

Abstract
INTRODUCTION
Studies suggest that nonsyndromic cleft lip and palate (NSCLP) is polygenic with variable penetrance, presenting a challenge in identifying all causal genetic variants. Despite relatively high prevalence of NSCLP among Amerindian populations, no large whole exome sequencing (WES) studies have been completed in this population.
OBJECTIVE
Identify candidate genes with rare genetic variants for NSCLP in a Honduran population using WES.
METHODS
WES was performed on two to four members of 27 multiplex Honduran families. Genetic variants with a minor allele frequency > 1% in reference databases were removed. Heterozygous variants consistent with dominant disease with incomplete penetrance were ascertained, and variants with predicted functional consequence were prioritized for analysis. Pedigree-specific p-values were calculated as the probability of all affected members in the pedigree being carriers, given that at least one is a carrier.
RESULTS
Preliminary results identified 3727 heterozygous rare variants; 1282 were predicted to be functionally consequential. Twenty-three genes had variants of interest in ≥3 families, where some genes had different variants in each family, giving a total of 50 variants. Variant validation via Sanger sequencing of the families and unrelated unaffected controls excluded variants that were sequencing errors or common variants not in databases, leaving four genes with candidate variants in ≥3 families. Of these, candidate variants in two genes consistently segregate with NSCLP as a dominant variant with incomplete penetrance: ACSS2 and PHYH.
CONCLUSION
Rare variants found at the same gene in all affected individuals in several families are likely directly related to NSCLP.
Keywords: Incomplete penetrance, craniofacial anomaly
INTRODUCTION
Orofacial clefts are the most common congenital craniofacial defect, with an average worldwide incidence of 1 to 7 in 1000 live births [Mossey et al., 2009]. The birth prevalence varies widely depending on geographic origin and ethnicity, with Asian and Amerindian populations having the highest reported prevalence, with rates as high as 1 in 500 [Dixon et al., 2011; Marazita, 2012]. Clefting has a number of physical features, but it is also associated with significant comorbidities including speech difficulties, malnutrition, hearing impairment, infection, and psychiatric disease. While orofacial clefts can be surgically corrected, treatment imposes a considerable financial burden on families and requires a multidisciplinary approach involving plastic surgery, maxillofacial surgery, otolaryngology, dentistry, speech therapy, and audiology [Wehby et al., 2010].
Cleft lip with or without cleft palate (CL/P) may occur as part of a syndrome (e.g. Down Syndrome, Van der Woude Syndrome) or as an isolated, nonsyndromic malformation. With the advent of the genomics era, many advances have been made in identifying causative mutations underlying syndromic forms of CL/P, such as Kabuki Syndrome [Ng et al. 2010]. However, the etiology of nonsyndromic CL/P (NSCLP), which constitute approximately 70% of NSCLP patients, has proven more difficult to characterize [Calzolari et al., 2007]. Genome-wide linkage and association studies have identified multiple loci thought to be associated or influencing risk of oral clefts, but these can explain only a fraction of cases [Marazita, 2012; Mangold et al., 2010]. Segregation analyses suggest the underlying genetics of NSCLP are quite complex, involving 2 to 15 genes of major effect in most populations, sometimes with a multifactorial background [Marazita, 2012]. Further adding to the complexity, recent studies suggest the spectrum of NSCLP likely includes subclinical morphological features such as orbicularis oris muscle defects and structural vertebral anomalies, consistent with variable penetrance [Weinberg et al. 2006]. In addition, twin studies and familial clustering analyses certainly support a genetic component, but environmental exposures and micronutrients are also likely to play a role [Mossey et al., 2009; Leslie, 2013].
Recent advances in genomic analysis include the ability to sequence every exon from a person’s DNA using whole exome capture and massively parallel DNA sequencing in a method known as whole exome sequencing (WES). Since protein coding regions harbor 85% of the known mutations responsible for disease-related traits but constitute only 1% of the human genome, this is a powerful and efficient tool for detecting pathogenic mutations [Choi et al. 2009]. Bureau et al. have recently applied WES to many families from around the world, looking for variants in a group of 348 previously identified candidate genes [Bureau et al, 2014]. However, none of these variants were found in more than one multiplex family and they did not include an Amerindian population.
Our aim is to use WES to identify rare genetic variants in any gene in a cohort of multiplex Amerindian families affected by NSCLP. We will focus on genes with variants that are predicted to be functional found in several families (3 or more). This approach should increase the likelihood that these genes are in fact related to NSCLP.
METHODS
Samples
This study was approved by the Institutional Review Board at Columbia University Medical Center (CUMC) and the local Institutional Review Board at Hospital Escuela in Tegucigalpa, Honduras. Subjects were recruited from patients presenting for treatment of CL/P at Hospital Escuela, a public hospital. The purpose of the study was explained to all subjects. Written informed consent was obtained from all adults, and parental written informed consent was obtained for all minors, as well as assent for those able to give it. Patients were examined to determine type of cleft and laterality along with a more general exam to exclude syndromic disease. Probands and their family members were interviewed to determine pedigree structure. DNA samples were obtained from blood samples obtained via venipuncture from both probands and their relatives. Twenty-eight families were selected because they included two or more family members affected with NSCLP. This gave a total of 191 individuals; 52 affected individuals and their families.
Genotyping
DNA samples were isolated from blood samples at CUMC using the Qiagen Flexigene DNA kit (Qiagen, Valencia, CA). DNA samples were sent to the University of Washington Center for Mendelian Genomics (UW-CMG) in Seattle, Washington for genotyping and sequencing, with the exception of families M32 and M33, which had been previously sequenced at Columbia University at an earlier date.
Sample Quality Control
DNA quantification, sex typing and molecular “fingerprinting” were performed using a high frequency, cosmopolitan genotyping assay. Samples were failed if the total amount, concentration, or integrity of DNA was too low, the fingerprint assay produced poor genotype data or gender inconsistencies were noted. One family, M10, was excluded due to inadequate DNA samples from the affected individuals.
