

Abstract
The objective of this study was to investigate the pH-dependent solubility and dissolution of weakly basic Biopharmaceutical Classification Systems (BCS) class II drugs, characterized by low solubility and high permeability, using carvedilol, a weak base with a pK a value of 7.8, as a model drug. A series of solubility and in vitro dissolution studies was carried out using media that simulate the gastric and intestinal fluids and cover the physiological pH range of the GI from 1.2 to 7.8. The effect of ionic strength, buffer capacity, and buffer species of the dissolution media on the solubility and dissolution behavior of carvedilol was also investigated. The study revealed that carvedilol exhibited a typical weak base pH-dependent solubility profile with a high solubility at low pH (545.1–2591.4 μg/mL within the pH range 1.2–5.0) and low solubility at high pH (5.8–51.9 μg/mL within the pH range 6.5–7.8). The dissolution behavior of carvedilol was consistent with the solubility results, where carvedilol release was complete (95.8–98.2% released within 60 min) in media simulating the gastric fluid (pH 1.2–5.0) and relatively low (15.9–86.2% released within 240 min) in media simulating the intestinal fluid (pH 6.5–7.8). It was found that the buffer species of the dissolution media may influence the solubility and consequently the percentage of carvedilol released by forming carvedilol salts of varying solubilities. Carvedilol solubility and dissolution decreased with increasing ionic strength, while lowering the buffer capacity resulted in a decrease in carvedilol solubility and dissolution rate.
KEY WORDS: buffer species, carvedilol, dissolution, ionic strength, solubility
INTRODUCTION
The dissolution of low-solubility and high-permeability, Biopharmaceutical Classification Systems (BCS) class II drugs, is rate-limiting to oral absorption (1–3). Weakly basic BCS class II drugs such as carvedilol (Fig. 1) exhibit a complex solubility pattern due to the pH gradient in the gastrointestinal (GI) fluid during transition from the stomach to the intestine. This class of drugs favors ionization and adequate solubilization in the stomach at low pH (4). However, reaching the intestinal environment (high pH), drug precipitation may occur due to its lower aqueous solubility (4, 5). Carvedilol, a nonselective beta blocker/alpha-1 blocker, is indicated in the treatment of mild to severe congestive heart failure and high blood pressure. It has a log P value of 3.8 with a basic pK a of 7.8 (6) and an acidic pK a of 15.0 (calculated using ChemAxon software). Carvedilol exhibits pH-dependent solubility (7). It dissolves in the acidic pH of the stomach, as it is presented in its ionized form. However, in basic pH, carvedilol may precipitate in the distal small intestine (6, 8, 9). Stillhart et al., for example, found that 78.2–91.8% of carvedilol precipitates in a crystalline or amorphous form under digestion condition (9). Due to its poor aqueous solubility, carvedilol exhibits very low bioavailability (10, 11). The oral absorption of carvedilol was predicted using GastroPlus™ simulation to be ~45.9% (6).
Fig. 1.
Chemical structure of carvedilol
In general, dissolution of BCS class II drugs is dependent on a wide variety of physiological factors. pH, ionic strength, and buffer capacity are three major characteristics of the GI fluids that can affect the rate of drug release (2, 12). Typical pH values in the stomach in the fasted state are within the range of 1.4–2.1. In the fed state, the pH of gastric fluid increases to 3.0–7.0 immediately following a meal, depending on its composition (1). The ionic strength of the gastric fluid is estimated to be in the range of 0.051–0.151 mol/L, with a median of 0.095 mol/L (13). The buffer capacity of human gastric fluids was found to be within the range from 0.013 to 0.019 M/ΔpH in the fasted state (14) and approximately 0.028 M/ΔpH in the fed state (15). On the other hand, the pH values in the small intestine gradually rise between the duodenum and ileum to a range of 5.5–8.3, depending on the location and fed and fasted states (1, 14, 16). This increase in pH is due to the neutralization of acid coming from the stomach with bicarbonate ions secreted by the pancreas (1, 12). The ionic strength of the intestinal fluid was found to be within the range of 0.070–0.166 mol/L (17). The buffer capacity values for human intestinal fluids in the fasted and fed states were reported to be 0.003 and 0.013 M/ΔpH, respectively (18). Kalantzi et al. measured the buffer capacity of human duodenal aspirates and found that the median buffer capacity in the fasted duodenum was 0.006 M/ΔpH, which was increased to 0.018–0.030 M/ΔpH in the fed state (15).
Since the physiology of GI fluids, such as pH, buffer capacity, and ionic strength, varies as a function of food intake and location within the gastrointestinal tract, studying the influence of these factors on the solubility and dissolution of the weakly basic BCS class II drugs in the GI environment is of considerable interest. The objective of the present work was therefore to study the pH-dependent solubility and dissolution behavior of the weakly basic BCS class II drug carvedilol in simulated gastric and intestinal fluids that cover the physiological pH range of GI fluids from pH 1.2 to pH 7.8. We also investigated the effect of buffer species, ionic strength, and buffer capacity on the solubility and dissolution behavior of carvedilol from Dilatrend® immediate-release tablets. Our study provides a better understanding of the solubility and in vitro dissolution behavior of weakly basic BCS class II drugs represented by carvedilol under the physiological pH range of GI fluids. Such understanding serves to improve our knowledge of the dissolution and precipitation behavior of these drugs in the intestine and provide guidance on the development of predictive dissolution methods with improved in vitro-in vivo correlations.
MATERIALS AND METHODS
Materials
Carvedilol powder (100.3% purity and 0.1% total impurities; Moehs Catalana, S. L., Barcelona, Spain) was provided as a gift from Hikma Pharmaceuticals (Amman, Jordan). No further confirmatory methods were done in our laboratory to substantiate the purity. Dilatrend® immediate-release (IR) tablets, 25 mg carvedilol (ROCHE Australia, lot # M2049B01), were purchased from the market. Sodium dihydrogen phosphate (NaH2PO4) and disodium hydrogen phosphate (Na2HPO4) were purchased from Sigma-Aldrich (Germany); potassium dihydrogen phosphate (KH2PO4), sodium hydroxide (NaOH), and sodium chloride (NaCl) were purchased from Riedel-de Haën (Germany); acetic acid (CH3COOH) was purchased from ACROS (USA); sodium acetate trihydrate (CH3COONa·3H2O) was purchased from Fluka (USA); and hydrochloric acid (HCl, 37% w/v) was purchased from GFS Chemicals (USA). Double-distilled water was obtained using a GFL water distilling apparatus (Germany). All chemical reagents were of analytical grade.
