

V-ATPases are fascinating molecular machines; cryo-EM is providing unprecedented insight into their structure and dynamics.
Keywords: Rotary ATPases, V-ATPase, Cryo-EM, cryoEM, structure, dynamics
Abstract
Electron cryomicroscopy (cryo-EM) has significantly advanced our understanding of molecular structure in biology. Recent innovations in both hardware and software have made cryo-EM a viable alternative for targets that are not amenable to x-ray crystallography or nuclear magnetic resonance (NMR) spectroscopy. Cryo-EM has even become the method of choice in some situations where x-ray crystallography and NMR spectroscopy are possible but where cryo-EM can determine structures at higher resolution or with less time or effort. Rotary adenosine triphosphatases (ATPases) are crucial to the maintenance of cellular homeostasis. These enzymes couple the synthesis or hydrolysis of adenosine triphosphate to the use or production of a transmembrane electrochemical ion gradient, respectively. However, the membrane-embedded nature and conformational heterogeneity of intact rotary ATPases have prevented their high-resolution structural analysis to date. Recent application of cryo-EM methods to the different types of rotary ATPase has led to sudden advances in understanding the structure and function of these enzymes, revealing significant conformational heterogeneity and characteristic transmembrane α helices that are highly tilted with respect to the membrane. In this Review, we will discuss what has been learned recently about rotary ATPase structure and function, with a particular focus on the vacuolar-type ATPases.
INTRODUCTION
Structural biology strives to construct models, ultimately at atomic resolution, that represent snapshots of biological macromolecules and to describe the ways in which these molecules move. Two traditional structure determination methods, x-ray crystallography and nuclear magnetic resonance (NMR) spectroscopy, have been widely used in this regard. Scattering of x-rays from well-ordered crystals of biological macromolecules results in diffraction patterns that can be used to calculate three-dimensional (3D) structures, often at atomic resolution. NMR spectroscopy examines macromolecules in the solution or solid state and provides quantitative information on the dynamics of molecules (1). Over the past six decades, these methods have provided our principal insights into the structure and dynamics of the macromolecules that make up cells. As a result of this type of knowledge, it is now possible to design small-molecule drug rationally in silico before experimental validation of their effects (2, 3). However, considerable challenges remain. More than a third of an organism’s genome typically encodes for membrane-associated or membrane-embedded proteins that are not often amenable to study using these techniques. In addition, large macromolecular assemblies (>300 kD) are often difficult to study by x-ray crystallography because of instability, conformational heterogeneity, and scarcity in the cell. Large asymmetric units in protein crystals can also make x-ray data collection difficult. Similarly, solution NMR spectroscopy of large proteins is complicated by slow tumbling of the molecules in solution, which degrades their NMR signal, and the large number of amino acid residues, which leads to overlapping peaks in spectra. Furthermore, x-ray crystallography and NMR spectroscopy often use isotopic labeling of heterologously expressed proteins. However, heterologous expression can be challenging for high–molecular- weight proteins.
Single-particle electron cryomicroscopy (cryo-EM) provides an alternative approach for large proteins and macromolecular complexes. For cryo-EM, macromolecules that are difficult to work with can be purified from natural sources to a purity and quantity that is significantly lower than what is required for x-ray crystallography or NMR spectroscopy, and no isotopic labeling is necessary during specimen preparation. In cryo-EM, purified assemblies are spread on a supporting substrate and frozen rapidly, producing a thin film of vitreous ice containing the molecules of interest in random orientations. The rapid freezing process helps to ensure that the native structures of molecules are largely preserved (4). After specimen preparation, structure determination by cryo-EM follows a well-defined path. Specimens are imaged with an electron microscope while being maintained at cryogenic temperature. The electron microscope uses a high-energy electron beam, typically 100 to 300 keV, and a limited total exposure of electrons (20 to 200 e−/Å2) to minimize radiation damage to the structure of the specimen. The resulting 2D images are approximately projections of the 3D structure of the specimen from the different randomly oriented views of the protein complexes (known as particles). These projection images are convoluted with the point-spread function of the microscope and have a low signal-to-noise ratio (SNR) because of the low-electron exposures needed to reduce radiation damage. To calculate a 3D structure of the protein, the relative orientations of the 2D projection images must be determined computationally so that different images can be combined, and the effects of the microscope point-spread function must be corrected. This process is intensely computational and requires iterative refinement of the protein structure.
Transmission electron microscopy is inherently an atomic resolution technique. The wavelength of a high-energy electron from a typical electron microscope is a few picometers, leading to a diffraction-limited resolution far beyond what is needed for atomic resolution biological studies (1 to 4 Å resolution). However, lens aberrations, even for aberration-corrected instruments, limit electron microscope resolution to ~0.5 Å for radiation-stable materials (5). The low-electron exposures needed to minimize radiation damage to biological specimens, primarily due to inelastic scattering of electrons (6), result in a low SNR in images. This low image SNR is the primary limit to resolution for cryo-EM. The SNR can be improved by averaging many particle images from the same view in a procedure known as 2D alignment and classification. However, the low SNR limits the accuracy with which images can be aligned. A second source of resolution loss is specimen movement due to instabilities in the microscope stage which are induced by interaction of the protein and vitreous ice with the electron beam. This movement results in “blurring” of cryo-EM images. The best resolutions for biological specimens have ranged from 1.9 Å for 2D crystals (7) to a recently obtained 2.2 Å map of β-galactosidase (8), the latter having relied on the advances discussed in this Review.
Multiple developments in cryo-EM hardware and image processing have resulted in significantly improved resolution in the past few years (9, 10). The introduction of direct detector device (DDD) cameras provided efficient detection of electrons along with fast readout rates that enable collection of movies instead of images. With movie output, it is now possible to correct computationally for some of the beam-induced movement of whole movie frames and individual particles (11–14). The resulting high-quality projection images also improve the determination of particle orientation during 3D map refinement. Another advance is the ability to separate computationally the different conformations of biological assemblies that exist simultaneously in solution, which would otherwise make the final 3D map an incoherent average of different structures (15). This procedure is known as 3D classification and, in combination with the aforementioned camera improvements, has helped push the resolution of the structures obtained by single-particle cryo-EM to atomic and near-atomic resolutions. Recent high-resolution structures determined by single-particle cryo-EM include the TRPV1 (16) and TRPA1 (17) ion channels, the ryanodine receptor (18–20), adenosine triphosphate (ATP)– and adenosine diphosphate (ADP)–bound N-ethylmaleimide–sensitive factor (21), the 20S proteasome (22), β-galactosidase (8), and the ribosome (23). 3D classification has made it possible to distinguish, at subnanometer resolution, multiple conformational states within a dynamic enzyme during its enzymatic cycle (24).
