

Ion channel subunits typically assemble in the endoplasmic reticulum. α-SNAP orchestrates a unique assembly and calcium selectivity of Orai1 subunits into functional multimers. Dynamic assembly of Orai1 and its dependence on α-SNAP could enable localization of calcium signals and regulation of rate and amount of calcium influx.
Abstract
Orai1 forms a highly calcium-selective pore of the calcium release activated channel, and α-SNAP is necessary for its function. Here we show that α-SNAP regulates on-site assembly of Orai1 dimers into calcium-selective multimers. We find that Orai1 is a dimer in resting primary mouse embryonic fibroblasts but displays variable stoichiometry in the plasma membrane of store-depleted cells. Remarkably, α-SNAP depletion induces formation of higher-order Orai1 oligomers, which permeate significant levels of sodium via Orai1 channels. Sodium permeation in α-SNAP–deficient cells cannot be corrected by tethering multiple Stim1 domains to Orai1 C-terminal tail, demonstrating that α-SNAP regulates functional assembly and calcium selectivity of Orai1 multimers independently of Stim1 levels. Fluorescence nanoscopy reveals sustained coassociation of α-SNAP with Stim1 and Orai1, and α-SNAP–depleted cells show faster and less constrained mobility of Orai1 within ER-PM junctions, suggesting Orai1 and Stim1 coentrapment without stable contacts. Furthermore, α-SNAP depletion significantly reduces fluorescence resonance energy transfer between Stim1 and Orai1 N-terminus but not C-terminus. Taken together, these data reveal a unique role of α-SNAP in the on-site functional assembly of Orai1 subunits and suggest that this process may, in part, involve enabling crucial low-affinity interactions between Orai1 N-terminus and Stim1.
INTRODUCTION
Store-operated calcium entry (SOCE) via calcium release–activated channels (CRACs) is induced in response to signaling via a variety of cell surface receptors and is crucial for many different cellular processes. However, the molecular underpinnings of the series of elaborately choreographed steps resulting in SOCE are far from clear. It is well established that receptor cross-linking–induced calcium store depletion is sensed by an endoplasmic reticulum (ER)–localized calcium sensor, Stim1, which undergoes structural changes and relocates to ER–plasma membrane (PM) junctional regions, where it interacts with Orai1 (or CRACM1). A number of proteins have been reported to play a role in facilitating relocation of Stim1 to ER-PM junctions, but the molecular steps that translate coentrapment of Stim1 and Orai1 into activation of SOCE remain unknown. Previously we reported that a cytosolic protein, α–soluble N-ethylmaleimide–sensitive factor (NSF) attachment protein (α-SNAP), directly binds Orai1 and Stim1 and is essential for SOCE activation (Miao et al., 2013). Regulation of SOCE by α-SNAP is independent of NSF binding and its role in membrane trafficking (Miao et al., 2013). Of importance, α-SNAP is not necessary for clustering of Orai1 with Stim1 in ER-PM junctions (Miao et al., 2013). Therefore understanding the precise mechanism of action of α-SNAP would reveal detailed molecular steps beyond Orai1-Stim1 entrapment that are necessary to activate SOCE.
The process of activation of CRAC is exceptionally slow and its permeation highly selective for calcium ions. Furthermore, unlike many ion channels, calcium selectivity and gating are not intrinsic properties of Orai1 but are believed to be imparted, at least in part, by an accessory protein, Stim1 (McNally et al., 2012). These studies suggest that Orai1 must acquire these attributes after its entrapment in ER-PM junctions by Stim1. Yet, others have suggested that Orai1 selectivity depends on its subunit count (Thompson and Shuttleworth, 2013), which is typically decided for ion channels during their assembly within the ER. It is therefore hard to reconcile these findings with our current understanding of activation of SOCE.
