is provided in Table S3). The 11 viruses from the 2007–2008 season were selected from the sample of 55 viruses to represent the wide range of the geographic diversity of these viruses.
Phylogenetic analysis
Phylogenetic analyses were performed using the Genetic Algorithm for Rapid Likelihood Inference (garli 0·96b7), based on General Time Reversible (GTR) + I + γ4 substitution model 28 and the tree analyzed in paup. 29
Results
Reduction in adamantane resistance prevalence: season 2006–2007
As a part of an ongoing strain surveillance activity, a total of 1451 A(H3N2) viruses circulating during 2006–2007 influenza season were tested for adamantane resistance. A pyrosequencing method was used to detect the presence of known markers of adamantane resistance in the M segment (M2 protein). The virus isolates originated from 46 countries with the majority from the USA, South America, and Asia (Table 1). The resistance‐conferring mutation S31N was detected in 73·3% of the viruses collected globally, which reflects a substantial decrease compared to the overall frequency reported for the previous 2005–2006 influenza season. 21 A decline in adamantane resistance was especially noticeable in several geographic areas such as Hong Kong and Canada (from 100% to 69·2%, and 100% to 30·1%, respectively). In the USA, the decrease in resistance between the two seasons was less pronounced [from 96·4% (n = 789) to 86·3% (n = 401)] and remained nearly unchanged for the isolates received from South America and Europe (mainly Russia and Ukraine). The re‐emergence of adamantane sensitive viruses in certain geographic areas in 2006–2007 prompted us to determine whether they arose from gene reassortment or from a reversion mutation in the M gene.
Table 1.
Prevalence of resistance to adamantanes among A(H3N2) viruses collected globally during the 2006–2007 influenza season (1 October 2006 through 30 September 2007)
| Geographic location | No. viruses tested | No. viruses resistant | Resistance (%) |
|---|---|---|---|
| Africa (total) | 32 | 7 | 21·9 |
| Asia (total) | 363 | 228 | 62·8 |
| China | 69 | 69 | 100 |
| Hong Kong (SAR) | 13 | 9 | 69·2 |
| Japan | 21 | 18 | 85·7 |
| Asia (other) | 159 | 76 | 47·8 |
| Europe (total) | 100 | 39 | 39·0 |
| North America (total) | 588 | 429 | 72·9 |
| Canada | 143 | 43 | 30·1 |
| Mexico | 44 | 40 | 90·9 |
| United States | 401 | 346 | 86·3 |
| South America (total) | 363 | 357 | 98·3 |
| Oceania (total) | 5 | 4 | 80·0 |
| Overall | 1451 | 1064 | 73·3 |
Data represents the number of total tested isolates compared with the number of resistant isolates. Africa: Egypt, Kenya, Madagascar, Mauritius, and Uganda; Other Asia: Bangladesh, India, Kazakhstan, Korea, Mongolia, Nepal, Taiwan, Thailand, and Vietnam; Europe: Belarus, Bulgaria, Croatia, Denmark, France, Greece, Italy, Russia, Spain, Ukraine and United Kingdom; South America: Argentina, Brazil, Chile, Costa‐Rica, Dominican Republic, El Salvador, French Guyana, Guadeloupe, Honduras, Panama, Peru, Trinidad‐Tobago, and Uruguay; Oceania: Australia and New Caledonia.
Phylogenetic relationships among viruses co‐circulating in 2006–2007
To test this hypothesis, full genomes of 100 viruses isolated globally from 2001 through 2007, including the 64 viruses sequenced during the present study and genome sequences of the 36 viruses available in GenBank, were subjected to phylogenetic analysis. Twenty‐six of the 54 viruses from the 2006–2007 season selected for analysis in this study were adamantane‐sensitive. Due to the segmented nature of influenza virus genome, phylogenetic relations among viruses were deduced based on the comprehensive analysis of phylogenetic trees constructed for individual genomic segments.
Genomic segments encoding the surface antigens HA and NA
HA segment. Examination of the HA phylogenetic tree (Figure 1A) revealed that viruses from 2006–2007 evolved from the N‐lineage (shown in red) and fell into four distinct genetic clades, designated A, B, C, and D. The HAs of all four clades are descendents of the N‐lineage keeping its characteristic marker S193F. The second marker, D225N, was also kept by the majority of 2006–2007 viruses; however, it was less conserved. The unique changes acquired by each clade are shown in (Figure 1A). The relatedness of the HA of these viruses did not correspond to their drug phenotype: viruses from genetically distant clades A and D were adamantane resistant (except three isolates from New Mexico), while viruses from clades B and C were drug‐sensitive (except two viruses from Canada).
Figure 1.
