

Abstract
Objective:
Fanconi anaemia (FA) is an inherited disease associated with congenital and developmental abnormalities resulting from the disruption of a multigenic DNA damage response pathway. This study aimed to define the MRI appearances of the brain in patients with FA in correlation with their genetic and clinical features.
Methods:
A review of the brain MRI in 20 patients with FA was performed. Pituitary size and frequencies of the radiological findings of individuals with FA and age-matched controls were determined.
Results:
Abnormalities were identified in 18 (90%) patients with FA, the commonest being a small pituitary (68%, p < 0.01 females and p < 0.001 males). In five cases (25%, p = 0.02), the pituitary morphology was also abnormal. Posterior fossa abnormalities were seen in six cases (30%, p = 0.01) including Chiari I malformation (n = 3), Dandy–Walker variant (n = 2) and cerebellar atrophy (n = 2). Six patients (30%, p = 0.01) had morphological structural variation of the corpus callosum (CC).
Conclusion:
The incidence of central nervous system (CNS) abnormalities in FA is higher than previously reported, with a midline predominance that points to impact in the early stages of CNS development. MRI brain imaging is important for endocrine assessment and pre-transplant evaluation and can make an important contribution to clinical decision-making.
Advances in knowledge:
The incidence of brain structural abnormalities in FA is higher than previously reported, with abnormalities of the posterior fossa, CC and pituitary being common. There is an association with gender and reduction in pituitary size which does not strongly correlate with biochemically evident endocrine abnormality.
INTRODUCTION
Fanconi anaemia (FA) is an inherited disease characterized by congenital and developmental abnormalities and cancer predisposition. FA results from the disruption of a DNA damage response pathway in which proteins encoded by at least 16 FA genes that have been identified to date (FANCA, B, C, D1, D2, E, F, G, I, J, L, M, N, O, P, Q).1 Mutations in FANCD1/BRCA2 cause FA in 3–5% of cases2 and link FA to inherited breast and ovarian cancer.3 On a cellular level, FA is characterized by hypersensitivity to cross-linking agents, such as mitomycin C (MMC), hypersensitivity to cytokines and a typical G2 arrest in the cell cycle, particularly in response to genotoxic stress.4 The clinical phenotype classically involves radial and thumb abnormalities, short stature and microcephaly. Clinical manifestations of FA can develop over time and involve many organ systems.5 Genotype/phenotype correlation can be variable, such that even siblings carrying the same mutations can display diverse phenotypic features.4,6,7 Given the high incidence of congenital abnormalities that can affect the skeleton, the urogenital system, the heart and the gastrointestinal tract, several imaging modalities are employed to assess the presence and severity of such abnormalities in FA.8 In addition to microcephaly, the central nervous system (CNS) in FA can be affected by structural abnormalities and also malignancies, which in most cases are medulloblastoma.2,9 Non-malignant CNS abnormalities have been reported to occur in FA with a frequency of approximately 8% according to published data.5,10,11 Imaging of the CNS of individuals with FA has mostly been carried out as part of an endocrine workup from the investigations of growth failure, and hence mainly concentrated on pituitary assessment,12 often using CT. Subtle pituitary abnormalities have been reported more frequently in FA,13 while other CNS structural abnormalities have been reported in individual cases only.14,15
To date, little has been published with respect to the incidence and patterns of findings on brain MRI in a large series of patients with FA. The purpose of this study was to define the spectrum and frequency of brain appearances using MRI in patients with FA and discuss the findings in a clinical and biological context.
METHODS AND MATERIALS
Patient identification
Institutional approval was given for the retrospective reporting of the anonymized imaging data as part of this study. Depending on age, patients or parents themselves gave informed consent for clinical imaging, with written consent given by patients in whom anonymized imaging has been reproduced in this study report. All patients with FA in our centre have regular clinical assessments in a specialist multidisciplinary clinic. This includes baseline followed by at least 4–6 monthly monitoring of growth and development, endocrine assessment and cancer surveillance. The patients included in this series represent the whole cohort of local, national or international patients with FA cared for at The Royal Manchester Children's Hospital for the purposes of clinical management and bone marrow transplant treatment. Detailed endocrine investigations are carried out for clinical concerns about growth or pubertal development with imaging, as required and indicated by any concerns raised by these clinical assessments.8
No large series of normal healthy children with MRI brain imaging was available to us. Consequently, corresponding age- and sex-matched controls were selected from patients scanned at our institution for routine assessment of headache or non-specific neurological symptoms and were discharged after normal clinical assessment, with a reported single normal MRI brain scan having been obtained. Whilst this does not reflect the normal population, it does give some information about the findings in FA in comparison with non-affected age- and sex-matched children.