Similarly, it is well accepted that Orai1 multimers form the pore of CRACs (Vig et al., 2006a, b; Vig and Kinet, 2007; Mignen et al., 2008b; Penna et al., 2008; Hou et al., 2012); however, the stoichiometry of Orai1 homomers under resting as well as store-depleted conditions remains debated (Ji et al., 2008; Mignen et al., 2008b; Penna et al., 2008; Madl et al., 2010; Hou et al., 2012; Thompson and Shuttleworth, 2013). A number of different approaches have been used to address this question, but a unifying model has yet to emerge. Using coexpression of varying numbers of covalently linked Orai1 concatamers, two groups previously showed that tetrameric Orai1 concatamers reconstitute currents that closely resemble native CRAC currents (Ji et al., 2008; Mignen et al., 2008b). Another group used a combination of fluorescence resonance energy transfer (FRET), single-molecule tracking, and brightness analysis to determine the stoichiometry of resting Orai1 as a tetramer (Madl et al., 2010). Chemical cross-linking followed by gel electrophoresis has also been used to determine the number of Orai1 subunits, but the conclusions have varied (Penna et al., 2008; Hou et al., 2012). Previously, two studies used single-molecule photobleaching to determine the subunit composition of CRACs (Ji et al., 2008; Penna et al., 2008). Both reported pure tetrameric assemblies of Orai1 in store-depleted cells, and one study found dimeric assemblies of Orai1 in resting cells (Penna et al., 2008). More recently, x-ray crystallography of Drosophila Orai lacking the N-terminal tail found that pure hexameric assemblies of Orai form the pore of CRACs (Hou et al., 2012). A subsequent study used concatamers of full-length human Orai1 and reported that hexameric concatamers of full-length human Orai1 form nonselective channels that display significant permeability to sodium, in contrast to the highly selective permeation of native CRACs to calcium (Thompson and Shuttleworth, 2013). A similar disagreement over stoichiometry resolved by different approaches occurred over a Staphylococcus aureus mechanosensitive channel (Mscl; Dorwart et al., 2010). Several factors in the experimental design could underlie the difference in conclusions of these studies. Therefore Orai1 stoichiometry in resting as well as store-depleted cells remains unestablished. Stim1 stoichiometry was also previously studied by a number of groups using a variety of approaches but remains unsettled (Hoover and Lewis, 2011; Li et al., 2011; McNally et al., 2012; Stathopulos et al., 2013).
We previously showed that α-SNAP depletion reduces the ratio of Stim1:Orai1 in ER-PM junctions without affecting Orai1 and Stim1 localization and translocation upon store depletion (Miao et al., 2013). To further understand the mechanism of action of α-SNAP in SOCE and determine the precise stoichiometry of Orai1 and Stim1 in the presence or absence of α-SNAP, here we developed a sparse single-molecule expression system by expressing Orai1 and Stim1 fusions with a highly monomeric photoconvertible fluorescent protein, mEos3.2 (Zhang et al., 2012), in Orai1−/− or Stim1−/− mouse embryonic fibroblasts (MEFs), respectively. Using this system, we performed single-molecule photobleaching of Orai1 and Stim1 in α-SNAP–depleted cells. Collectively our data suggest that α-SNAP enables a unique and dynamic process of on-site oligomerization of Orai1 subunits in the PM after store depletion. Further, single-particle tracking measurements of Orai1 mobility and FRET measurements between the N-versus C-terminus of Orai1 and Stim1 revealed discrete novel steps in the molecular interaction between Orai1 and Stim1 facilitated by α-SNAP. Our data suggest that these steps may also, in part, contribute to the functional assembly and selectivity of Orai1 multimers. Finally, we discuss the potential physiological implications of on-site functional assembly of Orai1 subunits in the localization of calcium signals and regulation of signal strength.
RESULTS
To analyze Orai1 stoichiometry in the presence or absence of α-SNAP, we adopted the single-molecule photobleaching approach with several modifications. This approach typically involves expression of a fusion protein with green fluorescent protein (GFP) or another fluorescent protein (FP) tag followed by gradual photobleaching of optically well resolved protein complexes in the plasma membrane using total internal reflection fluorescence microscopy (TIRFM) (Ulbrich and Isacoff, 2007; Ji et al., 2008; Penna et al., 2008; Nakajo et al., 2010; Simonson et al., 2010; Durisic et al., 2012; Hastie et al., 2013).