One hundred and ninety one subjects were genotyped for Illumina’s Human Core Exome BeadChip. Variants missing greater than five percent of genotypes were excluded, then PLINK v1.90 was used to confirm pedigree relationships using Mendelian error checking (http://pngu.mgh.harvard.edu/purcell/plink/) [Purcell et al., 2007]. These data were then used to estimate relationships using Kinship-based INference for Genome-wide association studies, an approach appropriate for admixed populations (http://people.virginia.edu/~wc9c/KING/) [Manichaikul et al., 2010]. No evidence for close, cryptic relationships across families was found. One family, M22, showed several inconsistencies between reported pedigrees and kinship estimates, and was excluded from analysis. Family M5 was excluded from analysis because one of the two affected individuals appeared unrelated to the rest of the family. Families M18 and M63 had respectively two and one unaffected individuals that appeared unrelated to the rest of the family, therefore these samples were excluded. Family M94 showed two samples were likely switched. Repeat blood samples were obtained from family members. We then tested three sites for which the two samples were each homozygous for different single nucleotide polymorphisms to confirm that the two samples had been switched. Based on these results, two to four subjects from each remaining family were selected for WES; at least two affected relatives were chosen. Where possible, an extra family member for variant phasing was included. In total, 59 individuals including 52 who were affected by NSCLP and seven of their relatives underwent WES.
Library Production and Exome Sequence Capture: Library construction and exome capture were performed in an automated process using Perkin-Elmer Janus II in 96-well plate format. One μg of genomic DNA was fragmented via acoustic sonication with Covaris, followed by end-polishing and A-tailing, then ligation of sequencing adaptors and PCR amplification with 8 bp barcodes for use in multiplexing. Roche/Nimblegen SeqCap EZ was used to perform exome capture. One μg of shotgun library was given 72 hours to hybridize to biotinylated capture probes, followed by binding to Streptavidin beads PCR amplification of fragments. Triplicate qPCR was used to determine library concentration and Agilent Bioanalyzer was used to confirm molecular weight distributions were 125 ± 15 bp. A Illumina HiSeq sequencer was used to perform massively parallel sequencing-by-synthesis with fluorescently labeled, reversibly terminating nucleotides.
Variant Calling: Base calls were generated on the HiSeq instrument, then Picard Extract Illumina Barcodes and IlluminaBasecallsToSam were used to create demultiplexed, unaligned BAM files. BAM files were aligned to a human reference (GRCh37/hg19) using the Burrows-Wheeler Aligner v0.6.2 [Li et al, 2009]. For quality control, read-pairs greater than two standard deviations different from the average library size were removed. Duplicate removal was performed with Picard MarkDuplicates v1.70. Additional post-processing was performed using GATK v1.6-11-g3b2fab9: insertion/deletion realignment by IndelRealigner, base qualities were recalibrated with TableRecalibration, and variant detection and genotyping were performed using the UnifiedGenotyper tool. Filtration Walker was used to identify lower quality areas and false positives. Variants with low quality scores (Q50), allelic imbalance (ABHet 0.75), long homopolymer runs (HRun> 3) and/or low quality by depth (QD < 5) were removed. For additional quality control, sequencing is continued until > 90% of the exome is sequenced at > 8X coverage and >80% of the exome is sequenced at > 20X coverage. All subjects sequenced at the UW CMG were called into a single multisample VCF for analysis.
Analyzing Called Variants: Variants that were monomorphic, had depth of coverage <6 or >500, or genotype quality <20 were excluded from analysis using VCFtools v0.1.12b [Danecek et al., 2011]. Variants were annotated using the Variant Effect Predictor tool v.72 (http://uswest.ensembl.org/info/docs/tools/vep/index.html) [McLauren et al., 2010]. Variants with mean allele frequency ≤1% in the UW CMG database, 1000 Genomes Project, or Exome Sequencing Project (ESP) reference populations were included for analysis [McVean et al., 2012; Exome variant server 2015] Gemini v0.11.0 was used to identify rare variants consistent with an autosomal dominant mode of inheritance giving 3237 variants [Paila et al., 2013]. Variants predicted to have medium or high impact by Gemini and non-synonymous variants with GERP scores ≥3 and/or PolyPhen2 scores ≤0.8 were prioritized, narrowing the list to 1282 variants [Adzhubei et al., 2010; Cooper et al., 2005]. Twenty-three genes with a total of 50 variants fitting these criteria in three or more families were included in our analysis. We define variants of interest as those that meet these criteria for predicted functional consequence. In some genes, one variant of interest was found in multiple families, in other genes different variants of interest were found in the same gene in different families. All 25 families not excluded had at least one initial variant of interest.
Confirmation with Sanger Sequencing: For most candidates, Sanger sequencing was performed for all affected individuals whose WES identified a variant of interest. If this variant of interest was confirmed in the affected family members, then additional unaffected members were sequenced. If a variant of interest appeared to be common in the population because it was seen in multiple founders, we sequenced Honduran control subjects without known CL/P in their families. We started with sequencing the same number of controls as cases for the family(s). If the variant of interest was as common in controls as cases, we assumed it was polymorphic in this population. If the rate was not clear after only a few samples, we sequenced up to 215 controls.
Primers were designed to amplify 400–600 base pair regions using Primer3 [Rozen et al., 1998]. Some variants proved difficult to amplify; in these cases a second set of nesting primers was designed. PCR products were sequenced by Macrogen. Chromatograms were read viewed using FinchTV (Version 1.4 http://www.geospiza.com/Products/finchtv.shtml).
We define candidate variants as those variants of interest passing this step of Sanger validation and screening out of common variants not seen in reference databases. Of the 50 variants of interest that were tested, only 14 variants passed this step of Sanger validation and screening out of common variants.
