

Abstract
The current studies entail systematic quality by design (QbD)-based development of stimuli-responsive gastroretentive drug delivery systems (GRDDS) of acyclovir using polysaccharide blends for attaining controlled drug release profile and improved patient compliance. The patient-centric quality target product profile was defined and critical quality attributes (CQAs) earmarked. Risk assessment studies, carried out through Ishikawa fish bone diagram and failure mode, effect, and criticality analysis, helped in identifying the plausible risks or failure modes affecting the quality attributes of the drug product. A face-centered cubic design was employed for systematic development and optimization of the concentration of sodium alginate (X 1) and gellan (X 2) as the critical material attributes (CMAs) in the stimuli-responsive formulations, which were evaluated for CQAs viz. viscosity, gel strength, onset of floatation, and drug release characteristics. Mathematical modeling was carried out for generation of design space, and optimum formulation was embarked upon, exhibiting formulation characteristics marked by excellent floatation and bioadhesion characteristics along with promising drug release control up to 24 h. Drug-excipient compatibility studies through FTIR and DSC revealed absence of any interaction(s) among the formulation excipients. In vivo pharmacokinetic studies in Wistar rats corroborated extension in the drug absorption profile from the optimized stimuli-responsive GR formulations vis-à-vis the marketed suspension (ZOVIRAX®). Establishment of in vitro/in vivo correlation (IVIVC) revealed a high degree of correlation between the in vitro and in vivo data. In a nutshell, the present investigations report the successful development of stimuli-responsive GRDDS of acyclovir, which can be applicable as a platform approach for other drugs too.
Electronic supplementary material
The online version of this article (doi:10.1208/s12249-015-0367-0) contains supplementary material, which is available to authorized users.
KEY WORDS: controlled release, gastroretention, in situ gelling, quality by design (QbD), smart polymers
INTRODUCTION
Development of oral controlled release products is invariably precluded by their inability to retain and localize the drug delivery system (DDS) within the desired region of the gastrointestinal tract. Considerable research, therefore, has poured into the plausibility of controlled and site-specific drug delivery to the gastrointestinal tract (1). Gastroretentive drug delivery systems (GRDDS), in this context, have been explored for maintaining the drug release characteristics within the “absorption window” ensuring optimal extent of oral bioavailability and decreasing the frequency of administration (2). However, majority of such systems are solid oral ones, possessing the obvious challenges for their administration in pediatric and geriatric patients leading eventually to poor patient compliance (3).
Of late, the stimuli-responsive systems have gained significant interest owing to their excellent site-specific drug delivery characteristics coupled with mucoadhesion properties (4). Such systems, primarily containing smart or stimuli-responsive polymers like polysaccharides, polyacrylic acids, polyanhydrides, polyethers, and polyesters, undergo transformation from “sol” to “gel” state, with change in various biological stimuli like pH, temperature, ionic content, and/or solvent composition (5). Among these, the polysaccharide-based materials such as gellan, xanthan, chitosan, pectin, cyclodextrin, and alginate derivatives possess tremendous potential in drug delivery owing to their cost-effectiveness, high drug loading capacity, controlled drug release characteristics, biocompatibility and biodegradability, and absence of systemic toxicity and long-term stability (6). Diverse applications of stimuli-responsive systems have been reported in literature for their promising controlled release profile of drug delivery through different routes of administration including oral, nasal, ocular, and vaginal (7). Applications of oral stimuli-responsive systems for site-specific delivery outweighs over GRDDS owing to their benefits like ease of administration of formulations in the “sol” state to the pediatric and geriatric patients, simple manufacturing, and above all, attainment of both floating and mucoadhesion characteristics leading to precise control of gastric retention (8).
Acyclovir, an analogue of purine nucleoside, is one of the most commonly used antiviral drugs recommended for the treatment of Herpes simplex, Varicella zoster, and Herpes zoster infections (9). It exhibits poor and inconsistent oral bioavailability (i.e., 15–30%) owing to low aqueous solubility (2.5 mg/mL) and lack of site-specific absorption in the gastric region. Recommended dosage schedule of 200–400 mg for two to five times a day tends to reduce the patient compliance and increase the overall cost of therapy. In this context, drug delivery strategies like mucoadhesive tablets (10), mucoadhesive microspheres (11,12), single-unit (13,14) and multiple-unit floating systems (15), and in situ gelling systems (16) have already been reported for reducing the dosing frequency, attaining controlled drug release profile and potentially improving the patient compliance of acyclovir, but yielding only limited fruition. This calls for developing an efficient stimuli-responsive GRDDS with potentially improved drug absorption characteristics and patient compliance.
Development of an impeccable stimuli-responsive GR formulation involves a number of formulation and process variables (17). Optimizing the formulation composition and process(es) involved during the manufacturing of such DDS using traditional one-factor-at-a-time (OFAT) approach is a herculean task, leading eventually to just workable solutions with maximal experimentation and expenditure of great deal of time, money, and effort (18). Systematic optimization of DDS employing quality by design (QbD) paradigms based on the salient principles of quality risk assessment (QRM) and design of experiments (DoE) has lately been popularized (19). These provide comprehensive understanding of the formulation system identifying plausible interaction(s) among the product and/or process-related factors to produce “the best” possible formulation systems (20).
The studies, therefore, were undertaken to develop the systematically optimized stimuli-responsive GRDDS of acyclovir employing natural and biodegradable, effective, and economical polymers, viz. sodium alginate and gellan, exhibiting pH-responsive sol to gel transformation characteristics. The prepared formulations were evaluated for biopharmaceutical performance through in vitro, ex vivo, and in vivo studies.
MATERIALS AND METHODS
Materials
Acyclovir was provided ex-gratis by M/s IPCA Laboratories Ltd., Mumbai, India. Various chemicals employed during the studies were procured from the respective suppliers, i.e., sodium alginate from M/s Signet Chemical Corporation, Mumbai, India; gellan from M/s Lab-Chem Ltd., Mumbai, India; gelatin from M/s SD Fine Chemicals Ltd., Mumbai, India; and calcium carbonate from M/s Universal Expo Chem., Mumbai, India. All other chemicals and reagents used were of analytical grade and were used as obtained.