We took advantage of the ability of mEos3.2 fluorescent protein (Zhang et al., 2012), referred to as mEos hereafter, to photoconvert from the green to the red form and performed single-subunit counting by spot photobleaching. We first used the 488-nm laser and green fluorescence of mEos-Orai1 to search and identify cells that exhibited a sparse, punctate distribution of Orai1 molecules in the PM using TIRFM. Subsequently we photoconverted all PM-mEos-Orai1 molecules to their red form using the 405-nm laser and initiated photobleaching of mEos red fluorescence using the 561-nm laser (Figure 1A). We generated MEFs from Orai1−/− mice and expressed mEos-Orai1 in these cells to ensure the absence of endogenous Orai1 and weak expression levels of transfected proteins. Therefore these cell lines were ideally suited for subunit counting of mEos-tagged Orai1 by photobleaching. The extended photobleaching time necessitated that cells be fixed with paraformaldehyde (PFA) to prevent lateral mobility of proteins during imaging. PFA fixation has been reported to cause store depletion and coclustering of Stim1 and Orai1 molecules (Demuro et al., 2011). Therefore we first analyzed resting cells transfected with mEos-Orai1 alone and fixed with PFA. We automated the identification of bleaching steps from images (Materials and Methods; Figure 1A) and constructed a histogram of the number of observed steps and their frequency of occurrence. mEos-Orai1 showed predominantly one or two bleaching steps (Figure 1B). A fraction of FPs is expected to be in dark state at any given time (Garcia-Parajo et al., 2001; Ulbrich and Isacoff, 2007; Durisic et al., 2014). Therefore we fitted these data to binomial distributions constructed using a combination of dimer and tetramer models in which the fractions of two species and the probability, p, of fluorescent state proteins was allowed to float (Materials and Methods). The best-fit models were obtained around p = 0.7, where resting cells expressing mEos-Orai1 were estimated to contain 90% Orai1 dimers (Figure 1B and Table 1). These data confirm a previous report that also observed resting-stage Orai1 as a dimer (Penna et al., 2008). A small fraction of three and four steps observed in resting mEos-Orai1–expressing cells could indicate tetramers resulting from PFA fixation–mediated partial store depletion as suggested previously (Demuro et al., 2011).
FIGURE 1:

Orai1 displays variable stoichiometry in resting and store-depleted cells. (A) Representative raw images of Orai1−/− MEF cell expressing mEos-Orai1. Top, sparse distribution of mEos-green and mEos-red fluorescence after photoconversion. Bottom, image mask after applying LoG filter and representative raw fluorescence trace from one spot (black line) and after Kalman averaging (red line). (B) Histogram of mEos-Orai1 bleaching steps obtained from resting Orai1−/− MEFs (black bars; 300 spots) and binomial fitting of experimental data to various models (colored bars) from Table 1. Representative examples of discrete fluorescence photobleaching steps (C) and histogram (D) obtained from store-depleted Orai1−/− MEFs expressing mEos-Orai1 and Stim1-myc (900 spots). Dark trace in C is the raw fluorescence signal overlaid with a red line from automatic step detection. (E) Binomial fitting of data (black bars) from D to various models (colored bars) from Table 2.
TABLE 1:
Binomial model fitting of Orai1 photobleaching in resting cells.
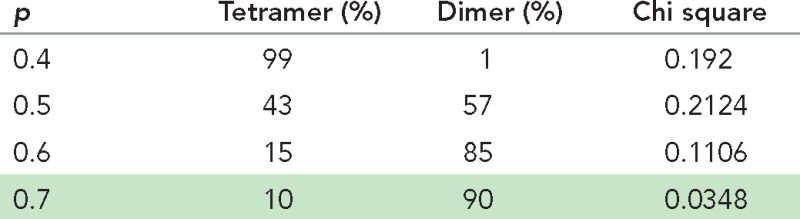 |
Binomial fitting of step photobleaching data from resting Orai1−/− MEFs expressing mEos-Orai1 alone (Figure 1B) to various models. Relative proportions of dimer/tetramer/hexamer species after binomial fitting at various p values and corresponding chi-square values indicate the goodness of fit of theoretical values to the observed data. The best fit is highlighted.