One patient (Patient 16) was previously published in a case report relating to pollicization of her index finger.16 This patient's neuroimaging has not yet been reported.
Patients were diagnosed with FA on clinical grounds if they demonstrated increased MMC sensitivity on peripheral lymphocytes, fibroblasts and/or immortalized Epstein Barr virus (EBV)-transformed lymphoblasts. The individual genetic defect underlying the diagnosis of FA was determined in most cases, by carrying out retroviral complementation analysis and/or sequencing.
Neuroimaging
MRI was performed as part of clinical assessment for the diagnostic workup of neurological or endocrine problems and routine baseline assessment according to our institutional FA imaging protocol. In two cases, brain MRI was carried out as an urgent diagnostic procedure for the management of neurological complications during haematopoietic stem cell transplantation (HSCT). All indications for scan at an individual patient level (including age at scan) are listed in Table 1.
Table 1.
Details of patients with FA, with respect to phenotypic features and clinical course, genetic and ethnic background, and MRI findings
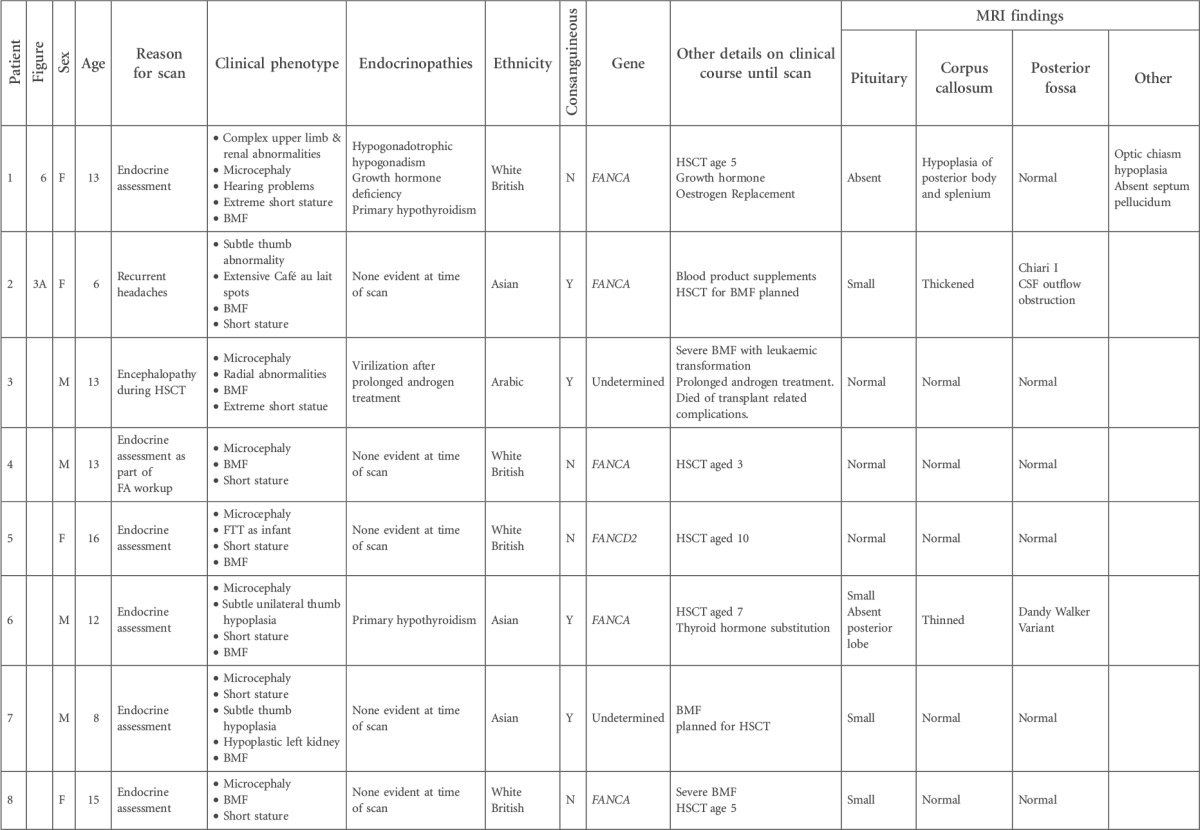
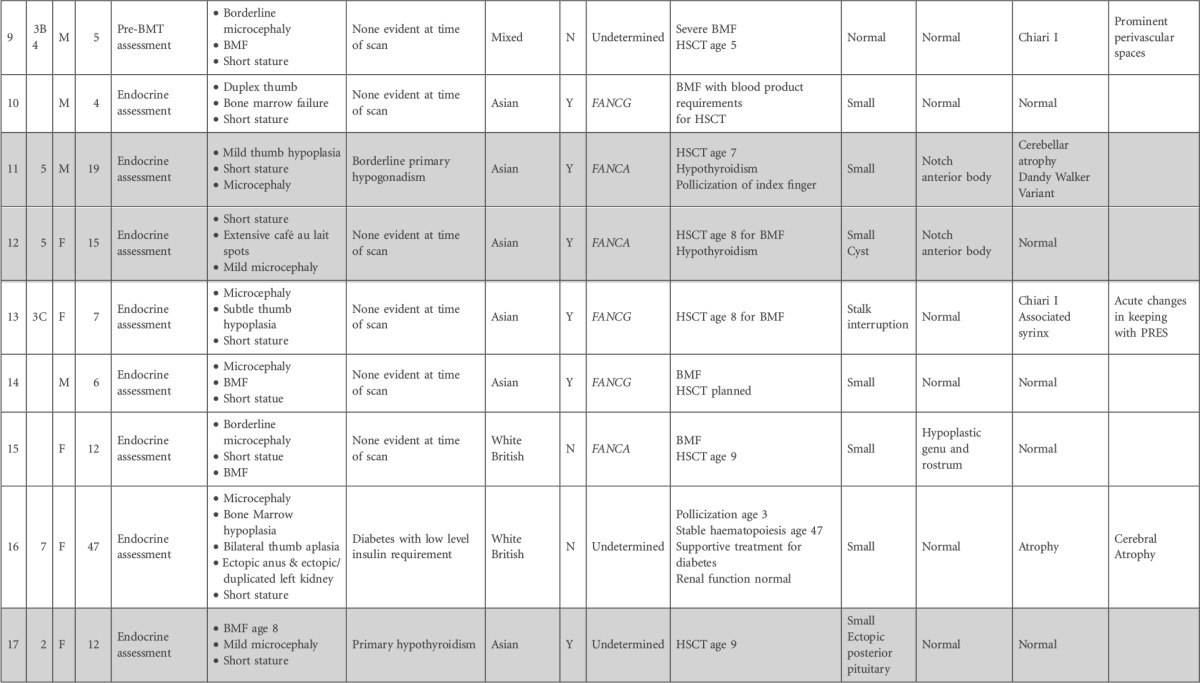
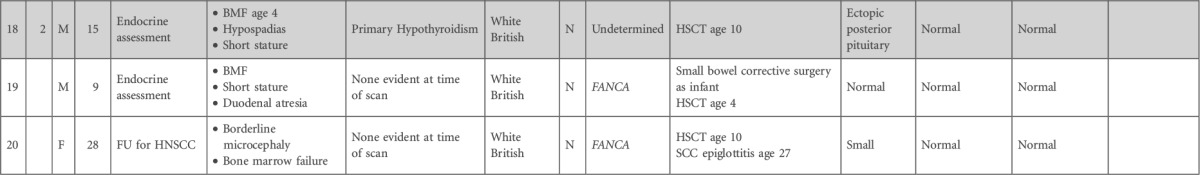
