

Abstract
The aim of the present study was to develop and evaluate a thermoresponsive depot system comprising of docetaxel-loaded cubosomes. The cubosomes were dispersed within a thermoreversible gelling system for controlled drug delivery. The cubosome dispersion was prepared by dilution method, followed by homogenization using glyceryl monooleate, ethanol and Pluronic® F127 in distilled water. The cubosome dispersion was then incorporated into a gelling system prepared with Pluronic® F127 and Pluronic® F68 in various ratios to formulate a thermoresponsive depot system. The thermoresponsive depot formulations undergo a thermoreversible gelation process i.e., they exists as free flowing liquids at room temperature, and transforms into gels at higher temperatures e.g., body temperature, to form a stable depot in aqueous environment. The mean particle size of the cubosomes in the dispersion prepared with Pluronic® F127, with and without the drug was found to be 170 and 280 nm, respectively. The prepared thermoresponsive depot system was evaluated by assessing various parameters like time for gelation, injectability, gel erosion, and in-vitro drug release. The drug-release studies of the cubosome dispersion before incorporation into the gelling system revealed that a majority (∼97%) of the drug was released within 12 h. This formulation also showed a short lag time (∼3 min). However, when incorporated into a thermoresponsive depot system, the formulation exhibited an initial burst release of ∼21%, and released only ∼39% drug over a period of 12 h, thus indicating its potential as a controlled drug delivery system.
KEY WORDS: controlled release, cubosomes, docetaxel, gelation, thermoresponsive gel
INTRODUCTION
Controlled drug delivery systems are typically designed to optimize the drug release profiles. Polymers, surfactants, and other such excipients are commonly employed in the preparation of modern controlled drug delivery systems. Specific surfactant and polymer systems are known to form supra-assemblies in situ and can be used to design systems for the delivery of active pharmaceutical ingredients (APIs) (1–4). These systems typically include liquid crystalline aggregates such as liposomes and cubosomes, or cross-linked gel networks such as hydrogels etc. Incorporation of drugs into the complex internal domains of these structures can facilitate a diffusion-controlled release of the drug into the surrounding external aqueous environment and may provide new ways to modify pharmacokinetic profiles using lipid-based systems (5–9). The success of these systems as controlled drug delivery systems depends significantly on several formulation and process variables. These factors include, but not limited to, the drug-loading capacity, physical-chemical compatibility of the drug and the vehicle, and the stability of the system (1).
Cubosomes have been described as bi-continuous cubic phase liquid crystals consisting of two separate, continuous, and non-intersecting hydrophilic regions divided by a lipid bilayer (1). Cubosomes possess several properties that make them amenable as potential carriers for drug delivery. Cubosomes are nanoparticles (or more accurately nanostructured particles) that exist in a self-assembled liquid-crystalline phase with a solid-like rheology. In recent years, there has been a growing interest among researchers in exploring the pharmaceutical applications of cubosomes and cubosome-based systems. With the recent developments in the technology, experience and expertise in the area of nano-pharmaceuticals, cubosome-based systems are being actively pursued as potential alternatives to now-common systems such as liposomes and niosomes (10,11). Cubosomes are made up of a binary system of monoolein and water, where the monoolein acts as a precursor for lipid bilayer, which divides the hydrophilic regions of cubic phases (10,12). This binary system can self-assemble into thermodynamically stable bi-continuous, cubic, liquid-crystalline phases (13).
The feasibility of cubosomes as carriers for parenteral, topical, and buccal delivery of drugs has been reported in the literature (14–19). Formulations based on this technology have been reported to have improved tolerance and drug bioavailability (20). The parenteral administration route is among the most common and efficient route for the delivery of active drug substances with poor bioavailability and a narrow therapeutic index. However, parenteral route, along with offering a rapid onset, is also associated with rapid decline in systemic drug levels. An effective treatment often requires maintaining systemic drug levels within the therapeutically effective concentration range for an extended period. This entails frequent injections, which may lead to patient discomfort and non-compliance.
Thus, a controlled drug delivery system comprising of cubosome-based thermoreversible gel formulation may provide a possible solution to improve therapeutic efficiency. A thermoreversible gel system essentially consists of a thermo-gelling polymer, the aqueous solution of which is a liquid at or below room temperature, and forms a gel at body temperature (∼37°C) (21). Such systems have been suggested for the delivery of cells or biopharmaceuticals that are susceptible to heat or organic solvents (21). Thermally induced gelling systems show thermoreversible sol/gel transitions and are characterized by a lower critical solution temperature (22–25). They are liquid at room temperature and produce a gel at and above the lower critical solution temperature (22–25). The incorporation of cubosomes into a thermoresponsive gel should increase drug loading while, in all probability, yielding a lower, more prolonged drug release compared with pure gel (26). Therefore, a system containing both thermoresponsive polymer and cubosome can combine the advantages of both systems, the thermos-gelling properties of polymer and the drug-carrying ability of the cubosome (26).
In the current study, the authors aimed to identify the feasibility of developing such a system for controlling the release of docetaxel. Docetaxel trihydrate (DTX) is an antineoplastic agent which acts by blocking the cellular mitotic and interphase functions (27,28). The anticancer activity of DTX is dependent on its concentration and duration of exposure. Although it is widely used in cancer chemotherapy, several problems associated with its clinical use remain unresolved. Due to its poor water-solubility, DXT is administered by a continuous intravenous delivery along with lipophilic solvents. This formulation has limited stability and is associated with significant vehicle-related toxicities (29–32).
The aim of our study was mainly divided into two components. First, the development of a controlled-release formulation of docetaxel consisting of cubosome as a carrier, loaded within a thermoresponsive gel system. Secondly, optimizing the concentrations of gel-forming agents that could potentially meet the criteria for typical thermoresponsive gel-based drug delivery systems. Figure 1 describes the general scheme of the work flow, as followed.
Fig. 1.
