

Abstract
To improve the solubility and oral bioavailability of erlotinib, a poorly water-soluble anticancer drug, solid self-emulsifying drug delivery system (SEDDS) was developed using solid inert carriers such as dextran 40 and Aerosil® 200 (colloidal silica). The preliminary solubility of erlotinib in various oils, surfactants, and co-surfactants was determined. Labrafil M2125CS, Labrasol, and Transcutol HP were chosen as the oil, surfactant, and co-surfactant, respectively, for preparation of the SEDDS formulations. The ternary phase diagram was evaluated to show the self-emulsifying area. The formulations were optimized using the droplet size and polydispersity index (PDI) of the resultant emulsions. Then, the optimized formulation containing 5% Labrafil M2125CS, 65% Labrasol, and 30% Transcutol was spray dried with dextran or Aerosil® and characterized for surface morphology, crystallinity, and pharmacokinetics in rats. Powder X-ray diffraction (PXRD) and differential scanning calorimetry (DSC) exhibited the amorphous form or molecular dispersion of erlotinib in the formulations. The pharmacokinetic parameters of the optimized formulations showed that the maximum concentration (C max) and area under the curve (AUC) of erlotinib were significantly increased, compared to erlotinib powder (p < 0.05). Thus, this SEDDS could be a promising method for enhancing the oral bioavailability of erlotinib.
KEY WORDS: bioavailability, erlotinib, SEDDS, spray drying
INTRODUCTION
Erlotinib, N-(3-ethynylphenyl)-6,7-bis(2-methoxyethoxy)-4-quinazolinamine, is a synthetic anilinoquinazoline derivative that selectively and reversibly inhibits the epidermal growth factor receptor (EGFR) tyrosine kinase, preventing autophosphorylation of tyrosine residues and thereby inhibiting further downstream signaling (1). Erlotinib has been indicated for treatment of patients with metastatic non-small cell lung cancer (NSCLC), and locally advanced, unresectable, or metastatic pancreatic cancer, in combination with gemcitabine with the approval of the US Food and Drug Administration (US-FDA). However, erlotinib belongs to the Biopharmaceutical Classification System (BCS) class II, which is characterized by low solubility and high permeability (log P of 2.7). This may be one of the reasons for low bioavailability and a large intra- and inter-patient variability in peak plasma concentration and area under the curve (AUC) after the same oral dose (2–4). For such compounds, the dissolution of the drug is the limiting step for absorption of the drug from the gastrointestinal (GI) tract.
For over a decade, self-emulsifying drug delivery systems (SEDDS) have been widely exploited to enhance the solubility, dissolution, and/or oral bioavailability of poorly water-soluble drugs (5–25). SEDDS are anhydrous, isotropic mixtures of oil(s), surfactant(s), the lipophilic drug, and co-surfactant(s) or co-solvent(s), which spontaneously form oil-in-water emulsions upon aqueous dilution with gentle agitation. However, although these systems have some advantages over emulsions, compared to solid dosage forms, there are still some practical drawbacks associated with the conventional (liquid) SEDDS, including storage instability and interaction with hard or soft capsules. Solidification of liquid SEDDS has been used increasingly in an effort to overcome the abovementioned limitations and combine the advantages of conventional lipid-based drug delivery systems (i.e., increased solubility and bioavailability) with those of solid dosage forms (e.g., high stability and reproducibility, ease of process control, and relatively low production cost) (26–34).
Therefore, the aim of this study was to develop solid SEDDS formulations as a potential tool to improve oral bioavailability of a poorly water-soluble drug, erlotinib, by spray drying method. In addition, the morphological analysis, solid state characterization, in vitro drug release, and in vivo pharmacokinetic (PK) study were reported to depict erlotinib-loaded SEDDS as a promising oral drug delivery system.
MATERIALS AND METHODS
Materials
Erlotinib was purchased from LC Labs (MA, USA). Polyglycolyzed glycerides (Labrafil M2125CS, Labrafil M1944CS, Capryol 90, Capryol PGMC, Labrafac Lipophile WL1349, Peceol, Plurol, Maisine 35-1, Labrasol, and Transcutol HP) were donated by Gattefosse (Saint-Priest Cedex, France). Polysorbate 20 (Tween 20), polysorbate 80 (Tween 80), and sorbitan monolaurate 20 (Span 20) were purchased from DC Chemical Co. (Seoul, South Korea). Polyoxyethylene 4 lauryl ether (Brij 30), sunflower seed oil, and castor oil were purchased from Sigma-Aldrich (St. Louis, MO, USA). Polyethylene glycol 400 (PEG 400) was purchased from Wako Pure Chemical Industries, Ltd (Tokyo, Japan). Dextran 40 (average molecular weight 40 kDa) and colloidal silica (Aerosil® 200) were acquired from Dong-A company (Seoul, South Korea). All other chemicals and reagents were of analytical grade and used without further purification.
Solubility Studies of Erlotinib
Solubility studies of erlotinib in various oils, surfactants, and co-surfactants were performed by adding an excess amount of erlotinib powder (about 200 mg) into a microtube (Axygen MCT-200-C) containing 1 mL of each vehicle alone (Labrafil M1944CS, Labrafil M2125CS, Labrafac Lipophile WL 1349, Peceol, sunflower seed oil, castor oil, Plurol, Maisine 35-1, Tween 20, Tween 80, Span 20, Brij 30, Capryol 90, Labrasol, Transcutol HP, PEG 400). The mixtures were vortexed for 30 s to facilitate solubilization and shaking (100 rpm) in a thermostatically controlled water bath at 37°C for 72 h. Then, the mixtures were centrifuged at 10,000×g for 10 min (Eppendorf, NY, USA) and the supernatants were filtered through 0.45-μm membrane filters (Whatman, UK) to remove undissolved erlotinib. The filtrates were suitably diluted with acetonitrile and the erlotinib concentrations were quantified by a validated HPLC method described below. A solubility study was also performed for solid SEDDS formulations in distilled water and phosphate buffer (pH 6.8) containing 0.1% Tween 80.