BMF, bone marrow failure; CSF, cerebrospinal fluid; FTT, failure to thrive; FU, follow-up; HNSCC, head and neck squamous cell carcinoma; HSCT, hematopoetic stem cell transplant; PRES, posterior reversible encephalopathy syndrome; SCC, squamous cell carcinoma.
Shaded cells indicate sibling pairs.
In all cases, imaging was performed on a 1.5-T Avanto scanner (Siemens, Forcheim, Germany). The sequences employed include axial T2 [turbo spin echo (TSE)/40,097] slice thickness 5 mm, field of view (FOV) 230 and matrix size 252 × 384. Coronal fluid attenuated inversion recovery (FLAIR) (TSE/9000/114), time to inversion 2500 ms, slice thickness 5 mm, FOV 230 and matrix size 280 × 320. T1 volume [gradient echo (GE)/1160/4.44], isotropic voxel size 1 mm3, FOV 230 and matrix size 292 × 256 with multiplanar report in all three orthogonal planes for assessment of grey/white matter differentiation and evidence of cortical dysplasia. We also undertook high-resolution imaging of the pituitary gland including a sagittal T1 (GE/14/4.76) slice thickness 1.25, FOV 230 and matrix size 192 × 256. Coronal T2 (TSE/3850/115) slice thickness 3 mm, FOV 210 and matrix size 358 × 512. Coronal T1 (spin echo/450/9) slice thickness 3 mm, FOV 190 and matrix size 218 × 256.