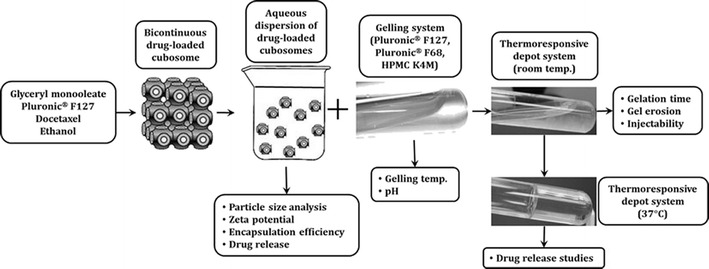
Schematic representation of the development of thermoresponsive gel formulations containing docetaxel-loaded cubosomes
MATERIALS AND METHODS
Materials
Docetaxel was obtained as a gift sample from Scino Pharmaceutical Pvt., Taiwan. Glyceryl monooleate was obtained from Otto Chemie., Mumbai (India). Pluronic® F127 (PF127) obtained from Research Lab Fine Chem Industries, Mumbai (India), and Pluronic® F68 (PF68) was obtained from Himedia Laboratory Pvt. Ltd. HPMC K4M was obtained from Raychem Lab. Chemical Pvt. Ltd., Chennai (India). Carbopol® 934P and methylcellulose were obtained from Loba Chemie Pvt. Ltd. Mumbai (India). Water was purified on a Milli-Q system obtained from a Millipore® synergy system (Millipore, Billerica, Massachusetts, USA). All other chemicals used were of analytical grade.
Preparation of Cubosomes
Bulk cubic phases were prepared by modifying the method suggested by Kojarunchitt et al. from a glyceryl monooleate and Pluronic® F127 melt, with or without drug (26). Briefly, glyceryl monooleate and Pluronic® F127 (9:1 w/w) were melted at 60°C and mixed until homogenous. The drug was loaded by first preparing a solution in ethanol. This drug solution was then added to a previously prepared homogeneous mixture of glyceryl monooleate and Pluronic® F127. To this mixture, 5.0 mL of ethanol was added as a hydrotropic solvent which helps bind the hydrophilic and lipophilic phases, to form a bi-continuous lipid bilayer.
To prepare the cubosome dispersion, the low-viscosity homogenous melt was either added drop-wise or injected into excess water, with continuous stirring on a magnetic stirrer such that the concentration of lipid in the sample was approximately 8–10% (w/w). The final volume was made up to 100.0 mL with distilled water (Table I). The samples were allowed to equilibrate at room temperature overnight. The samples were then homogenized for 2 min in a homogenizer at 1500–2000 rpm.
Table I.
Formulation of Cubosome Dispersion Using Pluronic® F127
| Cubosome dispersion formulation | Glyceryl monooleate (% w/w) | Pluronic F127 (% w/w) | Ethanol | DTX (% w/v) | Distilled water |
|---|---|---|---|---|---|
| CD 1 | 9 | 1 | 5 mL | – | q.s* |
| CD 2 | 9 | 1 | 5 mL | 2 | q.s |
*q.s. quantity sufficient to make 100 mL
Preparation of Gelling System
Poloxamer solutions were prepared according to the “cold method” previously described by Soga et al. with some modifications (33). Briefly, Pluronic® F127 and Pluronic® F68 were accurately weighed and solubilized in required volume of distilled water by continuous stirring, along with the addition of HPMC K4M. The dispersion was left to hydrate overnight (4–8°C) to obtain a uniform, glassy solution. After complete hydration of the polymers, the resultant solution was thoroughly mixed on a magnetic stirrer until a uniform and clear solution was obtained. The composition of different gelling systems prepared is shown in Table II.
Table II.
Composition of the Thermoresponsive Gelling Systems
| Gelling system | Pluronic® F127 (% w/v) | Pluronic® F68 (% w/v) | HPMC K4 M (% w/v) | Distilled water |
|---|---|---|---|---|
| G1 | 18 | 18 | 0.5 | q.s.* |
| G2 | 19 | 18 | 0.5 | q.s. |
| G3 | 20 | 18 | 0.5 | q.s. |
| G4 | 18 | 19 | 0.5 | q.s. |
| G5 | 19 | 19 | 0.5 | q.s. |
| G6 | 20 | 19 | 0.5 | q.s. |
| G7 | 20 | 20 | 0.5 | q.s. |
| G8 | 18 | 35 | 0.5 | q.s. |
| G9 | 18 | 38 | 0.5 | q.s. |
| G10 | 18 | 40 | 0.5 | q.s. |
| G11 | 17 | 35 | 0.5 | q.s. |
| G12 | 17 | 38 | 0.5 | q.s. |
*q.s. quantity sufficient to make 100 mL
Preparation of Thermoresponsive Depot System
Cubosome-containing thermoresponsive depot system was prepared by incorporation of previously prepared cubosome dispersion into the poloxamer gelling system. Final formulation was prepared by addition of cubosome dispersion (20 mg/mL) into the gelling system in the ratio of 1:2. The solution was then continuously stirred on a magnetic stirrer at 25°C for 24 h to get a homogenous mixture. The composition of different formulations prepared is shown in Table III.
Table III.
Thermoresponsive Depot Systems Containing DTX-Loaded Cubosomes
| Formulation | Gelling systema | Cubosome dispersion (mL) | Cubosome: gelling system | |
|---|---|---|---|---|
| PF127 | PF68 | |||
| F1 | 19% | 18% | 2 | 1:2 |
| F2 | 20% | 18% | 2 | 1:2 |
| F3 | 20% | 20% | 2 | 1:2 |
| F4 | 20% | 19% | 2 | 1:2 |
| F5 | 19% | 19% | 2 | 1:2 |
| F6 | 18% | 19% | 2 | 1:2 |
PF127 Pluronic® F127, PF68 Pluronic® F68
aQuantities indicate the % w/v composition of PF127 and PF68
Drug-Excipient Compatibility Studies
Thermal Analysis
The drug, the polymers, and their physical mixtures with DTX were analyzed by differential scanning calorimetry (DSC). Open pan DSC measurements were carried out using a DSC Q20 (TA Instruments Inc., New Castle, DE) with a sample size of approximately 5 mg weighed into each aluminum pans. Samples were heated at 10°C/min from 0 to 400°C. Nitrogen at a flow rate of 40 mL/min was used as a purge gas in DSC analyses. The results were analyzed using the Universal Analysis software version 4.5A, build 4.5.0.5 (TA Instruments, Inc., New Castle, DE, USA).
FTIR Spectroscopy
To determine any possible interactions, the physical mixtures of the drug and the polymers were analyzed using the Fourier transformed infrared (FTIR) spectroscopy. Briefly, the samples were dried in a hot air oven at 50°C for 2 h. The samples were compressed under pressure of 10 t/nm2 to prepare circular KBr disks. The samples were scanned in the range of 400 to 4000 cm−1. The shifts in the spectra of the drug in the presence of polymers and other components were investigated to determine physical interactions between the drug and the polymers, if any.