Statistical analysis
We define candidate genes as those with candidate variants in at least three families. For each candidate gene, for each pedigree in which at least one affected member carried a candidate variant in the candidate gene, we calculated the conditional probability that all affected pedigree members would carry the variant identical-by-descent, given that at least one affected member carried the variant, under the null hypothesis that candidate variants in the gene are not associated with affected status and the gene locus is not linked to any locus with an influence on the phenotype [Bureau et al., 2014]. These probabilities are shown in Table I. These calculations are valid when the known pedigree structures reflect all consanguinities and when the variants are sufficiently rare that in each pedigree, sharing is identical-by-descent from a single founder. Our validated pedigrees and variant filtering strategies are consistent with this assumption. For families in which all affected members carried a variant of interest identical-by-descent, this probability is a pedigree-specific p-value demonstrating the likelihood that this sharing within the family is due to chance alone.
Table I.
Family-specific p-values for candidate genes.
| gene | location of variant(s) | families | family p-values | overall p-value |
|---|---|---|---|---|
| 45 | 0.3333 | |||
| ACSS2 | chr20: 33509608 | 67 | 0.0667 | 7.18 × 10−4 |
| 94 | 0.0323 | |||
|
| ||||
| chr10:13325784 | 28 | 0.1428 | ||
| PHYH | chr10:13337497 | 45 | 0.3333 | 3.175 × 10−3 |
| chr10:13320305 | 46 | 0.0667 | ||
|
| ||||
| ♯ HKDC1 | chr10:71007342 | 32 | 0.00314 | * 5.462 × 10−4 |
| 36 | 0.0323 | |||
| 71 | 0.3333 | |||
| chr10:71020980 | 94 | 0.0323 | ||
|
| ||||
| chr13: 42245135 | 1 | 0.3333 | ||
| chr13:42189142 | 16 | 0.3333 | ||
| ♯ VWA8 | chr13: 42266081 | 32 | 0.00314 | * 4.333 × 10−3 |
| chr13:42306268 | 36 | 0.0323 | ||
| chr13:42385421 | 55 | 0.3333 | ||
does not meet criteria for significance because not found in all affected members of all families
alternative method for probability calculation in genes not meeting criteria
for significance as noted in methods section
Following along the lines of Bureau et al., we reported evidence for a candidate gene being associated with affected status only for candidate genes in which, for all candidate variants in the candidate gene found in any affected subject, the candidate variant was found in all affected members of the subject’s pedigree identical-by-descent. Because the families are all independent, the probability that this criterion is met is the product over families of the family-specific p-values. We computed the p-value as the product, over the pedigrees all of whose affected members carried a candidate variant in the candidate gene, of those conditional probabilities. These values are also shown in Table I.
Genes for which not all affected family members carried a candidate variant in any of the families with at least one affected family member carrying a candidate variant did not meet our criteria for significance. However, we received feedback that these genes were interesting nevertheless given the polygenic nature of NSCLP, and therefore we calculated an overall p-value appropriate for this situation. In that calculation, for each subset of the families, we computed the conditional probability that in exactly those families in the subset all affected families members were carriers given that at least one affected family member was a carrier, and took the sum of the resulting probabilities less than or equal to the probability corresponding to the observed subset to be the p-value. These values are also included in Table I.
The analysis presented here involves multiple tests, one for each candidate gene. However we do not present a p-value adjusted for multiple comparisons, but instead an estimated false discovery rate. This is because we are not making multiple comparisons in the service of testing the single hypothesis that there exists gene(s) where rare variants confer increased risk of NSCLP affected status, but rather scanning for genes in which there is evidence for rare variants conferring increased risk. To estimate the false discovery rate, we computed the conditional expectation, under the null hypothesis, of the proportion of candidate genes for which in all families in which at least one affected family member carried a candidate variant did all affected family members carry this variant identical-by-descent, and compared it to the observed proportion of such genes, according to the method of Benjamini and Hochberg [1995].
The expected proportion under the null hypothesis was 0.32, the actual proportion was 0.72, so that the estimated false discovery rate was 0.32(1−0.72)/0.72(1−.32) = 0.18.
RESULTS
Four genes, ACSS2, PHYH, HKDC1 and VWA8 qualified as candidate genes because they contained candidate variants (again defined as those variants consistent with autosomal dominant inheritance with variable penetrance meeting our criteria for predicted functional consequence and passing Sanger validation, see Methods section for further clarification) in 3 or more families. Details regarding these variants can be seen in Table II.
Table II.
Details for co-segregating candidate variants in candidate genes.
| gene | Chr | bp37 | ref | alt | rsID | transcript | impact | change | family(s) |
|---|---|---|---|---|---|---|---|---|---|
| ACSS2 | 20 | 33509608 | T | C | rs59088485 | ENST00000360596* | missense | p.Val496Ala | 45, 67, 94 |
| PHYH | 10 | 13320305 | A | AG | rs566116760 | ENST00000263038 | inframe | p.Asn337_Leu338insHis | 46 |
| AT | insertion | ||||||||
| PHYH | 10 | 13325784 | C | T | rs62619919 | ENST00000263038 | missense | p.Arg245Gln | 28 |
| PHYH | 10 | 13337497 | G | C | rs145404396 | ENSP00000263038* | missense | p.Arg82Gly | 45 |
| HKDC1 | 10 | 71007342 | C | T | rs201518882 | ENST00000354624 | missense | p.His420Tyr | 32,36,71 |
| HKDC1 | 10 | 71020980 | C | T | NA | ENST00000354624 | stop gain | p.Arg768Ter | 94 |
| VWA8 | 13 | 42189142 | C | T | rs73464952 | ENSP00000368612 | missense | p.Val1564Met | 16 |
| VWA8 | 13 | 42245135 | G | A | rs370112959 | ENSP00000368612 | stop gain | p.Arg1520Ter | 1 |
| VWA8 | 13 | 42266081 | A | G | NA | ENSP00000368612 | splice region | 32 | |
| VWA8 | 13 | 42306268 | CG | C | NA | ENSP00000281496 | frame shift | p.Thr817 | 55 |
| VWA8 | 13 | 42385421 | C | T | rs138075452 | ENSP00000281496 | missense | p.Arg668Gln | 36 |
Key:Chr = chromosome, bp37 = hg10/GRCh37 sequence position, ref = reference allele, alt = alternate allele
Note: Not cannonical transcript but transcript with highest PolyPhen 2 score
Candidate variants in ACSS2 and PHYH segregated with NSCLP in 3 or more families
One candidate variant was identified in ACSS2, and was seen in three different families. A single T>C missense variant in ACSS2 was identified in three families at chromosome (chr) 20:33509608. This variant, rs59088485, has a SIFT score of 0.01, PolyPhen2 score of 0.999 and a GERP score of 5.37 [Kumar et al., 2009]. This variant was found in the 1000 genomes database at a frequency of 0.0014, but in their Amerindian subpopulation at a frequency of 0.01. This variant is seen in the Exome Aggregation Consortium (ExAC) browser at a frequency of 0.001850 but is not seen in the ESP database [Exome Aggregation Consortium 2015]. Because this variant was observed at a relatively high frequency in the 1000 genomes Amerindian subpopulation, we genotyped it in our entire set of Honduran controls. We found 4 of 215 individuals were heterozygous for this variant, giving a population frequency of 0.0093. The evidence for co-segregation within the families carrying this variant can be seen in Figure 1. ACSS2 has a Residual Variation Intolerance Score (RVIS) of 22.7% based on the 1000 genomes project and 18.34% based on ExAC, meaning in the general population, this gene is among the 22% least tolerant of mutations [Petrovski et al., 2013]. The probability of both affected members of M45 sharing a single variant is p = 0.3333, for both affected members of M67 is p = 0.06667, and for M94 is p = 0.0323. This gives an overall p-value for all affected members of all three families sharing candidate variants in ACSS2 of p = 7.18 × 10−4.