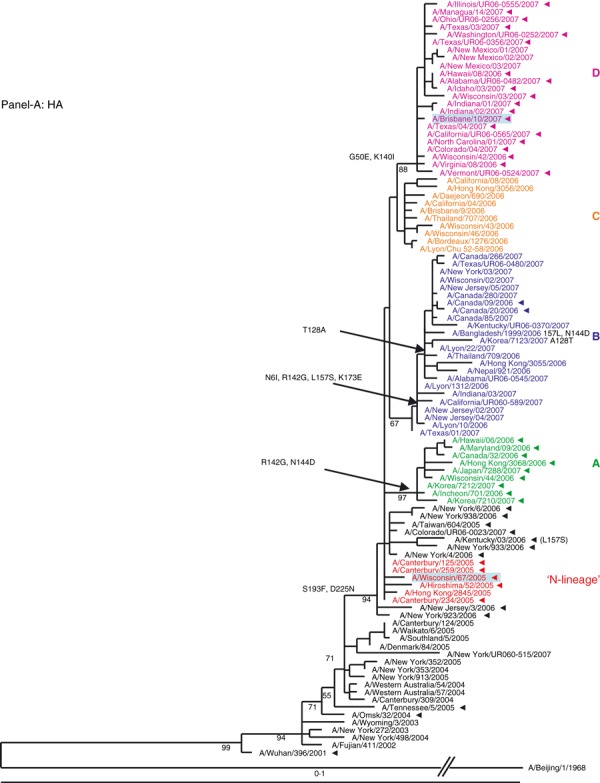
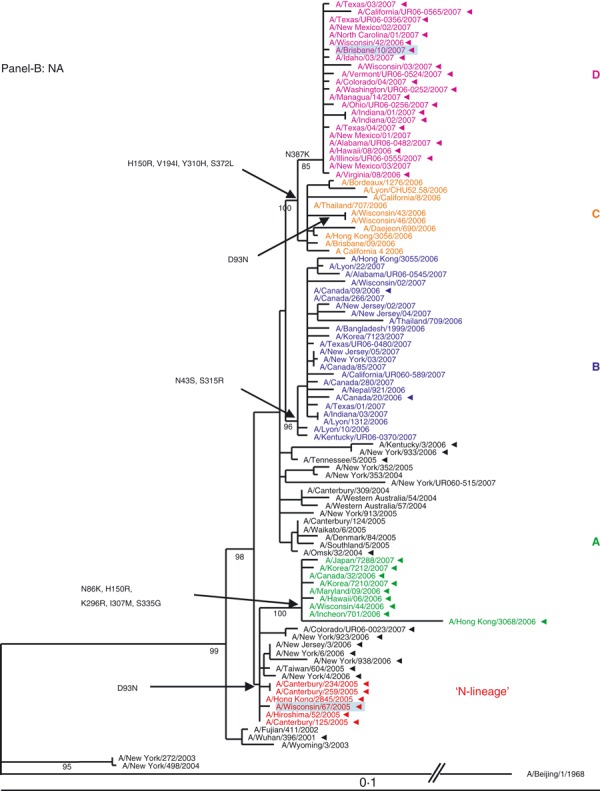
(A, B) Evolution of the surface antigens HA1 and NA genes of A(H3N2) viruses sampled during the 2006–2007 season. Phylogenetic tree was constructed from the HA1 genes of viruses sequenced during the study (n = 64) and 36 viruses with full genome sequences available in public domain. Trees were inferred using maximum likelihood available in garli 0·96b7 package. Using paup package bootstrap values, shown on tree branch nodes, were calculated from 100 replicates using of the data set to ensure robustness of the analysis (the results and parameters for each segments data set are available upon request). Phylogenetic trees throughout the manuscript were rooted with A/Beijing/1/1968, which was used as an out‐group. For clarity, the N‐lineage and four major genetic clades A–D are color‐coded in all figures according to their respective HA clade (N‐lineage – red, clade A – green, B – blue, C – orange, and D – purple). Characteristic amino acid changes are shown at the appropriate nodes. Vaccine strains for 2006–2007 and 2007–2008 seasons, A/Wisconsin/67/2005, and A/Brisbane/10/2007, respectively, are highlighted in light blue. Solid arrowheads indicate resistance to adamantanes.
NA segment. Similar to the HA, the phylogeny of the NA segment encoding the second surface antigen indicated that viruses of 2006–2007 season evolved from the N‐lineage and clustered into four distinct clades A, B, C, and D (Figure 1B). The