ROTARY ATPase FUNCTION
Rotary adenosine triphosphatases (ATPases) comprise a three-member family of energy-transducing enzymes that are found in organisms from bacteria to humans. The conserved function of these membrane-embedded heteromultimeric protein complexes is to couple the conversion of chemical energy, in the form of ATP, to an electrochemical potential across a biological membrane. To carry out this task, rotary ATPases have a soluble catalytic region responsible for ATP hydrolysis or synthesis and a membrane-embedded region responsible for ion transport. Vacuolar/archaeal-type ATPases (V/A-ATPases) are found in archaea and some species of eubacteria (25, 26) and can use either protons (27–29) or sodium ions (30–32) as coupling ions. V/A-ATPases function in both ATP synthesis and hydrolysis modes (33). For example, the V/A-ATPase in Thermus thermophilus operates as a proton-driven ATP synthase (34, 35), whereas in Gram-negative bacteria from the genera Enterococcus and Clostridium, the enzymes act as sodium pumps (36–38). F-type ATP synthases are found in bacterial plasma membranes, mitochondrial inner membranes, and chloroplast thylakoid membranes. These enzymes synthesize ATP powered by electrochemical membrane potentials, usually a proton-motive force (PMF). The PMF may be calculated by , where F is Faraday’s constant, R is the ideal gas constant, T is the temperature, Δψ is the transmembrane electrical potential, and ΔpH is the transmembrane pH gradient. For most energized membranes, both Δψ and ΔpH contribute to the PMF. However, in chloroplasts, the PMF is almost entirely due to ΔpH. Some F-type ATP synthases can also pump protons under specific cellular conditions (33). This feature of the enzyme allows bacteria to couple ATP metabolism and PMF with external stimuli such as oxygen levels. F-type ATP synthases can also transport sodium ions across the membrane as observed in Acetobacterium woodii (39) and Propionigenium modestum (40, 41). V-ATPases are eukaryotic enzymes responsible for acidification of intracellular organelles. In all eukaryotes, V-ATPases mediate diverse processes, such as secondary active transport (42), endosomal protein sorting (43), protein glycosylation in the Golgi (44), guanosine triphosphatase–mediated activation of the target of rapamycin complex 1 (TORC1) (45), and loading of secretory vesicles (42). In some specialized cells in higher eukaryotes, the V-ATPase is shuttled to the plasma membrane and mediates acidification of the extracellular environment. The subsequent decrease in external pH plays essential roles in processes such as bone demineralization by osteoclasts (46), sperm maturation and activation in the lumen of the reproductive tract (47), and proton secretion by kidney epithelial cells (48). These and other physiological roles of the V-ATPase are discussed in more detail elsewhere (49, 50). Eukaryotic V-ATPase function can be regulated by reversible dissociation of the catalytic V1 region and membrane-embedded VO regions (51, 52), a process that has not been observed in vivo in the V/A-ATPase or F-type ATP synthase. For the V-ATPase, dissociation of the V1 and VO regions has also not yet been observed in plants or mammals. However, suggested interaction of the mammalian V-ATPase with aldolase (53) and phosphofructokinase-1 (54) implies that its activity may be regulated by glucose metabolism in higher eukaryotes.
As discussed in the following section, ATP hydrolysis–dependent conformational changes in the V1 region are transmitted to the VO region, resulting in proton pumping against the concentration gradient. The dissociation of V1 and VO results in autoinhibition of the function of each of these regions. In humans, V-ATPase malfunction leads to multiple disorders. Osteoclast-mediated bone resorption is impaired by mutations in V-ATPase, which leads to osteopetrosis (46, 55–57). Similarly, mutations in V-ATPase subunits have been associated with renal tubular acidosis, resulting in impaired kidney function and also in deafness (58). Aberrant expression of different isoforms of the V-ATPase at the plasma membrane has been linked to the metastatic potential of cancer cells (59–62). Although more work must be done to elucidate the precise role of the V-ATPase in the complex environment of a human tumor, antibodies or small-molecule drugs that could specifically inhibit V-ATPase in an isoform- and location-specific manner would be invaluable reagents. Inhibition of the plasma membrane V-ATPase to limit bone resorption by osteoclasts to treat osteoporosis, and possibly to target extracellular acidification in tumor metastasis, is and intriguing possibility. Potential therapeutics would have to be extraordinarily specific to avoid significant toxicity. Consequently, isoform-specific antibody therapeutics limited to affecting the enzyme at the cell surface are particularly interesting.
STRUCTURAL STUDIES OF ROTARY ATPases
Rotary ATPases pose a significant challenge to traditional high-resolution structural biology techniques, such as NMR spectroscopy and x-ray crystallography, due to their intrinsic conformational heterogeneity and instability. Cryo-EM has proven to be a versatile technique for structural studies of these enzymes, initially at resolutions that revealed the shape and arrangement of subunits (63–68). With the advances made in detector design and image processing, recent cryo-EM maps of rotary ATPases have reached resolutions between 6.2 and 9.4 Å (24, 69–72) and have made it possible to distinguish different rotational states of the enzymes at subnanometer resolution (24, 70, 71). However, generation of higher-resolution maps has been hampered by the intrinsic flexibility of these rotary enzymes, which is necessary for their function (24). In the following section describing state-of-the-art knowledge of the structure and dynamics of rotary ATPases, we place the major emphasis on V-ATPase but mention other rotary ATPases where relevant. For consistency, we use the subunit nomenclature from the Saccharomyces cerevisiae V-ATPase.
Negative-stain EM (73–79), cryo-EM (24, 65–68, 70, 71, 80), and x-ray crystallography studies (81, 82) have elucidated similarities and differences in the subunit composition and arrangement of the different rotary ATPase (Fig. 1). The subunit composition of the S. cerevisiae V-ATPase is A3B3CDE3FG3Hac8c′c″de. Subunits with uppercase letter names reside in the soluble V1 region, whereas those with lowercase letter names are found in the membrane-embedded VO region. Pairs of A- and B-subunits are arranged like segments of an orange around the D-subunit of the central rotor (83), as expected from crystal structures of the related F1-ATPase (81). In addition to the central rotor (subunits D, F, d, c, c′, and c″), the V1 and VO regions are attached by three peripheral stalks (67, 72, 84), each composed of a heterodimer of subunits E and G (85). The peripheral stalks and central rotor extend toward the membrane to a collar structure that contains subunits H and C and the N-terminal domain of subunit a (64, 67). The peripheral stalks hold the a-subunit against the c-subunits, which are arranged into a ring within the lipid bilayer. The composition of the c-ring in S. cerevisiae is thought to be c8c′c″ because there are 10 subunits in the ring (86) and one copy of each of the c′- and c″-subunits (87). On the basis of cryo-EM maps, high-resolution crystal structures (88–91), and homology models (92–96), a mosaic model of yeast V-ATPase was generated (24), which lacked only the a- and e-subunits (Fig. 2). Two densities identified in the 6.9 Å resolution map of S. cerevisiae V-ATPase (24) could not be attributed to the a-subunit model and may represent the e-subunit and a previously unidentified V-ATPase subunit. A model for the a-subunit was subsequently produced by tracing the a-subunit sequence through the cryo-EM density map using constraints from evolutionary covariance, as described below (71). Cryo-EM of the insect V-ATPase from Manduca sexta revealed additional features that were not seen in the yeast V-ATPase, such as a density attached to the lumenal surface of the c-ring, which may be the protein Ac45 that is not found in yeasts (72).
Fig. 1. Subunit arrangement in three types of rotary ATPases.
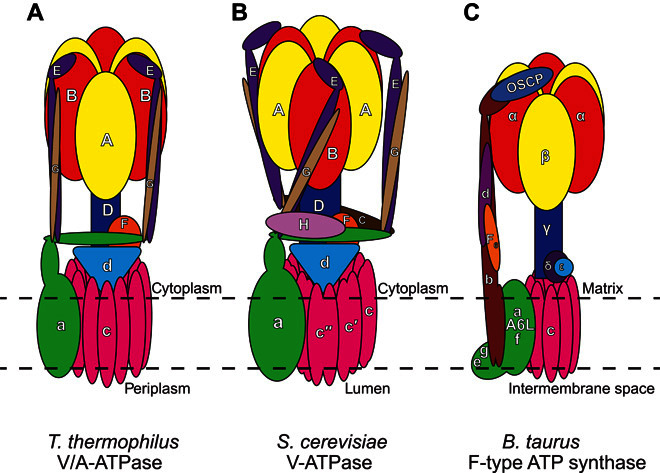
Schematic representations of T. thermophilus V/A-ATPase (A), S. cerevisiae V-ATPases (B), and B. taurus F-type ATP synthase (C) [adapted with permission from Schep et al. (71)]. OSCP, oligomycin sensitivity conferral protein.