Characterization of Cubosomes
Particle Size and Zeta Potential Analysis
The particle size analysis of the blank and the DTX-loaded cubosomes was carried out using photon correlation spectroscopy (PCS), with dynamic light scattering on a Zetasizer® nano (Model: Zen 3600, Malvern Instruments, Malvern, UK) equipped with a 5-mW helium neon laser with a wavelength output of 633 nm. The measurements were carried out at 25°C, at an angle of 90°, and a run time of at least 40–80 s. Water was used as a dispersant. The zeta potential was measured by Smoluchowski’s equation from the electrophoretic mobility of cubosomes (34). All measurements were performed in triplicate.
Entrapment Efficiency
The entrapment efficiency i.e., the DTX content encapsulated in cubosomes, was evaluated using a combination of methods described previously in the literature (35–37). Briefly, 1 mL of cubosomes containing DTX were added into the reservoir of Centricon® (Model: YM-100, Amicon, Millipore, Bedford, MA, USA). After centrifuging the cubosome dispersion at 15,000 rpm for 40 min, the filtrate containing free DTX was removed. The filtered dispersion was then diluted with methanol and analyzed for DTX content using HPLC. The HPLC analysis was used to compute the total concentration of DTX (C t), and the concentration of DTX contained in the filtrate after centrifugation (C f). The entrapment efficiency was calculated using the following equation:
Determination of In-Vitro DTX Release from Cubosomes
The DTX release from the cubosome dispersion was evaluated by measuring the diffusion of the drug across a cellophane membrane using Franz diffusion cell. The cell consisted of two compartments i.e., the donor compartment and the receptor compartment. A previously activated semipermeable membrane was placed between these two compartments. The dispersion formulation was added on to the donor compartment above the membrane. The receptor compartment contained 18 mL phosphate buffer saline solution (PBS, pH 7.4), maintained at 37 ± 0.5°C, as a release medium. At predetermined time intervals, aliquots of the release medium were withdrawn and replaced with an equal volume of fresh release medium. The drug concentrations in the release medium at various time intervals were analyzed using HPLC.
HPLC Analysis of DTX
DTX concentration was measured using HPLC analysis (SPD-10Avp Shimadzu pump, LC-10Avp Shimadzu UV-vis detector) at 230 nm as previously described by Loos et al. (38). Briefly, samples were chromatographed on a 4.6 mm × 250 mm reverse phase stainless steel column packed with 5 μm particles (Venusil XBP C-18, Agela, China) and eluted with a mobile phase consisting of acetonitrile/water (55:45, v/v) at a flow rate of 1 mL/min. The column temperature was maintained at room temperature. The samples were appropriately diluted with methanol and injected (20 μL) directly into the HPLC system without further treatment. The calibration of the peak area against concentration of DTX was found to be y = 11485x − 647.64 with r 2 = 0.9998 for the DTX concentration range of 1–40 μg/mL (where y: peak area and x: DTX concentration), and the limit of detection was found to be 0.02 μg/mL.
Characterization of Gelling System
Determination of Gelation Temperature by Tilting Method
The gelation temperature was determined using the method described earlier by Zaki et al. (39). Briefly, 2 mL aliquots of the polymer solution were transferred to a test tube and immersed in the water bath. The temperature of the water bath was increased slowly and allowed to equilibrate for 5 min at each new setting. The sample was then examined for gelation, which was said to have occurred when the meniscus would no longer move upon tilting at a 90° angle. Thermoreversible polymer-based liquid formulations, which provide in-situ gelling property at physiological temperatures, were developed with a goal of delaying the release of DTX from the depot system.
Determination of pH of the Gelling System
The pH of each formulation was determined by using a pH meter (Model: S220 SevenCompact™ pH/Ion, Mettler Toledo, USA). The pH meter was first calibrated using standard solutions of pH 4 and pH 9.2. The commonly observed pH range for in-situ gel formulations is 6.0–7.5 (40–43).
Characterization of Thermoresponsive Depot System
Determination of Gelation Time
The gelation time was determined by increasing the temperature of the formulations up to 37°C, and the time required by the formulations (containing different concentrations of the polymers) to form a stiff gel was recorded using a digital stopwatch.
In-Vitro Gel Erosion Study
The vials (inner diameter = 13.5 mm) containing approximately 1 g of prepared solution were placed in a water bath, maintained at a constant temperature of 37°C. After the formulations had transformed into gels, 1.5 mL PBS (pH 7.4), pre-warmed to 37°C, was carefully layered over the gel surface. At predetermined intervals, the entire release medium was removed, and the weight of the vial and the remaining gel was recorded. The percentage weight loss of the gel was calculated by dividing the decrease in the weight of the gel by the initial gel weight.
Gel Formation and Injectability Test
The injectability test of the prepared formulations was carried out using a 22-gauge needle and a syringe which is generally used for intramuscular administrations. Accurately measured formulation (2 mL) was injected into the release media (PBS, pH 7.4) maintained at a constant temperature of 37°C, and the ease of injection of the formulations through the needle was visually observed.
In-Vitro DTX Release Study from Depot Formulation
The “inverted cup” method, as described by Soderberg et al., was used with some modifications, to study the drug release from the depot formulations (44). Briefly, the formulation was introduced in an inverted cup, through a hole, by the means of a 1-mL syringe. The flow of the release medium around the sample was controlled by the stirring rate of the magnetic bar. The speed of the magnetic stirrer was maintained at 200 rpm. One thousand milliliters of PBS (pH 7.4) was used as a standard release medium, which filled the conical flask to the rim. The temperature of the system was maintained at 37 ± 0.5°C. One-milliliter samples of the release medium were drawn at hourly intervals up to 12 h. All the tests were performed in triplicate. The drug concentrations in the release medium were measured using HPLC analysis, as described above.
Statistical Analysis
All data were expressed as mean ± standard deviation (SD). The statistical analysis was carried out by a two-way analysis of variance (ANOVA) followed by Bonferroni post-test using GraphPad® Prism® software version 5.03 (San Diego, CA). The differences between means were considered to be significant if the P value was <0.05.
RESULTS
Drug-Excipient Compatibility Studies
Thermal Analysis
The DSC thermogram of pure DTX showed a sharp endothermic peak at ∼169°C, corresponding to DTX melting point (thermograms not shown). Additionally, the DSC thermograms of Pluronic® F127 and Pluronic® F68 revealed sharp endothermic peaks at temperatures of ∼58 and ∼56°C, respectively. HPMC K4M revealed broad, undefined endothermic peak over a temperature range of 20–80°C. Such broad endothermic peaks, mainly due to the dehydration process, are typically observed with predominantly amorphous polymers.