Drug Analysis
Erlotinib samples were analyzed using the Chromaster HPLC system (Hitachi, Tokyo, Japan) which is comprised of a diode array detector (model 5430), an autosampler (model 5210), a column oven (model 5310), and a pump system (model 5110). The column used was the Inersil® ODS-3 C18 column (GL Sciences Inc.; 5 μm, 4.6 × 250 mm). The mobile phase which is composed of acetonitrile and glycine buffer pH 9.0 (70:30, v/v) was eluted at the flow rate of 0.8 mL/min. Effluents were monitored at the wavelength of 331 nm.
Construction of a Pseudo-ternary Phase Diagram
A pseudo-ternary phase diagram was constructed for identification of the self-emulsifying region and microemulsion forming fields by visual observation. From the result of the solubility studies, various formulations in which Labrafil M2125CS was the oil phase (5–30% w/w), Labrasol was the surfactant (45–95% w/w), and Transcutol HP (0–50% w/w) was the co-surfactant were prepared. One hundred microliters of each formulation was added dropwise into 100 mL of distilled water and gently stirred (300 rpm) for 2 h at 37°C using a magnetic bar. The self-emulsifying process in distilled water was visually observed. The tendency of spontaneous emulsification was considered “good” when the droplets easily spread out in water and formed a fine emulsion without aggregation, and it was considered “bad” when there was poor or no emulsion formation with the immediate coalescence of oil droplets, especially when stirring was stopped (11,29,34). All studies were performed three times, and similar observations were made between repeats. The phase diagram was constructed using SigmaPlot® software (Systat Software Inc., CA, USA) to indicate the good self-emulsifying region.
Preparation of Erlotinib-Loaded Solid SEDDS
The liquid SEDDS formulations were prepared by dissolving erlotinib in mixtures of the oil, surfactant, and co-surfactant at room temperature. The resulting mixtures were vortexed until clear solutions were obtained. Particle size and polydispersity index (PDI) were measured using Zetasizer Nano ZS at a wavelength of 635 nm and at a scattering angle of 90° at 25°C. All measurements were performed three times and the values of z-average diameters were used.
Then, solid SEDDS formulations were prepared using a Büchi 190 nozzle-type mini-spray dryer (Flawil, Switzerland). Dextran 40 and colloidal silica (Aerosil® 200) were selected as the carriers for fabrication of solid SEDDS. Dextran 40 (5 g) was dissolved in 150 mL of water. In a similar experiment, Aerosil® 200 (5 g) was suspended in 150 mL of ethanol. Erlotinib was added to the optimized liquid SEDDS to make the final drug content of 5% (w/w) which can ensure the solubilization of erlotinib in the formulation as well as preserve the self-emulsifying ability of the formulation. Then, the drug-loaded liquid SEDDS (5 g) was dropped into the abovementioned solution/suspension with continuous stirring at 37°C for 30 min to obtain homogenous suspensions or emulsions. Each aqueous and ethanolic mixture was delivered to the pneumatic nozzle (diameter of 0.7 mm) at a flow rate of 3 mL/min by a peristaltic pump and spray dried at inlet temperatures of 130°C and 100°C and outlet temperatures of 70°C and 58°C, respectively. The air pressure was 4 kg/cm2, and aspiration was set at 80%, which indicated that the pressure of the aspirator filter vessel was −30 mbar. The direction of air flow was the same as that of the sprayed product (27,28,31,33,35). The physical mixtures of erlotinib and carriers were prepared by mixing erlotinib and each carrier with the same weight ratio in the solid SEDDS formulations.
Morphological Analysis
The morphologies of erlotinib powder and solid SEDDS formulations were visualized using a scanning electron microscope (S-4100, Hitachi, Japan). The samples were attached to a metal sample holder using double-sided adhesive tape and made electrically conductive by coating with platinum (6 nm/min) in a vacuum (6 Pa) using Hitachi Ion Sputter (E-1030, Tokyo, Japan) for 120 s at 15 mA.
Solid State Characterization
Thermal behaviors of the drug powder, dextran 40, colloidal silica, the physical mixtures, and solid SEDDS formulations were studied using a differential scanning calorimeter (DSC-Q200, TA Instruments, USA). Accurately weighed amounts of samples (about 2–3 mg) were crimped in aluminum pans. The DSC scans were recorded over the temperate range of 40°C to 180°C at a heating rate of 10°C/min, under a nitrogen purge with a flow rate of 50 mL/min.
In addition, the crystalline state properties of all abovementioned samples were assessed using an X’Pert PRO MPD diffractometer (PANalytical, Almelo, the Netherlands) with CuKα radiation (λ = 1.5406 Å) at ambient temperature. The operating current and voltage were 30 mA and 40 kV, respectively. Diffractograms were obtained in the angular range 2θ (diffraction angle) from 10° to 50° with a step size of 0.026°.
In Vitro Drug Release
In vitro drug release studies from erlotinib-loaded solid SEDDS formulations and drug powder were performed using an USP 32 dissolution apparatus 2 with 900 mL of phosphate buffer (pH 6.8) containing 0.1% Tween 80 as the dissolution medium at 37 ± 0.5°C, and the speed of the paddle was 50 rpm. Erlotinib-loaded solid SEDDS formulations (equivalent to 10 mg of erlotinib) and 10 mg of erlotinib powder were placed in a dissolution tester (Universal Scientific Co., Ltd, China). At specific time intervals, an aliquot (3 mL) of the sample was collected, filtered, and analyzed for the content of erlotinib using the HPLC method as mentioned above. An equivalent volume (3 mL) of fresh dissolution medium was added to compensate for the loss due to sampling.