Fig. 2. Rotational states of V-ATPase.

(A to C) Three cryo-EM maps from the S. cerevisiae V-ATPase representing three distinct conformational states of the enzyme. Two upper and lower panels show cross sections through the different conformations. Scale bar, 25 Å [adapted with permission from Zhao et al. (24)].
Each AB heterodimer provides a nucleotide-binding site and is found in a unique conformation at any point in the enzyme’s catalytic cycle. These conformations are tightly coupled to the content of the nucleotide-binding sites. ATP hydrolysis in the V1 region by the A3B3DF subcomplex causes rotation of the central rotor, including the c-ring. Rotation of the c-ring against the a-subunit in the VO region drives proton translocation across the membrane (97, 98). The contact between the a-subunit and c-ring is minimal, suggesting a dynamic interface (65–68). In the V-ATPase, proton translocation occurs from the cytoplasm to the lumenal side of the membrane during rotation of the c-ring (Fig. 1B). The dominant model for how the a-subunit allows proton translocation is that it provides two offset half-channels, each spanning half the thickness of the lipid bilayer (65, 99, 100). As the ring rotates, conserved mid-membrane glutamate residues on the c-subunits transition from an aqueous cytoplasmic half-channel to the middle of the lipid bilayer. Thermodynamic arguments suggest that these glutamate residues cannot be negatively charged in the lipid bilayer and must become protonated from the aqueous environment of the cytoplasmic half-channel as they move from one environment to the other (101). Rotation of the ring brings a protonated glutamate residue from a c-subunit out of the lipid bilayer and into a deprotonating lumenal half-channel, where interaction with a conserved and essential arginine residue of the a-subunit causes deprotonation and the net transport of a proton. The conserved arginine is likely essential for forcing the glutamate in the lumenal half-channel to become deprotonated, despite the lumenal half-channel being connected to a low-pH environment. Without this arginine residue, ATP hydrolysis and proton pumping become decoupled in both the F-type ATP synthase (102) and V-ATPase (103), consistent with this proposed role. Following the protonation and deprotonation events, the c-ring has been reset for the transport of the next proton. Perhaps the best evidence for the existence of these two half-channels comes from labeling experiments that detected aqueous channels in the Escherichia coli ATP synthase a-subunit (104–106). Cryo-EM studies (24, 68, 70–72) have revealed structures that could make up the half-channels as discussed below. Determining the path of protons through a rotary ATPase will likely require combining computational simulations with structural data that have sufficient resolution to determine amino acid side-chain positions and orientations. However, resolving the structure of the membrane-embedded VO region and the corresponding membrane-embedded regions of other rotary ATPases has proven extremely challenging. The domain arrangement of the VO region was first observed clearly in an 11 Å resolution cryo-EM map of the yeast V-ATPase (67). The α-helical arrangement of the c-ring and the a-subunit of the V-ATPase were recently visualized with a 6.9 Å resolution map of the S. cerevisiae V-ATPase (24), and 10 proton-carrying c-subunits were fit into the cryo-EM density. This stoichiometry sets the ATP/H+ ratio at 10 protons pumped for every 3 ATP molecules hydrolyzed for the yeast V-ATPase. With a Gibbs free energy for ATP hydrolysis of 57 kJ mol−1 at 30°C, an 3:10 ATP/H+ ratio sets the maximum pH gradient or voltage across the vacuolar membrane in S. cerevisiae to 3.0 units or 180 mV, respectively (24, 107), consistent with estimates of vacuolar pH (49). The c-ring is composed of an inner and an outer ring of transmembrane α helices. The outer part of the ring the observed to be in contact with two highly tilted transmembrane α helices of the a-subunit. A similarly tilted α-helical hairpin in the a-subunit was also observed in cryo-EM maps of the F-type mitochondrial ATP synthase from the colorless algae Polytomella sp. (69), the bovine mitochondrial ATP synthase (70), the T. thermophilus V/A-ATPase (71), and a crystal structure of the bacterial F-ATPase that unfortunately had low resolution for the membrane region (82). The similarity of the α-helical arrangement in the different rotary ATPases demonstrates structural conservation across all members of this family of enzymes. The two tilted α helices likely help form the two half-channels thought to be involved in proton translocation.
Numerous studies (103, 108–110) have suggested the presence of conserved residues in the a-subunit and the c-ring responsible for proton translocation. Atomic models that show the locations of these residues for the a-subunits from the S. cerevisiae V-ATPase, T. thermophilus V/A-ATPase, and Bos taurus F-ATP synthase were generated by tracing the amino acid sequence of the subunits in their corresponding 6 to 7 Å maps aided by distance constraints from evolutionary covariance analysis (70, 71). These models agree with biochemical data mapping the locations of residues of the membrane-embedded C-terminal half of the a-subunit (108, 111, 112). The three models suggest an evolutionarily conserved spatial organization of important residues within the membrane-embedded portion of the a-subunit (Fig. 3, A to C). In the model of the V-ATPase a-subunit, a conserved arginine residue resides within the highly tilted α-helical hairpin toward the lumenal side of the V-ATPase a-subunit, proximal to the conserved glutamate residues located in the middle of the outer helices of the c-subunits. The arginine residue is flanked on both sides by polar and charged residues that provide possible paths toward the cytosol and the lumen (Fig. 3D).
Fig. 3. Model of proton translocation via the a-subunit.
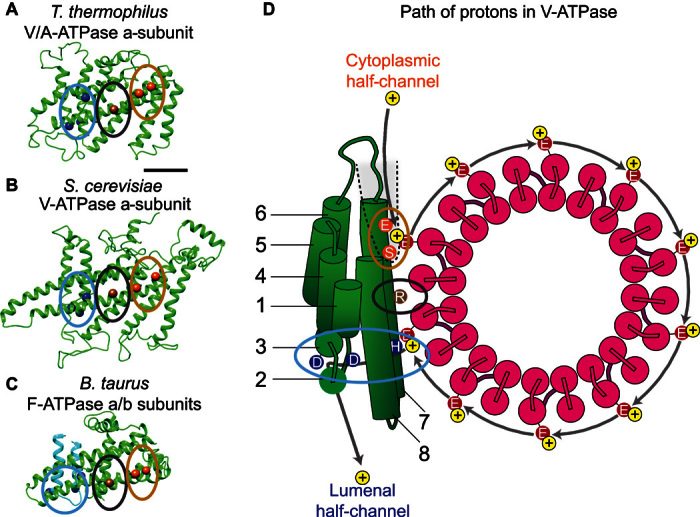
(A to C) A similar arrangement of residues is present in V/A-ATPase (A), V-ATPase (B), and F-type ATP synthase (C). Clusters of residues important for proton translocation are highlighted for the cytoplasmic half-channel (orange ellipse) and lumenal half-channel (blue ellipse). The conserved essential arginine residues are circled in black. (D) Schematic diagram of the half-channel model for proton translocation in the S. cerevisiae V-ATPase [adapted with permission from Schep et al. (71)].
The proposed model of the V-ATPase a-subunit from cryo-EM and evolutionary covariance also identified a 62-residue-long cytoplasmic loop that displays a high degree of covariance in a multiple sequence alignment of approximately 3000 sequences (71). This stretch of sequence displays the least similarity between the two different isoforms of the a-subunit in S. cerevisiae but contains a remarkable number of charged residues. Although a lack of conservation often indicates a lack of functional importance, evolutionary covariance analysis shows that this sequence varies between different sequences for the a-subunit in a remarkably coordinated way. Consequently, the proximity of this loop to the cytoplasmic half-channel, its net charge, and its isoform-dependent sequence suggest a functional role that differs in different subcellular compartments.