Figure 1.
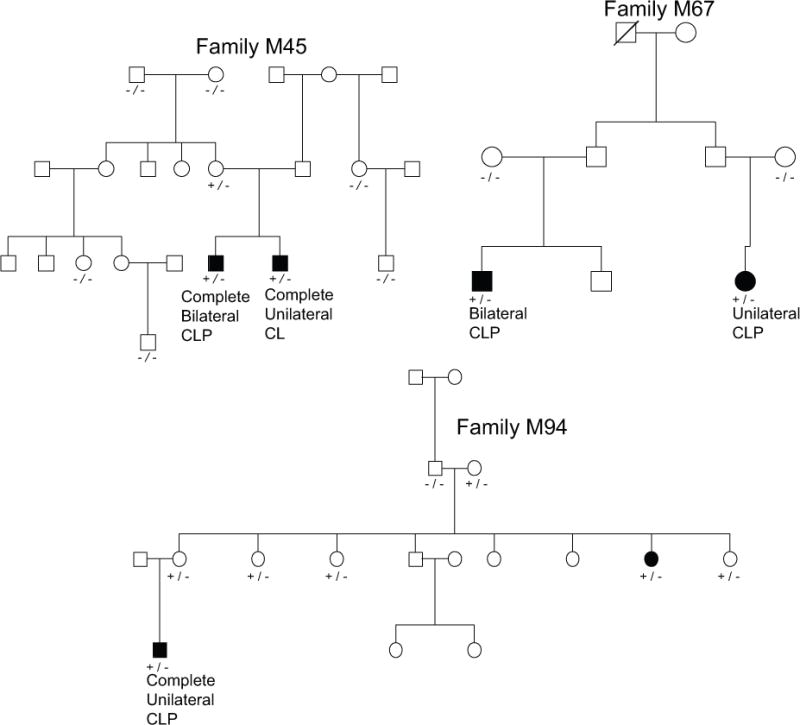
Evidence for cosegregation of variants in ACSS2. Key: −/− = homozygous reference genotype at candidate variant, +/− = heterozygous at candidate variant, +/+ homozygous for the alternate allele.
Three candidate variants in PHYH were identified, each in a different family. The evidence for co-segregation between NSCLP and these PHYH variants is shown in Figure 2. The family M46 variant is an A > AGAT insertion resulting in an inframe codon gain at chr10:13320305. This variant, rs566116760, does not have a SIFT or PolyPhen2 score, but has a GERP score of 5.67. It is not seen in the 1000 genomes project, but exists in the ESP database at a frequency of 0.002 and in ExAC at a frequency of 0.002. The family M28 variant in PHYH is a C>T missense mutation at chr10:13325784. This variant, rs62619919, has a SIFT score of 0.004, a PolyPhen score of 0.577 and a GERP score of 4.85. It is seen in the 1000 genomes project general population at a frequency of 0.0018, but not in the Amerindian subpopulation. It is seen in the ESP database at a frequency of 0.0045 and in ExAC at a frequency of 0.007841. The family M45 variant is a G>C missense and splice site variant at chr10:13337497. This variant, rs145404396, has a SIFT score of 0.02, PolyPhen2 score of 0.815 and GERP score of −10.5. It is seen in the 1000 genomes project at a frequency of 0.0018, and in the African subpopulation at a rate of 0.01. It is seen in in the ESP database at a frequency of 0.0016 and in the ExAC database at a frequency of 0.0004131. This gene has an RVIS score of 83.36% based on 1000 genomes or 72.37% based on ExAC, meaning in the general population, variants in this gene are in the 83.36% least tolerated compared to the rest of the genome. Family M46 has a p-value of 0.06667 for both affected members sharing a variant, family M28 has a p-value of 0.1428 and M45 has a p-value of 0.3333. This gives an overall p-value for all affected members of all three families sharing candidate variants in PHYH of 3.175 × 10−3.
Figure 2.