The DSC analysis of the physical mixtures, DTX: Pluronic® F127 (1:1), DTX: Pluronic® F68 (1:1), and DTX: HPMC K4M (1:1), revealed a negligible and a non-significant change in the thermal behavior of DTX in the presence of these polymers. Additionally, the melting signals (endotherm) of Pluronic® F127, and Pluronic® F68 were clearly distinguishable in the physical mixtures of the respective polymers with DTX. The absence of any other endothermic/exothermic event over the entire temperature range thus excluded any physical interaction or obvious incompatibility between the drug and the polymers. The DSC results thus indicated the suitability of these polymers to be used in the prepared formulations.
Fourier Transform Infrared (FTIR) Spectroscopy
The FTIR spectra for DTX, Pluronic® F127, Pluronic® F68 and HPMC K4M individually, the binary mixtures of the drug with individual polymers, as well as a mixture of the drug with all the polymers, are shown in Fig. 2. The FTIR analysis did not show any significant difference between the individual spectra and those obtained from their physical mixtures. The results obtained after the FTIR study thus indicated that there was no positive evidence for the interaction between docetaxel trihydrate and the used polymers.
Fig. 2.
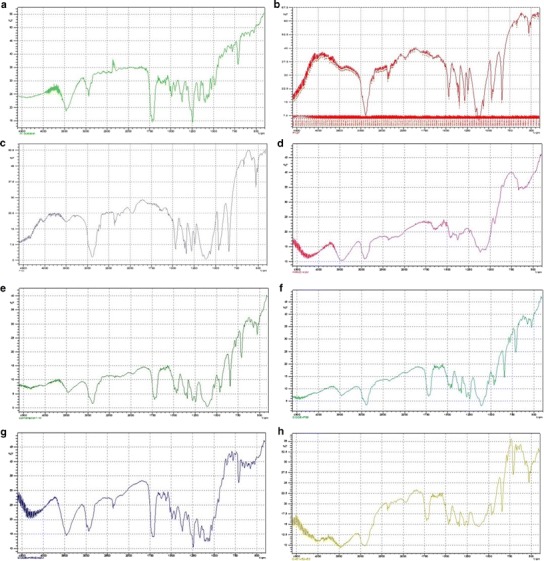
FTIR analysis of the drug and the polymers. a Docetaxel trihydrate, b Pluronic® F127, c Pluronic® F68, d HPMC K4M, e docetaxel trihydrate: Pluronic® F127 (50:50), f docetaxel trihydrate: Pluronic® F68 (50:50), g docetaxel trihydrate: HPMC K4M (50:50), h docetaxel trihydrate: Pluronic® F127: Pluronic® F68: HPMC K4M (25:25:25:25)
Characterization of Cubosome
Particle Size Determination
Smaller particles have higher surface area/volume ratio, which makes it easier for the encapsulated drug to be released from the cubosome via diffusion and surface erosion, and also have the added advantage for the drug-loaded cubosomes to penetrate into and permeate through the physiological drug barriers. It has been previously suggested that the larger particles (<5 mm) would be taken up via the lymphatics, and the smaller particles (<500 nm) can cross the membrane of epithelial cells through endocytosis (45,46). The mean particle size, polydispersity index (PDI), and the zeta potential values of cubosome dispersion prepared by Pluronic® F127, with (CD2) and without DTX (CD1), are shown in Table IV. The mean particle size of blank cubosomes (CD1) and DTX-loaded cubosomes (CD2) were found to be 184.9 ± 2.47 and 220.9 ± 3.02 nm, respectively. Thus, the DTX loading was found to only marginally increase the mean particle size of the cubosomes. Moreover, the low polydispersity index values, 0.162 ± 0.014 and 0.173 ± 0.019 for blank cubosomes (CD1) and DTX-loaded cubosomes (CD2), respectively, indicated a narrow particle size distribution (47). Zeta potential is another important index for the stability of the cubosomes. A high absolute value of zeta potential indicates a high electric charge on the surface of the cubosomes, which can cause strong repellent forces among particles and prevent aggregation of the cubosomes in a buffer solution (37,48). The zeta potential of blank cubosomes (CD1) and DTX-loaded cubosomes (CD2) were found to be −54.1 ± 1.57 and−47.4 ± 1.80 mV, respectively. Typically, a minimum zeta potential of greater than −30 mV is considered acceptable and indicative of a good physical stability (49).
Table IV.
Particle Size, Polydispersity Index, and Zeta Potential of Cubosomes Prepared with Pluronic F127
| Cubosome dispersion formulation | Particle size (nm) | Polydispersity index (PDI) | Zeta potential (mV) |
|---|---|---|---|
| CD 1 | 184.9 ± 2.47 | 0.162 ± 0.014 | −54.1 ± 1.57 |
| CD 2 | 220.9 ± 3.02 | 0.173 ± 0.019 | −47.4 ± 1.80 |
Values are mean ± std. dev. (n = 3)
Entrapment Efficiency
The drug entrapment efficiency is an important parameter for drug delivery systems. This is especially true for expensive drugs. The entrapment efficiency of the docetaxel-loaded cubosomes was found to be 94.74 ± 3.41 (% w/w), indicating that most of DTX was encapsulated in cubosomes.
In-Vitro DTX Release from Cubosome Dispersion
Figure 3 shows the release pattern of DTX from the cubosome dispersion (CD2) as evaluated using the Franz diffusion cell. The drug-loaded cubosomes exhibited an initial burst release (32.49%) within 1 h. This was followed by a controlled release of DTX from the cubosomes over a period of 12 h. At the end of 12 h, over 95% of DTX was released from the cubosomes. The results indicated the potential of cubosomes to be utilized in the controlled drug delivery systems.
Fig. 3.

The in-vitro release of DTX from cubosomes and from thermoresponsive depot formulations. All values are mean ± SD (n = 3). *p < 0.01, compared to DTX-loaded cubosomes
Characterization of Gelling System
The pH of all tested formulations was found to be in the range of pH 6.5–7.5.