ER calcium store depletion induces oligomerization and relocation of Stim1 to ER-PM junctions, which is believed to trap freely diffusing Orai1 molecules in the PM. Therefore we next analyzed the stoichiometry of Orai1 molecules in store-depleted Orai1−/− MEFs coexpressing mEos-Orai1 and Stim1-myc. Photobleaching of these cells showed a range of steps, up to a maximum of six (Figure 1, C and D). Previous studies reported either pure tetrameric or hexameric assemblies of Orai1, depending on the experimental approach (Ji et al., 2008; Penna et al., 2008; Demuro et al., 2011; Hou et al., 2012; Thompson and Shuttleworth, 2013). We fitted our data with binomial distribution models derived from tetramer only, hexamer only, or a mixture of dimer/tetramer/hexamer models. The best fit was observed with the model comprising dimers, tetramers, and hexamers, suggesting variable stoichiometric states of Orai1 in the PM of store-depleted cells (Figure 1E and Table 2).
TABLE 2:
Binomial model fitting of Orai1 photobleaching in store-depleted cells.
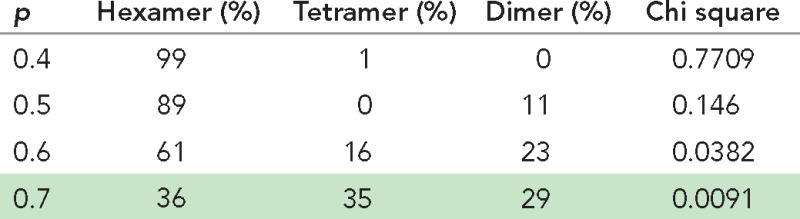 |
Binomial fitting of step photobleaching data from store-depleted cells coexpressing mEos-Orai1 and Stim1-myc (Figure 1D) to various models. Relative proportions of dimer/tetramer/hexamer species after binomial fitting at various p values and corresponding chi-square values indicate the goodness of fit of theoretical values to the observed data. The best fit is highlighted.
Next we sought to analyze Orai1 stoichiometry in α-SNAP–depleted cells. We treated Orai1−/− MEFs with α-SNAP or a scrambled (SCR)-sequence small interfering RNA (siRNA) conjugated with Cy5 fluorescent dye and confirmed depletion of α-SNAP protein using this siRNA by Western blot, as well as by measurement of SOCE (Figure 2, A and B).
FIGURE 2:

α-SNAP regulates functional assembly and calcium selectivity of Orai1 oligomers. (A) Representative Western blot showing depletion of α-SNAP in siRNA-treated cells. (B) Representative Fura-2 imaging plot showing defective SOCE in α-SNAP siRNA–treated cells. (C) Histogram of mEos-Orai1 bleaching steps obtained from siRNA-treated Orai1−/− MEFs expressing mEos-Orai1 and Stim1-myc (500–700 spots). (D) Binomial fitting of experimental data (red bars) from C to various models (colored bars) from Table 3. (E) Western blot showing chemically cross-linked Orai1 oligomers from SCR and α-SNAP siRNA–treated HEK 293 cells with stable and inducible expression of flag-Orai1 and Stim1-myc. (F) Representative SBFI imaging plot showing store-operated sodium influx in SCR or α-SNAP siRNA–treated WT or Orai1−/− MEF cells.
Because some recent studies suggested that α-SNAP inhibits AMP-activated protein kinase (AMPK) activity and mitochondrial biogenesis (Wang and Brautigan, 2013) and its inactivation could compromise cell viability (Naydenov et al., 2012a, b), we looked for these phenotypes in our cells. Annexin V and propidium iodide (PI) staining of siRNA-treated MEFs did not show any defects in viability at the time points used in our assays (Supplemental Figure S1). Cell cycle was also normal in α-SNAP–depleted cells (Supplemental Figure S2). Total as well as phospho-AMPK levels were similar in α-SNAP–depleted MEFs (Supplemental Figure S3), as well as in resting and receptor-stimulated, α-SNAP–hypomorphic hyh CD4 T-cells (Miao Bhojappa, Dani, Vig, et al., unpublished data). To analyze potential differences in mitochondrial mass, we stained α-SNAP–depleted cells with MitoTracker Green and found no difference in the gross morphology or total mitochondrial content (Supplemental Figure S4, A and B). The ultrastructural morphology and distribution of mitochondrial cristae was also intact in α-SNAP–depleted cells, as revealed by electron micrographs (Supplemental Figure S4C). Taken together, these data demonstrate that the health, viability, and mitochondria were unaffected in α-SNAP–depleted or –hypomorphic cells analyzed in our experiments.