Multiple studies have highlighted significant flexibility in the intact V-ATPase and its subunits. Initial evidence for the dynamic nature of rotary ATPases was seen in the variable conformation of individual subunits and subcomplexes in structures from x-ray crystallography and NMR spectroscopy (89–91, 113) and ion mobility–mass spectrometry studies (114). The V1 and VO regions were also observed in different relative orientations in both negative-stain and cryo-EM studies (115, 116). This flexibility was suggested to be essential for the high-efficiency coupling of ATP hydrolysis to proton pumping in the rotary enzymes (24). The recent cryo-EM observations of distinct rotational states of the intact S. cerevisiae V-ATPase (24) (Fig. 2) and related enzymes (70, 71) demonstrate the existence of flexibility in the enzyme structure (24). These V-ATPase and F-type ATP synthase structures were determined with their catalytic regions stopped in three different positions, related by ~120° rotations of the central rotor. Similarly, the membrane-embedded regions of the enzymes appeared to stop with integer rotational steps of the c-subunits in the c-ring. In these structures, coordinated flexibility in many of the subunits of the complex is necessary to accommodate the 3:10 symmetry mismatch between the soluble V1 and membrane-bound VO regions of the V-ATPase (24) and the 3:8 symmetry mismatch between the F1 and FO regions of the ATP synthase (70). On the other hand, the T. thermophilus V/A-ATPase, which has a 3:12 symmetry match for the V1 and VO regions, displays a more rigid transformation between the two rotational state structures determined (71). Some of the flexibility seen in the V- and F-type ATPase structures may be an artifact from examining complexes that are neither synthesizing nor hydrolyzing ATP. That is, when the central rotor is spinning at full speed, there may be less distortion of the enzyme than was seen in the inactive complexes studied. However, the ability of the enzymes to accommodate symmetry mismatch when stopped reveals flexibility in the structures, which likely smoothens the power transmission between their soluble and membrane-embedded motors. The observed distortions include not only the subunit-specific flexibility that allows the enzymes to accommodate their symmetry mismatch but also the long-range bending and flexing modes of the entire complexes (70).
FUTURE WORK
Despite the recent advances made in acquiring 6 to 7 Å resolution structures of rotary ATPase by cryo-EM, high-resolution structures are needed to understand the mechanism of proton translocation, proton specificity in the V-ATPase, and the interaction of rotary ATPases with drugs and antibodies. Significant conformational heterogeneity will continue to impede acquisition of atomic resolution cryo-EM maps unless better strategies are developed to computationally separate the different conformational states using large data sets. Nonuniform resolution within maps could be observed in 3D classification of rotational states of the V-ATPase, suggesting the presence of substates that require more stringent classification methods (24). Adoption of automated data acquisition methods can provide large data sets, which may increase the quality of the maps calculated for each individual conformational state and may aid further 3D classification. However, existing computational approaches will struggle with data sets that are orders of magnitude larger than those used at present. The use of nonhydrolyzable nucleotide analogs may also enable trapping of specific conformations, an approach that was used with great success in crystallographic studies of F1-ATPases (117). Alternatively, it may be possible to enhance the resolution of maps by stabilizing V-ATPase conformation using cross-linking agents (118), although a limitation in the resulting maps that should be remembered is that flexibility is an intrinsic property of rotary ATPases. Another factor impeding acquisition of a high-resolution map may be the detergent required to solubilize the complex. Detergent micelles, like protein, have a higher density than vitreous ice, increasing the background noise in images. This problem can be potentially circumvented by the use of nanodiscs (119), saposins (120), or the amphipathic polymers known as amphipols (16, 121), all of which allow for the removal of detergents from the buffers used in specimen preparation. The resulting improved image contrast will increase the accuracy of image alignment and the extraction of high-resolution information from the data. The third challenge in calculating a high-resolution cryo-EM map of the V-ATPase is the pseudosymmetric nature of the complex. Computer algorithms may confuse the different side views of the complex, which in low-SNR cryo-EM images may look quite similar. One approach to improving particle image alignment of pseudosymmetric particles may be to emphasize asymmetric features with attachment of antibodies (122). In addition, phase plates can improve resolution by providing higher SNRs for the low-resolution information used for image alignment and classification (123). Better contrast provided by further improved detective quantum efficiency in the next generation of DDD cameras, better particle motion correction algorithms (made possible by collection of “movies” instead of images), and stable specimen support grids made of gold rather than carbon (124, 125) can all aid the image analysis process, which would improve the potential to resolve multiple conformational states at high resolution.
V-ATPases have many interacting partners in cells (43, 45), a testament to their importance in cell physiology. Cryo-EM provides an ideal approach for the structural study of the V-ATPase “interactome.” The introduction of methods to allow cryo-EM maps to be calculated from rare populations of complexes from heterogeneous data sets could enable study of transient interactors of the V-ATPase. As mentioned earlier, the V-ATPase undergoes reversible disassembly, where the free V1 and VO regions adopt inhibited conformations preventing ATP hydrolysis by V1 and proton leakage by VO, respectively. How proton translocation is inhibited in the dissociated VO region is still unknown. A recent cryo-EM map of affinity-purified free VO subcomplex at 18 Å resolution (126) revealed movement of the N-terminal half of the a-subunit toward the expected position of the d-subunit, but higher-resolution information is needed to understand this inhibition process. Cryo-EM and analysis of evolutionary covariance have demonstrated an additional membrane-embedded subunit proximal to the C-terminal portion of the a-subunit (71). An atomic resolution map, in combination with biochemical and biophysical experiments, would aid in identification of this additional subunit. It was previously shown that the e-subunit can be removed from the detergent-purified V-ATPase by addition of a C-terminal tag without affecting in vitro ATP hydrolysis or proton pumping activity, which is consistent with it and the additional subunit being far from the interface of the a-subunit and c-ring (127). Precise mapping of the lumenal loop boundaries of the human VO region could allow generation of monoclonal antibodies. These reagents may demonstrate therapeutic potential for osteoporosis and could give valuable insight into the role of cell surface V-ATPases in cancer metastasis. Finally, identification of antibiotic-binding sites in the VO region could lead to the development of more potent and specific small-molecule inhibitors for use as chemical probes or even as therapeutics. Cryo-EM has markedly increased our insight into the structure, dynamics, and function of rotary ATPases across family branches and species. However, atomic resolution insight is necessary to understand the structure, function, and regulation of these remarkable macromolecular machines.