HA and NA tree topologies suggested similar evolutionary patterns for the two gene segments. Amino acid signature changes in each clade are indicated in Figure 1B. Based on the phylogenetic analyses of the HA and NA segments we established that both adamantane sensitive and resistant viruses of 2006–2007 season possessed HA and NA genes that evolved from the drug‐resistant N‐lineage, which dominated in the previous season (2005–2006).
The gene segments encoding the remaining (internal) genes
The phylogenetic trees for the internal genomic segments (M, PB2, PB1, PA, NP and NS) for all 100 analyzed viruses are provided in Figure 2A–F.
Figure 2.
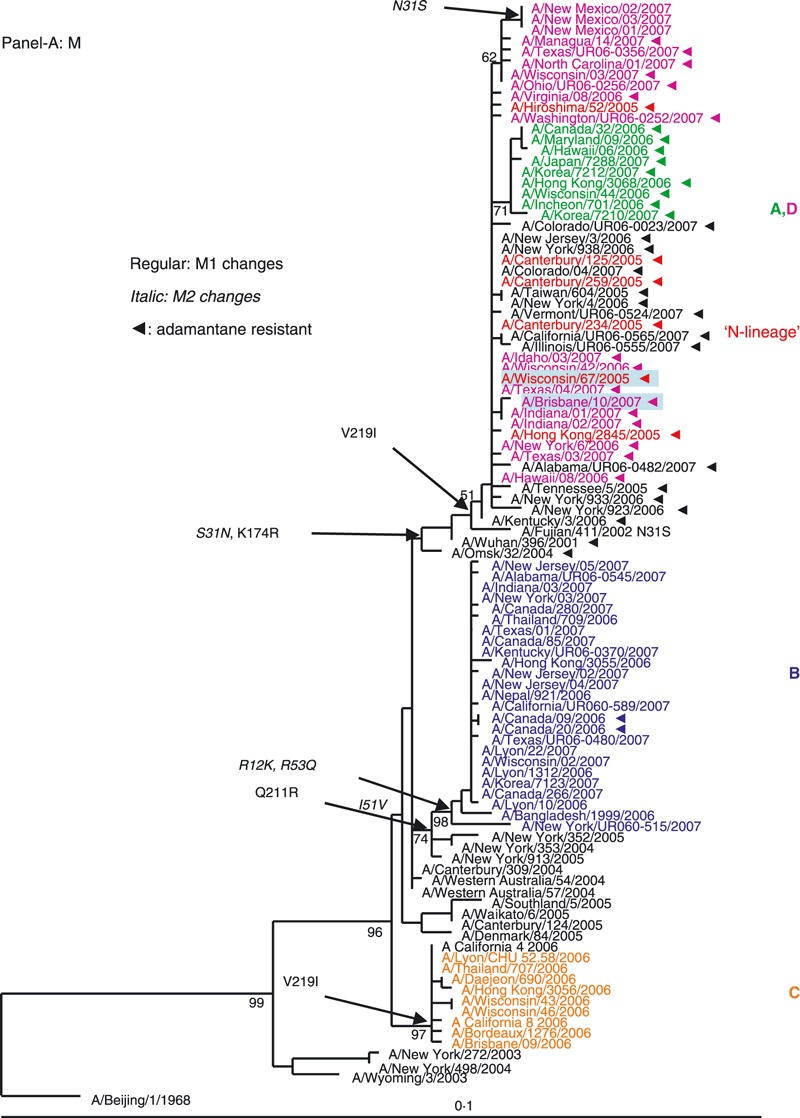
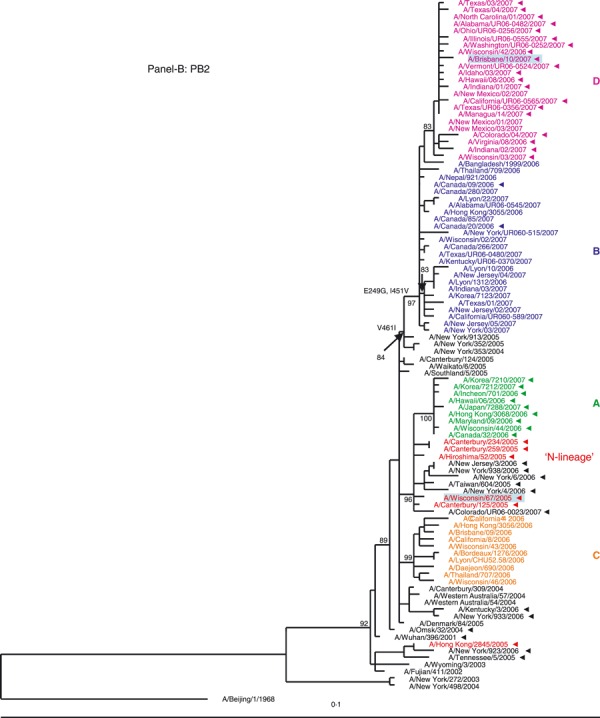
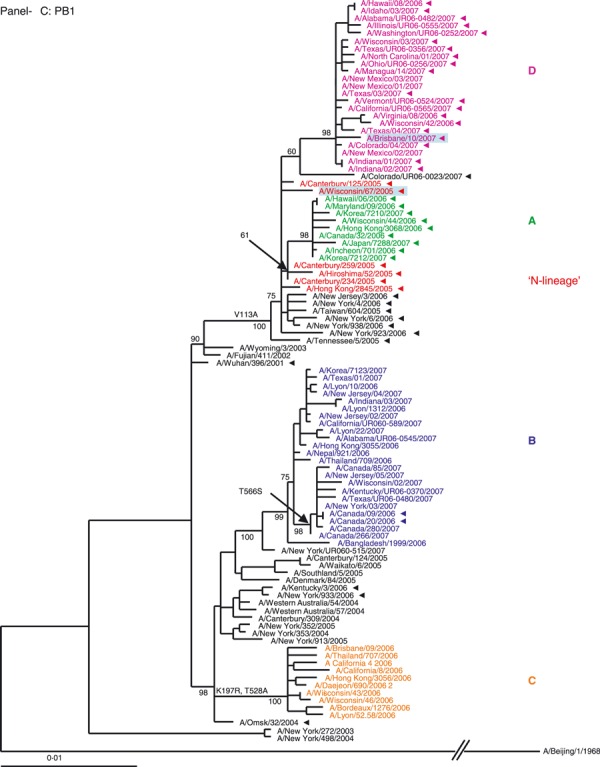
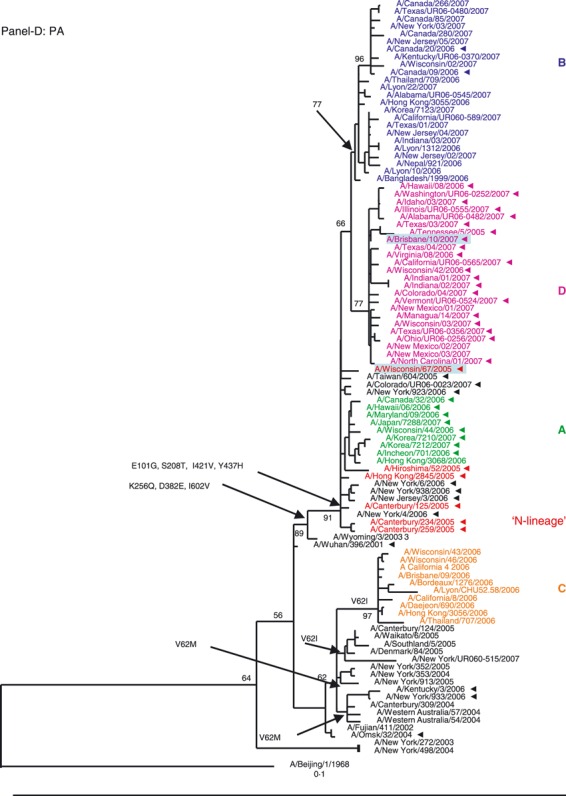
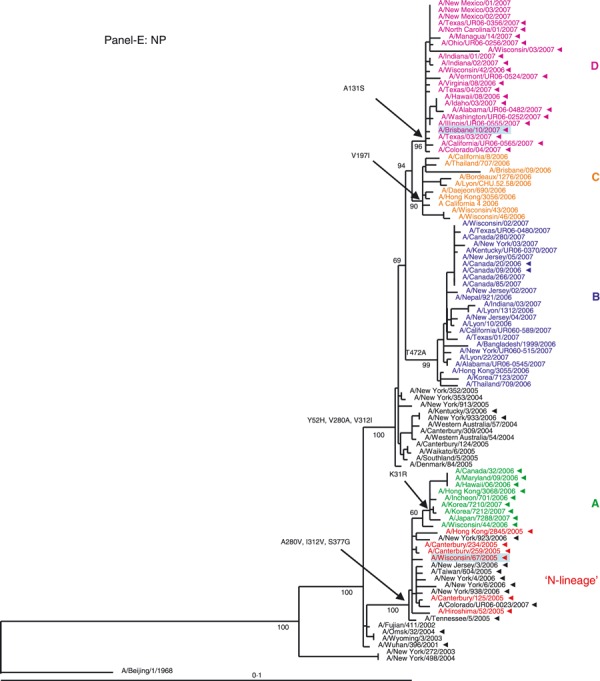
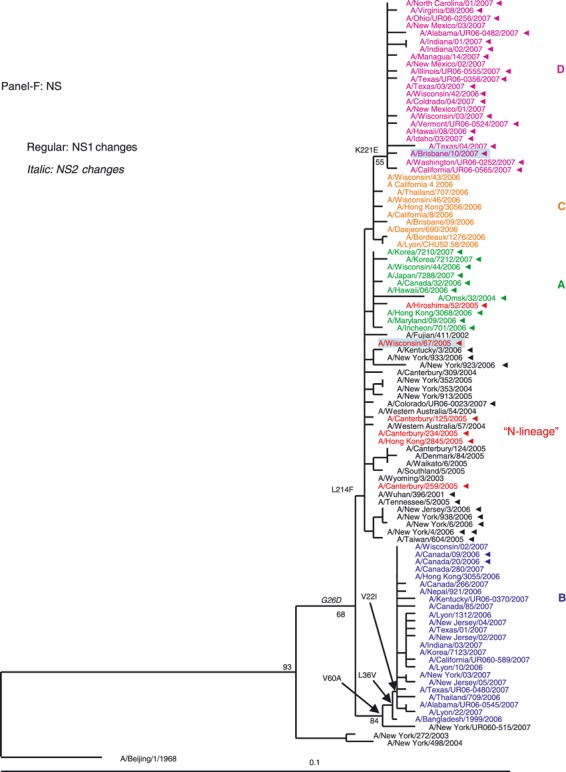
Phylogenetic analysis of the six internal gene segments of 2006–2007 A(H3N2). Phylogenetic trees were inferred and labeled as in Figure 1. Overall, the trees inferred from the internal gene segment revealed presence of the four major genetic clades, as obtained with the HA and NA segments (A–F). Noteworthy, M and PB1 phylogenies showed that clades B and D (A/Japan/7288/2007‐like and A/Brisbane/10/2006‐like), representing adamantane‐resistant viruses formed a single group; while viruses in groups B and C formed two distinct clades of adamantane‐sensitive viruses (A and C). See legend to Figure 1 for color codes.