Media Used for Solubility and Dissolution Studies
Simulated gastric and intestinal fluids were prepared and used in this study. Dissolution media that were used to simulate the gastric fluid are (a) 0.7% HCl (USP 38, pH 1.45), (b) simulated gastric fluid without pepsin (SGFsp, USP 38; pH 1.2), (c) blank fasted-state simulated gastric fluid (blank FaSSGF, pH 1.6) (19), and (d) blank fed-state simulated gastric fluid (blank FeSSGF, pH 5.0) (20). Dissolution media that were used to simulate the intestinal fluid are (a) simulated intestinal fluid without pancreatin (SIFsp, USP 26; pH 6.8), (b) 0.05 M phosphate buffer (International Pharmacopeia, IntPh3; pH 6.8) (21), (c) acetate buffer (USP 28, pH 4.5) that simulates the duodenal fluid, (d) blank fasted-state simulated intestinal fluid (blank FaSSIF, pH 6.5), and (d) blank fed-state simulated intestinal fluid (blank FeSSIF, pH 5.0) (22). In this study, the word “blank” refers to dissolution media prepared without the addition of enzymes, surfactants, or bile salts.
In addition to the standard compendial media, simulated intestinal fluid using phosphate buffers of pH 6.8 was prepared using different buffer concentrations (6.25, 12.5, 25, 50, and 100 mM) to obtain media with variable ionic strengths and buffer capacities. Phosphate buffer (100 mM, pH 6.8) was prepared according to the Sigma-Aldrich Buffer Reference Center. Double-distilled water of zero ionic strength and zero buffer capacity was used as a control. Phosphate buffers of pH 7.2 and 7.8 media were also prepared according to the Sigma-Aldrich Buffer Reference Center to simulate the pH during the fasted state of the proximal ileum (1, 23) and colon (24), respectively. The detailed chemical composition and physicochemical properties (pH, ionic strength, and buffer capacity) of the media used in this study are summarized in Tables I, II, III, and IV.
Table I.
Composition and Physicochemical Properties of 0.7% HCl, SGFsp, Blank FaSSGF, and Blank FeSSGF
| 0.7% HCl | SGFsp | Blank FaSSGF | Blank FeSSGF | |
|---|---|---|---|---|
| Composition | ||||
| HCl, 37% w/v (mM) | 83.7 | 83.7 | 35.9 | – |
| CH3COOH (mM) | – | – | – | 17.1 |
| CH3COONa·3H2O (mM) | – | – | – | 29.8 |
| NaCl (mM) | – | 34.2 | 34.2 | 237.0 |
| Double-distilled water (L) | 1.0 | 1.0 | 1.0 | 1.0 |
| Physicochemical properties | ||||
| pH | 1.5 ± 0.1 | 1.2 ± 0.1 | 1.6 ± 0.1 | 5.0 ± 0.1 |
| Ionic strength (mol/L) | 0.084 | 0.118 | 0.070 | 0.273 |
| Buffer capacity (M/∆pH) | – | – | – | 0.025 |
Table II.
Composition and Physicochemical Properties of Acetate Buffer, Blank FeSSIF, Blank FaSSIF, SIFsp, and 0.05 M Phosphate Buffer
| Acetate Buffer | Blank FeSSIF | Blank FaSSIF | SIFsp | 0.05 M Phosphate Buffer | |
|---|---|---|---|---|---|
| Composition | |||||
| KH2PO4 (mM) | – | – | – | 50.0 | 25 |
| Na2HPO4 (mM) | – | – | 28.4 | – | 25 |
| NaOH (mM) | – | 101.0 | 8.7 | 22.4 | – |
| CH3COOH (mM) | 28.0 | 144.1 | – | – | – |
| NaCl (mM) | – | 203.2 | 105.9 | – | – |
| CH3COONa·3H2O (mM) | 36.5 | – | – | – | – |
| Double-distilled water (L) | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 |
| Physicochemical properties | |||||
| pH | 4.5 ± 0.1 | 5.0 ± 0.1 | 6.5 ± 0.1 | 6.8 ± 0.1 | 6.8 ± 0.1 |
| Ionic strength (mol/L) | 0.037 | 0.304 | 0.200 | 0.072 | 0.100 |
| Buffer capacity (M/∆pH) | 0.034 | 0.130 | 0.120 | 0.034 | 0.023 |
Table III.
Composition and Physicochemical Properties of Phosphate Buffers of pH 6.8 of Various Concentrations (6.25, 12.5, 25, 50, and 100 mM)
| 6.25 mM | 12.5 mM | 25 mM | 50 mM | 100 mM | |
|---|---|---|---|---|---|
| Composition | |||||
| Na2HPO4 (mM) | 3.13 | 6.25 | 12.50 | 25.00 | 50.00 |
| NaH2PO4 (mM) | 3.13 | 6.25 | 12.50 | 25.00 | 50.00 |
| Double-distilled water (L) | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 |
| Physicochemical properties | |||||
| pH | 6.8 ± 0.1 | 6.8 ± 0.1 | 6.8 ± 0.1 | 6.8 ± 0.1 | 6.8 ± 0.1 |
| Ionic strength (mol/L) | 0.013 | 0.025 | 0.050 | 0.100 | 0.200 |
| Buffer capacity (M/∆pH) | 0.003 | 0.006 | 0.012 | 0.024 | 0.047 |
Table IV.
Composition and Physicochemical Properties of Phosphate Buffers pH 7.2 and 7.8
| Phosphate Buffer pH 7.2 | Phosphate Buffer pH 7.8 | |
|---|---|---|
| Composition | ||
| NaH2PO4 (mM) | 28.0 | 8.5 |
| Na2HPO4 (mM) | 72.0 | 91.5 |
| Double-distilled water (L) | 1.0 | 1.0 |
| Physicochemical properties | ||
| pH | 7.2 ± 0.1 | 7.8 ± 0.1 |
| Ionic strength (mol/L) | 0.244 | 0.283 |
| Buffer capacity (M/∆pH) | 0.058 | 0.037 |
Physicochemical Properties of the Media
The pH of the media was measured using a pH meter (Mettler Toledo, USA). The pH meter was calibrated before each measurement. The ionic strength of the solubility and dissolution media was calculated using Eq. (1) (25).
| 1 |
where
- I
Ionic strength
- J
Number of species in the solution
- ci
Molar concentration of ion i
- zi
Charge number of ion i
The summation symbol, Σ, indicates that the products of cz 2 terms of all the ionic species in the solution, from the first one to the Jth species, are to be added together.