Determination of Gelation Temperature by Tilting Method
The sol-to-gel transition end-point was determined by the “tilting method” (Fig. 4). The gelation temperature of the thermoresponsive gelling systems containing different ratios of the polymers were found to be in the range of 29–40°C. Specifically, the gelation temperature for the formulation F2 containing Pluronic® F127 (20%), Pluronic® F68 (18%), and HPMC K4M (0.5%) was found to be in the range of 36–38°C, which is considered appropriate for the in-situ gelling of the system (26,31,33,50–52).
Fig. 4.

Sol-to-gel transition of the dispersion. a Dispersion at 10°C and b dispersion at the gelation temperature (36–38°C)
Characterization of Thermoresponsive Depot System
Determination of Gelation Time
The time required by the prepared thermoresponsive depot formulations to form a stiff gel (sol-to-gel transition) in response to increasing temperature is listed in Table V. The temperature was increased from 10 to 37°C at the rate of 2°C/min. The temperature was then maintained at 37 ± 0.5°C, and the lag time of gelation for each formulation was determined. The formulations exhibited a wide range of gelation lag times. The formulation F2 was found to gel rapidly and demonstrated the shortest lag time of 3 min and 5 s.
Table V.
The Sol-To-Gel Lag Time for Different Formulations
| Formulation | Sol-to-gel lag timea (min) |
|---|---|
| F1 | 3.50 ± 0.12 |
| F2 | 3.05 ± 0.13 |
| F3 | 10.25 ± 0.46 |
| F4 | 20.48 ± 0.94 |
| F5 | 18.05 ± 0.87 |
| F6 | 18.20 ± 0.82 |
Values are mean ± std. dev. (n = 3)
aAll values are measured at 37 ± 0.5°C
In-Vitro Gel Erosion Study
The erosion of the gel from the prepared formulations was calculated by determining the gel loss (% w/w) at various time intervals, after the gel-containing vials were allowed to incubate at 37 ± 0.5°C. Table VI summarizes the rates of gel erosion (% w/w) at various time intervals. The formulations F1, F2, F3, F4, F5, and F6 exhibited 46.3%, 39.0%, 38.1%, 38.3%, 39.4%, and 41.0%, respectively, after 96 h. The extent of erosion was not found to be statistically significant between the formulations.
Table VI.
The In-Vitro Gel Erosion as a Function of Time
| Depot formulation | Gel lossa (% w/w) | ||||
|---|---|---|---|---|---|
| 12 h | 24 h | 48 h | 72 h | 96 h | |
| F1 | 7.5 ± 0.059 | 18.0 ± 1.34 | 25.5 ± 1.97 | 37.5 ± 3.08 | 46.3 ± 4.11 |
| F2 | 6.7 ± 0.48 | 15.2 ± 1.08 | 22.4 ± 1.85 | 30.3 ± 2.46 | 39.0 ± 3.18 |
| F3 | 5.2 ± 0.38 | 12.1 ± 0.87 | 20.2 ± 1.56 | 28 ± 2.14 | 38.1 ± 2.89 |
| F4 | 6.8 ± 0.51 | 16.2 ± 1.28 | 21.6 ± 1.72 | 28.0 ± 2.06 | 38.3 ± 2.71 |
| F5 | 7.2 ± 0.54 | 17.9 ± 1.33 | 23.0 ± 1.89 | 29.8 ± 2.11 | 39.4 ± 2.70 |
| F6 | 7.5 ± 0.60 | 18.3 ± 1.41 | 25.3 ± 1.92 | 32.0 ± 2.94 | 41.0 ± 3.74 |
Values are mean ± std. dev. (n = 3)
aAll values are measured at 37 ± 0.5°C
Gel Injectability Test
The parenteral administration of DTX requires passage of formulation through the needle for easy injectability. The injectability test carried out for prepared depot formulation showed that all the formulations successfully passed through a 22-gauge needle.
In-Vitro DTX Release Study from Thermoresponsive Depot Formulation
The cubosome dispersion-based depot formulations of DTX were injectable solutions at room temperature and turned into stiff gels rapidly at 37°C. The results from the in-vitro drug-release study are shown in Fig. 3. The release of DTX from cubosomes over a period of 12 h is compared with the DTX release from the depot formulations containing DTX-loaded cubosomes. The cubosomes exhibited a non-linear, albeit sustained release of DTX over a period of 12 h. At the end of 12 h, over 95% of DTX was released from the cubosomes. The formulation F2 showed an initial burst release of 21.48 ± 1.59%, followed by a controlled release of DTX for over 12 h. Formulations F3 and F5 showed initial burst release of 21.06 ± 1.67 and 20.77 ± 1.64%, respectively, and controlled the DTX release over 12 h. Whereas, formulation F1, F4, and F6 exhibited an initial burst release to 16.12 ± 1.04%, 16.80 ± 1.07%, and 15.63 ± 0.98%, respectively, and prolonged the release of DTX for over 12 h. At the end of 12 h, the DTX release ranged from 39 to 60% from these formulations.
DISCUSSION
A docetaxel-loaded thermoresponsive depot system for a controlled release was prepared. This depot system was evaluated for various parameters like gelation time, injectability, gel erosion (%), and in-vitro drug release. Formulations containing different concentrations of PF127 (18–20% w/v) were found to flow freely at ambient room temperature. The formulations containing a lower concentration of Pluronic® F127 (18% w/v) exhibited a higher gelation temperature, as determined by the test tube “tilting” method. On the other hand, the formulations containing a higher concentration of Pluronic® F127 (20% w/v) were observed to form a stable gel at lower temperature. The concentration of Pluronic® F68 was also found to have a significant effect on the gelation temperature of the gelling system. The formulations containing 18 or 19% w/v Pluronic® F68 demonstrated lower gelation temperatures compared to those containing 20–19% of Pluronic® F68.