Acknowledgments
Funding: M.T.M.-J. was supported by a Postdoctoral Fellowship from the Canadian Institutes of Health Research (CIHR) and J.L.R. holds a Canada Research Chair. This work was supported by operating grant MOP81294 from the CIHR (to J.L.R.). Author contributions: M.T.M.-J. and J.L.R. jointly wrote and revised the article. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
REFERENCES AND NOTES
- 1.Mittermaier A., Kay L. E., New tools provide new insights in NMR studies of protein dynamics. Science 312, 224–228 (2006). [DOI] [PubMed] [Google Scholar]
- 2.Shima F., Yoshikawa Y., Matsumoto S., Kataoka T., Discovery of small-molecule Ras inhibitors that display antitumor activity by interfering with Ras·GTP-effector interaction. Enzymes 34 (Pt. B), 1–23 (2013). [DOI] [PubMed] [Google Scholar]
- 3.Shima F., Yoshikawa Y., Ye M., Araki M., Matsumoto S., Liao J., Hu L., Sugimoto T., Ijiri Y., Takeda A., Nishiyama Y., Sato C., Muraoka S., Tamura A., Osoda T., Tsuda K.-i., Miyakawa T., Fukunishie H., Shimada J., Kumasaka T., Yamamoto M., Kataoka T., In silico discovery of small-molecule Ras inhibitors that display antitumor activity by blocking the Ras-effector interaction. Proc. Natl. Acad. Sci. U.S.A. 110, 8182–8187 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Knapek E., Dubochet J., Beam damage to organic material is considerably reduced in cryo-electron microscopy. J. Mol. Biol. 141, 147–161 (1980). [DOI] [PubMed] [Google Scholar]
- 5.Tanaka N., Present status and future prospects of spherical aberration corrected TEM/STEM for study of nanomaterials. Sci. Technol. Adv. Mater. 9, 014111 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baker L. A., Rubinstein J. L., Radiation damage in electron cryomicroscopy. Methods Enzymol. 481, 371–388 (2010). [DOI] [PubMed] [Google Scholar]
- 7.Gonen T., Cheng Y., Sliz P., Hiroaki Y., Fujiyoshi Y., Harrison S. C., Walz T., Lipid–protein interactions in double-layered two-dimensional AQP0 crystals. Nature 438, 633–638 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bartesaghi A., Merk A., Banerjee S., Matthies D., Wu X., Milne J. L. S., Subramaniam S., 2.2 Å resolution cryo-EM structure of β-galactosidase in complex with a cell-permeant inhibitor. Science 348, 1147–1151 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Smith M. T. J., Rubinstein J. L., Beyond blob-ology. Science 345, 617–619 (2014). [DOI] [PubMed] [Google Scholar]
- 10.Kühlbrandt W., The resolution revolution. Science 343, 1443–1444 (2014). [DOI] [PubMed] [Google Scholar]
- 11.Li X., Mooney P., Zheng S., Booth C. R., Braunfeld M. B., Gubbens S., Agard D. A., Chneg Y., Electron counting and beam-induced motion correction enable near-atomic-resolution single-particle cryo-EM. Nat. Methods 10, 584–590 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grant T., Grigorieff N., Measuring the optimal exposure for single particle cryo-EM using a 2.6 Å reconstruction of rotavirus VP6. Elife 4, e06980 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Scheres S. H., Beam-induced motion correction for sub-megadalton cryo-EM particles. Elife 3, e03665 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rubinstein J. L., Brubaker M. A., Alignment of cryo-EM movies of individual particles by optimization of image translations. J. Struct. Biol. 192, 188–195 (2015). [DOI] [PubMed] [Google Scholar]
- 15.Scheres S. H. W., Gao H., Valle M., Herman G. T., Eggermont P. P. B., Frank J., Carazo J.-M., Disentangling conformational states of macromolecules in 3D-EM through likelihood optimization. Nat. Methods 4, 27–29 (2007). [DOI] [PubMed] [Google Scholar]
- 16.Liao M., Cao E., Julius D., Cheng Y., Structure of the TRPV1 ion channel determined by electron cryo-microscopy. Nature 504, 107–112 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Paulsen C. E., Armache J. P., Gao Y., Cheng Y., Julius D., Structure of the TRPA1 ion channel suggests regulatory mechanisms. Nature 520, 511–517 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Efremov R. G., Leitner A., Aebersold R., Raunser S., Architecture and conformational switch mechanism of the ryanodine receptor. Nature 517, 39–43 (2014). [DOI] [PubMed] [Google Scholar]
- 19.Yan Z., Bai X.-c., Yan C., Wu J., Li Z., Xie T., Peng W., Yin C.-c., Li X., Scheres S. H. W., Shi Y., Yan N., Structure of the rabbit ryanodine receptor RyR1 at near-atomic resolution. Nature 517, 50–55 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zalk R., Clarke O. B., des Georges A., Grassucci R. A., Reiken S., Mancia F., Hendrickson W. A., Frank J., Marks A. R., Structure of a mammalian ryanodine receptor. Nature 517, 44–49 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhao M., Wu S., Zhou Q., Vivona S., Cipriano D. J., Cheng Y., Brunger A. T., Mechanistic insights into the recycling machine of the SNARE complex. Nature 518, 61–67 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Campbell M. G., Veesler D., Cheng A., Potter C. S., Carragher B., 2.8 Å resolution reconstruction of the Thermoplasma acidophilum 20S proteasome using cryo-electron microscopy. Elife 4, e06380 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Amunts A., Brown A., Bai X.-c., Llácer J. L., Hussain T., Emsley P., Long F., Murshudov G., Scheres S. H. W., Ramakrishnan V., Structure of the yeast mitochondrial large ribosomal subunit. Science 343, 1485–1489 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhao J., Benlekbir S., Rubinstein J. L., Electron cryomicroscopy observation of rotational states in a eukaryotic V-ATPase. Nature 521, 241–245 (2015). [DOI] [PubMed] [Google Scholar]
- 25.Mulkidjanian A. Y., Galperin M. Y., Makarova K. S., Wolf Y. I., Koonin E. V., Evolutionary primacy of sodium bioenergetics. Biol. Direct. 3, 13 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Müller V., Grüber G., ATP synthases: Structure, function and evolution of unique energy converters. Cell. Mol. Life Sci. 60, 474–494 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hochstein L. I., Stan-Lotter H., Purification and properties of an ATPase from Sulfolobus solfataricus. Arch. Biochem. Biophys. 295, 153–160 (1992). [DOI] [PubMed] [Google Scholar]
- 28.Inatomi K., Characterization and purification of the membrane-bound ATPase of the archaebacterium Methanosarcina barkeri. J. Bacteriol. 167, 837–841 (1986). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pisa K. Y., Weidner C., Maischak H., Kavermann H., Müller V., The coupling ion in the methanoarchaeal ATP synthases: H+ vs. Na+ in the A1Ao ATP synthase from the archaeon Methanosarcina mazei Gö1. FEMS Microbiol. Lett. 277, 56–63 (2007). [DOI] [PubMed] [Google Scholar]
- 30.Coskun Ü., Chaban Y. L., Lingl A., Müller V., Keegstra W., Boekema E. J., Grüber G., Structure and subunit arrangement of the A-type ATP synthase complex from the archaeon Methanococcus jannaschii visualized by electron microscopy. J. Biol. Chem. 279, 38644–38648 (2004). [DOI] [PubMed] [Google Scholar]
- 31.Lingl A., Huber H., Stetter K. O., Mayer F., Kellermann J., Müller V., Isolation of a complete A1AO ATP synthase comprising nine subunits from the hyperthermophile Methanococcus jannaschii. Extremophiles 7, 249–257 (2003). [DOI] [PubMed] [Google Scholar]