Methods
Defining the QTPP and CQAs
As the first step towards QbD-based product development of stimuli-responsive GR systems of acyclovir, the patient-centric quality target product profile (QTPP) was defined for accomplishing gastroretentive profile of drug delivery for maximal therapeutic benefits. In order to meet the QTPP, critical quality attributes (CQAs) of the stimuli-responsive GR formulations were identified viz. viscosity (η) and gel strength (Gs) (imperative for bioadhesion of the formulations), onset of floatation (F o) (indicative of gastroretentive potential of the formulations), time taken for 60% drug release (T 60%), and amount of drug released in 16 h (Q 16 h) (marker of drug release characteristics) (21). Table 1 of supplementary data summarizes the nuances of key elements of QTPP, while apt justifications of the CQAs selected for stimuli-responsive GR systems of acyclovir have been enlisted in Table 2 of supplementary data.
Formulation Optimization of Stimuli-Responsive GR Systems
Based on the preoptimization studies and risk assessment studies, the highly influential CMAs of stimuli-responsive GR systems were identified and systematically optimized employing face-centered cubic design (FCCD) with α = 1. The stimuli-responsive GR formulations were prepared by simple admixture method. Table I enlists the formulation composition of the sol form of stimuli-responsive GR system. Initially, calcium carbonate was dispersed in purified water (approx. 2 mL). Subsequently, the requisite quantities of sodium alginate, gellan, and gelatin (previously dissolved in 8 mL of hot water) were added followed by stirring on a magnetic stirrer. After uniform mixing, acyclovir (800 mg/10 mL) was dispersed in the resulting solution. Sodium alginate and gellan were selected as the vital polymers, since both of these are documented to possess pH-responsive gelling property, controlled drug release, and floatational characteristics, while gelatin was selected to synergistically increase the gel strength, and facilitate in regulation of drug release (22,23). Calcium carbonate was selected as a gas-forming agent, as it releases carbon dioxide in the presence of gastric fluid resulting in the formation of gel with floating characteristics (24). On the basis of various literature reports, the quantity of calcium carbonate used in the formulation was fixed at 300 mg (24,25), which is considerably lower than the permissible limit of 550 mg suggested by FDA (26) and much lower than the amount (i.e., 1250 mg) known to cause flatulence and/or systemic acidosis (27). A total of 13 different formulations were prepared employing sodium alginate (X 1) and gellan (X 2) as the CMAs at three different levels, i.e., low (−1), intermediate (0), and high (+1) levels, including quintuplicate studies at the center point (0,0) formulations. Table II summarizes an account of 13 experimental runs studied along with actual and coded values of the studied CMAs. All the prepared formulations were evaluated for various CQAs viz. η, G S, F O T 60%, and Q 16 h.
Table I.
Formulation Composition of Stimuli-Responsive Gastroretentive Formulation of Acyclovir
| Ingredient | Amount (mg/10 mL) |
|---|---|
| Acyclovir | 800 |
| Sodium alginate | 100–300 |
| Gellan | 50–100 |
| Gelatin | 20 |
| Calcium carbonate | 300 |
Table II.
Formulation Composition of Stimuli-Responsive Gastroretentive System Prepared as per Central Composite Design
| Formulation code | Trial no. | Coded factor levels | |
| Factor 1 | Factor 2 | ||
| F1 | 1 | −1 | −1 |
| F2 | 2 | −1 | 0 |
| F3 | 3 | −1 | 1 |
| F4 | 4 | 0 | −1 |
| F5* | 5 | 0 | 0 |
| F6 | 6 | 0 | 1 |
| F7 | 7 | 1 | −1 |
| F8 | 8 | 1 | 0 |
| F9 | 9 | 1 | 1 |
| Translation of coded values into actual units | |||
| Coded factor | Levels | Factor 1 sodium alginate (mg) | Factor 2 gellan (mg) |
| −1 | Low | 100 | 50 |
| 0 | Intermediate | 200 | 75 |
| 1 | High | 300 | 100 |
*Quintuplicate studies performed for center point formulation
Characterization of the Stimuli-Responsive GR Formulations
Rheology
Rheological measurements were carried out to determine the flow behavior of the stimuli-responsive GR formulations. The studies were performed using a rotational-type rheometer (Rheolab QC, M/s Anton Paar GmbH, Vienna, Austria) attached with a double gap spindle geometry (DG26) and a water jacket (C-LTD80/QC). A volume of 10 mL of the formulation was poured inside the hollow cylinder using a 5-mL syringe. The hollow spindle having Toolmaster™ was then placed inside the cylinder and snap-fitted to the instrument sensor. All the rheological measurements were conducted at pH 6.8 and 37 ± 0.2°C, with shear rate and shear stress maintained at 0 to 100 s−1 and 0 to 10 Pa, respectively. Data analysis was subsequently carried out using Rheoplus-32 software ver 3.40.
Gel Strength
The mechanical strength of the gel formed from the stimuli-responsive formulation was determined using Texture Analyzer (TA.XT.Plus Texture Analyzer, M/s Stable Microsystems, Surrey, UK) by placing the gelled formulation in a standard beaker below the probe. An analytical probe was then immersed into the sample. The Texture Analyzer was set to the “gelling strength test” mode or “compression” mode with a test speed of 1.0 mm/s. An acquisition rate of 50 points per second and a trigger force of 5 g were selected. An aluminium probe of 5-mm diameter was used for all the samples. The study was carried out at room temperature. The force required (kg) to penetrate the gel was measured as the gel strength.
In Vitro Floating
In vitro floating studies were carried out in USP apparatus II containing 900 mL of simulated gastric fluid (SGF, pH 1.2). An aliquot (1 mL) of the prepared sol formulation was poured in the medium, and time required for onset of floatation of the formulation in the upper one-third part of the vessel was visually observed and recorded as lag time in floatation.
In Vitro Drug Release
In vitro drug release studies of the stimuli-responsive GR formulations were carried out in triplicate, employing USP XXXIV paddle type (apparatus 2) using SGF (pH 1.2) with a volume of 900 mL as the dissolution medium at 50 rpm and at 37 ± 0.5°C. An aliquot (1 mL) of the prepared sol formulation was poured in the medium, and 5-mL samples were withdrawn periodically at suitable time intervals followed by replenishment with an equivalent volume of fresh dissolution medium. Samples were analyzed spectrophotometrically at 257 nm employing a UV–vis spectrophotometer 3000+ (M/s Labindia Instruments Pvt. Ltd., Mumbai, India). The raw data obtained from in vitro drug release studies were analyzed using ZOREL software. The software has the inbuilt provisions for applying the correction factor for volume and drug losses during sampling (Eq. 1) and calculating the values of amount of drug dissolved, percent release, rate of drug release, and log fraction released at varied times (28).
| 1 |
where C C = corrected concentration, C t = uncorrected absorbance, V s = sample volume, and V t = total volume of dissolution medium. Drug release data were fitted into Korsemeyer model for non-swellable compressed matrices, as described in Eq. (2) (29).
| 2 |
where M t is the amount of drug released at time “t”, M ∞ is the amount of drug released at an infinite time, K is the kinetic rate constant, and n is the release exponent.