M segment. The M segment encodes two proteins, the M1 (matrix) and the M2 (proton channel), the latter of which carries markers of adamantane resistance. The phylogenetic analysis of the M segment (Figure 2A) revealed that adamantane‐resistant viruses (A and D) clustered with the N‐lineage viruses and shared its characteristic markers in M1 and M2 proteins (Table S1). Phylogenetically, the closest ancestors of adamantane‐resistant viruses were A/Wuhan/396/2001‐like viruses that had circulated in prior seasons. The M genes of adamantane‐sensitive viruses of the 2006–2007 season formed two separate clades with clustering patterns similar to their corresponding HA and NA genes. The M segment of clade B viruses may have descended from viruses similar to those that circulated in New York in 2004–2005. Clade C drug‐sensitive viruses have likely acquired an M segment from pre‐existing sensitive viruses that were in circulation in Australia during 2004–2005, as previously reported. 25
There were two instances where the adamantane phenotype for a set of viruses was different from the remaining viruses in their clade. Within the adamantane resistant clade D, three isolates from New Mexico were sensitive. Based on sequence analysis, these isolates became adamantane sensitive through a reversion at amino acid 31 (N → S). Inversely, two Canadian isolates from the adamantane sensitive clade B acquired the resistance mutation S31N (Figure 2A). Clinical data indicate that the patients from whom these two viruses were collected were not treated with antiviral drugs.
With the exception of the New Mexico isolates, the 2006–2007 season sensitive viruses most likely acquired adamantane‐sensitivity via reassortment with drug sensitive viruses co‐circulating in prior seasons.
PB2 segment. The phylogeny inferred from the polymerase complex gene PB2 showed the 2006–2007 season isolates clustering into four distinct clades with high bootstrap value support (Figure 2B) with only clade A viruses clustering with the N‐lineage. The PB2 segment for clade B and D isolates may have descended from viruses similar to those that circulated in New York in 2004–2005, which share the amino acid change of V461I. Additionally, clades B and D acquired two replacements, E249G and I451V. The PB2 of clade C viruses are more likely to be descendants of viruses circulating prior to the N‐lineage (Figure 2B).
PB1 segment. The topology of the polymerase complex gene PB1 tree was similar to that of the M gene. Viruses forming PB1 clades A and D clustered with the N‐lineage whose ancestors were likely to be the A/Wuhan/396/2001‐like viruses that dominated during 2001–2003. Although both clades A and D were closely related to the N‐lineage PB1 clade (Figure 2C), clade D PB1 genes cluster somewhat apart from those of the N‐lineage and clade A viruses. The PB1 segment for clade B likely descended from viruses similar to those that circulated in New York in 2004–2005. The PB1 genes of clade C viruses are also likely to be descendants of viruses circulating prior to the appearance of the N‐lineage (Figure 2C).
PA segment. The phylogeny of the polymerase complex PA gene (Figure 2D) showed that the genes from clades A, B, and D all descended from the N‐lineage, sharing its seven characteristic markers. In contrast, the PA segment of viruses in clade C formed a separate grouping, which lacked all but one (Y437H) of those markers.
NP segment. Based on the NP phylogenetic tree, only viruses from clade A cluster with the N‐lineage (Figure 2E) having retained all its three characteristic markers A280V, I312V, and S377G (Table S1) with the additional change K31R. Clades B, C, and D are descendants of viruses circulating in seasons prior to the N‐lineage appearance. The viruses of clade C and D formed two sister taxa, but each with a unique replacement, V197I and A131S, respectively (Figure 2E).
NS segment. Phylogeny of the NS segment of 2006–2007 season viruses shows that clades A, C and D clustered with the N‐lineage and share the amino acid replacement L214F in their NS1. Viruses of clade B were easily distinguishable and formed a distinct clade with three shared replacements (Figure 2F).
Co‐circulating genotypes during the 2006–2007 season
The topologies of the constructed trees revealed a complex pattern of intra‐subtype reassortment among A(H3N2) viruses that circulated during the 2006–2007 season. A simplified diagram depicting the genome composition of the viruses circulating in 2006–2007 in relation to the N‐lineage is presented in Figure 3. The HA and NA segments of all four clades evolved from the N‐lineage. Overall, four major genotypes co‐circulated in the 2006–2007 season, corresponding to the four clades identified for the HA and NA genes.
Figure 3.