Pharmacokinetic Study
The protocols for the animal studies were approved by the Institutional Animal Ethical Committee, Yeungnam University, South Korea. Twelve Sprague–Dawley rats weighing 280 ± 20 g were divided into three groups (n = 4) and fasted overnight prior to the experiments with free access to water. Each rat was anesthetized with diethyl ether and the right femoral artery was cannulated using a polyethylene tube. The erlotinib-loaded solid SEDDS formulations and drug powder were orally administered to the rats in each group at the dosage of 20 mg/kg as erlotinib. Serial blood samples (0.3 mL) were collected from the artery at specified time intervals (0.5, 1, 2, 3, 4, 6, 8, 12, and 24 h). The blood samples were centrifuged at 10,000×g for 10 min. The supernatant layers were taken and stored frozen at −20°C until analysis.
Plasma samples (100 μL) were mixed with acetonitrile (100 μL), vortexed for 10 min, and then centrifuged at 10,000×g for 10 min. Twenty microliters of the supernatant layers were injected into the HPLC system using the same method mentioned above. The standard calibration curve showed excellent linearity (r = 0.999) in the concentration range of 0.1–10 μg/mL of erlotinib in plasma. The pharmacokinetic parameters, including maximum plasma concentration (C max), time taken for its occurrence (T max), area under the curve of plasma concentration–time profile from 0 to infinity (AUC0–∞), elimination half-life (t 1/2), and elimination rate constant (k el) were calculated using WinNonlin® software (CA, USA) using standard non-compartmental analysis.
Statistical Analysis
Student’s t test and analysis of variance (ANOVA) were performed for evaluation of significant differences between two and three groups, respectively (α = 0.05). Values were reported as the mean ± standard deviation (S.D.), and p < 0.05 was considered statistically significant.
RESULTS AND DISCUSSION
Selection of SEDDS Components
The SEDDS formulations were prepared to enhance the solubility and bioavailability of the drug via oral administration. Thus, each component used in the system should have high solubilizing capacity for the drug to obtain the optimum drug loading and to minimize the final dosing volume. For this reason, solubility studies of erlotinib in various vehicles were performed for selection of a suitable oil, surfactant, and co-surfactant for development of the optimal erlotinib-loaded SEDDS formulation. The solubility of erlotinib in various vehicles is shown in Table I. Erlotinib has the aqueous solubility of approximately 3 μg/mL, which was poorly water soluble. All vehicles increased the solubility of erlotinib. Among various oils tested, Labrafil M2125CS (linoleoyl polyoxyl-6 glycerides, hydrophilic-lipophilic balance (HLB) of 9) exhibited the highest solubility for erlotinib, which was selected as the oil. Among the surfactants and co-surfactants studied, Labrasol (caprylocaproyl polyoxyl-8 glycerides, HLB of 14) and Transcutol HP (diethylene glycol monoethyl ether, HLB of 4.2) exhibited the highest solubilizing capacity for the drug. In other studies, Labrasol was reported to enhance the intestinal absorption of drugs as well as exhibit high tolerance and low toxicity (5,36–38). Therefore, in this study, the aforementioned oil, surfactant, and co-surfactant were selected for formulation of erlotinib-loaded SEDDS.
Table I.
Solubility of Erlotinib in Various Vehicles
| Vehicle | Solubility (mg/mL) |
|---|---|
| Water | 3.24 ± 0.45 (×10−3) |
| Oils | |
| Labrafil M 2125 CS | 6.90 ± 0.38 |
| Labrafil M 1944 CS | 3.88 ± 0.13 |
| Capryol PGMC | 6.11 ± 0.38 |
| Labrafac Lipophile WL 1349 | 0.34 ± 0.01 |
| Peceol | 4.26 ± 0.57 |
| Plurol | 2.18 ± 0.19 |
| Maisine 35-1 | 1.97 ± 0.13 |
| Sunflower seed oil | 0.13 ± 0.03 |
| Castor oil | 0.53 ± 0.08 |
| Surfactants and co-surfactants | |
| Tween 20 | 21.90 ± 1.54 |
| Tween 80 | 12.31 ± 1.16 |
| Span 20 | 3.47 ± 0.15 |
| Brij 30 | 21.84 ± 3.04 |
| Capryol 90 | 20.95 ± 1.83 |
| Labrasol | 64.65 ± 5.01 |
| Transcutol HP | 96.89 ± 2.35 |
| PEG 400 | 84.42 ± 1.13 |
Each value represents the mean ± S.D. (n = 3)
PEG polyethylene glycol
Preparation of Solid SEDDS
A series of SEDDS formulations were prepared and their self-emulsifying properties were visually observed. A pseudo-ternary phase diagram was constructed in the absence of erlotinib for identification of the self-emulsifying regions and to optimize the concentration of oil, surfactant, and co-surfactant in the liquid SEDDS formulation (32,35). The phase diagram of the system containing Labrasol as the surfactant, Labrafil M2125CS as the oil, and Transcutol HP as the co-surfactant is shown in Fig. 1. As can be seen, the self-emulsifying region which is described by the black color accounts for 10% of the total area. Spontaneous emulsion formation was not efficient when the amount of surfactant was less than 50% in the liquid SEDDS. The efficiency of emulsification was good when the total concentration of the surfactant/co-surfactant was more than 75% w/w of the liquid SEDDS formulation. The formulations surrounding the self-emulsifying domain exhibited poor emulsion forming ability. It has been reported that incorporation of the drug into the liquid SEDDS may have some effects on self-emulsifying performance of the system. In our study, significant differences were found in self-emulsifying performance when compared with the corresponding formulations with 6% (w/w) of erlotinib. The reason may be to increase the solvent capacity of the formulations for the drug with intermediate log P value (2 < log P < 4), high content of hydrophilic surfactant and co-surfactant was used. When the formulations were dispersed into aqueous solutions, the hydrophilic substances had the tendency to separate from the oil to be dissolved in water phase, causing phase separation and partial drug precipitation (39).