The Van Slyke Eq. (2) (21) was used to calculate the buffer capacity of the media:
| 2 |
where
- β
Buffer capacity
- C
Total buffer concentration (sum of the molar concentrations of acid and salt)
- Ka
Acid dissociation constant
- [H3O+]
Molar concentration of the hydronium ion
Solubility Studies
The solubility of pure carvedilol was determined in all dissolution media. Briefly, excess amount of carvedilol powder was added to beakers containing 50 mL of the media. The beakers were placed in a shaking water bath rotating at 50 rpm and maintained at 37 ± 1°C. The pH of the solution was continuously checked using a calibrated pH meter (Mettler Toledo, USA) and adjusted, if necessary, to the original pH of the tested medium. A 5-mL sample was withdrawn after 1, 2, 4, 6, and 24 h. The samples were centrifuged at 3500 rpm for 15 min and the supernatant was diluted appropriately with the tested medium. The concentration of carvedilol in each sample was determined using UV spectrophotometry (Varian, Cary 50 UV/VIS spectrophotometer, USA) at 285 nm using a standard calibration curve of carvedilol prepared using the tested medium. The solubility of carvedilol in each medium was performed at least in triplicate with a standard deviation (SD) less than 10%. The ratio of solubility (C S) to drug concentration (C D), expressed as C S/C D, was calculated to represent the sink condition (S value). According to USP, an S value >3 is considered as sink condition (26). The percentage of carvedilol in the ionized form at various pH values was calculated based on Henderson-Hasselbalch using Eq. (3) (27).
| 3 |
where
- pH
pH value of the dissolution medium
- pKa
Negative logarithm of the dissociation constant (K a) of carvedilol
Estimation of Carvedilol in Dilatrend® Tablets
The assay of carvedilol was determined using UV-VIS spectrophotometry. Briefly, ten tablets were crushed and a powder mass equivalent to 25 mg carvedilol was transferred to a volumetric flask of 100 mL. Ten milliliters of double-distilled water, then 70 mL of methanol and 1 M HCl (9:1 v/v) were added to the flask. The flask was placed in a sonicator for 30 min. The volume was completed up to 100 mL using methanol and 1 M HCl (9:1 v/v). The sample was centrifuged at 3500 rpm for 15 min, and the supernatant was diluted appropriately with methanol and 1 M HCl (9:1 v/v). The concentration of carvedilol in each sample was determined using UV spectrophotometry (Varian, Cary 50 UV/VIS spectrophotometer, USA) at 285 nm using a standard calibration curve of carvedilol prepared in methanol and 1 M HCl (9:1 v/v). The uniformity of dosage units of Dilatrend® was performed in triplicate. The uniformity of Dilatrend® tablets has been demonstrated by weight variation.
Dissolution Studies
Dissolution tests were performed using a calibrated USP apparatus type II (Varian, VK 7000) fitted with an auto-sampling station consisting of a VK810 peristaltic pump and a VK750 digitally controlled heater/circulator. The dissolution apparatus was connected to a UV spectrophotometer (Cary 50, UV-VIS spectrophotometer). Paddles were rotating at 50 rpm. Dissolution was performed using 900 mL of dissolution medium, maintained at 37 ± 0.5°C. Samples were taken automatically at 5, 10, 15, 30, 45, 60, 75, 90, 105, 120, 150, 180, 210, and 240 min and passed through a 45-μm polyethylene filter (SUN-SRi, Rockwood, TN, USA). The sample volume was 15 mL, sufficiently enough to flush the filter before making the UV measurement. However, to evaluate carvedilol adsorption to polyethylene filters, a dissolution test of carvedilol powder in 0.7% HCl dissolution medium (USP 38, pH 1.45) was performed at a concentration similar to that found in Dilatrend® tablets and under similar dissolution conditions of those of Dilatrend® tablets. Filter tips were securely fitted at the end of the cannulas of three dissolution vessels, and the other three vessels were kept without filters. The UV absorption of filtered and unfiltered carvedilol solutions was measured at different time intervals. No significant loss of carvedilol was observed after filtration and the carvedilol recovery was >99.0%. The auto-sampling system pulls the sample into the flow cell, measures the absorbance of the sample, and then returns it to the vessel. Thus, no correction was made for the drug quantity removed during sample and no replacement of sample volume was needed. The absorbance was measured at 285 nm. To evaluate tablet excipient interference, a blend of table excipients consisting of sucrose, lactose, povidone, crospovidone, colloidal silicone dioxide, and magnesium stearate (Data Sheet, Dilatrend®, Roche) included in the Dilatrend® tablets was prepared. The dissolution test for tablet excipients (n = 6) was performed in 0.7% HCl dissolution medium (USP 38, pH 1.45) at concentrations similar to those found in Dilatrend® tablets and under similar dissolution conditions as those of Dilatrend® tablets. The absorbance of the excipient samples at 285 nm was reported to be zero, indicating no excipient interference. The amount of carvedilol released from Dilatrend® immediate-release tablets was calculated against standard calibration curves of carvedilol in each dissolution medium, and the percentage of carvedilol released versus time profiles were constructed. The pH of the dissolution media in each vessel was continuously monitored and recorded throughout the dissolution experiments. The difference in dissolution profiles was compared using the similarity factor (f 2) calculated using Eq. (4) (28).
| 4 |
where
- n
Number of sampling points
- Rj, Tj
Percent of the reference and test products dissolved at time point j, respectively
If the f 2 value was less than 50, then the dissolution profiles were considered significantly different. If f 2 was between 50 and 100, the dissolution profiles were considered similar.
RESULTS AND DISCUSSION
Solubility Studies
Carvedilol exhibits a high percentage of ionization along the physiological pH range of GI fluids ranging from 50 to 100% (Table V). The high percentage of ionized carvedilol (protonated free base) suggests the formation of salts with the anion of buffer systems, which may affect the solubility product of the weakly basic drug (10, 11, 29). Therefore, the selection of the anion of the buffer system (buffer species) may be important for the in vitro dissolution studies of carvedilol. As shown in Fig. 1, the NH group of carbazole in carvedilol is acidic of pK a of 15.0 (calculated using ChemAxon software) because the lone pair of electrons is involved in aromatization. Thus, the N–H bond of carbazole is weak; hydrogen is easily hydrolyzed as H+ (acidic proton). Contrarily, the aliphatic secondary amine is weakly basic of pK a = 7.8 (6) due to the electronegativity of the surrounding oxygen atoms and the bulkiness of attached alkyl groups harboring aromatic rings on two wings. Interestingly, the amphoteric characteristic (having both acidic and basic moieties) of carvedilol describes its ionization pattern. In acidic pH medium, the aliphatic NH is ionized forming a cationic center, whereas in basic pH medium, the carbazole NH is ionized forming an anionic center. Therefore, harboring a zwitterion characteristic demonstrates its solubility manner.
Table V.