Imaging was reviewed with anatomical assessment of the whole brain being undertaken at the reporting workstation (GE—Centricity RA 1000; GE Medical Systems, Milwaukee, WI) by two experienced dedicated paediatric neuroradiologists (SMS and NW) who both work at the Royal Manchester Children's Hospital covering a 6,000,000 person population. They routinely review 3000–3500 paediatric brain MRI scans per year. Each scan was reviewed independently by both the radiologists, and a consensus report was then derived from this following joint discussion and imaging re-review.
Pituitary gland assessments
Pituitary size was measured using the midline high-resolution sagittal imaging and the picture archiving and communication system (PACS) measurement callipers as per Tsunoda et al.17 As per previous studies,13,17 the overall pituitary size was deemed to be small if one or more standard deviation (SD) below the mean for the general population. In addition to pituitary size, data were collected on the shape of the gland, pituitary stalk and gland morphology including position of the posterior lobe T1 bright spot (all from the high-resolution pituitary imaging sequences). Independent measurements were made by both the observers, and the mean measurement was used for analysis.
Statistical analysis
Pituitary size measurements were grouped into patient and age-matched control groups by sex. A Student's t-test was used for bivariate analysis of pituitary height between groups and differences in biparietal head width between patients and controls. Fisher's exact test was used to test for independence of frequency of MRI apparent abnormalities in FA vs normal controls. The observed incidence of abnormalities in the FA cohort was compared with the estimated population rate obtained from the literature17,18 using a binomial test. p-values <0.05 were considered statistically significant.
RESULTS
MRI scans of 20 patients with FA were reviewed, alongside 20 age- and sex-matched controls. The 20 patients (including 3 sibling pairs) comprised 10 male (mean age, 10.7 years; age range, 4.5–19 years) and 10 female (mean age, 16.5 years; age range, 6.3–47 years) individuals with FA. Details regarding clinical features, genetics, indications for scanning and MRI findings are listed in Table 1. In only 2 of the 20 cases was the CNS MRI assessment normal, hence in the current series 18 of the 20 (90%, p < 0.001) patients had some kind of brain abnormality detected. The majority of these were pituitary abnormalities.
Cranial vault size
Whilst there was a tendency for a smaller biparietal head diameter in FA (mean diameter 123.87 mm) compared with the age- and sex-matched controls (mean diameter 128.44 mm), this difference did not reach statistical significance (p = 0.06).
Pituitary size
In one patient, there was complete absence of the pituitary gland. In 13 of the remaining 19 patients (68%, p < 0.001), a small-for-age pituitary was detected (Figure 1), defined as measuring >1 SD below the mean for age and sex.17 In male patients, there was a mean measurement of 3.97 mm (SD 0.79 mm) compared to control mean height of 5.23 mm (SD 0.76 mm), p < 0.0001. In female patients, the mean pituitary size was 3.96 mm (SD 1.9) compared to a mean size in controls of 5.9 mm (SD 0.68), p < 0.01.
Figure 1.

Pituitary height (vertical axis) in male (a) and female (b) subjects against age (horizontal axis). The difference in heights compared with controls is significant (p < 0.01 females and p < 0.0001 males).
Pituitary anatomy
Anatomical pituitary abnormalities beyond simple size reduction were seen in 5 of the 20 (25% p = 0.02) patients with FA. This included two male siblings aged 12 years 9 months (Patient 17) and 15 years (Patient 18), who both had an ectopic posterior pituitary with a strikingly similar MR appearance (Figure 2). Despite this, Patient 17 had a pituitary gland <1 SD below the mean for age, whilst the elder patient (Patient 18) had a normal-sized pituitary gland. Neither sibling had clinical or biochemical evidence of pituitary dysfunction at the time of imaging assessment. A second sibling pair both had normal pituitary morphology but small pituitaries for age. In addition to a small pituitary, they both had similar appearances of the corpus callosum (CC) but divergence in posterior fossa appearances (see “Other brain abnormalities” section and Figure 3). Clinical or biochemically evident pituitary dysfunction at the time of scan was evident in two patients with abnormal imaging, one of whom had complete absence of the posterior pituitary bright spot and the other complete absence of any detectable pituitary tissue. Finally, one patient (Patient 13) despite a small pituitary and complete interruption of the pituitary stalk, had no clinical or biochemical evidence of pituitary dysfunction at the time of imaging.
Figure 2.

Sagittal T1 midline images in one sibling pair (a, b). Both demonstrate ectopic posterior pituitary bright spot locations (white arrows). The imaging appearance of the ectopic posterior pituitary is remarkably similar. Despite this however, the size of the residual anterior pituitary in the sella (solid arrowheads) is different.