Fig. 1.

The pseudo-ternary phase diagram illustrating the nano-emulsion region
The rate and extent of drug release from the liquid SEDDS as well as the absorption and bioavailability of the drug are determined by the emulsion droplet size and size distribution (40–42). Therefore, the droplet size of the resultant emulsion is an essential factor in assessing self-emulsifying efficiency of the SEDDS formulation, and it was used as the main criterion for optimizing the SEDDS by monitoring the effect of various oil/surfactant and oil/surfactant/co-surfactant ratios (Fig. 2). When the surfactant concentration in the SEDDS formulations increased from 75% to 95%, the z-average particle size of the emulsions decreased (Fig. 2a). When the co-surfactant concentration increased from 0% to 20%, the z-average particle size increased. Continuing to increase the co-surfactant concentration, the z-average particle size decreased and reached a minimum value at 30% co-surfactant and then increased again (Fig. 2b). Therefore, the combination of Labrafil M2125CS/Labrasol/Transcutol HP at the weight ratio of 5/65/30 was selected as the optimal liquid SEDDS formulation for further study. Solid SEDDS formulations were prepared by spray drying the optimal drug-loaded liquid SEDDS formulation with solid carriers (Dextran 40, Aerosil® 200).
Fig. 2.
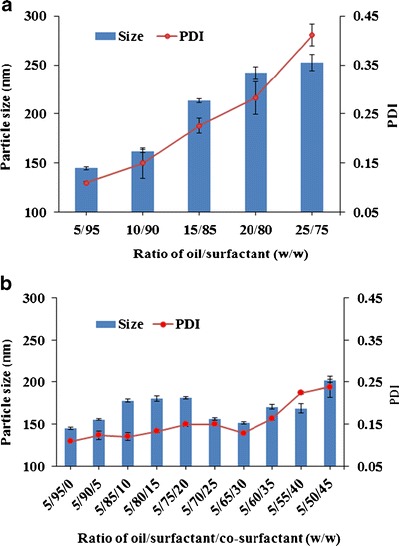
Effects of a oil/surfactant ratios and b oil/surfactant/co-surfactant ratios on the mean droplet size and PDI of resultant emulsions. These emulsions were formed by dropping 0.1-mL mixtures into 100 mL of distilled water. Each value represents the mean ± S.D. (n = 3)
Morphological Analysis
Scanning electron micrographs of erlotinib powder and the solid SEDDS formulations are shown in Fig. 3. Erlotinib powder (Fig. 3a) appeared as smooth-surfaced rectangular crystals. Dextran was described by our group as a smooth-surfaced structure (31). The SEDDS formulation was prepared with dextran (Fig. 3b) but appeared as spherical particles with irregular and crushed shapes. It is suggested that the liquid SEDDS may form a kind of microsized microcapsule with the hydrophilic carrier, dextran. Aerosil® 200 was reported to appear with a rough surface with porous particles (30,34). The solid SEDDS prepared with Aerosil® 200 (Fig. 3c) showed relatively smooth-surfaced particles, indicating that the liquid SEDDS may be coated or adsorbed inside the pores of Aerosil® 200.
Fig. 3.

Scanning electron micrographs (×3000): a erlotinib powder, b solid SEDDS prepared with dextran 40, and c solid SEDDS prepared with Aerosil® 200
Solid State Characterization
DSC is a commonly used thermoanalytical technique within the pharmaceutical field utilized in monitoring of endothermic processes (e.g., melting, phase transition, decomposition reactions) as well as exothermic processes (e.g., crystallization). It can be applied for determination of the drug/carrier interactions in the solid formulations. The DSC thermograms of pure erlotinib, the solid carriers, physical mixtures, and solid SEDDS formulations are shown in Fig. 4. Pure erlotinib (Fig. 4a) exhibited a small endothermic peak at about 105°C, which may have resulted from the water content in the hydrated form and a sharp endothermic peak at about 156°C, which corresponds to its melting point and indicates its crystalline nature (43). Dextran 40 (Fig. 4b) and Aerosil® 200 (Fig. 4e) showed no peaks over the entire temperature range scanned. The peak representing the melting point of erlotinib was shown with reduced intensity in the physical mixtures of erlotinib and carriers (Fig. 4c, f). However, this peak was absent in the SEDDS formulations prepared with dextran (Fig. 4d) and Aerosil® 200 (Fig. 4g), indicating that the drug may be in an amorphous form or molecularly dispersed in the carrier matrices (28,30,34).
Fig. 4.
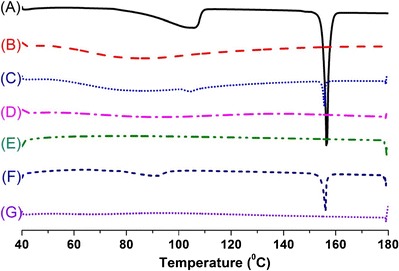
DSC thermograms of: (A) erlotinib, (B) dextran, (C) physical mixture of erlotinib and dextran, (D) solid SEDDS prepared with dextran 40, (E) Aerosil® 200, (F) physical mixture of erlotinib and Aerosil® 200, (G) solid SEDDS prepared with Aerosil® 200
The powder X-ray diffraction (PXRD) patterns are shown in Fig. 5. Erlotinib (Fig. 5a) had many sharp peaks at the diffraction angles (11.2°, 14.6°, 16.3°, 22.4°, 23.4°, 20.1°, 24.6°, 27.6°) showing a typical crystalline pattern. Dextran 40 (Fig. 5b) and Aerosil 200 (Fig. 5e) displayed no intrinsic peaks. The major characteristic peaks for the drug were still observed in patterns of both physical mixtures, especially at 24.6° and 27.6° (Fig. 5c, f). However, these distinctive peaks were absent in the patterns of the solid SEDDS formulations prepared with dextran (Fig. 5d) and Aerosil (Fig. 5g). Therefore, consistent with the DSC data, this result further confirmed the transformation of erlotinib into the amorphous state or molecular dispersion of erlotinib in the SEDDS formulations prepared with these carriers (28,34,35,44).