- 32.Pisa K. Y., Huber H., Thomm M., Müller V., A sodium ion-dependent A1AO ATP synthase from the hyperthermophilic archaeon Pyrococcus furiosus. FEBS J. 274, 3928–3938 (2007). [DOI] [PubMed] [Google Scholar]
- 33.Muench S. P., Trinick J., Harrison M. A., Structural divergence of the rotary ATPases. Q. Rev. Biophys. 44, 311–356 (2011). [DOI] [PubMed] [Google Scholar]
- 34.Yokoyama K., Akabane Y., Ishii N., Yoshida M., Isolation of prokaryotic V0V1-ATPase from a thermophilic eubacterium Thermus thermophilus. J. Biol. Chem. 269, 12248–12253 (1994). [PubMed] [Google Scholar]
- 35.Nakano M., Imamura H., Toei M., Tamakoshi M., Yoshida M., Yokoyama K., ATP hydrolysis and synthesis of a rotary motor V-ATPase from Thermus thermophilus. J. Biol. Chem. 283, 20789–20796 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Speelmans G., Poolman B., Abee T., Konings W. N., Energy transduction in the thermophilic anaerobic bacterium Clostridium fervidus is exclusively coupled to sodium ions. Proc. Natl. Acad. Sci. U.S.A. 90, 7975–7979 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kakinuma Y., Yamato I., Murata T., Structure and function of vacuolar Na+-translocating ATPase in Enterococcus hirae. J. Bioenerg. Biomembr. 31, 7–14 (1999). [DOI] [PubMed] [Google Scholar]
- 38.Boekema E. J., van Breemen J. F. L., Brisson A., Ubbink-Kok T., Konings W. N., Lolkema J. S., Biological motors: Connecting stalks in V-type ATPase. Nature 401, 37–38 (1999). [DOI] [PubMed] [Google Scholar]
- 39.Biegel E., Müller V., Bacterial Na+-translocating ferredoxin:NAD+ oxidoreductase. Proc. Natl. Acad. Sci. U.S.A. 107, 18138–18142 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dimroth P., von Ballmoos C., Meier T., Catalytic and mechanical cycles in F-ATP synthases. Fourth in the Cycles Review Series. EMBO Rep. 7, 276–282 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.von Ballmoos C., Cook G. M., Dimroth P., Unique rotary ATP synthase and its biological diversity. Annu. Rev. Biophys. 37, 43–64 (2008). [DOI] [PubMed] [Google Scholar]
- 42.Zhou Q., Petersen C. C. H., Nicoll R. A., Effects of reduced vesicular filling on synaptic transmission in rat hippocampal neurones. J. Physiol. 525 (Pt. 1), 195–206 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hurtado-Lorenzo A., Skinner M., El Annan J., Futai M., Sun-Wada G.-H., Bourgoin S., Casanova J., Wildeman A., Bechoua S., Ausiello D. A., Brown D., Marshansky V., V-ATPase interacts with ARNO and Arf6 in early endosomes and regulates the protein degradative pathway. Nat. Cell Biol. 8, 124–136 (2006). [DOI] [PubMed] [Google Scholar]
- 44.Corbacho I., Teixidó F., Olivero I., Hernández L. M., Dependence of Saccharomyces cerevisiae Golgi functions on V-ATPase activity. FEMS Yeast Res. 12, 341–350 (2012). [DOI] [PubMed] [Google Scholar]
- 45.Zoncu R., Bar-Peled L., Efeyan A., Wang S., Sancak Y., Sabatini D. M., mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H+-ATPase. Science 334, 678–683 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Blair H. C., Teitelbaum S. L., Ghiselli R., Gluck S., Osteoclastic bone resorption by a polarized vacuolar proton pump. Science 245, 855–857 (1989). [DOI] [PubMed] [Google Scholar]
- 47.Pietrement C., Sun-Wada G.-H., Da Silva N., McKee M., Marshansky V., Brown D., Futai M., Distinct expression patterns of different subunit isoforms of the V-ATPase in the rat epididymis. Biol. Reprod. 74, 185–194 (2006). [DOI] [PubMed] [Google Scholar]
- 48.Breton S., Brown D., Regulation of luminal acidification by the V-ATPase. Physiology 28, 318–329 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Maxson M. E., Grinstein S., The vacuolar-type H+-ATPase at a glance – more than a proton pump. J. Cell Sci. 127, 4987–4993 (2014). [DOI] [PubMed] [Google Scholar]
- 50.Cotter K., Stransky L., McGuire C., Forgac M., Recent insights into the structure, regulation, and function of the V-ATPases. Trends Biochem. Sci. 40, 611–622 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sumner J.-P., Dow J. A. T., Earley F. G. P., Klein U., Jäger D., Wieczorek H., Regulation of plasma membrane V-ATPase activity by dissociation of peripheral subunits. J. Biol. Chem. 270, 5649–5653 (1995). [DOI] [PubMed] [Google Scholar]
- 52.Kane P. M., Disassembly and reassembly of the yeast vacuolar H+-ATPase in vivo. J. Biol. Chem. 270, 17025–17032 (1995). [PubMed] [Google Scholar]
- 53.Lu M., Holliday L. S., Zhang L., Dunn W. A. Jr, Gluck S. L., Interaction between aldolase and vacuolar H+-ATPase: Evidence for direct coupling of glycolysis to the ATP-hydrolyzing proton pump. J. Biol. Chem. 276, 30407–30413 (2001). [DOI] [PubMed] [Google Scholar]
- 54.Su Y., Zhou A., Al-Lamki R. S., Karet F. E., The a-subunit of the V-type H+-ATPase interacts with phosphofructokinase-1 in humans. J. Biol. Chem. 278, 20013–20018 (2003). [DOI] [PubMed] [Google Scholar]
- 55.Frattini A., Orchard P. J., Sobacchi C., Giliani S., Abinun M., Mattsson J. P., Keeling D. J., Andersson A.-K., Wallbrandt P., Zecca L., Notarangelo L. D., Vezzoni P., Villa A., Defects in TCIRG1 subunit of the vacuolar proton pump are responsible for a subset of human autosomal recessive osteopetrosis. Nat. Genet. 25, 343–346 (2000). [DOI] [PubMed] [Google Scholar]
- 56.Li Y.-P., Chen W., Liang Y., Li E., Stashenko P., Atp6i-deficient mice exhibit severe osteopetrosis due to loss of osteoclast-mediated extracellular acidification. Nat. Genet. 23, 447–451 (1999). [DOI] [PubMed] [Google Scholar]
- 57.Väänänen H. K., Zhao H., Mulari M., Halleen J. M., The cell biology of osteoclast function. J. Cell Sci. 113 (Pt. 3), 377–381 (2000). [DOI] [PubMed] [Google Scholar]
- 58.Zhang J., Fuster D. G., Cameron M. A., Quiñones H., Griffith C., Xie X.-S., Moe O. W., Incomplete distal renal tubular acidosis from a heterozygous mutation of the V-ATPase B1 subunit. Am. J. Physiol. Renal Physiol. 307, F1063–F1071 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hinton A., Sennoune S. R., Bond S., Fang M., Reuveni M., Sahagian G. G., Jay D., MArtinez-Zaguilan R., Forgac M., Function of a subunit isoforms of the V-ATPase in pH homeostasis and in vitro invasion of MDA-MB231 human breast cancer cells. J. Biol. Chem. 284, 16400–16408 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sennoune S. R., Bakunts K., Martínez G. M., Chua-Tuan J. L., Kemir Y., Attaya M. N., Martínez-Zaguilán R., Vacuolar H+-ATPase in human breast cancer cells with distinct metastatic potential: Distribution and functional activity. Am. J. Physiol. Cell Physiol. 286, C1443–C1452 (2004). [DOI] [PubMed] [Google Scholar]
- 61.Capecci J., Forgac M., The function of vacuolar ATPase (V-ATPase) a subunit isoforms in invasiveness of MCF10a and MCF10CA1a human breast cancer cells. J. Biol. Chem. 288, 32731–32741 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sennoune S. R., Martinez-Zaguilan R., Plasmalemmal vacuolar H+-ATPases in angiogenesis, diabetes and cancer. J. Bioenerg. Biomembr. 39, 427–433 (2007). [DOI] [PubMed] [Google Scholar]
- 63.Rubinstein J. L., Walker J. E., Henderson R., Structure of the mitochondrial ATP synthase by electron cryomicroscopy. EMBO J. 22, 6182–6192 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Muench S. P., Huss M., Song C. F., Phillips C., Wieczorek H., Trinick J., Harrison M. A., Cryo-electron microscopy of the vacuolar ATPase motor reveals its mechanical and regulatory complexity. J. Mol. Biol. 386, 989–999 (2009). [DOI] [PubMed] [Google Scholar]