Figure 3.

Axial T2 (a), coronal fluid attenuated inversion recovery (b) and sagittal T1 (c) in Patient 1 with colpocephaly (dashed arrows) resultant from partial agenesis of the corpus callosum (CC) (solid arrowhead). The optic chiasm is small (open arrowhead), and the pituitary gland is absent (solid arrow). Features are in keeping with septo-optic dysplasia and partial agenesis of the CC.
Posterior fossa abnormalities
Posterior fossa abnormalities were seen in 6 of the 20 patients with FA (30%, p = 0.02) with none seen in the control group. This included three Chiari I malformations in patients with FA (Figure 4a–c, p = 0.001, assuming a rate of 1% in the general population18). Chiari I malformations were defined as the lower most extent of the cerebellar tonsils >5 mm below the foramen magnum. In Patient 2, investigations were for recurrent headaches and phase-contrast MR cerebrospinal fluid (CSF) flow studies confirmed CSF outflow obstruction at the level of the foramen magnum resultant from the Chiari I malformation. This patient also had an abnormally thickened but intact CC (Figure 4a). Another case of Chiari I malformation (Patient 9) had an associated prominent conglomeration of perivascular spaces in the posterior left centrum-semi-ovale (Figure 5). The final case of Chiari I malformation (Patient 13) was imaged for acute encephalopathy during HSCT and had features on neuroimaging in keeping with posterior reversible encephalomyelitis syndrome. The Chiari I malformation was identified incidentally, as was a spinal cord syrinx and an abnormal CC thinned in the posterior body and splenium (Figure 4c).
Figure 4.

Three patients with Chiari I malformations (white solid arrows). Note also the difference in callosal morphology with a thickened callosum in (a), normal morphology in (b) and thinned posterior body/splenium in (c) (arrowheads). Patient in (c) also had functional cerebrospinal fluid outflow obstruction (demonstrated by PCMRI imaging) and a spinal cord syrinx (dashed arrows c).
Figure 5.

Patient 9 who on midline imaging is seen to have a Chiari I malformation. Axial T1 (a), coronal fluid attenuated inversion recovery (b) and Sagittal T1 (c) demonstrate a conglomeration of very prominent perivascular spaces (solid white arrows). There is no associated gliosis or abnormal cortical formation seen.
In two further cases of posterior fossa abnormality, a Dandy–Walker variant was detected (p < 0.001, assuming an incidence of 1 in 25,000–30,000 as defined by the NIH incidence rate for rare diseases). This was defined as a midline CSF containing posterior fossa cyst communicating with the fourth ventricle through a defect caused by partial agenesis of the inferior cerebellar vermis. One of these cases was in a child from a sibling pair (Patients 11 and 12). Both had abnormal notching of the anterior body of the CC but divergence of their posterior fossa appearances with only Patient 11 having a Dandy–Walker variant and cerebellar hemisphere atrophy (Figure 6). The other case of Dandy–Walker variant (Patient 6) had an absent posterior pituitary bright spot and a small pituitary gland, but normal cerebellar hemisphere volumes. In both cases, there were no associated clinical symptoms such as ataxia or other motor dysfunctions at the time of imaging.
Figure 6.

Sibling pair [Patient 11 (a) and Patient 12 (b)]. Arrowheads demonstrate abnormal notching of the anterior body of the corpus callosum at a similar site. However, there is divergence of their posterior fossa appearances with only Patient 12 having a Dandy–Walker variant (solid arrow) and cerebellar hemisphere atrophy.
Furthermore, one patient (Patient 16) demonstrated cerebellar and supratentorial volume loss in keeping with global cerebral and cerebellar atrophy. Her imaging is discussed further below.
Other brain abnormalities
Abnormalities or variants of the CC were detected in 6 of the 20 FA cases (30% p = 0.02), again not identified in the control group. These included a morphologically intact but thickened CC in two patients, both of which had small pituitaries and one a functional Chiari I malformation. There was partial agenesis of the CC associated with absence of the pituitary gland and an absent septum pellucidum in one case (Patient 1, Figure 3). In addition, in this case, there was a small optic chiasm. This combination of features being radiologically in keeping with septo-optic dysplasia with an associated callosal abnormality.
In three families, we were able to compare findings in siblings affected by FA. We noted striking similarities between the siblings for some of the findings, even in the presence of differences in the imaging appearances otherwise, as discussed above. For example, sibling patients 11 and 12 both of whom had an almost identical notch on the anterior body of the CC. One of these siblings had an associated Dandy–Walker variant and cerebellar atrophy whilst the other had a normal cerebellum (Figure 6).