Percentage of Ionized and Unionized Carvedilol (pK a 7.8) at Various pH Values Calculated Using Eq. (3)
| pH | Ionized carvedilol (%) | Unionized carvedilol (%) |
|---|---|---|
| 1.2 | 100 | 0 |
| 1.45 | 100 | 0 |
| 1.6 | 100 | 0 |
| 4.5 | 99.95 | 0.05 |
| 5.0 | 99.84 | 0.16 |
| 6.5 | 95.23 | 4.77 |
| 6.8 | 90.91 | 9.09 |
| 7.2 | 79.92 | 20.08 |
| 7.8 | 50 | 50 |
Carvedilol drug substance (solid state) is stable when exposed to heat (80°C for 14 days), heat and relative humidity (RH) (40°C and 75% RH for 14 days), and UV-VIS light (1.3 × 106 lx h). Degradation products were found to be less than 0.05% under these conditions. In addition, it has been shown that carvedilol solution is stable in acidic (0.1 N HCl, 60°C for 4 h), basic (0.1 N NaOH, 60°C for 4 h), UV-VIS light (1.3 × 106 lx h, water), and heat (60°C, water for 4 h), where no individual degradation product >0.06% was generated under these stress conditions (30). In another study (31), no significant degradation products were observed after exposure of carvedilol, both as solid state and methanolic solutions, to acid (1 N HCl solution, 90°C for 6 h), neutral (water, 90°C for 6 h), and photolytic (UVC-254 nm 30-W lamp, 25°C for 7 days) conditions, carried out according to ICH guideline Q1A (R2). However, gradual degradation was observed in basic conditions (1 N NaOH, 90°C for 6 h).
Several studies used the same experimental method that was used in this work for assessing the solubility of carvedilol in different dissolution media (hydrochloric acid solution, pH 1.2; acid phthalate buffer, pH 3.0; acetate buffer, pH 4.5; and phosphate buffers, pH 5.8, 6.8, 7.4) and in water at 37 ± 0.5°C until equilibrium was reached for 24 or 48 h (32–34). Samples were not protected from light during solubility analysis. The solid phase of the in situ salt formed during the solubility studies was not identified in this work.
The saturation solubility of carvedilol in all dissolution media is given in Table VI. Carvedilol with a pK a of 7.8 exhibits the typical pH-dependent solubility profile of a weak base with a high solubility in dissolution media of low pH, and low solubility in the dissolution media of high pH. It is worth noting that the saturation solubility of carvedilol was achieved within 1 h in the acidic media (pH 1.2–5.0). However, in the basic media (pH 6.5–7.8), the saturation solubility of carvedilol was achieved after 24 h. At pH 6.8, the solubility of carvedilol in phosphate buffers 25, 50, and 100 mM was ~37% less compared to phosphate buffers 6.25 and 12.5 mM. Lower solubility of carvedilol in buffers at high salt concentrations might be related to the higher ionic strength values of the dissolution media, where it has been shown that solubility decreases with an increase in ionic strength (35, 36). Furthermore, although more than 90% of carvedilol was in its protonated form at pH 6.8 (Table V), the formation of the less soluble carvedilol phosphate salt, as reported by Loftsson (10), significantly reduces the solubility of carvedilol in these dissolution media. Carvedilol shows a very low solubility at pH 7.2. At pH 7.8, the solubility dropped by 90% compared to its solubility at pH 6.8. The solubility of carvedilol in double-distilled water of zero ionic strength and zero buffer capacity was also reduced by ~45% compared to its solubility at pH 6.8.
Table VI.
Saturation Solubility (C S) and Relative Sink Conditions (S Value) of Carvedilol in Dissolution Media. Data are Presented as the Mean ± SD (n = 3)
| Dissolution media | pH | Solubility (C S, μg/mL) | S value (C S/C D a) |
|---|---|---|---|
| 0.7% HCl | 1.45 | 545.1 ± 5.0 | 19.6 |
| SGFsp | 1.2 | 832.0 ± 66.0 | 29.9 |
| Blank FaSSGF | 1.6 | 2398.6 ± 40.9 | 86.3 |
| Blank FeSSGF | 5.0 | 995.6 ± 20.5 | 35.8 |
| Acetate buffer | 4.5 | 2591.4 ± 35.8 | 93.2 |
| Blank FaSSIF | 6.5 | 51.6 ± 5.5 | 1.9 |
| Blank FeSSIF | 5.0 | 830.7 ± 17.4 | 29.9 |
| SIFsp | 6.8 | 51.1 ± 3.0 | 1.8 |
| Phosphate buffer (IntPh3) | 6.8 | 51.9 ± 5.8 | 1.9 |
| Phosphate buffer 6.25 mM | 6.8 | 52.5 ± 4.2 | 1.9 |
| Phosphate buffer 12.5 mM | 6.8 | 53.5 ± 1.1 | 1.9 |
| Phosphate buffer 25 mM | 6.8 | 31.8 ± 1.9 | 1.1 |
| Phosphate buffer 50 mM | 6.8 | 31.5 ± 1.0 | 1.1 |
| Phosphate buffer 100 mM | 6.8 | 34.3 ± 0.7 | 1.2 |
| Phosphate buffer 100 mM | 7.2 | 19.1 ± 1.5 | 0.7 |
| Phosphate buffer 100 mM | 7.8 | 5.8 ± 0.5 | 0.2 |
| Double-distilled water | 6.5 | 27.9 ± 3.9 | 1.0 |
a C D = 27.8 μg/mL
Therefore, our solubility studies illustrate that carvedilol is more soluble in acidic media. This means that the basicity of the aliphatic NH, rather than the acidity of the carbazole NH, accounts for its solubility. Thus, the basic strength of the aliphatic NH is higher than the acidic strength of the carbazole NH and consequently drives its solubility in acetate buffer (pH 4.5). Surprisingly, the solubility in acidic media is preferred particularly at pH 1.6 and 4.5 in fasting condition. The higher pH led to a decrease in solubility, as shown in pH 6.8 phosphate buffers. Altogether, the acidic pH medium is preferred for complete solubility of carvedilol.
The calculated solubility (C S) to dose (C D) ratio (S value) for carvedilol resulted in values >3 in all acidic media, which indicates that they provided sink conditions. However, the S value was <3 in the basic media, indicating no sink condition (Table VI). The poor solubility of carvedilol in the small intestine with no sink condition could account for the precipitation of carvedilol in the small intestine.
Carvedilol Content in Dilatrend® Tablets
Carvedilol content in Dilatrend® tablets was 99.7 ± 2.2% (n = 3), and the uniformity of Dilatrend® tablets was between 98.5 and 100.5% (n = 10).
Dissolution Studies
As shown in Fig. 2, the rate of release of carvedilol from Dilatrend® tablets in acidic dissolution media (0.7% HCl, SGFsp, blank FaSSGF, and blank FeSSGF) was rapid and nearly superimposable (f 2 > 50) due to their low pH. Dissolution results were also consistent with carvedilol solubility data (Table VI). Therefore, carvedilol is expected to dissolve quickly and completely in both the fasted- and fed-state conditions of the stomach.