Optimization Data Analysis and Validation of QbD
The optimization data analysis was carried out after evaluating the stimuli-responsive GR formulations for various CQAs like η, G S, F O, T 60%, and Q 16 h. Mathematical modeling was carried out by employing second-order quadratic model for identifying interaction(s) among the studied CMAs. Only the significant polynomial coefficients as per the t test were considered in framing the polynomial equation. One-way analysis of variance (ANOVA) was carried out for analyzing the model fitting parameters by model p value, coefficient of correlation (r 2), and lack of fit. The response surface analysis was carried out employing 3D response surface plots and 2D contour plots. The prognosis of optimum formulation was conducted in two stages, i.e., constructing a feasible knowledge space followed by exhaustive grid search to predict the optimized formulation. Also, the numerical optimization was carried out using desirability function by “trading off” of various CQAs, as per the selected acceptance criteria, i.e., maximization of T 60%, Q 16 h, Gs, and minimization of F o and η, respectively.
Validation of the QbD methodology was carried out by selecting eight confirmatory check-point formulations from feasibility and grid search region. The validation formulations were evaluated for various CQAs, and the observed responses were compared with the predicted ones. Linear correlation plots were constructed between the observed and predicted responses, forcing the line through the origin. Further, the percent prediction error (bias) was also calculated with respect to the observed responses, and residual plots were drawn between the observed responses and the percent bias.
Sol to Gel Transformation Studies
An aliquot (2 mL each) of the optimized stimuli-responsive GR formulation was placed in each of the test tubes containing solutions with pH ranging between 2 and 7 at room temperature. Tubes were left undisturbed for 1 h so as to check the “sol” to “gel” transformation of the formulation. The upper surface of the formulation was visually observed by tilting and inverting the test tubes to check complete transformation of sol form to gel state (30).
Drug-Excipient Compatibility Studies
Fourier Transform Infrared Spectroscopy
The Fourier transform infrared (FTIR) spectroscopy was performed to characterize the possible interactions between the drug and excipients, if any. The FTIR spectra of pure drug and its physical mixture with each excipient viz. sodium alginate, gellan, calcium carbonate, and gelatin were recorded in KBr disc over the range 4000–400 cm−1 using an FTIR spectrophotometer (M/s Perkin Elmer, MA, USA).
Differential Scanning Calorimetry
The differential scanning calorimetry (DSC) studies were carried out to investigate the thermodynamic compatibility of physical mixture of the drug with each excipient based on their melting point. The DSC thermograms of pure drug and its physical mixture with each of the excipients viz. sodium alginate, gellan, calcium carbonate, and gelatin were also recorded. Approximately 3–5 mg of the samples were transferred in an aluminium pan and heated at a rate of 10°C.min−1 up to 300°C under nitrogen environment at a flow rate of 20 mL.min−1. Thermal analyses of DSC thermograms were conducted using the Q Series Thermal Advantage DSC software (DSC Q20, M/s TA Instruments, DE, USA).
Ex Vivo Gastroretention Studies
Ex vivo gastroretention potential of the optimized formulation was determined by oral administration of formulation mixed with methylene blue dye. The Wistar rats were sacrificed by cervical dislocation, and stomach was excised followed by dissection at an interval of 3 and 6 h after oral administration to check the retention of the formulation in the stomach.
In Vivo Pharmacokinetic Studies
In vivo parallel pharmacokinetic studies of the stimuli-responsive GR formulation were performed as per the protocol approved by the Institutional Ethical Committee of Panjab University, Chandigarh, India. Twelve healthy unisex Wistar rats (weighing 250–300 g) were employed for the current studies and kept under standard laboratory conditions at 25 ± 2°C and 55 ± 5% RH with free access to standard diet and tap water ad libitum. Prior to experimentation, the animals were divided into two groups, each containing six animals. Group I was administered with marketed oral suspension (Zovirax®, M/s GlaxoSmithKline, New Delhi, India), and group II was administered with optimized stimuli-responsive GR formulation orally using a stomach sonde needle for rats. All the animal groups received formulation containing a dose equivalent to 80 mg of acyclovir. Animals were anesthetized, and blood samples were collected in heparinized tubes by retro-orbital puncture at predetermined time intervals. Plasma was separated by centrifugation at 10,000 rpm (5590×g) for 10 min and stored at −80°C until analyzed using HPLC. The detail experimental protocol regarding the development and validation of HPLC method has been given in supplementary material text, Section 1, while the information on preparation of bioanalytical samples has been mentioned in supplementary material text, Section 2.
Computer-based pharmacokinetic data analysis and modeling on plasma drug concentration versus time data was carried out employing Win-Nonlin version 5.0 (M/s Pharsight, CA, USA). The files were created with the plasma concentration data along with other pertinent information. The data were fitted in 1-Compartment Body Model (1-CBM), and various pharmacokinetic parameters like maximum plasma concentration (C max) and the corresponding time (t max), area under the curve (AUC0–24 h) and the total area under the curve (AUC0−∞), absorption rate constant (K a), and elimination rate constant (K) were computed using Wagner–Nelson method and their statistical validity ratified. Statistical validity of the results were discerned on the basis of minimization of various model fitness parameters like Akaike information criterion (AIC), Schwartz criterion (SC), sum of squares of residuals (SSR), and maximization of Pearsonian correlation (R) and model selection criteria (MSC). The experimental results were statistically analyzed by two-way analysis of variance (ANOVA) using GraphPad Prism software ver 5.0 (M/s GraphPad Software Inc., CA, USA) with the statistical significance set at 5%.
In Vitro/In Vivo Correlation
Level A correlations were attempted between the in vivo pharmacokinetic parameter and the in vitro dissolution parameter for optimized stimuli-responsive GR formulation and the marketed oral suspension. For exploring the level A in vitro/in vivo correlation (IVIVC), a fraction of drug absorbed in vivo at various time points obtained using modified Wagner–Nelson method were correlated with fraction of drug release in vitro at the corresponding time points.