Our series also included the MR assessment of a 47-year-old female with FA. Her CNS imaging revealed evidence of structural abnormalities of the CC with abnormal thinning of the anterior body, which was likely congenital as seen in the CC abnormalities in the other patients with FA. However, in this case, there was also MRI evidence of cerebral- and cerebellar-atrophy–associated cerebral white matter signal changes in keeping with a microvascular angiopathy beyond what would be expected in a patient of 47 years with no cardiovascular risk factors (Figure 7). In addition, she too had a small pituitary gland, but this was not associated with abnormal pituitary biochemistry.
Figure 7.

Axial T2 (a, b) and coronal fluid attenuated inversion recovery (c) in female patient with FA. There is MRI evidence of cerebral and cerebellar atrophy with associated cerebral and cerebellar white matter signal changes in keeping with microvascular angiopathy beyond what would be expected in a patient of 47 years with no cardiovascular risk factors.
DISCUSSION
While previous case reports and small series have identified abnormalities on MRI of the brain in FA, particularly with respect to pituitary imaging,11–14 no larger series of FA-associated MRI finding have been reported. To address this, we have retrospectively analysed the MRI findings in our cohort of patients with FA. The spectrum of findings includes pituitary abnormalities which, whilst mainly affecting the size as previously recognized,13 also include variable structural pituitary abnormalities. These findings are important since delineation of structural abnormalities involving the pituitary is typically part of a comprehensive endocrine workup in children with growth disorders and aids decision-making with respect to hormone substitution and monitoring.
We identified abnormalities of the CC in 30% (p = 0.02) and the posterior fossa in 30% (p = 0.02). In the general paediatric population, the incidence of Chiari I malformations is of the order of 1%;18 in our series, it was found to be 3 of the 20 cases (15%). Indeed, in one case, a Chiari I malformation was associated with a spinal cord syrinx (Patient 13; Figure 4c). The high frequency of posterior fossa abnormalities in FA warrants screening to identify cases requiring early neurosurgical intervention particularly in the presence of attributable symptoms. Importantly, awareness of individual structural abnormalities such as prominent perivascular spaces is helpful for the management of patients during HSCT when neuroimaging can be essential for the management of acute neurological complications.
Therefore, as CNS imaging is important for the long-term clinical management of growth and pituitary dysfunction and for the acute management of neurological complications during HSCT we have now adopted MRI of the CNS as standard practice at workup for the management of FA.
In sibling pairs, we found a striking resemblance of some abnormalities, but divergence of other findings. Normal structural imaging of the CNS was found in the minority of our patients and implicates an important role of the FA pathway for normal CNS development. The distribution of abnormalities with midline defects points to impact of the FA defect in the early stages of CNS development. Our cohort included three sibling pairs and allowed for the first time the comparison of CNS imaging in siblings with the identical underlying genetic defect. While divergent phenotypic features have been reported in siblings with the same genetic defect,7 this has not been reported specifically for CNS abnormalities. Intriguingly, while we identified a striking similarity of findings with respect to ectopically positioned pituitary and characteristic CC in two sibling pairs (Figure 2), we also discovered diversity of imaging features, including a Dandy–Walker abnormality in one sibling from another pair despite a radiologically similar CC appearance (Figure 6). This illustrates the relevance of the underlying genetic defect at different stages of CNS development, under the potential influence of other, possible environmental factors.
One patient with a severe Fanconi phenotype (Patient 1) with respect to other features of FA also had the most CNS abnormalities. Within the spectrum of the classic clinical phenotype with microcephaly, short stature and bone marrow failure, we found mainly midline abnormalities including pituitary and CC structural abnormalities; a radiological appearance in keeping with septo-optic dysplasia and associated partial agenesis of the CC. Overall these findings would support MRI as part of a routine assessment of FA, in particular in patients with a more severe clinical phenotype. Moreover, our radiological discovery of clinically covert features, including a Chiari I malformation and prominent perivascular spaces, indicates that this imaging modality can detect anomalies in a significant proportion of FA patients with more subtle clinical phenotypes.
The frequent detection of CC abnormalities is another feature of FA that is shared with alcohol embryopathy,19,20 in addition to microcephaly, and other features of FA such as red cell macrocytosis. Although of limited significance in terms of clinical management of FA, this intriguing finding is biologically plausible given the role of the FA pathway in counteracting the toxic effects of endogenously produced aldehydes.20,21 Furthermore, this implies the possibility of aldehyde metabolism and toxicity in early brain development, for which the intact FA/BRCA (breast cancer related tumour suppression gene) pathway is important. Further investigations into the relevance of the FA/BRCA pathway in terms of brain development and initiation of malignant transformation in the CNS would be important.