Fig. 2.
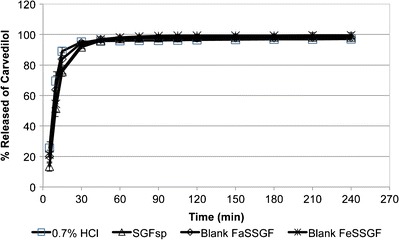
Dissolution profiles of carvedilol from Dilatrend® tablets in 0.7% HCl (pH 1.45, n = 6), SGFsp (pH 1.2, n = 6), blank FaSSGF (pH 1.6, n = 6), and blank FeSSGF (pH 5.0, n = 6). Data are represented as the mean ± SD
Carvedilol release was complete and approached a plateau level within 60 min, achieving an average of 95.8, 97.2, 97.4, and 98.2% in 0.7% HCl, SGFsp, blank FaSSGF, and blank FeSSGF, respectively. Although blank FeSSGF exhibited higher pH values compared to 0.7% HCl, SGFsp, and blank FaSSGF (5.0 vs. 1.5, 1.2, and 1.6, respectively), the dissolution of carvedilol was complete. This might be attributed to the presence of acetate buffer in blank FeSSGF, which forms a water-soluble acetate salt (10). Due to the low pH value of the dissolution media, the influence of ionic strength and buffer capacity was not prominent, indicating the important role of pH in determining the percentage of carvedilol released in acidic media.
The buffer composition of the dissolution media can nonetheless have an effect on the dissolution of drugs as seen with blank FeSSGF, where the presence of acetate enhances the dissolution rate. As shown in Fig. 3, the rate of carvedilol release in acetate buffer was fast, with ~39.2% of carvedilol released within 5 min and almost completely dissolved (97.4%) within the first 15 min. In acetate buffer (pH 4.5), 99.95% of carvedilol is in its protonated form (Table V). Loftsson et al. (10) have reported that the protonated base would form a water-soluble salt with the anionic form of acetic acid, resulting in increased dissolution. Therefore, the dissolution rate of carvedilol was clearly dependent on pH and buffer species of the dissolution media.
Fig. 3.
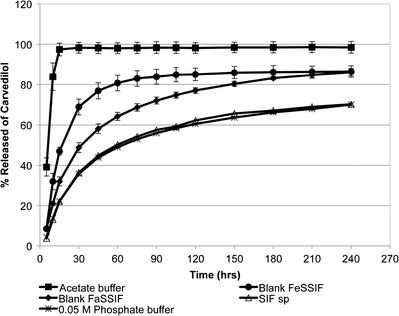
Dissolution profiles of carvedilol from Dilatrend® tablets in acetate buffer (pH 4.5, n = 6), blank FeSSIF (pH 5.0, n = 4), blank FaSSIF (pH 6.5, n = 3), SIFsp (pH 6.8, n = 3), and 0.05 M phosphate buffer (pH 6.8, n = 3). Data are represented as the mean ± SD
When comparing the dissolution profiles of blank FaSSIF (pH 6.5) and blank FeSSIF (pH 5.0), the dissolution rate in blank FeSSIF was significantly faster (f 2 = 39.7). After 240 min, however, the percentage of carvedilol released was equal (~86%) in both media. The increase in the dissolution rate of carvedilol in blank FeSSIF can be explained by the lower pH value of this medium compared to the blank FaSSIF (5.0 vs. 6.5). In addition, the presence of acetate buffer in blank FeSSIF would enhance carvedilol dissolution as has been reported by Loftsson et al. (10). Unlike blank FaSSIF, the presence of NaCl and phosphate species resulted in the formation of the less soluble hydrochloride and phosphate carvedilol salts, thus reducing the rate of carvedilol dissolution in blank FaSSIF (10).
In simulated intestinal fluids, the amount of carvedilol released was significantly reduced to 70.2 and 70.0% in the dissolution media SIFsp and 0.05 M phosphate buffer (IntPh3), respectively. The dissolution profiles of carvedilol in SIFsp and 0.05 M phosphate buffer (IntPh3) were superimposable (f 2 > 50), indicating that the difference in the species composition of the two buffers (KH2PO4 and NaOH in SIFsp vs. KH2PO4 and Na2HPO4 in 0.05 M phosphate buffer (IntPh3)) had no influence on the rate of carvedilol release. Since the pH and ionic strength of the two buffers were equivalent (Table II), dissolution profiles were found to be similar. Although the buffer capacity of the 0.05 M phosphate buffer (IntPh3) was slightly higher than that of SIFsp, no difference in the dissolution profiles was observed. This is consistent with a previous study by Dressman et al. who found that SIFsp could be used interchangeably with 0.05 M phosphate buffer (IntPh3) in the dissolution testing of immediate-release dosage forms (37). Overall, acetate buffer and blank FeSSIF allowed for higher dissolution rate than blank FaSSIF, SIFsp, and 0.05 M phosphate buffer (IntPh3).
In general, while weak bases exhibit high dissolution rate in the stomach, they tend to supersaturate and precipitate when entering the small intestine. For carvedilol, less precipitation is expected in the duodenum and fed-state intestinal fluid as apparent from the dissolution results of carvedilol in acetate buffer pH 4.5 and blank FeSSIF. Nonetheless, SIFsp and 0.05 M phosphate buffer (IntPh3) remain commonly used as compendial dissolution media (7, 11, 38–42).
To better understand the dissolution behavior of carvedilol in the intestinal fluid and to adequately predict its precipitation upon entry into the small intestine, the dissolution of carvedilol tablets in phosphate buffers of different ionic strengths and buffer capacities was tested. Figure 4a compares the dissolution profiles of carvedilol from Dilatrend® tablets in phosphate buffers (pH 6.8) of different buffer concentrations (6.25, 12.5, 25, 50, and 100 mM). Phosphate buffers vary in their ionic strengths and buffer capacities (Table III). The dissolution profile of carvedilol in double-distilled water of zero ionic strength and zero buffer capacity was used as a reference. The ionic strength of phosphate buffers ranged from 0.013 to 0.200 mol/L, which covers the typical values of ionic strength of the human intestinal fluid (0.070–0.166 mol/L) (17). Similarly, the buffer capacity of the phosphate buffers ranged from 0.003 to 0.047 M/ΔpH, which approximates the buffer capacity of the human intestinal fluid (0.003–0.030 M/ΔpH) (15, 18).
Fig. 4.