To investigate the stoichiometry of Orai1 subunits, we expressed mEos-Orai1 along with Stim1-myc in siRNA-treated, store-depleted Orai1−/− MEF cells fixed and analyzed by photobleaching. We found a significant increase in the fraction of four, five, and six mEos-Orai1 photobleaching events from α-SNAP–depleted cells compared with cells treated with SCR siRNA (Figure 2C). Binomial fitting of these data indicated an increase in the fraction of Orai1 hexamers, with a concomitant decrease in the dimer and tetramer fractions (Figure 2D and Table 3). To confirm these observations, we used chemical cross-linking in HEK 293 cells stably transfected with Flag-Orai1 and Stim1-myc (Miao et al., 2013). Briefly, we treated the cells with SCR or α-SNAP siRNA, induced the expression of Orai1 and Stim1, store depleted, and lysed the cells. We collected the membrane fraction by ultracentrifugation and performed protein cross-linking using the amine reactive cross-linker, BS3, as described previously (Hou et al., 2012). SDS–PAGE followed by Western blotting identified cross-linked Flag-Orai1 protein bands corresponding to dimer, tetramer, and hexamer in control cells (Figure 2E). α-SNAP siRNA–treated cells showed a distinct increase in the proportion of higher-order oligomers of Orai1, particularly hexamers.
TABLE 3:
Binomial model fitting of Orai1 photobleaching in α-SNAP–depleted cells.
 |
Binomial fitting of mEos-Orai1 step photobleaching data from store-depleted cells coexpressing Stim1-myc and treated with α−SNAP siRNA (Figure 2C). Relative proportions of dimer/tetramer/hexamer species after binomial fitting at various p values and corresponding chi-square values indicate the goodness of fit of theoretical values to the observed data, with the best fit highlighted.
Previously hexameric concatamers of Orai1 have been shown to form a nonselective pore with significant sodium permeation (Thompson and Shuttleworth, 2013). To determine whether formation of higher-order Orai1 oligomers in α-SNAP–depleted cells is accompanied with changes in sodium permeation, we loaded α-SNAP–depleted wild-type (WT) MEFs with sodium-binding benzofuran isophthalate (SBFI; a ratiometric sodium indicator dye) and measured sodium influx in response to thapsigargin (TG)-mediated store depletion. Remarkably, α-SNAP–deficient cells showed distinct permeability to sodium upon store depletion (Figure 2F). To determine whether sodium permeation was specific to disrupted Orai1 oligomerization and assembly, we depleted α-SNAP expression in Orai1−/− MEFs. No sodium permeation was observed in these cells (Figure 2F). To determine whether change in SBFI fluorescence was specific to sodium influx, we exchanged extracellular sodium with an organic monovalent cation, N-methyl-d-glucamine, and found that sodium influx was abrogated. Taken together, these data demonstrate that assembly of Orai1 dimers into calcium-selective multimeric channels is orchestrated on site in a α-SNAP–dependent manner.
We next investigated whether α-SNAP depletion affects the oligomerization of Stim1. To address this question, we generated and used a Stim1−/− MEF cell line expressing Stim1-mEos3.2 along with Orai1-myc. We observed a maximum of eight distinct photobleaching steps for Stim1-mEos spots in store-depleted Stim1−/− MEFs (Figure 3A). Of importance, Stim1 photobleaching events from SCR or α-SNAP siRNA–treated cells did not show any appreciable difference in the total number or distribution of steps, suggesting that unlike, Orai1, Stim1 oligomerization is not modulated by α-SNAP (Figure 3B).