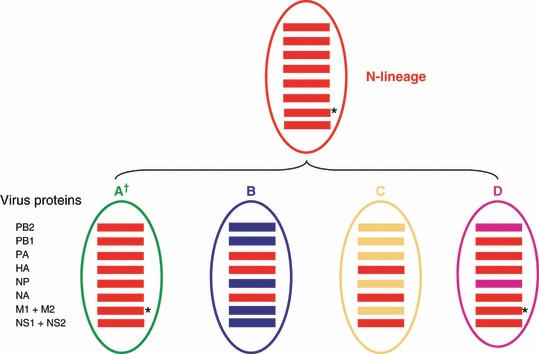
Schematic representation of four genotypes in A(H3N2) viruses co‐circulating in 2006–2007 season. Genetic analysis of individual segments revealed that the 2006–2007 belonged to one of four gene constellations. Viruses of genotypes B (blue) and C (orange) are sensitive to adamantanes, whereas viruses in genotypes A (green) and D (purple) are adamantane‐resistant. The N‐lineage viruses are colored in red. Genetic groups are defined based on the HA and NA gene segment phylogenies.
Viruses of genotype A appear to be direct descendants of the N‐lineage viruses being closely related to them in all gene segments (Figure 3). Genotype A viruses kept all 17 characteristic markers of the N‐lineage, 25 including three markers in the surface antigens HA and NA and the marker of adamantane resistance in M2 (Table S1). Additionally, viruses in genotype A acquired the change K31R in their NP gene.
In contrast, the genesis of the three remaining genotypes B, C, and D was due to independent reassortment events resulting in different constellations of the internal gene segments. Genotype B is the result of reassortment between the N‐lineage and adamantane‐sensitive viruses that circulated during 2004–2005 season, with only the PA gene inherited from the N‐lineage. Viruses from genotype C inherited only the NS gene from the N‐lineage viruses. The remaining internal gene segments were likely acquired from adamantane sensitive viruses that circulated prior to the N‐lineage (Figure 3). Genotype D viruses acquired the majority of their internal genes (except the PB2 and NP) from the N‐lineage viruses, including the adamantane resistance marker‐bearing M gene. Its PB2 and NP segments were most likely acquired via reassortment with pre‐existing viruses similar to those that circulated in Australia, New York, and New Zealand in 2004–2005.
HA gene evolution and adamantane resistance in A(H3N2) viruses that circulated in early season of 2007–2008
While the current study was in progress, it became apparent that the subsequent season of 2007–2008 was marked by global dominance (∼100%) of the adamantane‐resistant viruses. 30 To gain insight into their HA and NA genes evolution, a set of viruses (n = 55) collected in nine countries from 4 October 2007 through 4 April 2008 was chosen for phylogenetic analysis (Figure 4A,B). Our data revealed that clade D viruses remained dominant in the 2007–2008 season.
Figure 4.
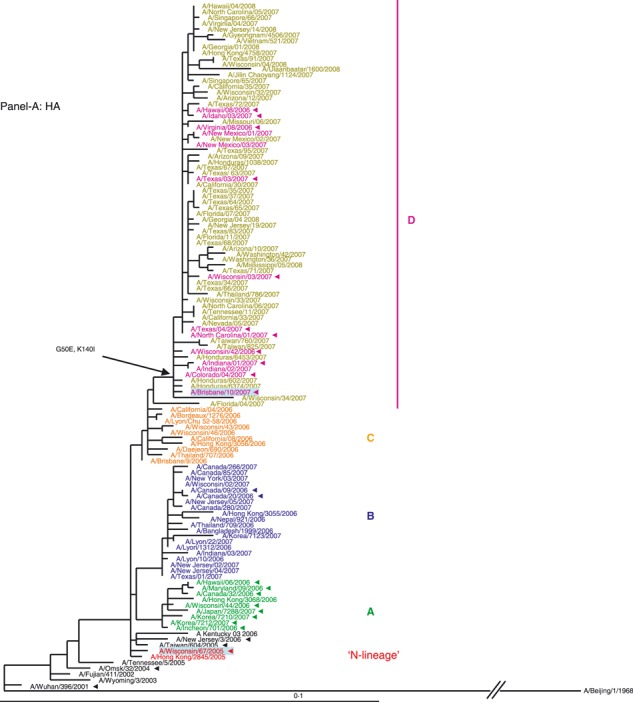
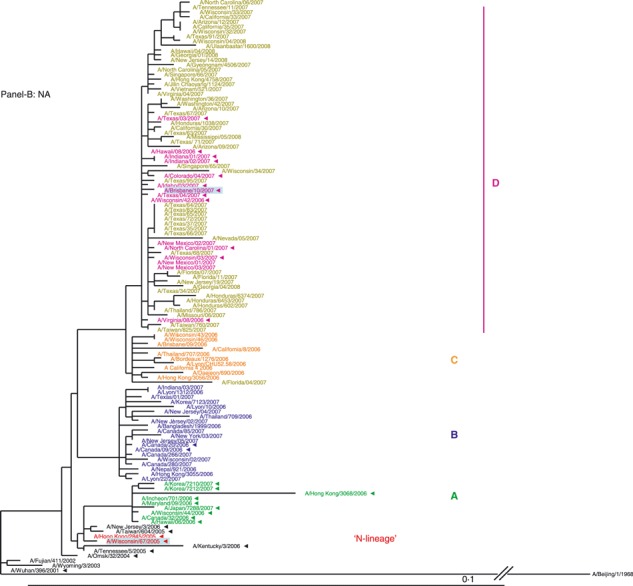
Phylogenetic trees were constructed from 119 viruses, including 55 HA1 (A) and NA (B) gene sequences of viruses sampled globally from October 2007 through April 2008 and showed that the genotype D remained dominant in circulation in the 2007–2008 influenza season. Trees were generated and labeled as in Figure 1, and strain designations of the 2007–2008 viruses were colored in olive for distinction.
To further investigate the genomic composition of the 2007–2008 viruses, complete genome sequencing was performed for 11 viruses from the 2007–2008 season. Phylogenetic analysis revealed that they all belong to genotype D, with the exception of A/Florida/04/2007, which belongs to genotype C (phylogenetic trees not shown). The data showed no evidence of further reassortment events within the adamantane‐resistant viruses during the 2007–2008 season. Similar to the viruses from genotype D from the previous season, they retained 13 markers characteristic of the N‐lineage suggesting that the four ‘lost’ markers were not essential for 2007–2008 viruses fitness and spread. The characteristic changes for the newer dominant A(H3N2) genotype D viruses are included in Table S2.