- 65.Lau W. C. Y., Rubinstein J. L., Structure of intact Thermus thermophilus V-ATPase by cryo-EM reveals organization of the membrane-bound VO motor. Proc. Natl. Acad. Sci. U.S.A. 107, 1367–1372 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Baker L. A., Watt I. N., Runswick M. J., Walker J. E., Rubinstein J. L., Arrangement of subunits in intact mammalian mitochondrial ATP synthase determined by cryo-EM. Proc. Natl. Acad. Sci. U.S.A. 109, 11675–11680 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Benlekbir S., Bueler S. A., Rubinstein J. L., Structure of the vacuolar-type ATPase from Saccharomyces cerevisiae at 11-Å resolution. Nat. Struct. Mol. Biol. 19, 1356–1362 (2012). [DOI] [PubMed] [Google Scholar]
- 68.Lau W. C. Y., Rubinstein J. L., Subnanometre-resolution structure of the intact Thermus thermophilus H+-driven ATP synthase. Nature 481, 214–218 (2012). [DOI] [PubMed] [Google Scholar]
- 69.Allegretti M., Klusch N., Mills D. J., Vonck J., Kühlbrandt W., Davies K. M., Horizontal membrane-intrinsic α-helices in the stator a-subunit of an F-type ATP synthase. Nature 521, 237–240 (2015). [DOI] [PubMed] [Google Scholar]
- 70.Zhou A., Rohou A., Schep D. G., Montgomery M. G., Walker J. E., Grigorieff N., Rubinstein J. L., Structure and conformational states of the bovine mitochondrial ATP synthase by cryo-EM. Elife 4, e10180 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schep D. G., Zhao J., Rubinstein J. L., Models for the a subunits of the Thermus thermophilus V/A-ATPase and Saccharomyces cerevisiae V-ATPase enzymes by cryo-EM and evolutionary covariance. Proc. Natl. Acad. Sci. U.S.A. 113, 3245–3250 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rawson S., Phillips C., Huss M., Tiburcy F., Wiezoreck H., Trinck J., Harrison M. A., Muench S. P., Structure of the vacuolar H+-ATPase rotary motor reveals new mechanistic insights. Structure 23, 461–471 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chan T. L., Greenawalt J. W., Pedersen P. L., Biochemical and ultrastructural properties of a mitochondrial inner membrane fraction deficient in outer membrane and matrix activities. J. Cell Biol. 45, 291–305 (1970). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bernal R. A., Stock D., Three-dimensional structure of the intact Thermus thermophilus H+-ATPase/synthase by electron microscopy. Structure 12, 1789–1798 (2004). [DOI] [PubMed] [Google Scholar]
- 75.Vonck J., Pisa K. Y., Morgner N., Brutschy B., Müller V., Three-dimensional structure of A1A0 ATP synthase from the hyperthermophilic archaeon Pyrococcus furiosus by electron microscopy. J. Biol. Chem. 284, 10110–10119 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Domgall I., Venzke D., Lüttge U., Ratajczak R., Böttcher B., Three-dimensional map of a plant V-ATPase based on electron microscopy. J. Biol. Chem. 277, 13115–13121 (2002). [DOI] [PubMed] [Google Scholar]
- 77.Diepholz M., Börsch M., Böttcher B., Structural organization of the V-ATPase and its implications for regulatory assembly and disassembly. Biochem. Soc. Trans. 36, 1027–1031 (2008). [DOI] [PubMed] [Google Scholar]
- 78.Diepholz M., Venzke D., Prinz S., Batisse C., Flörchinger B., Rössele M., Svergun D. I., Böttcher B., Féthière J., A different conformation for EGC stator subcomplex in solution and in the assembled yeast V-ATPase: Possible implications for regulatory disassembly. Structure 16, 1789–1798 (2008). [DOI] [PubMed] [Google Scholar]
- 79.Zhang Z., Zheng Y., Mazon H., Milgrom E., Kitagawa N., Kish-Trier E., Heck A. J. R., Kane P. M., Wilkens S., Structure of the yeast vacuolar ATPase. J. Biol. Chem. 283, 35983–35995 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lau W. C. Y., Baker L. A., Rubinstein J. L., Cryo-EM structure of the yeast ATP synthase. J. Mol. Biol. 382, 1256–1264 (2008). [DOI] [PubMed] [Google Scholar]
- 81.Abrahams J. P., Leslie A. G. W., Lutter R., Walker J. E., Structure at 2.8 Â resolution of F1-ATPase from bovine heart mitochondria. Nature 370, 621–628 (1994). [DOI] [PubMed] [Google Scholar]
- 82.Morales-Rios E., Montgomery M. G., Leslie A. G. W., Walker J. E., Structure of ATP synthase from Paracoccus denitrificans determined by X-ray crystallography at 4.0 Å resolution. Proc. Natl. Acad. Sci. U.S.A. 112, 13231–13236 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhang Z., Charsky C., Kane P. M., Wilkens S., Yeast V1-ATPase: Affinity purification and structural features by electron microscopy. J. Biol. Chem. 278, 47299–47306 (2003). [DOI] [PubMed] [Google Scholar]
- 84.Kitagawa N., Mazon H., Heck A. J. R., Wilkens S., Stoichiometry of the peripheral stalk subunits E and G of yeast V1-ATPase determined by mass spectrometry. J. Biol. Chem. 283, 3329–3337 (2008). [DOI] [PubMed] [Google Scholar]
- 85.Lee L. K., Stewart A. G., Donohoe M., Bernal R. A., Stock D., The structure of the peripheral stalk of Thermus thermophilus H+-ATPase/synthase. Nat. Struct. Mol. Biol. 17, 373–378 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zhao J., Brubaker M. A., Benlekbir S., Rubinstein J. L., Description and comparison of algorithms for correcting anisotropic magnification in cryo-EM images. J. Struct. Biol. 192, 209–215 (2015). [DOI] [PubMed] [Google Scholar]
- 87.Hirata R., Graham L. A., Takatsuki A., Stevens T. H., Anraku Y., VMA11 and VMA16 encode second and third proteolipid subunits of the Saccharomyces cerevisiae vacuolar membrane H+-ATPase. J. Biol. Chem. 272, 4795–4803 (1997). [DOI] [PubMed] [Google Scholar]
- 88.Drory O., Frolow F., Nelson N., Crystal structure of yeast V-ATPase subunit C reveals its stator function. EMBO Rep. 5, 1148–1152 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sagermann M., Stevens T. H., Matthews B. W., Crystal structure of the regulatory subunit H of the V-type ATPase of Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U.S.A. 98, 7134–7139 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Basak S., Lim J., Manimekalai M. S. S., Balakrishna A. M., Grüber G., Crystal and NMR structures give insights into the role and dynamics of subunit F of the eukaryotic V-ATPase from Saccharomyces cerevisiae. J. Biol. Chem. 288, 11930–11939 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Oot R. A., Huang L.-S., Berry E. A., Wilkens S., Crystal structure of the yeast vacuolar ATPase heterotrimeric EGChead peripheral stalk complex. Structure 20, 1881–1892 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Numoto N., Hasegawa Y., Takeda K., Miki K., Inter-subunit interaction and quaternary rearrangement defined by the central stalk of prokaryotic V1-ATPase. EMBO Rep. 10, 1228–1234 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Arai S., Saijo S., Suzuki K., Mizutani K., Kakinuma Y., Ishizuka-Katsura Y., Ohsawa N., Terada T., Shirouzu M., Yokoyama S., Iwata S., Yamato I., Murata T., Rotation mechanism of Enterococcus hirae V1-ATPase based on asymmetric crystal structures. Nature 493, 703–707 (2013). [DOI] [PubMed] [Google Scholar]