FIGURE 3:

Regulation of Orai1 assembly and selectivity by α-SNAP is independent of Stim1 levels. (A) Representative photobleaching steps obtained from Stim1-mEos–expressing, store- depleted Stim1−/− MEF cells. (B) Histogram showing number of Stim1-mEos photobleaching steps vs. normalized frequency from store-depleted Stim1−/− MEFs treated with SCR or α-SNAP siRNA (600–700 spots). (C, D) Box-and-whisker plots of constitutive calcium (C) and sodium influx (D) measured from SCR or α-SNAP siRNA–treated Orai1−/− MEF cells transfected with Orai1-SS (40–70 cells). Top and bottom box boundaries represent 25th and 75th percentile values, respectively, the median is represented by a horizontal line within the box, and whiskers denote extreme 25% quartile values of the data. Student’s t-test p < 0.0001 (C) and 0.0175 (D).
Increasing the number of Stim1-Orai1–activating region (SOAR/CAD) domains bound to Orai1 C-terminus has been shown to enhance the amplitude of CRAC currents and impart calcium selectivity to Orai1 channels (Li et al., 2011; McNally et al., 2012). Therefore we wondered whether expression of an Orai1-SOAR-SOAR (Orai1-SS) construct could bypass the requirement for α-SNAP in orchestrating Orai1 assembly and calcium selectivity. Remarkably, we found that constitutive calcium influx via Orai1-SS was strongly inhibited in α-SNAP–depleted cells and sodium influx was significantly higher (Figure 3, C and D). Taken together, these data demonstrate that α-SNAP regulates the stoichiometry and calcium selectivity of Orai1 channels independently of Stim1 levels.
Because α-SNAP regulates Orai1 assembly and selectivity, we next sought to probe potential ultrastructural changes in Stim:Orai clusters with or without α-SNAP and determine the precise localization of α-SNAP with respect to Orai1 and Stim1. Using the photoblinking ability of mEos and antibody dye pairs, we performed single-color and multicolor subdiffraction single-molecule localization microscopy (STORM/PALM). Single-color live STORM of Stim1-mEos3.2 showed redistribution of tubular Stim1 into circular clusters upon store depletion (Supplemental Video S1). Compared to conventional TIRF images (Figure 4A), mEos-Orai1 and Stim1-myc were observed to colocalize within small circular as well as elongated tubular structures (Figure 4, B and C). Of interest, we found α-SNAP to be tightly coassociated with Orai1 and Stim1 localizations in both types of structures, suggesting a consistent and active role of α-SNAP in regulating the assembly of Orai1 subunits after store depletion (Figure 4, D and E). However, α-SNAP–depleted cells did not show significant differences in the Orai/Stim STORM ultrastructures compared with those obtained from control cells. Similarly, transmission electron microscopy on α-SNAP and SCR RNAi–treated, store-depleted HEK cells showed no obvious difference in the size of cortical ER or its distance from the plasma membrane (Figure 4F). These data demonstrate that α-SNAP tightly coassociates with Orai1 and Stim1 at an ultrastructural level and its depletion does not cause any gross structural alterations within ER-PM junctions.
FIGURE 4:

α-SNAP coclusters with Orai1 and Stim1 at nanoscale resolution inside CRAC clusters. (A) Conventional fluorescence and (B) STORM image of Orai1 punctate fluorescence seen in store-depleted MEF cells expressing mEos-Orai1 and Stim1-myc. (C) Representative colocalization of Orai1 and Stim1 in tubular clusters. (D, E) Colocalization of α-SNAP in tubular as well as circular Orai1 and Stim1 clusters. (F) TEM images of ER-PM junctions from HEK 293 cells treated with SCR or α-SNAP siRNA.