In contrast to results obtained in this study, Wei et al. reported that addition of pluronic® F68 into Pluronic® F127 based thermoresponsive gels decreases sol-gel transition temperature (52). Vadnere et al. had previously demonstrated that this was due to higher hydrophilicity of the Pluronic® F68 than that of the Pluronic® F127, which could disrupt the hydration shells around the hydrophobic portion of Pluronic® F127 molecules (51). Klouda et al. also explained the changes in properties of Pluronic® F127 molecule after a change in concentration and temperature of the Pluronic® solution system (50). This was the main reason for selecting the concentrations of Pluronic® F127 (20% w/v) and Pluronic® F68 (18% w/v) for further study and formulation of the depot system. The depot formulation showed a rapid sol-to-gel transition at temperatures close to physiological temperature i.e., 37 ± 0.5°C. The prepared formulations showed a range of lag time for gelation. Among the formulations tested, the formulation containing 20% of Pluronic® F127 and 18% of Pluronic® F68 exhibited the fastest sol-to-gel transition i.e., a lower lag time of 3 min 5 s. The results of injectability test carried out for prepared depot formulation showed that the formulations were able to pass easily from a 22-gauge needle. The in-vitro gel erosion study showed that the formulation containing 20% Pluronic® F127 and 18% Pluronic® F68 had a relatively higher gel strength (only 39.0% of gel erosion after 96 h). The formulations containing lower concentration of Pluronic® F127 (19%) and Pluronic® F68 (18%) exhibited lower gel strength (46.3% gel erosion after 96 h). The results indicate that the gel strength of the system enhances when the concentration of Pluronic® F127 was gradually increased, along with the addition of 0.5% HPMC K4M which act as a gelling agent and tends to improve the gel strength. The concentration of HPMC K4M was chosen by studying the effect of various concentrations of HPMC K4M (0.3–1.0%) on the gel behavior, gel erosion, and gel stability in aqueous environment. The concentration of HPMC K4M when increase from 0.3 to 1.0%, the stability and gel strength were also found to be increased. However, the gelling system containing 1.0% HPMC K4M resulted in a turbid formulation, while that containing 0.3% HPMC K4M showed the formation of a transparent glassy solution with lower gel strength. Thus, a concentration of 0.5% HPMC K4M was found to be optimal for the prepared thermoresponsive formulations; this concentration resulted in a gel with sufficient gel strength without any noticeable turbidity.
The in-vitro drug release studies on cubosome dispersions, carried out using the Franz diffusion cell, revealed that loading of DTX into cubosome considerably slowed the release of DTX, extending up to 12 h. Further reduction of release was observed from the drug-loaded cubosomes, when incorporated into the thermoresponsive depot system. The prepared thermoresponsive depot formulation exhibited a controlled release of the drug compared to the drug release from cubosome dispersion alone. These findings were expected, and can be attributed to an increase in the number of barriers to the passive diffusion of the drug. The drug loaded in cubosomes has to cross only one barrier of cubosome. However, when incorporated into a thermoresponsive gelling system, cubosomes act as a drug-reservoir surrounded by a protective layer of gelling system which enabled drug release over a prolonged time, i.e., over 12 h. The formulation of thermoresponsive depot system containing cubosomes and the gelling system (prepared using a combination of Pluronic® F127 (20%) and Pluronic® F68 (18%)) showed an initial burst release (21.48 ± 1.59%) within the first hour and extended the release of of DTX over 12 h. Only 39.83 ± 3.27% of the drug was found to be released from this formulation at the end of 12 h. The results thus indicate the feasibility of the formulations based on cubosome-thermoresponsive gel system, as a promising delivery system for controlled release of docetaxel and similar drugs.
CONCLUSIONS
A simple process, based on the dispersion and homogenization of glyceryl monooleate and Pluronic® in water, leads to the formulation of cubosomes. The incorporation of the drug-loaded cubosomes into a thermoresponsive gelling system resulted in a slower and a prolonged drug release. According to the results of this study, a thermoresponsive depot system based on Pluronic® (F127 and F68), containing cubosomal DTX, can be developed for a controlled drug delivery.
The cubosome-containing thermoresponsive depot formulation was found to be free flowing at ambient temperature and formed a depot gel at body temperature. Cubosomes may not only provide the means for substantially increased lipophilic drug loading but it may also act as a reservoir for sustained drug release. Therefore, this system containing the drug-loaded cubosomes incorporated in a thermoresponsive gelling system can be effectively used as controlled-release depot formulations. Nevertheless, further specialized studies such as detailed microscopic characterization of cubosome formation, as well as the drug loading in cubosomes, are required to further our understanding of this system as an alternative, parenteral-controlled drug delivery system.
References
- 1.Garg G, Saraf S, Saraf S. Cubosomes: an overview. Biol Pharm Bull. 2007;30(2):350–3. doi: 10.1248/bpb.30.350. [DOI] [PubMed] [Google Scholar]
- 2.Lynch ML, Ofori-Boateng A, Hippe A, Kochvar K, Spicer PT. Enhanced loading of water-soluble actives into bicontinuous cubic phase liquid crystals using cationic surfactants. J Colloid Interface Sci. 2003;260(2):404–13. doi: 10.1016/S0021-9797(02)00016-4. [DOI] [PubMed] [Google Scholar]
- 3.Xing Y, Wang C, Han P, Wang Z, Zhang X. Acetylcholinesterase responsive polymeric supra-amphiphiles for controlled self-assembly and disassembly. Langmuir. 2012;28(14):6032–6. doi: 10.1021/la300612k. [DOI] [PubMed] [Google Scholar]
- 4.Zhu Z, Reed WF. Enhanced surfactant supramicellar assembly by hydrophobic dopants. Langmuir. 2013;29(33):10376–82. doi: 10.1021/la402107z. [DOI] [PubMed] [Google Scholar]
- 5.Burrows R, Collett JH, Attwood D. The release of drugs from monoglyceride-water liquid crystalline phases. Int J Pharm. 1994;111(3):283–93. doi: 10.1016/0378-5173(94)90351-4. [DOI] [Google Scholar]
- 6.Dong YD, Larson I, Hanley T, Boyd BJ. Bulk and dispersed aqueous phase behavior of phytantriol: effect of vitamin E acetate and F127 polymer on liquid crystal nanostructure. Langmuir. 2006;22(23):9512–8. doi: 10.1021/la061706v. [DOI] [PubMed] [Google Scholar]
- 7.Drummond CJ, Fong C. Surfactant self-assembly objects as novel drug delivery vehicles. Curr Opin Colloid Interface Sci. 1999;4(6):449–56. doi: 10.1016/S1359-0294(00)00020-0. [DOI] [Google Scholar]
- 8.Fong W-K, Hanley T, Boyd BJ. Stimuli responsive liquid crystals provide ‘on-demand’ drug delivery in vitro and in vivo. J Control Release. 2009;135(3):218–26. doi: 10.1016/j.jconrel.2009.01.009. [DOI] [PubMed] [Google Scholar]
- 9.Shah JC, Sadhale Y, Chilukuri DM. Cubic phase gels as drug delivery systems. Adv Drug Deliv Rev. 2001;47(2–3):229–50. doi: 10.1016/S0169-409X(01)00108-9. [DOI] [PubMed] [Google Scholar]
- 10.Bei D, Marszalek J, Youan BB. Formulation of dacarbazine-loaded cubosomes-part I: influence of formulation variables. AAPS PharmSciTech. 2009;10(3):1032–9. doi: 10.1208/s12249-009-9293-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kumar MS, Balaji A. Review on Liposome Based Nanocarriers. Indo Am J Pharm Res. 2013;3(6):4372–82. [Google Scholar]
- 12.Larsson K. Two cubic phases in monoolein-water system. 1983;304:664-. doi:10.1038/304664c0.