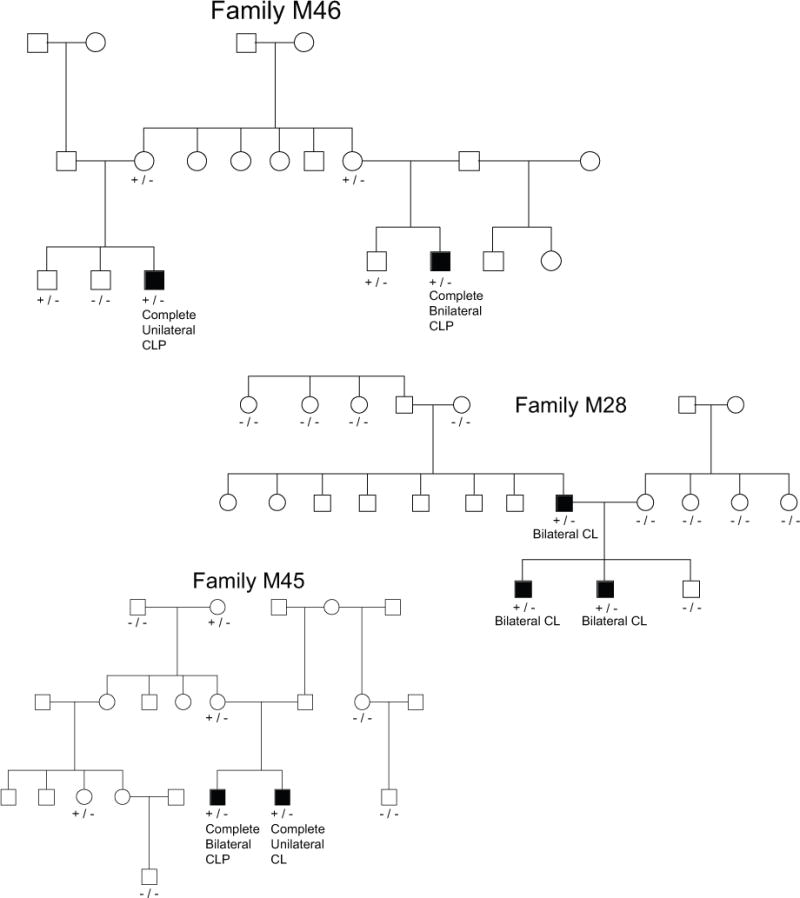
Evidence for cosegregation of variants in PHYH. Key: −/− = homozygous reference genotype at candidate variant, +/− = heterozygous at candidate variant, +/+ homozygous for the alternate allele.
Candidate variants identified in VWA8 and HKDC1 did not consistently segregate with NSCLP
Two candidate variants were identified in HKDC1, with one candidate variant found in three families, and the other found in only one family. The pedigrees carrying variants in HKDC1 can be seen in Figure 3. The first variant is a C>T missense mutation at chr10:71007342. This variant, rs201518882, has a SIFT score of 0.02, PolyPhen2 score of 0.654, and GERP score of 4.84. It is not seen in the 1000 genomes database. It is seen in the ESP database at a frequency of 0.00007 and the ExAC database at a frequency of 0.000648. The family 94 variant in HKDC1 is a C>T stop gain at chr10:71020980. This change does not have SIFT or PolyPhen2 scores, but it has a GERP score of 2.79. It is not seen in the 1000 genomes database nor the ESP database. It is seen in the ExAC database at a frequency of 8.243 × 10−6. HKDC1 has an RVIS score of 70.88% based on 1000 genomes or 32.29% based on ExAC, meaning that in the general population, this gene is in either the 70% or 32% least tolerant of variation, depending on the reference data set chosen. For family M36, the p-value for both affected individuals carrying a single mutation is p = 0.0323, for M71 it is p = 0.33333 and for M94 it is also p = 0.0323. All affected family members in all four families did not carry candidate variants in HKDC1, therefore this gene did not meet our strictest criterion for significance. However, we calculated the probability that out of four families with at least one member carrying a candidate variant in HKDC1, in at least three of these families all affected members did carry the candidate variant, as described in the methods section, which came to 5.462 × 10−4.
Figure 3.
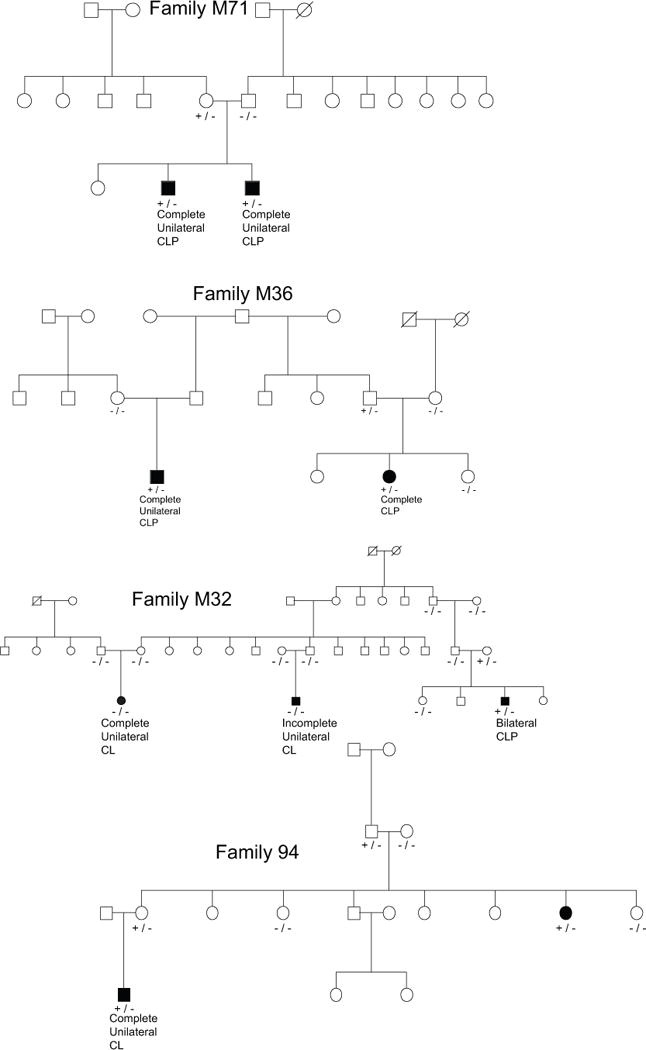
Evidence for cosegregation of variants in HKDC1. Key: −/− = homozygous reference genotype at candidate variant, +/− = heterozygous at candidate variant, +/+ homozygous for the alternate allele.