We also included the brain MRI of one of the oldest recorded patients with FA in the literature.17,18 The findings of clinically silent changes, that are normally encountered in much older patients where there is an age-related vascular change, is in keeping with FA being a disease of accelerated ageing.4 All the patients reported here had a single MRI scan at an age as indicated in Table 1. To monitor further changes associated with ageing, it would be interesting to repeat MRI scanning at regular intervals. This is not currently thought to be necessary from a clinical management perspective, but would be of academic interest.
FA is an uncommon and clinically variable disease. Whilst the cohort reported here is the largest in the current literature to date, the full spectrum of FA-associated abnormalities might not be fully included. In particular, changes associated with BRCA2 mutations (which are frequently associated with medulloblastoma2) might have additional or specific abnormalities which would be important to recognize.
This MRI study of incidence and patterns of brain abnormalities in FA demonstrates that CNS abnormalities are more common than previously recognized, pointing to an important role of an intact FA/BRCA pathway for normal CNS developments. Our study implies a role for the FA/BRCA pathway in normal brain development and also the ageing process of the CNS, which we are currently hoping to demonstrate in mouse models. Clinically, CNS imaging is helpful in the management of endocrine manifestations and complications during HSCT and as such should become a routine investigation for the workup of patients with FA.
FUNDING
This research was funded by the National Institute for Health Research, Leukaemia Lymphoma Research UK and the Kay Kendal Leukaemia Fund and facilitated by the Manchester Biomedical Research Centre and the NIHR Greater Manchester: Clinical Research Network.
Contributor Information
Stavros M Stivaros, Email: stavros.stivaros@manchester.ac.uk.
Robert Alston, Email: robert.d.alston@manchester.ac.uk.
Neville B Wright, Email: Neville.Wright@cmft.nhs.uk.
Kate Chandler, Email: Kate.Chandler@cmft.nhs.uk.
Denise Bonney, Email: Denise.Bonney@cmft.nhs.uk.
Robert F Wynn, Email: Robert.Wynn@cmft.nhs.uk.
Andrew M Will, Email: Andrew.Will@cmft.nhs.uk.
Maqsood Punekar, Email: Maqsood.Punekar@lthtr.nhs.uk.
Sean Loughran, Email: Sean.Loughran@cmft.nhs.uk.
John-Paul Kilday, Email: John-Paul.Kilday@cmft.nhs.uk.
Detlev Schindler, Email: schindler@biozentrum.uni-wuerzburg.de.
REFERENCES
- 1.Kottemann MC, Smogorzewska A. Fanconi anaemia and the repair of Watson and Crick DNA crosslinks. Nature 2013; 493: 356–63. doi: 10.1038/nature11863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Meyer S, Tischkowitz M, Chandler K, Gillespie A, Birch JM, Evans DG. Fanconi anaemia, BRCA2 mutations and childhood cancer: a developmental perspective from clinical and epidemiological observations with implications for genetic counselling. J Med Genet 2014; 51: 71–5. doi: 10.1136/jmedgenet-2013-101642 [DOI] [PubMed] [Google Scholar]
- 3.Foulkes WD. Inherited susceptibility to common cancers. N Engl J Med 2008; 359: 2143–53. doi: 10.1056/NEJMra0802968 [DOI] [PubMed] [Google Scholar]
- 4.Neveling K, Endt D, Hoehn H, Schindler D. Genotype-phenotype correlations in Fanconi anemia. Mutat Res 2009; 668: 73–91. doi: 10.1016/j.mrfmmm.2009.05.006 [DOI] [PubMed] [Google Scholar]
- 5.Tischkowitz M, Dokal I. Fanconi anaemia and leukaemia—clinical and molecular aspects. Br J Haematol 2004; 126: 176–91. doi: 10.1111/j.1365-2141.2004.05023.x [DOI] [PubMed] [Google Scholar]
- 6.Faivre L, Guardiola P, Lewis C, Dokal I, Ebell W, Zatterale A, et al. Association of complementation group and mutation type with clinical outcome in fanconi anemia. European Fanconi Anemia Research Group. Blood 2000; 96: 4064–70. [PubMed] [Google Scholar]