Fig. 5.
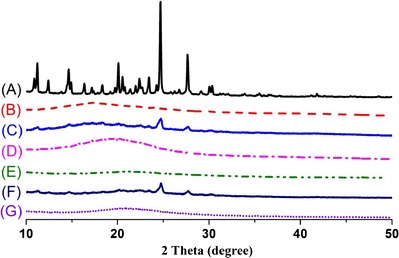
Powder X-ray diffraction patterns of: (A) erlotinib, (B) dextran, (C) physical mixture of erlotinib and dextran 40, (D) solid SEDDS prepared with dextran 40, (E) Aerosil® 200, (F) physical mixture of erlotinib and Aerosil® 200, (G) solid SEDDS prepared with Aerosil® 200
Dissolution Study
In vitro drug release study was performed for solid SEDDS formulations and erlotinib powder, and their release profiles are shown in Fig. 6. The drug powder showed a low dissolution percentage because of its hydrophobic nature and low aqueous solubility. The solid SEDDS formulations prepared with each solid carrier exhibited a significantly faster release rate and higher dissolution percentage than those of erlotinib powder (p < 0.05). In particular, both SEDDS formulations reached the maximum release percentages after 15–30 min. However, the release percentages were less than 100% due to the partial precipitation in the dissolution medium. In addition, the solid SEDDS formulation prepared with Aerosil 200 showed higher dissolution percentage than the one prepared with dextran 40, which might have resulted from the higher solubility of the former formulation than that of the latter one (Table II).
Fig. 6.
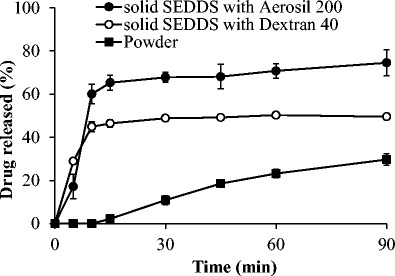
Dissolution profiles of erlotinib from powder (black square), solid SEDDS prepared with dextran 40 (white circle), and solid SEDDS prepared with Aerosil® 200 (black circle) in phosphate buffer pH 6.8. Each value represents the mean ± S.D. (n = 3)
Table II.
Solubility of Erlotinib in Solid SEDDS Formulations
| Solubility of erlotinib (mg/mL) | ||
|---|---|---|
| pH 6.8 | Distilled water | |
| Solid SEDDS prepared with dextran 40 | 10.86 ± 1.73 | 11.04 ± 1.38 |
| Solid SEDDS prepared with Aerosil 200 | 19.22 ± 2.15 | 18.45 ± 1.57 |
Each value represents the mean ± S.D. (n = 3)
SEDDS self-emulsifying drug delivery system
Pharmacokinetic Study
The plasma concentration–time profiles following oral administration of solid SEDDS formulations and drug powder are shown in Fig. 7. The drug powder group showed the lowest average drug plasma concentration due to its low solubility and dissolution rate compared to solid SEDDS formulations. In particular, two solid SEDDS formulations exhibited faster absorption than the powder which can be seen from the significant difference in plasma drug concentrations even from 0.5-h time point. In addition, the solid SEDDS formulation prepared with Aerosil® 200 had higher drug plasma concentrations than the solid SEDDS formulation prepared with dextran.
Fig. 7.
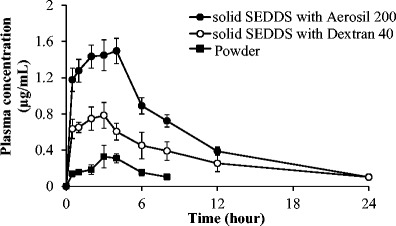
Plasma concentration–time profiles of erlotinib after oral administration of powder and solid SEDDS formulations in rats. Each value represents the mean ± S.D. (n = 4). Erlotinib powder (black square), solid SEDDS prepared with dextran 40 (white circle), and solid SEDDS prepared with Aerosil® 200 (black circle)
The pharmacokinetic parameters of erlotinib from these formulations and drug powder are summarized in Table III. Both solid SEDDS formulations showed significantly higher AUC and C max compared to erlotinib powder (p < 0.05). Specifically, the AUC0–∞ ((9.06 ± 2.02 h·μg)/mL) and C max (0.85 ± 0.12 μg/mL) values of solid SEDDS prepared with dextran were approximately 2.1- and 2.4-fold higher than those of erlotinib powder, respectively. The AUC0–∞ ((15.26 ± 0.66 h · μg)/mL) and C max (1.52 ± 0.15 μg/mL) values of solid SEDDS prepared with Aerosil 200 were approximately 3.5- and 4.2-fold higher than those of erlotinib powder, respectively. Therefore, both formulations augmented the oral bioavailability of erlotinib. In addition, as can be inferred from Table III, the relative standard deviation of C max and AUC0–∞ values of erlotinib powder were much larger than those of solid SEDDS formulations. Thus, the solid SEDDS not only improved the oral bioavailability but decreased the inter-subject variability.
Table III.