Discussion
The 2006–2007 A(H3N2) influenza season saw the co‐circulation of four distinct HA and NA clades which emerged due to antigenic drift from the predominant group of viruses (N‐lineage) seen in the previous season. As deduced from whole genome sequence analysis, these four clades represented four different genotypes that had evolved through reassortment and/or mutations in the genes coding for non‐glycosylated proteins. The co‐circulation of these genotypes coincided with a noticeable reduction in resistance to adamantanes seen in A(H3N2) global surveillance when compared to the previous 2005–2006 season. 16 , 26 Recently, Furuse et al. 31 reported that A(H3N2) viruses that viruses that circulated in Japan during 2006–2007 acquired adamantane sensitivity marker through reassortment.
Gene reassortment within the same influenza virus subtype is an integral part of influenza evolution and has been described in human, 3 , 25 , 32 , 33 in avian, 34 , 35 , 36 in swine, 37 , 38 and in the 1918 pandemic influenza viruses. 4 , 39 In the 2005–2006 season, reassortment led to the genesis of a dominant genotype (N‐lineage 25 ) which had acquired an M gene from a lineage with a mutation conferring adamantane resistance. Analysis of individual segment phylogenies revealed three different patterns of reassortment between the N‐lineage and genotypes circulating more widely prior to the 2005–2006 season, while the fourth genotype, A, retained all gene segments from the N‐lineage.
Among the 2006–2007 genotypes, two retained the M gene from the adamantane‐resistant lineage while the others acquired their M gene from sensitive viruses that had circulated in prior seasons. These findings provide clear evidence that the drop in adamantane resistance was the direct result of reassortment events rather than reversion of the previously dominant resistant phenotype. The striking difference in the frequencies of resistance to adamantanes among isolates collected from different regions and countries of the world was largely due to the distribution of the genotypes circulating. For example, the level of adamantane resistance in Europe was 39·0% while in the USA it was as high as 86·3%. This reflects the dominance of genotypes B and C in Europe and genotypes A and D in the USA. Of note, full genome sequences of 2006–2007 isolates from Africa, Australia, and Europe are very limited in public databases. However, the prototype viruses in lineages C and D (A/Brisbane/09/2006 A/Brisbane/10/2007, respectively) are from Australia and analysis of HA and NA of viruses currently circulating in this region indicate their close relatedness to the D lineage (Garten RJ, unpublished data). Similarly, genome analysis of several viruses isolated in France fell into the lineage B and C (1, 2).
In addition to differences in the geographic distribution of genotypes during the 2006–2007 season, there were also temporal variations. Viruses of genotypes A, B, and C appeared early in the Northern hemisphere season, and from December of 2006, were progressively replaced by the genotype D that represented a new antigenic variant of H3N2 viruses, A/Brisbane/10/2007‐like viruses. By the beginning of the next Southern hemisphere season, nearly all circulating viruses were from genotype D. Only a small fraction of viruses from genotype A remained in circulation until June of 2007. These data suggest that antigenic drift might have provided a selective advantage to genotype D viruses for its global spread. A/Brisbane/10/2007, the prototype of the D‐lineage, was selected as the H3 component of the influenza vaccine since 2008 (WHO, 2008), and replaced A/Wisconsin/67/2005 that belongs to the N‐lineage.
It is worth noting that analysis of amino acid sequences of the surface glycoproteins from the 2006–2007 season viruses revealed presence of several changes at residues previously associated with antigenic positive selection. 40 These include R142G in the HA of A and B lineages, N144D in lineage A, and G50E and K140I in the D lineage. The latter lineage of viruses also acquired a unique change in NA (N387K) which is located in a region previously described as an antigenic site by Fanning et al. 41
After the 2005–2006 season, Simonsen et al. 25 speculated that the N‐lineage genome composition somehow provided a fitness advantage and therefore adamantane resistant viruses of this genotype might dominate in future seasons. The subsequent co‐circulation of adamantane sensitive viruses, followed by appearance and increasing dominance of the adamantane resistant D‐lineage in the 2006–2007 season highlights the unpredictable nature of A(H3N2) evolution. Recent studies by Nelson et al. 42 suggest that the majority of currently circulating adamantane resistant A(H3N2) viruses descended from a single introduction that was first seen in Hong Kong.
Further analysis of viruses collected globally during the 2007–2008 Northern hemisphere season showed that viruses from genotype D have persisted and continue to dominate. Since their emergence in late 2006, genotype D viruses have acquired few genetic changes, and undergone no new reassortment events.
Despite the apparent antigenic advantage contributing to the fitness and dominance of this genotype, the continuing evolution of A(H3N2) viruses through reassortment remains highly plausible. If this genome fitness is maintained during antigenic evolution of H3N2 viruses, resistance to adamantanes could become fixed in circulating A(H3N2) viruses. The fact that there is no direct evidence that drug usage is required to maintain drug‐resistance would favor such a possibility. Fixation of the adamantane‐conferring resistance mutation S31N in swine influenza viruses in Europe lends credence to this possible scenario. 43 It should be noticed, however, that antigenic pressure among pigs is less intense than in the human population.
Although reassortment was responsible for the appearance of adamantane sensitive viruses in 2006–2007, there were also less‐frequent reversion events that led to circulation of adamantane sensitive viruses. Conversely, adamantane resistance was acquired through mutation, S31N, of the M gene in a small number of viruses from adamantane‐sensitive lineages. These observations underscore the degree of complexity of the mechanisms underlying the dynamics of adamantane resistance in seasonal influenza A viruses. Resistance to adamantanes was also reported in some groups of avian influenza A(H5N1) viruses isolated from humans and poultry in various countries. 44 , 45 , 46
This study demonstrates the importance of full genome sequencing and continued monitoring of resistance to available antivirals as well as the need to develop new anti‐influenza drugs and therapeutic approaches.
Addendum
V Deyde and R Garten: designed study, analyzed data, and wrote manuscript. V Deyde and T Sheu: performed antiviral analysis and genome sequencing. C Smith, A Myrick, and J Barnes: performed genome sequencing and analysis. X Xu, M Shaw, A Klimov, and L Gubareva: designed study, analyzed data, and critically revised manuscript.