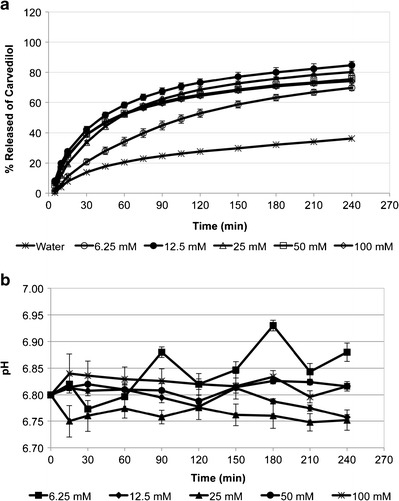
a Dissolution profiles of carvedilol from Dilatrend® tablets in phosphate buffers of pH 6.8 of various concentrations (6.25, 12.5, 25, 50, and 100 mM), and in double-distilled water. Data are represented as the mean ± SD (n = 5). b pH profiles during dissolution studies of carvedilol from Dilatrend® tablets in phosphate buffers of pH 6.8 of various concentrations (6.25, 12.5, 25, 50, and 100 mM). Data are represented as the mean ± SD (n = 5)
The rate of carvedilol release was slowest in double-distilled water with only 36.2% of carvedilol released at 240 min. Due to the high pH value of the phosphate buffers (pH 6.8), no complete dissolution was observed for carvedilol, where only 69.6, 84.7, 80.3, 75.5, and 74.1% of carvedilol was released in 6.25, 12.5, 25, 50, and 100 mM buffers, respectively. Of the buffers, carvedilol release was lowest in 6.25 mM phosphate buffer (69.6% after 240 min). The dissolution profile of carvedilol in 6.25 mM was significantly different from its dissolution in 12.5, 25, 50, and 100 mM (f 2 < 50). This might be due to the low buffer capacity of the 6.25 mM phosphate buffer (0.003 M/ΔpH). It has been found that lowering the buffer capacity of the dissolution media decreased drug solubility and hence the dissolution rate (43, 44).
When the ionic strength increased in phosphate buffers 12.5, 25, 50, and 100 mM, the amount of drug released was slightly decreased in the order 12.5 mM (84.7%), 25 mM (80.3%), 50 mM (75.5%), and 100 mM (74.1%), after 240 min. Nonetheless, no significant difference was observed in their dissolution profiles (f 2 > 50). These results were in agreement with Kincl et al. who studied the rate of diclofenac release from lipophilic matrix tablets in phosphate buffers with various ionic strengths (36). Kincl et al. found that the rate of diclofenac release decreased with increasing ionic strength. However, there was no significant difference between drug release profiles in phosphate buffers with high ionic strength (0.14–0.51 mol/L). The dissolution data were also in agreement with the solubility results. The almost equal solubilities of carvedilol in phosphate buffers 25, 50, and 100 mM seem to be consistent with their dissolution profiles, where no significant difference was observed in carvedilol dissolution behavior (f 2 > 50). Despite equal solubility of carvedilol in phosphate buffers 6.25 and 12.5 mM, carvedilol exhibited higher dissolution rate in 12.5 mM than 6.25 mM. This might be attributed to the relatively higher buffer capacity of 12.5 mM compared to 6.25 mM. No significant difference was observed (f 2 > 50) in the dissolution profiles of carvedilol in phosphate buffers 12.5, 25, 50, and 100 mM; SIFsp; and 0.05 M phosphate buffer (IntPh3).
To confirm that the buffer capacity of the phosphate buffers, particularly those of low concentrations (6.25, 12.5, and 25 mM), was maintained throughout the dissolution experiment, the pH of the buffers was measured and is illustrated in Fig. 4b. The results show that the initial pH value of 6.80 was maintained throughout the dissolution experiments, with a maximum deviation of ±0.05 pH units/min from the initial pH value. Therefore, the results of the pH measurements confirm that phosphate buffers 12.5, 25, 50, and 100 mM maintained their buffer capacities. In phosphate buffer 6.25 mM, pH values deviated from the initial pH value reaching a maximum value of 6.93, indicating that the initial pH value was less precisely maintained throughout the dissolution experiment.
To further investigate the influence of pH on the rate of carvedilol release, dissolution studies were performed in phosphate buffer 100 mM of pH 7.2 and 7.8. Carvedilol release in these buffers was compared to its release in phosphate buffer 100 mM of pH 6.8 (Fig. 5). At pH 6.8, the maximum amount of carvedilol released was about 74.1% at 240 min. At pH 7.2, the dissolution rate was slower, where only 43.6% of carvedilol was released after 240 min. The dissolution rate of carvedilol was slowest at pH 7.8, where only 15.9% of carvedilol was released after 240 min. These results suggest that carvedilol exhibits poor dissolution in the distal small intestine and colon, making it a poor candidate for controlled-release dosage forms (11). The dissolution results of carvedilol in these media were consistent with their solubility data (Table VI).
Fig. 5.
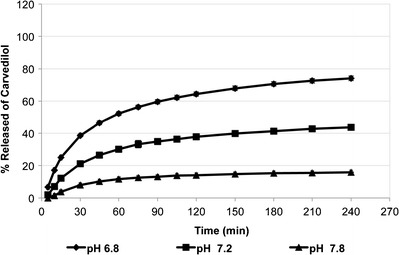
Dissolution profiles of carvedilol from Dilatrend® tablets in phosphate buffers 100 mM of pH 7.2 (n = 3) and pH 7.8 (n = 3). The dissolution profile of carvedilol in phosphate buffer 100 mM of pH 6.8 (n = 5) was added to the figure for comparison. Data are represented as the mean ± SD
CONCLUSIONS
This work focuses on studying the pH-dependent solubility and dissolution behavior of the weakly basic BCS class II model drug carvedilol which has a pK a value of 7.8. The solubility and percentage of carvedilol released in the in vitro dissolution studies were highest in media simulating the gastric fluid with low pH values. However, in media simulating the intestinal fluid, the solubility and percentage of carvedilol released were reduced dramatically, suggesting precipitation in the small intestine. It is evident that the solubility and dissolution of carvedilol are strongly dependent on pH. Carvedilol solubility and dissolution decreased with increasing ionic strength. Lowering buffer capacity resulted in a decrease in carvedilol solubility and hence dissolution rate. Buffer species significantly influenced the solubility and dissolution rate of carvedilol.
ACKNOWLEDGMENTS
This project was financially supported by the Deanship of Academic Research and Graduate Studies at Al-Zaytoonah University of Jordan.