Five candidate variants were identified in VWA8, each in only one family. However, these variants did not consistently and completely segregate with CL/P. The pedigrees carrying variants in VWA8 can be seen in Figure 4. The family M16 variant is a C>T missense mutation at chr13:42189142 with SIFT score of 0.05, PolyPhen score of 0.99 and GERP score of 6.08. The family M1 variant is a G>A stop gain at chr13: 42245135 and GERP score of 4.73. The variant found in family M32 is an A>G splice region variant at chr13: 42266081 with GERP score of 7.42, but it is only carried by two of three affected individuals. Within family M55, a frame shift insertion of a G at chr13:42306268 with GERP score of 1.88 was found in one affected individual but not in her affected sister. Lastly, family M36 contains a C>T missense variant at chr13:42385421 with SIFT score of 0.05, PolyPhen2 score of 0.99 and GERP score of 5.37. The p-value for both members of M16 carrying the same variant is 0.3333, for M1 is 0.3333, and for M36 is 0.0323. Because not all affected family members in all families where a candidate variant in VWA8 was found were positive for that candidate variant, this gene did not meet our strictest criteria for significance. We did, however, calculate the probability that all affected individuals in three or more families would carry candidate variants in VWA8 given that at least one affected individual in five families carry candidate variants, which came out to 4.333 × 10−3.
Figure 4.
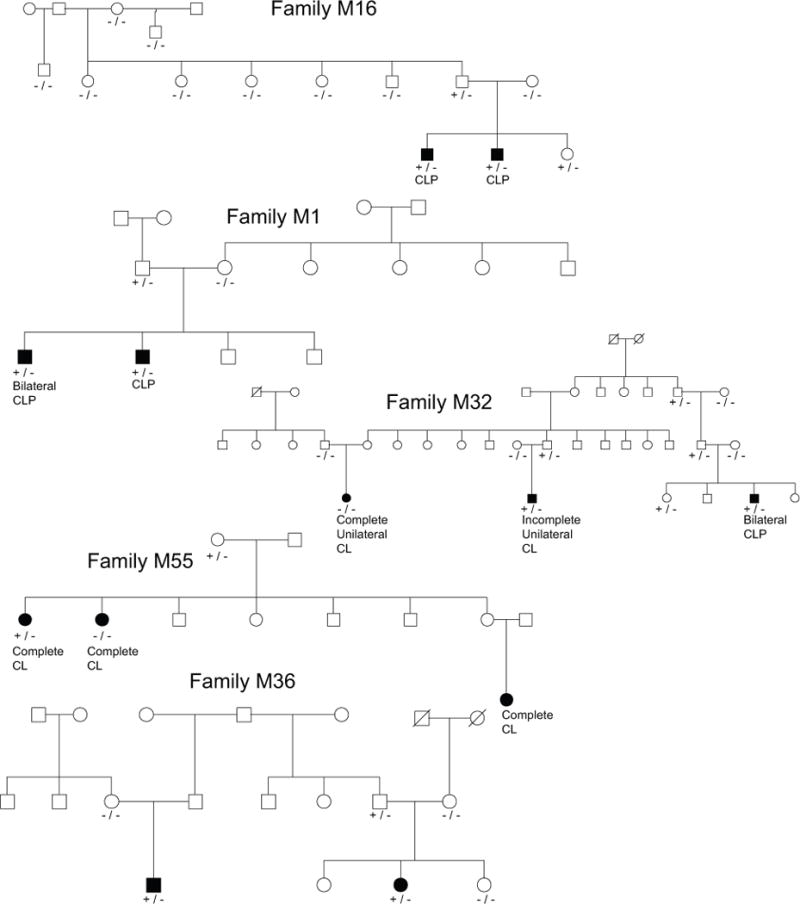
Evidence for cosegregation of variants in VWA8. Key: −/− = homozygous reference genotype at candidate variant, +/− = heterozygous at candidate variant, +/+ homozygous for the alternate allele.
After eliminating variants failing Sanger validation or common polymorphisms on further testing, only one candidate variant in a single family was left in the genes LRBA, NEFH and DENND4B, therefore they did not qualify as candidate genes. Four variants were initially called in LRBA. Two of these failed Sanger validation and another was found to be a common polymorphism. Because the remaining candidate variant was found in only one family (M71), we excluded it from further analysis. Furthermore, the two affected members of M71 are full siblings, so have a high likelihood of sharing any variant due to chance alone (p-value = 0.3333). Two variants were called in NEFH, one of which was found to be a common polymorphism. The verified candidate variant was found in only one family, M55. Similarly, two variants were called in DENND4B, one of which was found to be a common polymorphism. The verified candidate variant was found in only one family, M55. Because these two variants were each found in only one family, we excluded them from further analysis. In addition, the affected individuals in family M55 where candidate variants in both NEFH and DENND4B were found are brothers; they therefore have a high likelihood of sharing any variant due to chance alone (p=0.3333).
Variants identified in LFNG, DARS, ATP6V1D, PRKCSH, DCP1B, ASB10, MAGEF1 and MAGI1, were common polymorphisms in difficult to sequence areas, as verified by sequencing Honduran control subjects unaffected by NSCLP. Four variants were initially identified in GIGYF2, three of which failed Sanger validation, and the last of which was a common polymorphism. Five variants were initially identified in GOLGA2, four of which failed Sanger validation, and the last was a common polymorphism. Two variants were identified in EPHB6, HOMEZ and C9orf156; in each case, one failed Sanger validation, and the other was a common polymorphism. Three variants were initially identified in DHX34; two failed Sanger validation and the third was a common variant. Two variants in HSPBP1 both failed Sanger validation. Two variants were initially called in ZIC2, one of which was discovered to be a common variant. We were unable to get clean sequencing results for the second ZIC2 variant, despite trying several different methodologies. We sequenced controls, and had the same difficulties with the region. We therefore believe this was a likely sequencing error. In addition, this variant was only initially identified by whole exome sequencing in one affected individual in each of two families.