- 13.Fontell K, Mandell L, Ekwall P. Some isotropic mesophases in systems containing amphiphilic compounds. Acta Chem Scand. 1968;22:3209–23. doi: 10.3891/acta.chem.scand.22-3209. [DOI] [Google Scholar]
- 14.Boyd BJ, Whittaker DV, Khoo S-M, Davey G. Hexosomes formed from glycerate surfactants—formulation as a colloidal carrier for irinotecan. Int J Pharm. 2006;318(1–2):154–62. doi: 10.1016/j.ijpharm.2006.03.010. [DOI] [PubMed] [Google Scholar]
- 15.Engstrom S, Ericsson B, Landh T. A cubosome formulation for intravenous administration of somatostatin. Proc Int Symp Control Rel Bioact Mater. 1996;23(2):89–90. [Google Scholar]
- 16.Leesajakul W, Nakano M, Taniguchi A, Handa T. Interaction of cubosomes with plasma components resulting in the destabilization of cubosomes in plasma. Colloids Surf B: Biointerfaces. 2004;34(4):253–8. doi: 10.1016/j.colsurfb.2004.01.010. [DOI] [PubMed] [Google Scholar]
- 17.Lopes L, Ferreira D, de Paula D, Garcia MT, Thomazini J, Fantini MA, et al. Reverse hexagonal phase nanodispersion of monoolein and oleic acid for topical delivery of peptides: in vitro and in vivo skin penetration of cyclosporin A. Pharm Res. 2006;23(6):1332–42. doi: 10.1007/s11095-006-0143-7. [DOI] [PubMed] [Google Scholar]
- 18.Swarnakar N, Jain V, Dubey V, Mishra D, Jain NK. Enhanced oromucosal delivery of progesterone via hexosomes. Pharm Res. 2007;24(12):2223–30. doi: 10.1007/s11095-007-9409-y. [DOI] [PubMed] [Google Scholar]
- 19.Esposito E, Cortesi R, Drechsler M, Paccamiccio L, Mariani P, Contado C, et al. Cubosome dispersions as delivery systems for percutaneous administration of indomethacin. Pharm Res. 2005;22(12):2163–73. doi: 10.1007/s11095-005-8176-x. [DOI] [PubMed] [Google Scholar]
- 20.Han S, Shen JQ, Gan Y, Geng HM, Zhang XX, Zhu CL, et al. Novel vehicle based on cubosomes for ophthalmic delivery of flurbiprofen with low irritancy and high bioavailability. Acta Pharmacol Sin. 2010;31(8):990–8. doi: 10.1038/aps.2010.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee J, Joo MK, Oh H, Sohn YS, Jeong B. Injectable gel: poly(ethylene glycol)–sebacic acid polyester. Polymer. 2006;47(11):3760–6. doi: 10.1016/j.polymer.2006.03.109. [DOI] [Google Scholar]
- 22.Eliaz RE, Kost J. Characterization of a polymeric PLGA-injectable implant delivery system for the controlled release of proteins. J Biomed Mater Res. 2000;50(3):388–96. doi: 10.1002/(SICI)1097-4636(20000605)50:3<388::AID-JBM13>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 23.Eliaz RE, Szoka FC., Jr Robust and prolonged gene expression from injectable polymeric implants. Gene Ther. 2002;9(18):1230–7. doi: 10.1038/sj.gt.3301786. [DOI] [PubMed] [Google Scholar]
- 24.Haglund BO, Joshi R, Himmelstein KJ. An in situ gelling system for parenteral delivery. J Control Release. 1996;41(3):229–35. doi: 10.1016/0168-3659(96)01333-8. [DOI] [Google Scholar]
- 25.Shively ML, Coonts BA, Renner WD, Southard JL, Bennett AT. Physico-chemical characterization of a polymeric injectable implant delivery system. J Control Release. 1995;33(2):237–43. doi: 10.1016/0168-3659(94)00097-E. [DOI] [Google Scholar]
- 26.Kojarunchitt T, Hook S, Rizwan S, Rades T, Baldursdottir S. Development and characterisation of modified poloxamer 407 thermoresponsive depot systems containing cubosomes. Int J Pharm. 2011;408(1–2):20–6. doi: 10.1016/j.ijpharm.2011.01.037. [DOI] [PubMed] [Google Scholar]
- 27.Bissery MC. Preclinical pharmacology of docetaxel. Eur J Cancer. 1995;31A(Suppl 4):S1–6. doi: 10.1016/0959-8049(95)00357-O. [DOI] [PubMed] [Google Scholar]
- 28.Trudeau ME. Docetaxel: a review of its pharmacology and clinical activity. Can J Oncol. 1996;6(1):443–57. [PubMed] [Google Scholar]
- 29.Beijnen JH, Beijnenbandhoe AU, Dubbelman AC, Vangijn R, Underberg WJM. Chemical and physical stability of etoposide and teniposide in commonly used infusion fluids. J Parenter Sci Technol. 1991;45(2):108–12. [PubMed] [Google Scholar]
- 30.S-S F, Huang G. Effects of emulsifiers on the controlled release of paclitaxel (Taxol®) from nanospheres of biodegradable polymers. J Control Release. 2001;71(1):53–69. doi: 10.1016/S0168-3659(00)00364-3. [DOI] [PubMed] [Google Scholar]
- 31.Nie S, Hsiao WL, Pan W, Yang Z. Thermoreversible Pluronic® F127-based hydrogel containing liposomes for the controlled delivery of paclitaxel: in vitro drug release, cell cytotoxicity, and uptake studies. Int J Nanomedicine. 2011;6:151–66. doi: 10.2147/IJN.S15057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yeh M-K, Coombes AGA, Jenkins PG, Davis SS. A novel emulsification-solvent extraction technique for production of protein loaded biodegradable microparticles for vaccine and drug delivery. J Control Release. 1995;33(3):437–45. doi: 10.1016/0168-3659(94)00123-C. [DOI] [Google Scholar]
- 33.Soga O, van Nostrum CF, Fens M, Rijcken CJF, Schiffelers RM, Storm G, et al. Thermosensitive and biodegradable polymeric micelles for paclitaxel delivery. J Control Release. 2005;103(2):341–53. doi: 10.1016/j.jconrel.2004.12.009. [DOI] [PubMed] [Google Scholar]