Stability Studies
Stability studies of the optimized formulation were carried out at refrigerated conditions (5 ± 1°C) for 6 months. The samples were packaged in air-tight glass amber-colored bottles and evaluated for drug content, dissolution performance, gel strength, viscosity, and onset of floatation at predetermined time points, 0, 1, 2, 3 and 6 months, respectively.
RESULTS AND DISCUSSION
Characterization of Stimuli-Responsive GR Systems
Rheological Studies
All the formulations prepared as per the experimental design exhibited non-Newtonian rheological behavior, characterizing a typical pseudoplastic flow. The values of sol viscosity ranged between 45.6 and 493.3 mPas at pH 6.8, which was the inherent pH of the prepared formulations. The viscosity of stimuli-responsive GR systems increased linearly with increase in the content of each of the polymers, with more prominent influence of sodium alginate owing to the entanglement of a polysaccharide backbone in the presence of gastric fluid and plausible cross-linking, leading eventually to the enhancement in rheological properties of the formulations (31). Gellan, however, being cationic in nature, partially influences the viscosity of in situ gel formulation owing to its gel-forming nature at the alkaline pH (32).
Gel Strength Measurement
The gel strength of different formulations prepared as per the experimental design was found to range between 0.163 and 1.412 kg (Figure 1 of supplementary data). The gel strength exhibited a linear increasing trend with increase in the concentration of each polymer. Interestingly, the effect of gellan was more prominent on the gel strength vis-à-vis sodium alginate, which can be attributed to the superior rheological and gelling properties of the former (32).
In Vitro Floating Studies
The in vitro floating studies revealed that the prepared stimuli-responsive GR formulations exhibited onset of floatation ranging between 3 and 13 min. The onset of floatation time increased quite significantly with increase in the concentration of each polymer, ostensibly owing to the swelling or hydration of the polymer hydrocolloid particles in the presence of gastric fluid. It has already been documented in literature that the balance between polymer swelling and water acceptance is a vital factor to ensure floatation of the formulations (33). The generation of carbon dioxide gas due to the presence of calcium carbonate as gas-generating agent in the dosage form helped in floatation of in situ gel in the gastric medium. Also, the polymer hydration and gelation properties contributed in attaining the floatation characteristics of the formulation.
In Vitro Drug Release Studies
Figure 1 illustrates the in vitro dissolution profile of all the formulations (F1–F9) prepared as per the CCD. The values of Q 16 h for the prepared GR floating system ranged between 57.24 and 99.98%. Drug release profiles from the formulations portrayed a linear decreasing trend with an increase in the concentration of sodium alginate and gellan. The formulations containing higher concentrations of sodium alginate (F7–F9) revealed precise control of drug release as compared to the formulations with medium (F4–F6) to lower (F1–F3) levels of sodium alginate. The T 60% values exhibited an increasing trend with an increase in the levels of either of the polymers. The values of T 60% were selected to represent the extension in drug release profile, as it was found to be a much more discriminating attribute in comparison to other possible parametric options like T 70%, T 80%, and T 90%. Likewise, selection of Q 16 h was undertaken to investigate whether any significant amount of drug unreleased would remain in the polymer system as captive or not. Evaluation of drug release kinetics of the prepared formulations as per the Korsmeyer–Peppas equation indicated that the values of release rate exponent (n) ranged between 0.228 and 0.551, connoting a Fickian to quasi-Fickian mechanism of drug release (34). Table 3 of supplementary data enlists the details on various dissolution and kinetic parameters of the prepared formulations. The dissolution parameters obtained in the data were found to be in consonance with the release kinetics parameter, where lower values of “n” at higher concentrations of polymers owe to sustained drug release profile revealing the Fickian diffusion mechanism and vice versa.
Fig. 1.
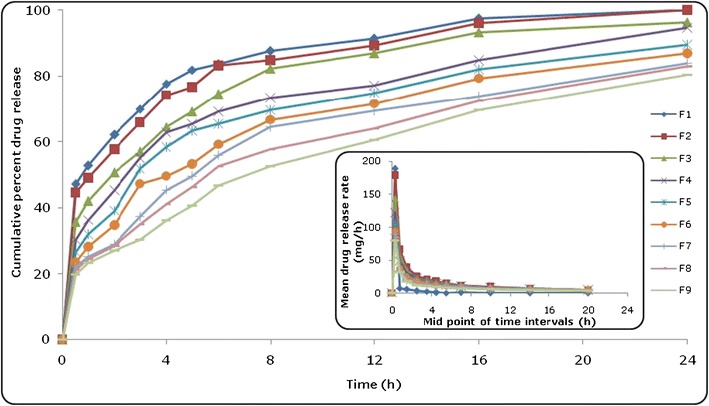
In vitro drug release profiles of formulations (F1–F9) prepared as per the experimental design. The inset depicts the mean drug release rate versus mid-point of time intervals
Response Surface Analysis
The coefficients of the polynomial equation, generated as per Eq. (3) using multiple linear regression analysis (MLRA) for all the CQAs, i.e., η, G s, F o, t 60%, and Q 16 h, revealed excellent goodness of fit to the data, with the values of r 2 ranging between 0.990 and 1.000 (p < 0.001 in each case).
| 3 |
where Y stands for the response variables (i.e., CQAs) investigated for the prepared formulation, β 0–β 7 represents the coefficients of polynomial equations generated for each response variable, and X 1 and X 2 are the CMAs.
Figure 2a–e depicts the 3D response surface plots for various CQAs. Figure 2a illustrates the 3D response surface plot for viscosity, where a sharp ascending trend was observed with an increase in the concentration of sodium alginate at the low levels of gellan. However, an increase in the concentration of gellan shows a relatively negligible influence on the viscosity of formulation. This may be due to the swellable nature of the hydrocolloid (i.e., sodium alginate), which upon contacting with acidic gastric fluid leads to the formation of “gel” phase. Likewise, the response surface plot in Fig. 2b demonstrated an identical relationship between the amount of sodium alginate and gellan on the gel strength of stimuli-responsive GR formulations. In the response surface plot portrayed in Fig. 2c for onset of floatation, a linear increasing trend was observed with an increase in the alginate and gellan fractions. Like viscosity and gel strength, alginate was found to exert greater influence on the onset of floatation with respect to gellan. The faster onset of floatation was observed at lower levels of both sodium alginate and gellan. The 3D response surface plot in Fig. 2d depicts an increasing trend for T 60% with augmentation in the values of sodium alginate and gellan fractions. However, the influence of sodium alginate on T 60% was observed to be more prominent, as is evident from the sharp inclining pattern of increase in T 60%. The maximum values for T 60% were observed at the higher levels of both the polymers. Conversely, the response surface plot depicted in Fig. 2e revealed a linearly descending trend for Q 16 h with an increase in the concentration of sodium alginate and gellan. Minimum value of Q 16 h was observed at the highest levels of sodium alginate and gellan.