Pharmacokinetic Parameters of Erlotinib in Rats After Oral Administration of Erlotinib Powder and the SEDDS Formulations
| Parameters | Powder | SEDDS prepared with dextran 40 | SEDDS prepared with Aerosil® 200 |
|---|---|---|---|
| T max (h) | 3.75 ± 0.50 | 3.00 ± 0.82 | 3.75 ± 0.50 |
| C max (μg/mL) | 0.36 ± 0.12 | 0.85 ± 0.12* | 1.52 ± 0.15*,** |
| AUC0-∞ (h·μg/mL) | 4.34 ± 1.20 | 9.06 ± 2.02* | 15.26 ± 0.66*,** |
| k el (h−1) | 0.04 ± 0.02 | 0.09 ± 0.03 | 0.11 ± 0.01 |
| t 1/2 (h) | 18.89 ± 7.09 | 7.85 ± 1.99 | 6.37 ± 0.17 |
Each value represents the mean ± S.D. (n = 4)
SEDDS self-emulsifying drug delivery system, C max maximum concentration, T max time for the occurrence of C max, AUC area under the curve, k el elimination rate constant, t 1/2 elimination half-life
*p < 0.05 compared with erlotinib powder; **p < 0.05 compared with SEDDS prepared with dextran 40
CONCLUSION
In this study, solid SEDDS formulations of erlotinib were prepared by spray drying method, using dextran 40 and Aerosil 200 as solid carriers. DSC and PXRD analyses suggested that erlotinib may be in the amorphous form or molecularly dispersed in the solid SEDDS formulations. In dissolution study, the solid SEDDS formulations had faster in vitro release rates than pure drug powder. In vivo pharmacokinetic study in rats showed that solid SEDDS formulations provided significantly enhanced bioavailability of erlotinib compared to the pure drug powder, indicating that the self-emulsification performance of liquid SEDDS is still preserved after solidification. Therefore, this solid self-emulsifying drug delivery system could provide a promising oral solid dosage form for the poorly water-soluble drug, erlotinib.
Acknowledgments
This study was supported by a grant from the Medical Cluster R&D Support Project of Daegu Gyeongbuk Medical Innovation Foundation, Republic of Korea (2013) (No. HT13C0011).
Contributor Information
Chul Soon Yong, Phone: +82-53-810-2812, Email: csyong@ynu.ac.kr.
Jong Oh Kim, Phone: +82-53-810-2813, Email: jongohkim@yu.ac.kr.
References
- 1.van Erp NP, Gelderblom H, Guchelaar H-J. Clinical pharmacokinetics of tyrosine kinase inhibitors. Cancer Treat Rev. 2009;35(8):692–706. doi: 10.1016/j.ctrv.2009.08.004. [DOI] [PubMed] [Google Scholar]
- 2.Hidalgo M, Siu LL, Nemunaitis J, Rizzo J, Hammond LA, Takimoto C, et al. Phase I and pharmacologic study of OSI-774, an epidermal growth factor receptor tyrosine kinase inhibitor, in patients with advanced solid malignancies. J Clin Oncol. 2001;19(13):3267–3279. doi: 10.1200/JCO.2001.19.13.3267. [DOI] [PubMed] [Google Scholar]
- 3.Scheffler M, Di Gion P, Doroshyenko O, Wolf J. Clinical pharmacokinetics of tyrosine kinase inhibitors. Clin Pharmacokinet. 2011;50(6):371–403. doi: 10.2165/11587020-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 4.Tan AR, Yang X, Hewitt SM, Berman A, Lepper ER, Sparreboom A, et al. Evaluation of biologic end points and pharmacokinetics in patients with metastatic breast cancer after treatment with erlotinib, an epidermal growth factor receptor tyrosine kinase inhibitor. J Clin Oncol. 2004;22(15):3080–3090. doi: 10.1200/JCO.2004.08.189. [DOI] [PubMed] [Google Scholar]
- 5.Kommuru T, Gurley B, Khan M, Reddy I. Self-emulsifying drug delivery systems (SEDDS) of coenzyme Q10: formulation development and bioavailability assessment. Int J Pharm. 2001;212(2):233–246. doi: 10.1016/S0378-5173(00)00614-1. [DOI] [PubMed] [Google Scholar]
- 6.Borhade V, Nair H, Hegde D. Design and evaluation of self-microemulsifying drug delivery system (SMEDDS) of tacrolimus. AAPS PharmSciTech. 2008;9(1):13–21. doi: 10.1208/s12249-007-9014-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kale AA, Patravale VB. Design and evaluation of self-emulsifying drug delivery systems (SEDDS) of nimodipine. AAPS PharmSciTech. 2008;9(1):191–196. doi: 10.1208/s12249-008-9037-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Date AA, Nagarsenker M. Design and evaluation of self-nanoemulsifying drug delivery systems (SNEDDS) for cefpodoxime proxetil. Int J Pharm. 2007;329(1–2):166–172. doi: 10.1016/j.ijpharm.2006.08.038. [DOI] [PubMed] [Google Scholar]
- 9.Villar AMS, Naveros BC, Campmany ACC, Trenchs MA, Rocabert CB, Bellowa LH. Design and optimization of self-nanoemulsifying drug delivery systems (SNEDDS) for enhanced dissolution of gemfibrozil. Int J Pharm. 2012;431(1):161–175. doi: 10.1016/j.ijpharm.2012.04.001. [DOI] [PubMed] [Google Scholar]
- 10.Mandawgade SD, Sharma S, Pathak S, Patravale VB. Development of SMEDDS using natural lipophile: application to β-artemether delivery. Int J Pharm. 2008;362(1):179–183. doi: 10.1016/j.ijpharm.2008.06.021. [DOI] [PubMed] [Google Scholar]