- 94.Iwata M., Imamura H., Stambouli E., Ikeda C., Tamakoshi M., Nagata K., Makyio H., Hankamer B., Barber J., Yoshida M., Yokoyama K., Iwata S., Crystal structure of a central stalk subunit C and reversible association/dissociation of vacuole-type ATPase. Proc. Natl. Acad. Sci. U.S.A. 101, 59–64 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Srinivasan S., Vyas N. K., Baker M. L., Quiocho F. A., Crystal structure of the cytoplasmic N-terminal domain of subunit I, a homolog of subunit a, of V-ATPase. J. Mol. Biol. 412, 14–21 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Murata T., Yamato I., Kakinuma Y., Structure and mechanism of vacuolar Na+-translocating ATPase from Enterococcus hirae. J. Bioenerg. Biomembr. 37, 411–413 (2005). [DOI] [PubMed] [Google Scholar]
- 97.Peskin C. S., Odell G. M., Oster G. F., Cellular motions and thermal fluctuations: The Brownian ratchet. Biophys. J. 65, 316–324 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ait-Haddou R., Herzog W., Brownian ratchet models of molecular motors. Cell Biochem. Biophys. 38, 191–214 (2003). [DOI] [PubMed] [Google Scholar]
- 99.Vik S., Antionio B. J., A mechanism of proton translocation by F1F0 ATP synthases suggested by double mutants of the a subunit. J. Biol. Chem. 269, 30364–30369 (1994). [PubMed] [Google Scholar]
- 100.Junge W., Lill H., Engelbrecht S., ATP synthase: An electrochemical transducer with rotatory mechanics. Trends Biochem. Sci. 22, 420–423 (1997). [DOI] [PubMed] [Google Scholar]
- 101.MacCallum J. L., Bennett W. F. D., Tieleman D. P., Distribution of amino acids in a lipid bilayer from computer simulations. Biophys. J. 94, 3393–3404 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Cain B. D., Simoni R. D., Proton translocation by the F1F0ATPase of Escherichia coli. Mutagenic analysis of the a subunit. J. Biol. Chem. 264, 3292–3300 (1989). [PubMed] [Google Scholar]
- 103.Kawasaki-Nishi S., Nishi T., Forgac M., Arg-735 of the 100-kDa subunit a of the yeast V-ATPase is essential for proton translocation. Proc. Natl. Acad. Sci. U.S.A. 98, 12397–12402 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Angevine C. M., Fillingame R. H., Aqueous access channels in subunit a of rotary ATP synthase. J. Biol. Chem. 278, 6066–6074 (2003). [DOI] [PubMed] [Google Scholar]
- 105.Steed P. R., Fillingame R. H., Aqueous accessibility to the transmembrane regions of subunit c of the Escherichia coli F1F0 ATP synthase. J. Biol. Chem. 284, 23243–23250 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Angevine C. M., Herold K. A. G., Fillingame R. H., Aqueous access pathways in subunit a of rotary ATP synthase extend to both sides of the membrane. Proc. Natl. Acad. Sci. U.S.A. 100, 13179–13183 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.D. G. Nicholls, S. J. Ferguson, Bioenergetics (Academic Press, London, 2002). [Google Scholar]
- 108.Toei M., Toei S., Forgac M., Definition of membrane topology and identification of residues important for transport in subunit a of the vacuolar ATPase. J. Biol. Chem. 286, 35176–35186 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Leng X.-H., Manolson M. F., Liu Q., Forgac M., Site-directed mutagenesis of the 100-kDa subunit (Vph1p) of the yeast vacuolar (H+)-ATPase. J. Biol. Chem. 271, 22487–22493 (1996). [DOI] [PubMed] [Google Scholar]
- 110.Leng X.-H., Manolson M., Forgac M., Function of the COOH-terminal domain of Vph1p in activity and assembly of the yeast V-ATPase. J. Biol. Chem. 273, 6717–6723 (1998). [DOI] [PubMed] [Google Scholar]
- 111.Wang Y., Toei M., Forgac M., Analysis of the membrane topology of transmembrane segments in the C-terminal hydrophobic domain of the yeast vacuolar ATPase subunit a (Vph1p) by chemical modification. J. Biol. Chem. 283, 20696–20702 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Leng X. H., Nishi T., Forgac M., Transmembrane topography of the 100-kDa a subunit (Vph1p) of the yeast vacuolar proton-translocating ATPase. J. Biol. Chem. 274, 14655–14661 (1999). [DOI] [PubMed] [Google Scholar]
- 113.Stewart A. G., Lee L. K., Donohoe M., Chaston J. J., Stock D., The dynamic stator stalk of rotary ATPases. Nat. Commun. 3, 687 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Zhou M., Politis A., Davies R. B., Liko I., Wu K.-J., Stewart A. G., Stock D., Robinson C. V., Ion mobility–mass spectrometry of a rotary ATPase reveals ATP-induced reduction in conformational flexibility. Nat. Chem. 6, 208–215 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Gregorini M., Wang J., Xie X.-S., Milligan R. A., Engel A., Three-dimensional reconstruction of bovine brain V-ATPase by cryo-electron microscopy and single particle analysis. J. Struct. Biol. 158, 445–454 (2007). [DOI] [PubMed] [Google Scholar]
- 116.Song C. F., Papachristos K., Rawson S., Huss M., Wieczorek H., Paci E., Trinick J., Harrison M. A., Muench S. P., Flexibility within the rotor and stators of the vacuolar H+-ATPase. PLOS One 8, e82207 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Walker J. E., The ATP synthase: The understood, the uncertain and the unknown. Biochem. Soc. Trans. 41, 1–16 (2013). [DOI] [PubMed] [Google Scholar]
- 118.Stark H., GraFix: Stabilization of fragile macromolecular complexes for single particle Cryo-EM. Methods Enzymol. 481, 109–126 (2010). [DOI] [PubMed] [Google Scholar]
- 119.Schuler M. A., Denisov I. G., Sligar S. G., Nanodiscs as a new tool to examine lipid–protein interactions. Methods Mol. Biol. 974, 415–433 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Frauenfeld J., Löving R., Armache J.-P., Sonnen A. F.-P., Guettou F., Moberg P., Zhu L., Jegerschöld C., Flayhan A., Briggs J. A. G., Garoff H., Löw C., Cheng Y., Nordlund P., A saposin-lipoprotein nanoparticle system for membrane proteins. Nat. Methods 13, 345–351 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kleinschmidt J. H., Popot J.-L., Folding and stability of integral membrane proteins in amphipols. Arch. Biochem. Biophys. 564, 327–343 (2014). [DOI] [PubMed] [Google Scholar]
- 122.Wu S., Avila-Sakar A., Kim J., Booth D. S., Greenberg C. H., Rossi A., Liao M., Li X., Alian A., Griner S. L., Juge N., Yu Y., Mergel C. M., Chaparro-Riggers J., Strop P., Tampé R., Edwards R. H., Stroud R. M., Craik C. S., Cheng Y., Fabs enable single particle cryoEM studies of small proteins. Structure 20, 582–592 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Danev R., Buijsse B., Khoshouei M., Plitzko J. M., Baumeister W., Volta potential phase plate for in-focus phase contrast transmission electron microscopy. Proc. Natl. Acad. Sci. U.S.A. 111, 15635–15640 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Russo C. J., Passmore L. A., Ultrastable gold substrates for electron cryomicroscopy. Science 346, 1377–1380 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Bharat T. A. M., Russo C. J., Löwe J., Passmore L. A., Scheres S. H. W., Advances in single-particle electron cryomicroscopy structure determination applied to sub-tomogram averaging. Structure 23, 1743–1753 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Couoh-Cardel S., Milgrom E., Wilkens S., Affinity purification and structural features of the yeast vacuolar ATPase Vo membrane sector. J. Biol. Chem. 290, 27959–27971 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Bueler S. A., Rubinstein J. L., Vma9p need not be associated with the yeast V-ATPase for fully-coupled proton pumping activity in vitro. Biochemistry 54, 853–858 (2015). [DOI] [PubMed] [Google Scholar]