Fig. 2.
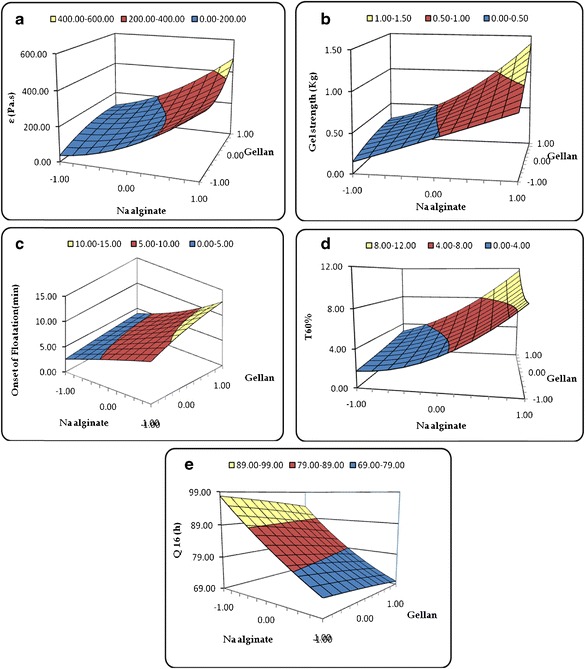
3D response surface plots depicting the influence of CMAs, i.e., amounts of sodium alginate and gellan on the CQAs, a viscosity (η), b gel strength (G s), c onset of floatation (F o), d time taken for 60% drug release (T 60%), and e amount of drug released in 16 h (Q 16 h)
Search for Optimum Formulation and Validation of QbD
Search for the optimized formulation was carried out with the help of feasibility search/numerical optimization desirability function by “trading off” various CQAs to attain the desired goals, i.e., maximization of values of Q 16 h, T 60%, and Gs and minimizing the values of η and F O, to obtain the desirability function close to 1. The optimized formulation contained sodium alginate (104 mg) and gellan (97 mg), exhibited η of 117.04 Pas, G s of 0.291 kg, F o of 4.1 min, T 60% of 2.88 h, and Q 16 h of 93.16%, respectively. Figure 3 depicts the design space showing the optimal region and optimized formulation, respectively.
Fig. 3.
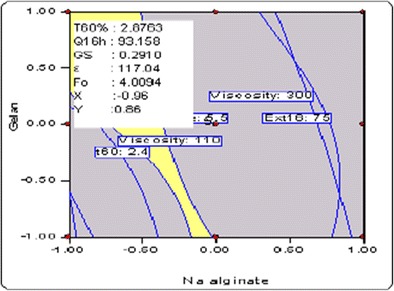
Overlay plot depicting the design space region and optimized formulation
Validation of the QbD methodology was accomplished by preparing eight check-point formulations and comparison of their observed responses with those predicted ones. The percent prediction error (i.e., percent bias) for the CQAs varied between −6.38 and 3.33%, with overall mean ± SEM as −0.134 ± 2.193, with higher values of r 2 ranging between 0.982 and 0.999, thus ratifying excellent goodness of fit of the data (p < 0.001 in each case) (Figure 2 of supplementary data). The corresponding residual plots were found to be quite regulated with a relatively narrow, uniform, and random scatter around zero axis, indicating high degree of prognosis of the QbD approach.
Sol to Gel Transformation Studies
On keeping the optimized formulation in contact with the solutions of different pH, the meniscus was visually observed by inversion or tilting of the test tubes. It was observed that the “sol” form of the formulations got immediately transformed into the “gel” state below pH 4 with viscosities of 125.2, 152.1, 178.6, and 182.3 Pas for pH 4, 3, 2, and 1.2, respectively (Fig. 4). Absence of gel formation at alkaline pH can be attributed to the dissociation of calcium carbonate as its constituent ions, which inhibit the gelling of sodium alginate and gellan. Interestingly, this phenomenon is favorable, as the formulation is intended to remain in the “sol” form during its sojourn in the oral cavity and esophagus and gets transformed to the gel state after reaching the gastric environment (35).
Fig. 4.

Sol to gel transformation studies at different pH conditions
Ex Vivo Gastroretention Studies
Visual observation of the rat stomach following administration of “sol” form of the optimized stimuli-responsive GR formulation (without drug) containing a marker dye indicated that approximately 86% of the soft gel remained after 3 h of administration and 73% gel remained after 6 h of administration (Fig. 5). Maintenance of the integrity of the gel over this time period probably resulted in the prolongation of drug release from the gel matrix within the region of absorption window, as is evident from the absence of color indication of the dye in the intestine. This confirmed the gastroretentive potential of the optimized stimuli-responsive GR formulation, as it gelled and floated instantaneously in the pH conditions of the stomach.
Fig. 5.
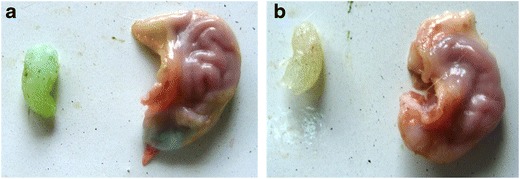
Excised rat stomach administered with in situ gelling GR formulation containing methylene blue after 3 h (a) and after 6 h (b)
Drug-Excipient Compatibility Studies
Fourier Transform Infrared Spectroscopy
Figure 6 portrays the overlay FTIR spectra of pure drug and its physical mixtures with excipients viz. sodium alginate, gellan, gelatin, and calcium carbonate. The IR spectrum of acyclovir shows characteristic absorption bands at 3522.2 cm−1 (indicating aromatic C–H stretching), 3297.9 cm−1 (indicating methyl C–H stretching), 1538.8 and 1575.3 cm−1 (indicating C = ring stretching), 1287.7 cm−1 (indicating asymmetric C–O–C stretching), 3440.0 cm−1 (aliphatic tertiary N–H stretch), 1232.4 cm−1 and 1216.5 cm−1 (aliphatic tertiary C–N stretch), and 1483.3 cm−1 (CH2 bend). However, the IR spectra of physical mixtures of drug with each of the excipients did not reveal any shift(s) in the peaks, indicating absence of any interaction(s) among them.