REFERENCES
- 1.Dressman JB, Amidon GL, Reppas C, Shah VP. Dissolution testing as a prognostic tool for oral drug absorption: immediate release dosage forms. Pharm Res. 1998;15(1):11–22. doi: 10.1023/A:1011984216775. [DOI] [PubMed] [Google Scholar]
- 2.Galia E, Nicolaides E, Horter D, Lobenberg R, Reppas C, Dressman JB. Evaluation of various dissolution media for predicting in vivo performance of class I and II drugs. Pharm Res. 1998;15(5):698–705. doi: 10.1023/A:1011910801212. [DOI] [PubMed] [Google Scholar]
- 3.Sheng JJ, Kasim NA, Chandrasekharan R, Amidon GL. Solubilization and dissolution of insoluble weak acid, ketoprofen: effects of pH combined with surfactant. Eur J Pharm Sci. 2006;29(3–4):306–14. doi: 10.1016/j.ejps.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 4.Kostewicz ES, Wunderlich M, Brauns U, Becker R, Bock T, Dressman JB. Predicting the precipitation of poorly soluble weak bases upon entry in the small intestine. J Pharm Pharmacol. 2004;56(1):43–51. doi: 10.1211/0022357022511. [DOI] [PubMed] [Google Scholar]
- 5.Carlert S, Palsson A, Hanisch G, von Corswant C, Nilsson C, Lindfors L, et al. Predicting intestinal precipitation—a case example for a basic BCS class II drug. Pharm Res. 2010;27(10):2119–30. doi: 10.1007/s11095-010-0213-8. [DOI] [PubMed] [Google Scholar]
- 6.Tsume Y, Mudie DM, Langguth P, Amidon GE, Amidon GL. The Biopharmaceutics Classification System: subclasses for in vivo predictive dissolution (IPD) methodology and IVIVC. Eur J Pharm Sci. 2014;57:152–63. doi: 10.1016/j.ejps.2014.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kukec S, Dreu R, Vrbanec T, Srcic S, Vrecer F. Characterization of agglomerated carvedilol by hot-melt processes in a fluid bed and high shear granulator. Int J Pharm. 2012;430(1–2):74–85. doi: 10.1016/j.ijpharm.2012.03.041. [DOI] [PubMed] [Google Scholar]
- 8.Mahmoud EA, Bendas ER, Mohamed MI. Preparation and evaluation of self-nanoemulsifying tablets of carvedilol. AAPS PharmSciTech. 2009;10(1):183–92. doi: 10.1208/s12249-009-9192-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stillhart C, Duerr D, Kuentz M. Toward an improved understanding of the precipitation behavior of weakly basic drugs from oral lipid-based formulations. J Pharm Sci. 2014;103(4):1194–203. doi: 10.1002/jps.23892. [DOI] [PubMed] [Google Scholar]
- 10.Loftsson T, Vogensen SB, Desbos C, Jansook P. Carvedilol: solubilization and cyclodextrin complexation: a technical note. AAPS PharmSciTech. 2008;9(2):425–30. doi: 10.1208/s12249-008-9055-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Planinsek O, Kovacic B, Vrecer F. Carvedilol dissolution improvement by preparation of solid dispersions with porous silica. Int J Pharm. 2011;406(1–2):41–8. doi: 10.1016/j.ijpharm.2010.12.035. [DOI] [PubMed] [Google Scholar]
- 12.Tsume Y, Langguth P, Garcia-Arieta A, Amidon GL. In silico prediction of drug dissolution and absorption with variation in intestinal pH for BCS class II weak acid drugs: ibuprofen and ketoprofen. Biopharm Drug Dispos. 2012;33(7):366–77. doi: 10.1002/bdd.1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lindahl A, Ungell AL, Knutson L, Lennernas H. Characterization of fluids from the stomach and proximal jejunum in men and women. Pharm Res. 1997;14(4):497–502. doi: 10.1023/A:1012107801889. [DOI] [PubMed] [Google Scholar]
- 14.Bergstrom CAS, Holm R, Jorgensen SA, Andersson SBE, Artursson P, Beato S, et al. Early pharmaceutical profiling to predict oral drug absorption: current status and unmet needs. Eur J Pharm Sci. 2014;57:173–99. doi: 10.1016/j.ejps.2013.10.015. [DOI] [PubMed] [Google Scholar]
- 15.Kalantzi L, Goumas K, Kalioras V, Abrahamsson B, Dressman JB, Reppas C. Characterization of the human upper gastrointestinal contents under conditions simulating bioavailability/bioequivalence studies. Pharm Res. 2006;23(1):165–76. doi: 10.1007/s11095-005-8476-1. [DOI] [PubMed] [Google Scholar]
- 16.Garbacz G, Kolodziej B, Koziolek M, Weitschies W, Klein S. A dynamic system for the simulation of fasting luminal pH-gradients using hydrogen carbonate buffers for dissolution testing of ionisable compounds. Eur J Pharm Sci. 2014;51:224–31. doi: 10.1016/j.ejps.2013.09.020. [DOI] [PubMed] [Google Scholar]
- 17.Johnson JL, Holinej J, Williams MD. Influence of ionic-strength on matrix integrity and drug release from hydroxypropyl cellulose compacts. Int J Pharm. 1993;90(2):151–9. doi: 10.1016/0378-5173(93)90151-5. [DOI] [Google Scholar]
- 18.Persson EM, Gustafsson AS, Carlsson AS, Nilsson RG, Knutson L, Forsell P, et al. The effects of food on the dissolution of poorly soluble drugs in human and in model small intestinal fluids. Pharm Res. 2005;22(12):2141–51. doi: 10.1007/s11095-005-8192-x. [DOI] [PubMed] [Google Scholar]
- 19.Vertzoni M, Dressman J, Butler J, Hempenstall J, Reppas C. Simulation of fasting gastric conditions and its importance for the in vivo dissolution of lipophilic compounds. Eur J Pharm Biopharm. 2005;60(3):413–7. doi: 10.1016/j.ejpb.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 20.Jantratid E, Janssen N, Reppas C, Dressman JB. Dissolution media simulating conditions in the proximal human gastrointestinal tract: an update. Pharm Res. 2008;25(7):1663–76. doi: 10.1007/s11095-008-9569-4. [DOI] [PubMed] [Google Scholar]
- 21.Arndt M, Chokshi H, Tang K, Parrott NJ, Reppas C, Dressman JB. Dissolution media simulating the proximal canine gastrointestinal tract in the fasted state. Eur J Pharm Biopharm. 2013;84(3):633–41. doi: 10.1016/j.ejpb.2013.01.010. [DOI] [PubMed] [Google Scholar]
- 22.Klein S, Wempe MF, Zoeller T, Buchanan NL, Lambert JL, Ramsey MG, et al. Improving glyburide solubility and dissolution by complexation with hydroxybutenyl-beta-cyclodextrin. J Pharm Pharmacol. 2009;61(1):23–30. doi: 10.1211/jpp/61.01.0004. [DOI] [PubMed] [Google Scholar]
- 23.Klein S, Rudolph M, Dressman J. Drug release characteristics of different mesalazine products using USP apparatus 3 to simulate passage through the GI tract. Dissolut Technol. 2002;9:6–12. doi: 10.14227/DT090402P6. [DOI] [Google Scholar]
- 24.Marques MRC, Loebenberg R, Almukainzi M. Simulated biological fluids with possible application in dissolution testing. Dissolut Technol. 2011;18(3):15–28. doi: 10.14227/DT180311P15. [DOI] [Google Scholar]
- 25.Solon K, Flores-Alsina X, Mbarnba CK, Volcke EIP, Tait S, Batstone D, et al. Effects of ionic strength and ion pairing on (plant-wide) modelling of anaerobic digestion. Water Res. 2015;70:235–45. doi: 10.1016/j.watres.2014.11.035. [DOI] [PubMed] [Google Scholar]
- 26.Jamzad S, Fassihi R. Role of surfactant and pH on dissolution properties of fenofibrate and glipizide—a technical note. AAPS PharmSciTech. 2006;7(2):E33-E. [DOI] [PMC free article] [PubMed]
- 27.Buffered and isotonic solutions. In: Sinko PJ, editor. Martin’s physical pharmacy and pharmaceutical sciences. 6 ed. Philadelphia, PA: Lippincott Williams and Wilkins; 2011. p. 164.