Previous single-particle tracking studies using PAGFP-Orai1 suggested that after store depletion, Orai1 mobility becomes subdiffusive due to interactions with Stim1 (Wu et al., 2014). Whether additional proteins contribute to the subdiffusive behavior of Orai1 within ER-PM junctions either along with or independently of Stim1 remains unexplored. We investigated the diffusion of mEos-Orai1 by performing live single-particle mobility analysis in HEK 293 cells (Materials and Methods; Supplemental Video S2). Analysis of the diffusion coefficients (Figure 5A) and mean square displacement of single-molecule trajectories as a function of time delay (Figure 5B) showed that similar to PAGFP-Orai1 (Wu et al., 2014), mEos-Orai1 molecules diffuse freely in resting cells, whereas in Stim1-coexpressing, store-depleted cells, mEos-Orai1 mobility becomes significantly slower and confined (Figure 5, A and B).
FIGURE 5:

α-SNAP stabilizes Orai1-Stim1 interactions within ER-PM junctions. (A, B) Cumulative distribution of (A) diffusion coefficients or (B) mean square displacement (MSD) vs. time delay, Δt, of mEos-Orai1 obtained from live single-particle imaging in HEK 293 cells coexpressing Stim1-myc. Open circles are data from resting cells, and filled circles are data from store-depleted cells. (C–G) Single-molecule diffusion analysis of mEos-Orai1 from store- depleted HEK 293 cells treated with SCR (black) or α-SNAP (red) siRNA. (C, D) Cumulative distribution of diffusion coefficients (C) and MSD vs. Δt (D). (E, F) Representative TIRF images from siRNA-treated, store-depleted cells expressing mEos-Orai1+Stim1-myc overplayed with single-particle trajectories obtained for mEos-Orai1 diffusion. Trajectories are coded according to their diffusion coefficient. (G) Histogram of distance traveled by mEos-Orai1 particles in E and F. (H) Box-and-whisker plot representing FRET analysis between C-terminally tagged Orai1 (Orai1-CFP) and Stim1-YFP (C-C FRET) or N-terminally tagged Orai1 (CFP-Orai1) and Stim1-YFP (N-C FRET) performed on SCR (black) or α-SNAP (red) siRNA–treated, store-depleted cells. Eleven cells and 100–120 junctional regions per group.
To determine whether α-SNAP depletion affects Orai1 diffusion, we analyzed the mobility of mEos-Orai1 in α-SNAP or SCR siRNA–treated, store-depleted cells coexpressing Stim1-myc. Surprisingly, Orai1 displayed significantly faster and less constrained diffusion in α-SNAP–depleted cells (Figure 5, C and D) than the subdiffusive behavior of mEos-Orai1 in SCR siRNA–treated cells (Figure 5, C and D). An overlay of individual molecule trajectories, color coded according to their diffusion coefficients, onto the store-depleted TIRF images indicated that faster-moving Orai1 molecules from α-SNAP–depleted cells were still localized within the area of PM that forms junctional contacts with cortical ER upon store depletion (Figure 5, E and F) but sampled a longer/wider distance when compared with SCR siRNA-treated cells (Figure 5G).
Orai1 is known to interact with Stim1 via its C-terminal as well as N-terminal ends (Park et al., 2009; McNally and Prakriya, 2012; Zheng et al., 2013). Therefore enhanced Orai1 diffusion in the absence of α-SNAP could reflect unstable interactions between Orai1 oligomers and Stim1. We performed FRET analysis between Orai1 C-terminus–tagged cyan fluorescent protein (Orai1-CFP) and Stim1–yellow fluorescent protein (YFP), as well as between Orai1 N terminus–tagged CFP (CFP-Orai1) and Stim1-YFP. Distinct FRET was observed between CFP-Orai1 and Stim1-YFP, as well as between Orai1-CFP and Stim1-YFP (Figure 5H). Remarkably, α-SNAP–depleted cells showed normal interaction between Orai1-CFP and Stim1-YFP; however, CFP-Orai1 to Stim1-YFP FRET was significantly reduced, demonstrating that α-SNAP facilitates low-affinity interaction between Orai1 N-terminus and Stim1, which is known to be crucial for CRAC function (Palty and Isacoff, 2016).