Fig. 6.
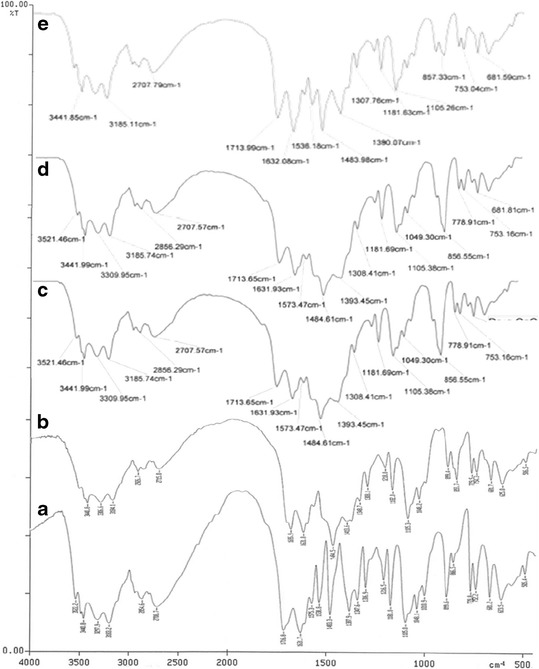
FTIR spectra: a pure drug, b drug + sodium alginate and gellan, c drug + calcium carbonate, d drug + gelatin, e physical mixture of all excipients with drug
Differential Scanning Calorimetry
Figure 7 illustrates the DSC thermograms of pure drug and its physical mixture with each of the studied excipients. The DSC thermogram of pure drug showed a single fusion endothermic peak at 246.83°C. Likewise, other DSC thermograms of physical mixture of drug with individual excipients did not exhibit any significant change in the melting point of drug, thus connoting a lack of any significant interaction(s) among them.
Fig. 7.
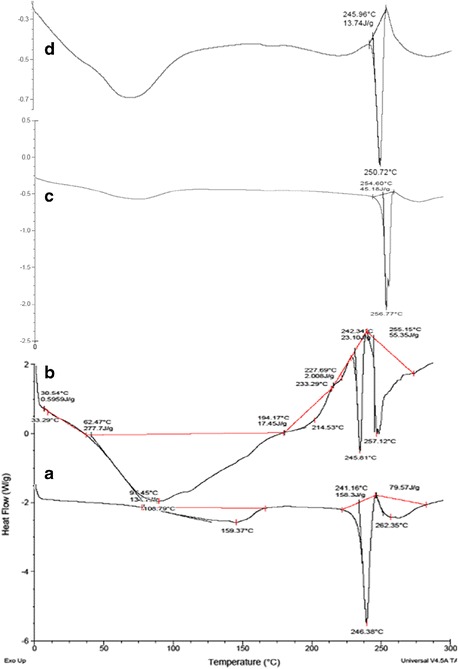
DSC thermograms: a pure drug, b drug + sodium alginate and gellan, c drug + calcium carbonate, d physical mixture of all excipients with drug
In Vivo Pharmacokinetic Studies
Figure 8 portrays the plasma concentration time profiles of acyclovir with mean ± SD values of marketed suspension (ZOVIRAX®) and optimized stimuli-responsive GR formulation. The pharmacokinetic profiles showed highly significant statistical difference (p < 0.05) in the plasma concentration at all the studied time points. The data analysis was carried out using the biexponential 1-CBM kinetics without lag time for calculation of the pharmacokinetic parameters. Table III vividly illustrates various pharmacokinetic parameters, i.e., C max, T max, AUC24 h, K a, and C max/AUC24 h ratio of the optimized stimuli-responsive GR formulation and the marketed formulation. Among all the studied pharmacokinetic parameters, maximal change (i.e., nearly 3.2-fold extension) was observed in T max with respect to the marketed formulation (p < 0.001). Further, the extent of drug absorption was also found to be significantly enhanced with 22% improvement in AUC24 h (p < 0.001). However, there was no statistically significant change observed in case of pharmacokinetic parameters like C max and C max/AUClast ratio.
Fig. 8.
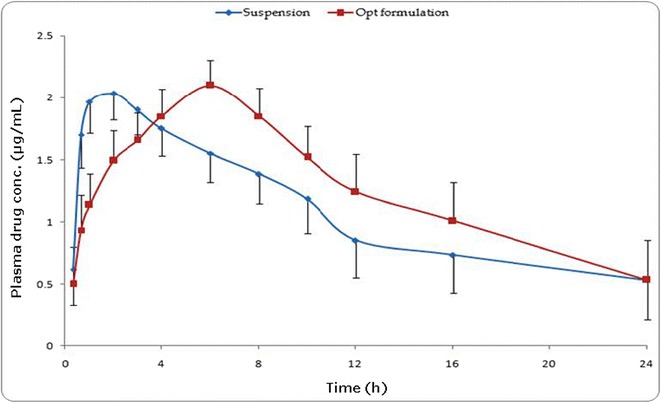
Graph showing concentration of drug in rat plasma at various time points following administration of the optimized stimuli-responsive formulation and marketed oral suspension (Zovirax®)
Table III.
Pharmacokinetic Parameters Obtained after Oral Administration of Various Formulations of Acyclovir*
| Formulations | Pharmacokinetic parameters | ||||
|---|---|---|---|---|---|
| C max | AUC24 h | C max/AUC24 h | K a | T max | |
| (μg.mL−1) | (μg.h.mL−1) | (h−1) | (h−1) | (h) | |
| Marketed oral suspension (ZOVIRAX®) | 2.05 ± 0.22 | 32.88 ± 13.00 | 0.069 ± 0.03 | 2.41 ± 0.86 | 1.93 ± 0.15 |
| Optimized stimuli-responsive GRDDS | 2.11 ± 0.20 | 40.22 ± 17.71 | 0.060 ± 0.03 | 0.42 ± 0.08 | 6.13 ± 0.70 |
GRDDS gastroretentive drug delivery systems
*Data represented as mean ± SD (n = 6)
The relatively high magnitude of T max and improved AUC24 h of the drug from the stimuli-responsive GR formulations vis-à-vis the marketed formulation construed controlled drug release profile and enhanced drug bioavailability from the former. This was further supported by significant reduction in the value of K a revealing the extension in the drug absorption (p < 0.01). Appreciable improvement in the systemic bioavailability of the optimized formulation can be attributed to the increase in gastric residence time vis-à-vis the marketed formulation. Moreover, this construed improvement in the biopharmaceutical performance of the developed formulation.