DISCUSSION
Segregation of CL/P with rare variants predicted to be damaging within a family provides evidence that the gene in which the variants are found may be directly related to NSCLP. Identification of likely damaging variants in the same gene in all affected members of several multiplex families strengthens that evidence.
Previous studies suggest a biologically plausible relationship between these genes and CL/P. Several previous studies have uncovered evidence that variants in ACSS2 may be related to NSCLP. Loikkanen et al. showed ACSS2 is differentially expressed in specific tissues at discrete times during embryogenesis, suggesting that this gene is likely involved in embryonic development [Loikkanen et al., 2002; Smith et al., 2014]. They also demonstrated ACSS2 is specifically expressed in mouse facial tissue during development. ACSS2 interacts with several genes previously identified by Jugessur et al. as genes possibly related to clefting: ALDH1A1, ATIC, CTH, DARS, MTHFD1, CBS, and LPL [Jugessur et al., 2009; Kalathur et al., 2013]. PHYH has been associated with rhizomelic chondrodysplasia punctata, which can include craniofacial anomalies such as micrognathia and high arched palate [Jansen et al., 1997; Barr et al., 1993] PHYH has been shown to interact with PEX7, a previously identified gene possibly linked to clefting [Jugessur et al., 2009; Kalathur et al., 2013]. PEX7 has also been implicated in rhizomelic chondrodysplasia punctate [Braverman et al., 1997].
This analysis focused on the probability that a rare, damaging variant in the same gene would be shared by two or more distantly related individuals affected by NSCLP in two or more families. Historically, most papers using this approach have not attempted to calculate p-values for such data. We have chosen to use a method based on Bureau et al. to calculate pedigree specific P-values. We then calculated the probability that the specific multiplex families included in our study would share damaging variants in a given gene in a methodology that is a variation on their method. The assumptions underlying the calculation of p-values included that there is only one founder mutation per family. This assumption is more certain because the frequencies of the alleles in the population are very low and founders are not so substantially related as to be likely to share the mutation identical by descent. Bureau et al. suggested that variants with frequencies of 2% or less were suitable for the calculations, and our candidate variants meet this criterion.
By only including genes with likely damaging variants in at least three families, it is possible that we excluded additional causal genes. By prioritizing only those variants that appeared to be autosomal dominant with high or medium predicted impact and GERP scores ≥3 and/or Polyphen2 scores ≤0.8, it is also possible that we have excluded causal variants. However, we focused on this subset of variants in order to make the best use of limited resources.
Sanger sequencing demonstrated that unaffected family members also carry these likely damaging variants, suggesting incomplete penetrance. This is not unexpected, as the distribution of affected relatives in our pedigrees clearly requires incomplete penetrance for any simple Mendelian mode of inheritance. While the variant we identified in ACSS2 is relatively common (with a rate of 0.01) the rate of NSCLP in this population is around 0.002. This would mean about 1 in 5 individuals with the variant would need to have NSCLP, which appears plausible given this incomplete penetrance and the distribution of NSCLP in the observed pedigrees.
HKDC1 is mutated in only one affected individual in family M32 and therefore did not meet our strict inclusion criteria; however this individual has a more severe phenotype than other affected individuals in the family, with bilateral complete cleft lip and palate as opposed to unilateral cleft lip only. This can be interpreted as evidence for HKDC1 being involved in NSCLP, but suggests some genetic heterogeneity within and between families.
As shown by our p-values, any gene with variants predicted to be damaging that segregate with NSCLP in more than one family is likely due to a biological relationship between this gene and NSCLP. Therefore, the two genes in our study meeting this criteria, PHYH and ACSS2, warrant further follow up and inclusion in future studies investigating genetic causes of non-syndromic cleft lip and palate.
Acknowledgments
Sequencing was provided by the University of Washington Center for Mendelian Genomics (UW-CMG) and was funded by the National Human Genome Research Institute and the National Heart, Lung and Blood Institute grant 1U54HG006493 to Drs. Debbie Nickerson, Jay Shendure and Michael Bamshad. The authors would like to thank the Honduran Medical Institute, Inc. for funding support. The authors would like to thank the Exome Aggregation Consortium and the groups that provided exome variant data for comparison. A full list of contributing groups can be found at http://exac.broadinstitute.org/about.