- 11.Balakrishnan P, Lee BJ, Oh DH, Kim JO, Lee YI, Kim DD, et al. Enhanced oral bioavailability of coenzyme Q10 by self-emulsifying drug delivery systems. Int J Pharm. 2009;374:66–72. doi: 10.1016/j.ijpharm.2009.03.008. [DOI] [PubMed] [Google Scholar]
- 12.Eman Atef AAB. Formulation and in vitro and in vivo characterization of a phenytoin self-emulsifying drug delivery system (SEDDS) Eur J Pharm Sci. 2008;35:257–263. doi: 10.1016/j.ejps.2008.07.004. [DOI] [PubMed] [Google Scholar]
- 13.Dixit AR, Rajput SJ, Patel SG. Preparation and bioavailability assessment of SMEDDS containing valsartan. AAPS Pharmscitech. 2010;11(1):314–321. doi: 10.1208/s12249-010-9385-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.S-x C, S-f N, Li L, Wang C-g, Pan W-s, J-p S. Preparation and evaluation of self-microemulsifying drug delivery system containing vinpocetine. Drug Dev Ind Pharm. 2009;35(5):603–611. doi: 10.1080/03639040802488089. [DOI] [PubMed] [Google Scholar]
- 15.Zhang P, Liu Y, Feng N, Xu J. Preparation and evaluation of self-microemulsifying drug delivery system of oridonin. Int J Pharm. 2008;355(1):269–276. doi: 10.1016/j.ijpharm.2007.12.026. [DOI] [PubMed] [Google Scholar]
- 16.Balakumar K, Raghavan CV, Abdu S. Self nanoemulsifying drug delivery system (SNEDDS) of rosuvastatin calcium: design, formulation, bioavailability and pharmacokinetic evaluation. Colloids Surf B: Biointerfaces. 2013;112:337–343. doi: 10.1016/j.colsurfb.2013.08.025. [DOI] [PubMed] [Google Scholar]
- 17.Gupta S, Chavhan S, Sawant KK. Self-nanoemulsifying drug delivery system for adefovir dipivoxil: design, characterization, in vitro and ex vivo evaluation. Colloids Surf A Physicochem Eng Asp. 2011;392(1):145–155. doi: 10.1016/j.colsurfa.2011.09.048. [DOI] [Google Scholar]
- 18.Elnaggar YS, El-Massik MA, Abdallah OY. Self-nanoemulsifying drug delivery systems of tamoxifen citrate: design and optimization. Int J Pharm. 2009;380(1):133–141. doi: 10.1016/j.ijpharm.2009.07.015. [DOI] [PubMed] [Google Scholar]
- 19.Fahmy UA, Ahmed OA, Hosny KM. Development and evaluation of avanafil self-nanoemulsifying drug delivery system with rapid onset of action and enhanced bioavailability. AAPS PharmSciTech. 2014:1–6 [DOI] [PMC free article] [PubMed]
- 20.Dwivedi P, KHATIK R, Khandelwal K, Srivastava R, TANEJA I, Raju KSR, et al. Self-nanoemulsifying drug delivery system (SNEDDS) for oral delivery of arteether: pharmacokinetics, toxicity and antimalarial activity in mice. RSC Adv. 2014.
- 21.Tran TH, Guo Y, Song D, Bruno RS, Lu X. Quercetin‐containing self‐nanoemulsifying drug delivery system for improving oral bioavailability. J Pharm Sci. 2014;103(3):840–852. doi: 10.1002/jps.23858. [DOI] [PubMed] [Google Scholar]
- 22.Mahmoud DB, Shukr MH, Bendas ER. In vitro and in vivo evaluation of self-nanoemulsifying drug delivery systems of cilostazol for oral and parenteral administration. Int J Pharm. 2014. [DOI] [PubMed]
- 23.Khan AW, Kotta S, Ansari SH, Sharma RK, Ali J. Self-nanoemulsifying drug delivery system (SNEDDS) of the poorly water-soluble grapefruit flavonoid Naringenin: design, characterization, in vitro and in vivo evaluation. Drug Deliv. 2014;(0):1–10. [DOI] [PubMed]
- 24.Singh G, Pai RS. Optimized self-nanoemulsifying drug delivery system of atazanavir with enhanced oral bioavailability: in vitro/in vivo characterization. Expert Opin Drug Deliv. 2014;(0):1–10. [DOI] [PubMed]
- 25.Bandyopadhyay S, Katare O, Singh B. Optimized self nano-emulsifying systems of ezetimibe with enhanced bioavailability potential using long chain and medium chain triglycerides. Colloids Surf B: Biointerfaces. 2012;100:50–61. doi: 10.1016/j.colsurfb.2012.05.019. [DOI] [PubMed] [Google Scholar]
- 26.Mohd AB, Sanka K, Bandi S, Diwan PV, Shastri N. Solid self-nanoemulsifying drug delivery system (S-SNEDDS) for oral delivery of glimepiride: development and antidiabetic activity in albino rabbits. Drug Deliv. 2014;(0):1–10. [DOI] [PubMed]
- 27.Kang JH, Oh DH, Oh Y-K, Yong CS, Choi H-G. Effects of solid carriers on the crystalline properties, dissolution and bioavailability of flurbiprofen in solid self-nanoemulsifying drug delivery system (solid SNEDDS) Eur J Pharm Biopharm. 2012;80(2):289–297. doi: 10.1016/j.ejpb.2011.11.005. [DOI] [PubMed] [Google Scholar]
- 28.Seo YG, Kim DH, Ramasamy T, Kim JH, Marasini N, Oh Y-K, et al. Development of docetaxel-loaded solid self-nanoemulsifying drug delivery system (SNEDDS) for enhanced chemotherapeutic effect. Int J Pharm. 2013;452(1):412–420. doi: 10.1016/j.ijpharm.2013.05.034. [DOI] [PubMed] [Google Scholar]