- 28.Costa P, Manuel J, Lobo S. Modeling and comparison of dissolution profiles. Eur J Pharm Sci. 2001;13(2):123–33. doi: 10.1016/S0928-0987(01)00095-1. [DOI] [PubMed] [Google Scholar]
- 29.Takacs-Novak K, Szoke V, Voelgyi G, Horvath P, Ambrus R, Szabo-Revesz P. Biorelevant solubility of poorly soluble drugs: rivaroxaban, furosemide, papaverine and niflumic acid. J Pharmaceut Biomed. 2013;83:279–85. doi: 10.1016/j.jpba.2013.05.011. [DOI] [PubMed] [Google Scholar]
- 30.Beattie K, Phadke G, Novakovic J. Carvedilol. Profiles of drug substances, excipients, and related methodology. 2013;38:113–57. doi: 10.1016/B978-0-12-407691-4.00004-6. [DOI] [PubMed] [Google Scholar]
- 31.Lanzanova FA, Argenta D, Arend MZ, Brum Junior L, Cardoso SG. LC and LC-MS evaluation of stress degradation behavior of carvedilol. J Liq Chromatogr R T. 2009;32(4):526–43. doi: 10.1080/10826070802671481. [DOI] [Google Scholar]
- 32.Chakraborty S, Shukla D, Jain A, Mishra B, Singh S. Assessment of solubilization characteristics of different surfactants for carvedilol phosphate as a function of pH. J Colloid Interface Sci. 2009;335(2):242–9. doi: 10.1016/j.jcis.2009.03.047. [DOI] [PubMed] [Google Scholar]
- 33.Yuvaraja K, Khanam J. Enhancement of carvedilol solubility by solid dispersion technique using cyclodextrins, water soluble polymers and hydroxyl acid. J Pharmaceut Biomed. 2014;96:10–20. doi: 10.1016/j.jpba.2014.03.019. [DOI] [PubMed] [Google Scholar]
- 34.Zhang Y, Zhi Z, Li X, Gao J, Song Y. Carboxylated mesoporous carbon microparticles as new approach to improve the oral bioavailability of poorly water-soluble carvedilol. Int J Pharm. 2013;454(1):403–11. doi: 10.1016/j.ijpharm.2013.07.009. [DOI] [PubMed] [Google Scholar]
- 35.Chahiyan H, Gharib F, Farajtabar A. Thermodynamic studies on solubility and protonation constant of acetaminophen at different ionic strengths and various temperatures. J Mol Liq. 2014;199:137–42. doi: 10.1016/j.molliq.2014.08.033. [DOI] [Google Scholar]
- 36.Kincl M, Meleh M, Veber M, Vrecer F. Study of physicochemical parameters affecting the release of diclofenac sodium from lipophilic matrix tablets. Acta Chim Slov. 2004;51(3):409–25. [Google Scholar]
- 37.Stippler E, Kopp S, Dressman JB. Comparison of US Pharmacopeia simulated intestinal fluid TS (without pancreatin) and phosphate standard buffer pH 6.8, TS of the international pharmacopoeia with respect to their use in in vitro dissolution testing. Dissolut Technol. 2004;11:6–10. doi: 10.14227/DT110204P6. [DOI] [Google Scholar]
- 38.Fadda HM, Merchant HA, Arafat BT, Basit AW. Physiological bicarbonate buffers: stabilisation and use as dissolution media for modified release systems. Int J Pharm. 2009;382(1–2):56–60. doi: 10.1016/j.ijpharm.2009.08.003. [DOI] [PubMed] [Google Scholar]
- 39.Piao Z-Z, Choe J-S, Oh KT, Rhee Y-S, Lee B-J. Formulation and in vivo human bioavailability of dissolving tablets containing a self-nanoemulsifying itraconazole solid dispersion without precipitation in simulated gastrointestinal fluid. Eur J Pharm Sci. 2014;51:67–74. doi: 10.1016/j.ejps.2013.08.037. [DOI] [PubMed] [Google Scholar]
- 40.Raju V, Murthy KVR. Development and validation of new discriminative dissolution method for carvedilol tablets. Indian J Pharm Sci. 2011;73(5):527–36. doi: 10.4103/0250-474X.99000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shono Y, Jantratid E, Dressman JB. Precipitation in the small intestine may play a more important role in the in vivo performance of poorly soluble weak bases in the fasted state: case example nelfinavir. Eur J Pharm Biopharm. 2011;79(2):349–56. doi: 10.1016/j.ejpb.2011.04.005. [DOI] [PubMed] [Google Scholar]
- 42.Wagner C, Jantratid E, Kesisoglou F, Vertzoni M, Reppas C, Dressman JB. Predicting the oral absorption of a poorly soluble, poorly permeable weak base using biorelevant dissolution and transfer model tests coupled with a physiologically based pharmacokinetic model. Eur J Pharm Biopharm. 2012;82(1):127–38. doi: 10.1016/j.ejpb.2012.05.008. [DOI] [PubMed] [Google Scholar]
- 43.Corrigan OI, Devlin Y, Butler J. Influence of dissolution medium buffer composition on ketoprofen release from ER products and in vitro-in vivo correlation. Int J Pharm. 2003;254(2):147–54. doi: 10.1016/S0378-5173(03)00004-8. [DOI] [PubMed] [Google Scholar]
- 44.Ozturk SS, Palsson BO, Dressman JB. Dissolution of ionizable drugs in buffered and unbuffered solutions. Pharm Res. 1988;5(5):272–82. doi: 10.1023/A:1015970502993. [DOI] [PubMed] [Google Scholar]