In Vitro/In Vivo Correlation
Level A IVIVC was attempted between the fractions of drug dissolved and absorbed at respective time points. Figure 9 summarizes the statistical parameters of level A IVIVC employing different curve fitting approaches. Higher magnitude of r 2 was observed for the stimuli-responsive GR formulations indicating a linear relationship, while the marketed formulation showed non-linear model fitting between the in vitro dissolution and in vivo absorption parameters. The results confirmed the prevalence of IVIVC with the prepared stimuli-responsive GR formulation.
Fig. 9.
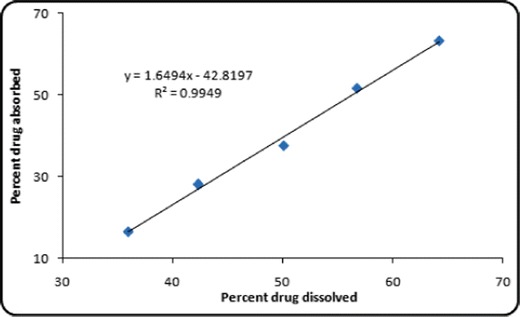
Level A IVIVC for the optimized stimuli-responsive GR systems
Acyclovir as a BCS class IV drug exhibit incomplete gastrointestinal absorption owing to inadequate permeability and lipophilicity (36). Thus, dissolution-limited or permeability-limited absorption is considered to be a major rate-governing step. Since the optimized stimuli-responsive GR formulation of acyclovir released the drug in a sustained manner, the in vivo absorption was able to keep a linear pace with the in vitro dissolution construing that the drug absorption behaved linearly with the dissolution of the drug. The marketed suspension, however, incomplete drug absorption was occurred plausibly owing to the poor retentivity of the dosage form within the gastric region for drug absorption. Accordingly, as was anticipated, IVIVC could not be attempted due to the rapid drug dissolution and, hence, inadequate number of data points.
Stability Studies
The stability studies of the optimized formulations revealed a lack of any significant difference in various physical and dissolution parameters like drug content, viscosity, gel strength, onset of floatation, and in vitro drug release over a period of 6 months at long-term stability. This corroborated high degree of physiochemical stability of the stimuli-responsive formulation over the periods of storage.
CONCLUSIONS
The present studies successfully vouch the use of rational QbD-oriented approach for the development of optimized stimuli-responsive GR system of acyclovir employing a simple and cost-effective approach for improving the patient compliance coupled with oral bioavailability improvement. Preformulation and risk assessment studies facilitated the selection of sodium alginate and gellan as the CMAs for formulation development employing the central composite design. The optimized formulation fulfilled maximum requisites of low viscosity “sol” form, lower onset of floatation, higher gel strength, and higher regulation of drug release rate. Further, the in vitro gastroretention studies and in vivo pharmacokinetic studies in rats indicated a significant extension of drug absorption rate along with improved oral bioavailability vis-à-vis the marketed formulation. The promising outcomes of the current studies ratified successful development of the optimized stimuli-responsive GR formulation of acyclovir for improving its patient compliance and oral bioavailability. The results can be rationally extrapolated to other BCS class II/IV drugs.
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
(DOCX 726 kb)
Acknowledgments
Conflict of Interest
The authors declare that they have no competing interests.
References
- 1.Davis SS. Formulation strategies for absorption windows. Drug Discov Today. 2005;10(4):249–57. doi: 10.1016/S1359-6446(04)03351-3. [DOI] [PubMed] [Google Scholar]
- 2.Kotreka UK, Adeyeye MC. Gastroretentive floating drug-delivery systems: a critical review. Crit Rev Ther Drug Carrier Syst. 2011;28(1):47–99. doi: 10.1615/CritRevTherDrugCarrierSyst.v28.i1.20. [DOI] [PubMed] [Google Scholar]
- 3.Gupta H, Bhandari D, Sharma A. Recent trends in oral drug delivery: a review. Recent Pat Drug Deliv Formul. 2009;3(2):162–73. doi: 10.2174/187221109788452267. [DOI] [PubMed] [Google Scholar]
- 4.Prajapati VD, Jani GK, Khutliwala TA, Zala BS. Raft forming system-an upcoming approach of gastroretentive drug delivery system. J Control Release. 2013;168(2):151–65. doi: 10.1016/j.jconrel.2013.02.028. [DOI] [PubMed] [Google Scholar]
- 5.Madan M, Bajaj A, Lewis S, Udupa N, Baig JA. In situ forming polymeric drug delivery systems. Indian J Pharm Sci. 2009;71(3):242–51. doi: 10.4103/0250-474X.56015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bawa P, Pillay V, Choonara YE, du Toit LC. Stimuli-responsive polymers and their applications in drug delivery. Biomed Mater. 2009;4(2):022001. doi: 10.1088/1748-6041/4/2/022001. [DOI] [PubMed] [Google Scholar]
- 7.Madhugiri P, Venkatesh PKL, Kumar TMP, Shivakumar HG. In situ gels based drug delivery systems. Curr Drug Therapy. 2014;6(3):213–22. [Google Scholar]
- 8.Patel RB, Chauhan MA, Shah AL, Suthar SJ, Patel MP, Patel JK. Floating in situ gel: new trends in controlled and sustained gastroretentive drug delivery system. Res J Pharm Tech. 2012;5(7):889–93. [Google Scholar]
- 9.Mehta P. Acyclovir. Pediatr Infect Dis. 2013;5(4):178–80. doi: 10.1016/j.pid.2013.11.004. [DOI] [Google Scholar]
- 10.Ruiz-Caro R, Gago-Guillan M, Otero-Espinar FJ, Veiga MD. Mucoadhesive tablets for controlled release of acyclovir. Chem Pharm Bull. 2012;60(10):1249–57. doi: 10.1248/cpb.c12-00324. [DOI] [PubMed] [Google Scholar]
- 11.Tao Y, Lu Y, Sun Y, Gu B, Lu W, Pan J. Development of mucoadhesive microspheres of acyclovir with enhanced bioavailability. Int J Pharm. 2009;378(1–2):30–6. doi: 10.1016/j.ijpharm.2009.05.025. [DOI] [PubMed] [Google Scholar]