- 29.Kim DW, Kang JH, Oh DH, Yong CS, Choi H-G. Development of novel flurbiprofen-loaded solid self-microemulsifying drug delivery system using gelatin as solid carrier. J Microencapsul. 2012;29(4):323–330. doi: 10.3109/02652048.2011.651497. [DOI] [PubMed] [Google Scholar]
- 30.Balakrishnan P, Lee B-J, Oh DH, Kim JO, Hong MJ, Jee J-P, et al. Enhanced oral bioavailability of dexibuprofen by a novel solid self-emulsifying drug delivery system (SEDDS) Eur J Pharm Biopharm. 2009;72(3):539–545. doi: 10.1016/j.ejpb.2009.03.001. [DOI] [PubMed] [Google Scholar]
- 31.Kim GG, Poudel BK, Marasini N, Lee DW, Hiep TT, Yang KY, et al. Enhancement of oral bioavailability of fenofibrate by solid self-microemulsifying drug delivery systems. Drug Dev Ind Pharm. 2013;39(9):1431–1438. doi: 10.3109/03639045.2012.719903. [DOI] [PubMed] [Google Scholar]
- 32.Sermkaew N, Ketjinda W, Boonme P, Phadoongsombut N, Wiwattanapatapee R. Liquid and solid self-microemulsifying drug delivery systems for improving the oral bioavailability of andrographolide from a crude extract of Andrographis paniculata. Eur J Pharm Sci. 2013;50(3):459–466. doi: 10.1016/j.ejps.2013.08.006. [DOI] [PubMed] [Google Scholar]
- 33.Marasini N, Tran TH, Poudel BK, Choi H-G, Yong CS, Kim JO. Statistical modeling, optimization and characterization of spray-dried solid self-microemulsifying drug delivery system using design of experiments. Chem Pharm Bull. 2013;61(2):184–193. doi: 10.1248/cpb.c12-00823. [DOI] [PubMed] [Google Scholar]
- 34.Kim DW, Kwon MS, Yousaf AM, Balakrishnan P, Park JH, Kim DS, et al. Comparison of a solid SMEDDS and solid dispersion for enhanced stability and bioavailability of clopidogrel napadisilate. Carbohydrate Polymers. 2014. [DOI] [PubMed]
- 35.Oh DH, Kang JH, Kim DW, Lee B-J, Kim JO, Yong CS, et al. Comparison of solid self-microemulsifying drug delivery system (solid SMEDDS) prepared with hydrophilic and hydrophobic solid carrier. Int J Pharm. 2011;420(2):412–418. doi: 10.1016/j.ijpharm.2011.09.007. [DOI] [PubMed] [Google Scholar]
- 36.Prasad Y, Puthli S, Eaimtrakarn S, Ishida M, Yoshikawa Y, Shibata N, et al. Enhanced intestinal absorption of vancomycin with Labrasol and D-α-tocopheryl PEG 1000 succinate in rats. Int J Pharm. 2003;250(1):181–190. doi: 10.1016/S0378-5173(02)00544-6. [DOI] [PubMed] [Google Scholar]
- 37.Hu Z, Tawa R, Konishi T, Shibata N, Takada K. A novel emulsifier, Labrasol, enhances gastrointestinal absorption of gentamicin. Life Sci. 2001;69(24):2899–2910. doi: 10.1016/S0024-3205(01)01375-3. [DOI] [PubMed] [Google Scholar]
- 38.Rama Prasad Y, Minamimoto T, Yoshikawa Y, Shibata N, Mori S, Matsuura A, et al. In situ intestinal absorption studies on low molecular weight heparin in rats using Labrasol as absorption enhancer. Int J Pharm. 2004;271(1):225–232. doi: 10.1016/j.ijpharm.2003.11.013. [DOI] [PubMed] [Google Scholar]
- 39.Pouton CW. Lipid formulations for oral administration of drugs: non-emulsifying, self-emulsifying and ‘self-microemulsifying’ drug delivery systems. Eur J Pharm Sci. 2000;11:S93–S98. doi: 10.1016/S0928-0987(00)00167-6. [DOI] [PubMed] [Google Scholar]
- 40.Constantinides PP, Scalart J-P, Lancaster C, Marcello J, Marks G, Ellens H, et al. Formulation and intestinal absorption enhancement evaluation of water-in-oil microemulsions incorporating medium-chain glycerides. Pharm Res. 1994;11(10):1385–1390. doi: 10.1023/A:1018927402875. [DOI] [PubMed] [Google Scholar]
- 41.Tarr BD, Yalkowsky SH. Enhanced intestinal absorption of cyclosporine in rats through the reduction of emulsion droplet size. Pharm Res. 1989;6(1):40–43. doi: 10.1023/A:1015843517762. [DOI] [PubMed] [Google Scholar]
- 42.Parmar N, Singla N, Amin S, Kohli K. Study of cosurfactant effect on nanoemulsifying area and development of lercanidipine loaded (SNEDDS) self nanoemulsifying drug delivery system. Colloids Surf B: Biointerfaces. 2011;86(2):327–338. doi: 10.1016/j.colsurfb.2011.04.016. [DOI] [PubMed] [Google Scholar]
- 43.Muralidhara RD, Parthasaradhi RB, Raji RR, Rathnakar RK, Srinivasa RT. A novel hydrated form of erlotinib free base and a process for preparation of erlotinib hydrochloride polymorph form a substantially free of polymorph form b. Google Patents; 2010.
- 44.Chudasama A, Shah B, Patel V, Nivsarkar M, Vasu K, Shishoo C. Development of self emulsifying drug delivery system of itraconazole for oral delivery: formulation and pharmacokinetic consideration. J Pharm Investig. 2015;45(3):271–283. doi: 10.1007/s40005-015-0172-5. [DOI] [Google Scholar]