Acknowledgements
We thank Henrietta Hall, Teresa Wallis, Angie Foust, Amanda Balish, Wendy Sessions, Gregory Kosher, and Jan Mabry, from the Influenza Division, Centers for Disease Control and Prevention, for their active involvement in virus surveillance activities, Dr Carolyn Bridges from the Influenza Division, Centers for Disease Control and Prevention, for critical review of the manuscript, the National, State Laboratories, and Influenza Centers, the WHO Collaborating Centers for submitting their specimens for testing. Tiffany Sheu is grateful to the Oak Ridge Institute for Science and Education (ORISE) for providing her fellowship.
Funding source
This work was funded by the US Centers for Disease Control and Prevention.
Disclaimer
The findings and conclusions of this report are those of the authors and do not necessarily represent the views of the Centers for Disease Control and Prevention.
Conflict of interest statement
We declare that we have no conflict of interest.
Supporting information
Table S1. Characteristic amino acid changes in the season 2006‐07 A(H3N2) viruses in comparison to those of the N‐lineage
Table S2. Characteristic amino acid changes in the D‐genotype of A(H3N2) viruses that dominated in late 2006‐07 through the season 2007‐08 in comparison to those of the N‐lineage
Table S3. A(H3N2) viruses sequenced in the present study
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Supporting info item
References
- 1. Fitch WM, Leiter JM, Li XQ, Palese P. Positive Darwinian evolution in human influenza A viruses. Proc Natl Acad Sci USA 1991; 88:4270–4274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Fitch WM, Bush RM, Bender CA, Cox NJ. Long term trends in the evolution of H(3) HA1 human influenza type A. Proc Natl Acad Sci USA 1997; 94:7712–7718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Holmes EC, Ghedin E, Miller N et al. Whole‐genome analysis of human influenza A virus reveals multiple persistent lineages and reassortment among recent H3N2 viruses. PLoS Biol 2005; 3:e300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nelson MI, Viboud C, Simonsen L et al. Multiple reassortment events in the evolutionary history of H1N1 influenza A virus since 1918. PLoS Pathog 2008; 4:e10000126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hay AJ, Zambon MC, Wolstenholme AJ, Skehel JJ, Smith MH. Molecular basis of resistance of influenza A viruses to amantadine. J Antimicrob Chemother 1986; 18(Suppl B):19–29. [DOI] [PubMed] [Google Scholar]
- 6. Wang C, Takeuchi K, Pinto LH, Lamb RA. Ion channel activity of influenza A virus M2 protein: characterization of the amantadine block. J Virol 1993; 67:5585–5594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Aoki FY. Amantadine and rimantadine; in Nicholson KG, Webster RG, Hay AJ. (eds): Textbook of Influenza. Oxford, UK: Blackwell Science, 1998: 457–476. [Google Scholar]
- 8. Belshe RB, Smith MH, Hall CB, Betts R, Hay AJ. Genetic basis of resistance to rimantadine emerging during treatment of influenza virus infection. J Virol 1988; 62:1508–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Klimov AI, Rocha E, Hayden FG, Shult PA, Roumillat LF, Cox NJ. Prolonged shedding of amantadine‐resistant influenzae A viruses by immunodeficient patients: detection by polymerase chain reaction‐restriction analysis. J Infec Dis 1995; 172:1352–1355. [DOI] [PubMed] [Google Scholar]
- 10. Boivin G, Goyette N, Bernatchez H. Prolonged excretion of amantadine‐resistant influenza A Virus quasi species after cessation of antiviral therapy in an immunocompromised patient. Clin Infect Dis 2002; 34:E23–E25. [DOI] [PubMed] [Google Scholar]
- 11. Hayden FG, Belshe RB, Clover RD, Hay AJ, Oakes MG, Soo W. Emergence and apparent transmission of rimantadine‐resistant influenza A virus in families. N Engl J Med 1989; 321:1696–1702. [DOI] [PubMed] [Google Scholar]
- 12. Belshe RB, Burk B, Newman F, Cerruti RL, Sim IS. Resistance of influenza A virus to amantadine and rimantadine: results of one decade of surveillance. J Infect Dis 1989; 159:430–435. [DOI] [PubMed] [Google Scholar]
- 13. Prud’homme IT, Zoueva O, Weber JM. Amantadine susceptibility in influenza A virus isolates: determination methods and lack of resistance in a Canadian sample, 1991–94. Clin Diagn Virol 1997; 8:41–51. [DOI] [PubMed] [Google Scholar]
- 14. Ziegler T, Hemphill ML, Ziegler ML et al. Low incidence of rimantadine resistance in field isolates of influenza A viruses. J Infect Dis 1999; 180:935–939. [DOI] [PubMed] [Google Scholar]
- 15. Public Health Agency of Canada (PHAC) . National and international surveillance for antiviral resistance. Can Commun Dis Rep 2006; 32:2–3. [Google Scholar]
- 16. Pabbaraju K, Ho KC, Wong S et al. Adamantane resistance in circulating human influenza A viruses from Alberta, Canada (1970–2007). Antiviral Res 2008; 79:81–86. [DOI] [PubMed] [Google Scholar]
- 17. Bright RA, Medina MJ, Xu X et al. Incidence of adamantane resistance among influenza A (H3N2) viruses isolated worldwide from 1994 to 2005: a cause for concern. Lancet 2005; 366:1175–1181. [DOI] [PubMed] [Google Scholar]
- 18. Bright RA, Shay DK, Shu B, Cox NJ, Klimov AI. Adamantane resistance among influenza A viruses isolated early during the 2005–2006 influenza season in the United States. JAMA 2006; 295:891–894. [DOI] [PubMed] [Google Scholar]
- 19. Barr IG, Hurt AC, Iannello P, Tomasov C, Deed N, Komadina N. Increased adamantane resistance in influenza A(H3) viruses in Australia and neighbouring countries in 2005. Antiviral Res 2006; 73:112–117. [DOI] [PubMed] [Google Scholar]
- 20. Barr IG, Hurt AC, Deed N, Iannello P, Tomasov C, Komadina N. The emergence of adamantane resistance in influenza A(H1) viruses in Australia and regionally in 2006. Antiviral Res 2007; 75:173–176. [DOI] [PubMed] [Google Scholar]