References
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. Nat Methods. 2010;7(4):248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barr DG, Kirk JM, al Howasi M, Wanders RJ, Schutgens RB. Rhizomelic chondrodysplasia punctata with isolated DHAP-AT deficiency. Arch Dis Child. 1993;68(3):415–417. doi: 10.1136/adc.68.3.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. Journal of the Royal Statistical Society. 1995;57(1):289–300. [Google Scholar]
- Braverman N, Steel G, Obie C, Moser A, Moser H, Gould SJ, Valle D. Human PEX7 encodes the peroxisomal PTS2 receptor and is responsible for rhizomelic chondrodysplasia punctata. Nature Genet. 1997;15:369–376. doi: 10.1038/ng0497-369. [DOI] [PubMed] [Google Scholar]
- Bureau A, Parker M, Ruczinski I, Taub MA, et al. Whole exome sequencing of distant relatives in multiplex families implicates rare variants in candidate genes for oral clefts. Genetics. 2014;197:1039–1044. doi: 10.1534/genetics.114.165225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calzolari E, Pierini A, Astolfi G, Bianchi F, Neville AJ, Rivieri F. Associated anomalies in multi-malformed infants with cleft lip and palate: An epidemiologic study of nearly 6 million births in 23 EUROCAT registries. American journal of medical genetics. 2007;143A:528–537. doi: 10.1002/ajmg.a.31447. [DOI] [PubMed] [Google Scholar]
- Choi M, Scholl UI, Ji W, et al. Genetic diagnosis by whole exome capture and massively parallel DNA sequencing. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:19096–19101. doi: 10.1073/pnas.0910672106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper GM, Stone EA, Asimenos G, NISC Comparative Sequencing Program. Green ED, Batzoglou S, Sidow A. Distribution and intensity of constraint in mammalian genomic sequence. Genome Research. 2005;15:901–913. doi: 10.1101/gr.3577405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, Handsaker RE, Lunter G, Marth GT, Sherry ST, McVean G, Durbin R, 1000 Genomes Project Analysis Group Bioinformatics. 2011;27(15):2156–8. doi: 10.1093/bioinformatics/btr330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon MJ, Marazita ML, Beaty TH, Murray JC. Cleft lip and palate: understanding genetic and environmental influences. Nature reviews Genetics. 2011;12:167–178. doi: 10.1038/nrg2933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Exome Aggregation Consortium (ExAC) Cambridge, MA: (URL: http://exac.broadinstitute.org) [April 2015] [Google Scholar]
- Exome Variant Server, NHLBI GO Exome Sequencing Project (ESP) Seattle, WA: (URL: http://evs.gs.washington.edu/EVS/) [April 2015] [Google Scholar]
- Jansen GA, Mihalik SJ, Watkins PA, Moser HW, Jakobs C, Heijmans HS, Wanders RJ. Phytanoyl-CoA hydroxylase is not only deficient in classical Refsum disease but also in rhizomelic chondrodysplasia punctata. J Inherit Metab Dis. 1997;20(3):444–6. doi: 10.1023/a:1005379406639. [DOI] [PubMed] [Google Scholar]
- Jugessur A, Shi M, Gjessing HK, Lie RT, Wilcox AJ, Weinberg CR, et al. Genetic Determinants of Facial Clefting: Analysis of 357 Candidate Genes Using Two National Cleft Studies from Scandinavia. PLoS ONE. 2009;4(4):e5385. doi: 10.1371/journal.pone.0005385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalathur RKR, Pinto JP, Hernández-Prieto MA, Machado RSR, Almeida D, Chaurasia G, Futschik ME. 2–13. UniHI 7: an enhanced database for retrieval and interactive analysis of human molecular interaction networks. Nucl Acids Res. doi: 10.1093/nar/gkt1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4(7):1073–81. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- Leslie EJ, Marazita ML. Genetics of cleft lip and cleft palate. American journal of medical genetics Part C, Seminars in medical genetics. 2013;163:246–258. doi: 10.1002/ajmg.c.31381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler Transform. Bioinformatics. 2009;25:1754–60. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loikkanen I1, Haghighi S, Vainio S, Pajunen A. Expression of cytosolic acetyl CoA synthetase gene is developmentally regulated. Mech Dev. 2002;115(1–2):139–41. doi: 10.1016/s0925-4773(02)00097-7. [DOI] [PubMed] [Google Scholar]
- Mangold E, Ludwig KU, Birnbaum S, et al. Genome-wide association study identifies two susceptibility loci for nonsyndromic cleft lip with or without cleft palate. Nature genetics. 2010;42:24–26. doi: 10.1038/ng.506. [DOI] [PubMed] [Google Scholar]
- Manichaikul A, Mychaleckyj JC, Rich SS, Daly K, Sale M, Chen WM. Robust relationship inference in genome-wide association studies. Bioinformatics. 2010;26(22):2867–2873. doi: 10.1093/bioinformatics/btq559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marazita ML. The evolution of human genetic studies of cleft lip and cleft palate. Annual review of genomics and human genetics. 2012;13:263–283. doi: 10.1146/annurev-genom-090711-163729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaren W, Pritchard B, Rios D, Chen Y, Flicek P, Cunningham F. Deriving the consequences of genomic variants with the Ensembl API and SNP Effect Predictor. Bioinformatics. 2010;26(16):2069–70. doi: 10.1093/bioinformatics/btq330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McVean, et al. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mossey PA, Little J, Munger RG, Dixon MJ, Shaw WC. Cleft lip and palate. Lancet. 2009;374:1773–1785. doi: 10.1016/S0140-6736(09)60695-4. [DOI] [PubMed] [Google Scholar]
- Ng SB, Bigham AW, Buckingham KJ, et al. Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nature genetics. 2010;42:790–793. doi: 10.1038/ng.646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paila U, Chapman BA, Kirchner R, Quinlan AR. GEMINI: Integrative Exploration of Genetic Variation and Genome Annotations. PLoS Comput Biol. 2013;9(7):e1003153. doi: 10.1371/journal.pcbi.1003153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrovski S, Wang Q, Heinzen EL, Allen AS, Goldstein DB. Genic Intolerance to Functional Variation and the Interpretation of Personal Genomes. PLOS Genetics. 2013 doi: 10.1371/journal.pgen.1003709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, Maller J, Sklar P, de Bakker PIW, Daly MJ, Sham PC. PLINK: a toolset for whole-genome association and population-based linkage analysis. American Journal of Human Genetics. 2007;81 doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozen S, Skaletsky HJ. Primer3. 1998 doi: 10.1385/1-59259-192-2:365. Code available at http://www-genome.wi.mit.edu/genome_software/other/primer3.html. [DOI] [PubMed]
- Smith CM, Finger JH, Hayamizu TF, McCright IJ, Xu J, Berghout J, Campbell J, Corbani LE, Forthofer KL, Frost PJ, Miers D, Shaw DR, Stone KR, Eppig JT, Kadin JA, Richardson JE, Ringwald M. The mouse Gene Expression Database (GXD): 2014 update. Nucleic Acids Res. 2014;42(D1):D818–D824. doi: 10.1093/nar/gkt954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehby GL, Cassell CH. The impact of orofacial clefts on quality of life and healthcare use and costs. Oral diseases. 2010;16:3–10. doi: 10.1111/j.1601-0825.2009.01588.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg SM, Neiswanger K, Martin RA, et al. The Pittsburgh Oral-Facial Cleft Study: Expanding the Cleft Phenotype. left Palate–Craniofacial Journal. 2006;43(1) doi: 10.1597/04-122r1.1. [DOI] [PubMed] [Google Scholar]