- 7.Koc A, Pronk JC, Alikasifoglu M, Joenje H, Altay C. Variable pathogenicity of exon 43del (FAA) in four Fanconi anaemia patients within a consanguineous family. Br J Haematol 1999; 104: 127–30. doi: 10.1046/j.1365-2141.1999.01156.x [DOI] [PubMed] [Google Scholar]
- 8.De Kerviler E, Guermazi A, Zagdanski AM, Gluckman E, Frija J. The clinical and radiological features of Fanconi’s anaemia. Clin Radiol 2000; 55: 340–5. doi: 10.1053/crad.2000.0445 [DOI] [PubMed] [Google Scholar]
- 9.Offit K, Levran O, Mullaney B, Mah K, Nafa K, Batish SD, et al. Shared genetic susceptibility to breast cancer, brain tumors, and Fanconi anemia. J Natl Cancer Inst 2003; 95: 1548–51. doi: 10.1093/jnci/djg072 [DOI] [PubMed] [Google Scholar]
- 10.Giampietro PF, Adler-Brecher B, Verlander PC, Pavlakis SG, Davis JG, Auerbach AD. The need for more accurate and timely diagnosis in Fanconi anemia: a report from the International Fanconi Anemia Registry. Pediatrics 1993; 91: 1116–20. [PubMed] [Google Scholar]
- 11.Pavlakis SG, Frissora CL, Giampietro PF, Davis JG, Gould RJ, Adler-Brecher B, et al. Fanconi anemia: a model for genetic causes of abnormal brain development. Dev Med Child Neurol 1992; 34: 1081–4. doi: 10.1111/j.1469-8749.1992.tb11420.x [DOI] [PubMed] [Google Scholar]
- 12.Giri N, Batista DL, Alter BP, Stratakis CA. Endocrine abnormalities in patients with Fanconi anemia. J Clin Endocrinol Metab 2007; 92: 2624–31. doi: 10.1210/jc.2007-0135 [DOI] [PubMed] [Google Scholar]
- 13.Sherafat-Kazemzadeh R, Mehta SN, Care MM, Kim MO, Williams DA, Rose SR; Fanconi Anemia Comprehensive Care Center. Small pituitary size in children with Fanconi anemia. Pediatr Blood Cancer 2007; 49: 166–70. doi: 10.1002/pbc.21148 [DOI] [PubMed] [Google Scholar]
- 14.Deepak Amalnath S, Subramanian R, Swaminathan RP, Indumathi N. Incidental detection of Chiari malformation in Fanconi anaemia. Br J Haematol 2012; 158: 154. doi: 10.1111/j.1365-2141.2012.09195.x [DOI] [PubMed] [Google Scholar]
- 15.Akiyama K, Koh K, Mori M, Sekinaka Y, Seki M, Arakawa Y, et al. Association between Chiari malformation and bone marrow failure/myelodysplastic syndrome. Br J Haematol 2013; 163: 411–2. doi: 10.1111/bjh.12495 [DOI] [PubMed] [Google Scholar]
- 16.Stivaros SM, Punekar M, Chandler K, Rost I, Schindler D, Meyer S. Pollicization of the index finger in Fanconi anaemia: appearances and functionality 40 years after the intervention. Br J Haematol 2014; 166: 807. doi: 10.1111/bjh.12996 [DOI] [PubMed] [Google Scholar]
- 17.Tsunoda A, Okuda O, Sato K. MR height of the pituitary gland as a function of age and sex: especially physiological hypertrophy in adolescence and in climacterium. AJNR Am J Neuroradiol 1997; 18: 551–4. [PMC free article] [PubMed] [Google Scholar]
- 18.Aitken LA, Lindan CE, Sidney S, Gupta N, Barkovich AJ, Sorel M, et al. Chiari type I malformation in a pediatric population. Pediatr Neurol 2009; 40: 449–54. doi: 10.1016/j.pediatrneurol.2009.01.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wozniak JR, Muetzel RL, Mueller BA, McGee CL, Freerks MA, Ward EE, et al. Microstructural corpus callosum anomalies in children with prenatal alcohol exposure: an extension of previous diffusion tensor imaging findings. Alcohol Clin Exp Res 2009; 33: 1825–35. doi: 10.1111/j.1530-0277.2009.01021.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Langevin F, Crossan GP, Rosado IV, Arends MJ, Patel KJ. Fancd2 counteracts the toxic effects of naturally produced aldehydes in mice. Nature 2011; 475: 53–8. doi: 10.1038/nature10192 [DOI] [PubMed] [Google Scholar]
- 21.Rosado IV, Langevin F, Crossan GP, Takata M, Patel KJ. Formaldehyde catabolism is essential in cells deficient for the Fanconi anemia DNA-repair pathway. Nat Struct Mol Biol 2011; 18: 1432–4. doi: 10.1038/nsmb.2173 [DOI] [PubMed] [Google Scholar]