- 12.Kyada C, Ranch K, Shah D. Optimization of mucoadhesive microspheres of acyclovir by applying 32 full factorial design. J Drug Deliv Sci Technol. 2014;24(1):61–8. doi: 10.1016/S1773-2247(14)50009-3. [DOI] [Google Scholar]
- 13.El Gamal SS, Naggar VF, Allam AN. Optimization of acyclovir oral tablets based on gastroretention technology: factorial design analysis and physicochemical characterization studies. Drug Dev Ind Pharm. 2011;37(7):855–67. doi: 10.3109/03639045.2010.546404. [DOI] [PubMed] [Google Scholar]
- 14.Kharia AA, Hiremath SN, Singhai AK, Omray LK, Jain SK. Design and optimization of floating drug delivery system of acyclovir. Indian J Pharm Sci. 2010;72(5):599–606. doi: 10.4103/0250-474X.78527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bhosale UV, Devi K, Choudhary S. Multiunit floating drug delivery system of acyclovir: development, characterization and in vitro-in vivo evaluation of spray-dried hollow microspheres. J Drug Deliv Sci Technol. 2012;22(6):548–54. doi: 10.1016/S1773-2247(12)50095-X. [DOI] [Google Scholar]
- 16.Chaudhary B, Verma S. Preparation and evaluation of novel in situ gels containing acyclovir for the treatment of oral herpes simplex virus infections. Sci World J. 2014;2014:1–7. doi: 10.1155/2014/280928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vora C, Patadia R, Mittal K, Mashru R. Risk based approach for design and optimization of stomach specific delivery of rifampicin. Int J Pharm. 2013;455(1–2):169–81. doi: 10.1016/j.ijpharm.2013.07.043. [DOI] [PubMed] [Google Scholar]
- 18.Singh B, Kumar R, Ahuja N. Optimizing drug delivery systems using systematic "design of experiments." Part I: fundamental aspects. Crit Rev Ther Drug Carrier Syst. 2005;22(1):27–105. doi: 10.1615/CritRevTherDrugCarrierSyst.v22.i1.20. [DOI] [PubMed] [Google Scholar]
- 19.Singh B, Kapil R, Nandi M, Ahuja N. Developing oral drug delivery systems using formulation by design: vital precepts, retrospect and prospects. Expert Opin Drug Deliv. 2011;8(10):1341–60. doi: 10.1517/17425247.2011.605120. [DOI] [PubMed] [Google Scholar]
- 20.Yu LX, Amidon G, Khan MA, Hoag SW, Polli J, Raju GK, et al. Understanding pharmaceutical quality by design. AAPS J. 2014;16(4):771–83. doi: 10.1208/s12248-014-9598-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Venkatesh MP, Liladhar PK, Kumar TMP, Shivakumar HG. In situ gels based drug delivery systems. Curr Drug Ther. 2014;6(3):213–22. doi: 10.2174/157488511796392004. [DOI] [Google Scholar]
- 22.James HP, John R, Alex A, Anoop KR. Smart polymers for the controlled delivery of drugs—a concise overview. Acta Pharm Sin B. 2014;4(2):120–7. doi: 10.1016/j.apsb.2014.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hamcerencu M, Desbrieres J, Popa M, Riess G. Original stimuli-sensitive polysaccharide derivatives/N-isopropylacrylamide hydrogels. Role of polysaccharide backbone. Carbohydr Polym. 2012;89(2):438–47. doi: 10.1016/j.carbpol.2012.03.026. [DOI] [PubMed] [Google Scholar]
- 24.Choi BY, Park HJ, Hwang SJ, Park JB. Preparation of alginate beads for floating drug delivery system: effects of CO(2) gas-forming agents. Int J Pharm. 2002;239(1–2):81–91. doi: 10.1016/S0378-5173(02)00054-6. [DOI] [PubMed] [Google Scholar]
- 25.Abou Youssef NA, Kassem AA, El-Massik MA, Boraie NA. Development of gastroretentive metronidazole floating raft system for targeting Helicobacter pylori. Int J Pharm. 2015;486(1–2):297–305. doi: 10.1016/j.ijpharm.2015.04.004. [DOI] [PubMed] [Google Scholar]
- 26.www.accessdata.fda.gov/scripts/cder/iig/getiigWEB.cfm. Accessed on 12 June 2015.
- 27.https://dailymed.nlm.nih.gov/dailymed/archives/fda. Accessed on 17 June 2015.
- 28.Singh B, Kaur T, Singh S. Correction of raw dissolution data for loss of drug during sampling. Indian J Pharm Sci. 1997;59:196–9. [Google Scholar]
- 29.Korsmeyer RW, Gurny R, Doelker EM, Buri P, Peppas NA. Mechanism of solute release from porous hydrophilic polymers. Int J Pharm. 1983;15:25–35. doi: 10.1016/0378-5173(83)90064-9. [DOI] [PubMed] [Google Scholar]
- 30.Li S, Lin S. Effect of HPMC and Carbopol on the release and floating properties of gastric floating drug delivery system using factorial design. Int J Pharm. 2003;253:13–22. doi: 10.1016/S0378-5173(02)00642-7. [DOI] [PubMed] [Google Scholar]
- 31.Zhang H, Wang H, Wang J, Guo R, Zhang Q. The effect of ionic strength on the viscosity of sodium alginate solution. Polym Adv Technol. 2001;12:740–5. doi: 10.1002/pat.97. [DOI] [Google Scholar]
- 32.Nitta Y, Nishinari K. Gelation and gel properties of polysaccharides gellan gum and tamarind xyloglucan. J Biol Macromol. 2005;5(3):47–52. [Google Scholar]
- 33.Baumgartner S, Kristl J, Vreer F, Vodopivec P, Zorko B. Optimisation of floating matrix tablets and evaluation of their gastric residence time. Int J Pharm. 2000;195:125–35. doi: 10.1016/S0378-5173(99)00378-6. [DOI] [PubMed] [Google Scholar]
- 34.Korsmeyer R, Gurny R, Doelker E, Buri P, Peppas N. Mechanisms of potassium chloride release from compressed, hydrophilic, polymeric matrices: effect of entrapped air. J Pharm Sci. 1983;72:1189–91. doi: 10.1002/jps.2600721021. [DOI] [PubMed] [Google Scholar]
- 35.Nagarwal R, Srinatha A, Pandit J. In situ forming formulation: development, evaluation, and optimization using 33 factorial design. AAPS PharmSciTech. 2009;10:977–84. doi: 10.1208/s12249-009-9285-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Amidon G, Lennernas H, Shah V, Crison J. A theoretical basis for a biopharmaceutical drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm Res. 1995;12:413–20. doi: 10.1023/A:1016212804288. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOCX 726 kb)