- 21. Deyde VM, Xu X, Bright RA et al. Surveillance of resistance to adamantanes among influenza A(H3N2) and A(H1N1) viruses isolated worldwide. J Infect Dis 2007; 196:249–257. [DOI] [PubMed] [Google Scholar]
- 22. Saito R, Li D, Suzuki H. Amantadine‐resistant influenza A (H3N2) virus in Japan, 2005–2006. N Engl J Med 2007a; 356:312–313. [DOI] [PubMed] [Google Scholar]
- 23. Saito R, Li D, Suzuki Y et al. High prevalence of amantadine‐resistance influenza A (H3N2) in six prefectures, Japan, in the 2005–2006 season. J Med Virol 2007b; 79:1569–1576. [DOI] [PubMed] [Google Scholar]
- 24. Centers for Disease Control and Prevention (CDC) . High levels of adamantane resistance among influenza A(H3N2) viruses and interim guidelines for use antiviral agents. United States, 2005–2006 influenza season. MMWR Morb Mortal Wkly Rep 2006; 55:44–46. [PubMed] [Google Scholar]
- 25. Simonsen L, Viboud C, Grenfell BT et al. The genesis and spread of reassortment human influenza A/H3N2 viruses conferring adamantane resistance. Mol Biol Evol 2007; 24:1811–1820. [DOI] [PubMed] [Google Scholar]
- 26. Deyde VM, Garten R, Sheu TG, Gubareva LV, Klimov AI. Letter to the editor: reply to Saito et al. . J Infect Dis 2008; 197:632–633. [Google Scholar]
- 27. Nelson MI, Edelman L, Spiro DJ et al. Molecular epidemiology of A/H3N2 and A/H1N1 influenza virus during a single epidemic season in the United States. PLoS Pathog 2008a; 4:e1000133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zwickl DJ. Genetic Algorithm Approaches for the Phylogenetic Analysis of Large Biological Sequence Datasets Under the Maximum Likelihood Criterion. PhD dissertation. The University of Texas at Austin, 2006. [Google Scholar]
- 29. Swafford D. PAUP* Phylogenetic Analysis Using Parsimony (*and other methods). Sunderland (MA): Sinauer Associates, 2003. [Google Scholar]
- 30. Centers for Disease Control and Prevention (CDC) . Influenza activity – United States and worldwide, 2007–08 season. Morb Mortal Wkly Rep 2008; 57:692–697. [PubMed] [Google Scholar]
- 31. Furuse Y, Suzuki A, Kamigaki T, Shimizu M, Fuji N, Oshitani HJ. Reversion of influenza A (H3N2) virus from amantadine resistant to amantadine sensitive by further reassortment in Japan during the 2006‐to‐2007 influenza season. Clini Microb 2009; 47:841–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Barr IG, Komadina N, Hurt AC et al. An influenza A(H3) reassortant was epidemic in Australia and New Zealand in 2003. J Med Virol 2005; 76:391–397. [DOI] [PubMed] [Google Scholar]
- 33. Rambaut A, Pybus OG, Nelson MI, Viboud C, Taubenberger JK, Holmes EC. The genomic and epidemiological dynamics of human influenza A virus. Nature 2008; 453:615–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dung NT, Vinh NT, Vijaykrishna D et al. Multiple sublineages of influenza A virus (H5N1), Vietnam, 2005–2007. Emerg Infect Dis 2008; 14:632–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Monne I, Joannis TM, Fusaro A et al. Reassortant avian influenza virus (H5N1) in poultry, Nigeria, 2007. Emerg Infect Dis 2008; 14:637–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Song MS, Oh TK, Moon HJ et al. Ecology of H3 avian influenza viruses in Korea and assessment of their pathogenic potentials. J Gen Virol 2008; 89:949–957. [DOI] [PubMed] [Google Scholar]
- 37. Yasuda J, Shortridge KF, Shimizu Y, Kida H. Molecular evidence for a role of domestic ducks in the introduction of avian H3 influenza viruses to pigs in Southern China, where the A/Hong Kong/68 (H3N2) strain emerged. J Gen Virol 1991; 72:2007–2010. [DOI] [PubMed] [Google Scholar]
- 38. Yu H, Hua RH, Zhang Q et al. Genetic evolution of swine influenza A (H3N2) viruses in China from 1970 to 2006. J Clin Microbiol 2008; 46:1067–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Vana G, Westover KM. Origin of the 1918 Spanish influenza virus: a comparative genomic analysis. Mol Phylogenet Evol 2008; 47:1100–1110. [DOI] [PubMed] [Google Scholar]
- 40. Bush RM, Fitch WM, Bender CA, Cox NJ. Positive selection on the H3 hemagglutinin gene of human influenza virus A. Mol Biol Evol 1999b; 16:1457–1465. [DOI] [PubMed] [Google Scholar]
- 41. Fanning TG, Reid AH, Taubenberger JK. Influenza A virus neuraminidase: regions of the protein potentially involved in virus‐host interactions. Virol 2000; 276:417–423. [DOI] [PubMed] [Google Scholar]
- 42. Nelson MI, Simonsen L, Viboud C, Miller MA, Holmes EC. The origin and global emergence of adamantane resistant A/H3N2 influenza viruses. Virol 2009; 388:270–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. De Jong JC, Smith DJ, Lapedes AS et al. Antigenic and genetic evolution of swine influenza A (H3N2) viruses in Europe. J Virol 2007; 81:4315–4322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Cheung CL, Rayner JM, Smith GJ et al. Distribution of amantadine‐resistant H5N1 avian influenza variants in Asia. J Infect Dis 2006; 193:1626–1629. [DOI] [PubMed] [Google Scholar]
- 45. Hurt AC, Selleck P, Komadina N, Shaw R, Brown L, Barr IG. Susceptibility of highly pathogenic A(H5N1) avian influenza viruses to the neuraminidase inhibitors and adamantanes. Antiviral Res 2007; 73:228–231. [DOI] [PubMed] [Google Scholar]
- 46. He G, Qiao J, Dong C, He C, Zhao L, Tian Y. Amantadine‐resistance among H5N1 avian influenza viruses isolated in Northern China. Antiviral Res 2008; 77:72–76. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Characteristic amino acid changes in the season 2006‐07 A(H3N2) viruses in comparison to those of the N‐lineage
Table S2. Characteristic amino acid changes in the D‐genotype of A(H3N2) viruses that dominated in late 2006‐07 through the season 2007‐08 in comparison to those of the N‐lineage
Table S3. A(H3N2) viruses sequenced in the present study
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Supporting info item