- 34.Sze A, Erickson D, Ren L, Li D. Zeta-potential measurement using the Smoluchowski equation and the slope of the current–time relationship in electroosmotic flow. J Colloid Interface Sci. 2003;261(2):402–10. doi: 10.1016/S0021-9797(03)00142-5. [DOI] [PubMed] [Google Scholar]
- 35.Chung H, Kim J, Um JY, Kwon IC, Jeong SY. Self-assembled “nanocubicle” as a carrier for peroral insulin delivery. Diabetologia. 2002;45(3):448–51. doi: 10.1007/s00125-001-0751-z. [DOI] [PubMed] [Google Scholar]
- 36.Gao K, Sun J, Liu K, Liu X, He Z. Preparation and characterization of a submicron lipid emulsion of docetaxel: submicron lipid emulsion of docetaxel. Drug Dev Ind Pharm. 2008;34(11):1227–37. doi: 10.1080/03639040802005057. [DOI] [PubMed] [Google Scholar]
- 37.Zhang Z, Feng S-S. The drug encapsulation efficiency, in vitro drug release, cellular uptake and cytotoxicity of paclitaxel-loaded poly(lactide)–tocopheryl polyethylene glycol succinate nanoparticles. Biomaterials. 2006;27(21):4025–33. doi: 10.1016/j.biomaterials.2006.03.006. [DOI] [PubMed] [Google Scholar]
- 38.Loos WJ, Verweij J, Nooter K, Stoter G, Sparreboom A. Sensitive determination of docetaxel in human plasma by liquid–liquid extraction and reversed-phase high-performance liquid chromatography. J Chromatogr B Biomed Sci Appl. 1997;693(2):437–41. doi: 10.1016/S0378-4347(97)00089-3. [DOI] [PubMed] [Google Scholar]
- 39.Zaki NM, Awad GA, Mortada ND, Abd ElHady SS. Enhanced bioavailability of metoclopramide HCl by intranasal administration of a mucoadhesive in situ gel with modulated rheological and mucociliary transport properties. Eur J Pharm Sci. 2007;32(4–5):296–307. doi: 10.1016/j.ejps.2007.08.006. [DOI] [PubMed] [Google Scholar]
- 40.Chenite A, Chaput C, Wang D, Combes C, Buschmann MD, Hoemann CD, et al. Novel injectable neutral solutions of chitosan form biodegradable gels in situ. Biomaterials. 2000;21(21):2155–61. doi: 10.1016/S0142-9612(00)00116-2. [DOI] [PubMed] [Google Scholar]
- 41.Mandal S, Thimmasetty MKMJ, Prabhushankar GL, Geetha MS. Formulation and evaluation of an in situ gel-forming ophthalmic formulation of moxifloxacin hydrochloride. Int J Pharm Investig. 2012;2(2):78–82. doi: 10.4103/2230-973X.100042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ruel-Gariépy E, Leroux J-C. In situ-forming hydrogels—review of temperature-sensitive systems. Eur J Pharm Biopharm. 2004;58(2):409–26. doi: 10.1016/j.ejpb.2004.03.019. [DOI] [PubMed] [Google Scholar]
- 43.Srividya B, Cardoza RM, Amin PD. Sustained ophthalmic delivery of ofloxacin from a pH triggered in situ gelling system. J Control Release. 2001;73(2–3):205–11. doi: 10.1016/S0168-3659(01)00279-6. [DOI] [PubMed] [Google Scholar]
- 44.Söderberg L, Dyhre H, Roth B, Björkman S. The “inverted cup”—a novel in vitro release technique for drugs in lipid formulations. J Control Release. 2006;113(1):80–8. doi: 10.1016/j.jconrel.2006.03.015. [DOI] [PubMed] [Google Scholar]
- 45.LeFevre ME, Vanderhoff JW, Laissue JA, Joel DD. Accumulation of 2-micron latex particles in mouse Peyer’s patches during chronic latex feeding. Experientia. 1978;34(1):120–2. doi: 10.1007/BF01921939. [DOI] [PubMed] [Google Scholar]
- 46.Savic R, Luo L, Eisenberg A, Maysinger D. Micellar nanocontainers distribute to defined cytoplasmic organelles. Science. 2003;300(5619):615–8. doi: 10.1126/science.1078192. [DOI] [PubMed] [Google Scholar]
- 47.Rogošić M, Mencer HJ, Gomzi Z. Polydispersity index and molecular weight distributions of polymers. Eur Polym J. 1996;32(11):1337–44. doi: 10.1016/S0014-3057(96)00091-2. [DOI] [Google Scholar]
- 48.Mu L, Feng SS. Vitamin E TPGS used as emulsifier in the solvent evaporation/extraction technique for fabrication of polymeric nanospheres for controlled release of paclitaxel (Taxol (R)) J Control Release. 2002;80(1–3):129–44. doi: 10.1016/S0168-3659(02)00025-1. [DOI] [PubMed] [Google Scholar]
- 49.Freitas C, Müller RH. Effect of light and temperature on zeta potential and physical stability in solid lipid nanoparticle (SLN™) dispersions. Int J Pharm. 1998;168(2):221–9. doi: 10.1016/S0378-5173(98)00092-1. [DOI] [Google Scholar]
- 50.Klouda L, Mikos AG. Thermoresponsive hydrogels in biomedical applications. Eur J Pharm Biopharm. 2008;68(1):34–45. doi: 10.1016/j.ejpb.2007.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vadnere M, Amidon G, Lindenbaum S, Haslam JL. Thermodynamic studies on the gel-sol transition of some pluronic polyols. Int J Pharm. 1984;22(2–3):207–18. doi: 10.1016/0378-5173(84)90022-X. [DOI] [Google Scholar]
- 52.Wei G, Xu H, Ding PT, Li SM, Zheng JM. Thermosetting gels with modulated gelation temperature for ophthalmic use: the rheological and gamma scintigraphic studies. J Control Release. 2002;83(1):65–74. doi: 10.1016/S0168-3659(02)00175-X. [DOI] [PubMed] [Google Scholar]