Collectively these data demonstrate that α-SNAP, a cytosolic protein, forms an integral component of the active CRAC supramolecular complex and orchestrates functional assembly and calcium selectivity of the Orai1 pore within ER-PM junctions, in part, by enabling a crucial low-affinity functional interaction between the Orai1 N-terminus and Stim1 C-terminus.
DISCUSSION
We showed that Orai1, the pore-forming subunit of CRACs, displays a unique, on-site stoichiometric assembly into optimally functional and calcium-selective multimers and this process is dependent on α-SNAP.
Most ion channels are multimeric and require specific homomeric or heteromeric assembly of pore subunits as well as correct positioning within the oligomer for optimal function. The pore subunits typically assemble within the ER either with or without the help of accessory proteins. Some may undergo additional posttranslational modifications or functional refolding at or close to the actual site of activation (Gurnett and Campbell, 1996; Colledge and Froehner, 1998; Green, 1999). However, to our knowledge, on-site stoichiometric assembly of pore-forming subunits is highly unusual. This added layer of regulation to the functional assembly of Orai1 may be necessary inside cells, given its unusual mechanism of activation, which requires physical interaction with an ER-resident protein, Stim1. Because Stim1 is sensitive to diverse stimuli as well as cellular stress, the functional assembly of Orai1 oligomers within ER could lead to nonspecific store depletion at unwanted sites within ER or during membrane transport. Furthermore, it is reasonable to speculate that stimulus-specific, on-site organization of different pore assemblies could serve as one of the means to regulate the absolute rate and level of calcium influx and translate the strength of stimulus at the cell surface into different biological responses.
Previous studies proposed that heteromeric assemblies of Orai1 and Orai3 form arachidonate-regulated channels (Mignen et al., 2008a). Gating and permeation of Orai1 and Orai3 heteromeric assemblies has also been shown to be quite different compared with Orai1 homomeric assemblies (Mignen et al., 2008a; Demuro et al., 2011). Whether these heteromeric Orai assemblies form within the ER or on-site, however, in a stimulus-specific manner remains unestablished. Because α-SNAP can bind all three Orais (Miao et al., unpublished data), one possibility is that α-SNAP enables stimulus-dependent subunit selection, assembly, and contacts with Stim1.
It is remarkable that constitutive calcium influx mediated by Orai1-SS is dependent on α-SNAP even though the soluble CAD/SOAR domain can restore SOCE in α-SNAP–deficient cells (Miao et al., 2013). Some previous studies suggested that Orai1-SS is self-sufficient in activating large, calcium-selective currents, whereas other studies showed a requirement for binding of Stim1 to the N-terminal tail of Orai1-SS for its optimal activation (Park et al., 2009; McNally et al., 2012; Palty and Isacoff, 2016). Intriguingly, the binding affinity of Stim1 to Orai1 N-terminal tail is far lower than its interaction with Orai1 C-terminal tail (Park et al., 2009), and the structure of Orai1 with an intact N-terminus is unknown (Hou et al., 2012). The faster mobility of Orai1 assemblies within ER-PM junctions of α-SNAP–depleted cells suggests that in the absence of α-SNAP, the contacts between Stim1 and Orai1 are purely mediated by the Orai1 C-terminal tail and are unstable by themselves. These interactions may be sufficient for coentrapment of the two proteins within ER-PM junctions but not for their optimal and productive engagement.
The molecular mechanisms that enable the crucial low-affinity interaction between Stim1 and Orai1 N-terminus in the presence of higher-affinity interacting domains such as Orai1 C-terminus are unknown. One possibility is that α-SNAP enables refolding/unfolding of the Orai1 N-terminus to allow stable interactions with Stim1, which in turn orchestrates stoichiometric assembly of Orai1 dimers and calcium selectivity via the Orai1 N-terminus. Indeed, a faint association between α-SNAP and Orai1 N-terminus was observed (Miao et al., 2013).
In a second scenario, Orai1 channel mobility could depend on its activity in a manner similar to TRPV1 (Senning and Gordon, 2015). Indeed, in this case, selective calcium permeation would be expected to retard Orai1 diffusion, and, in α-SNAP–deficient cells, nonselective permeation of sodium via Orai1 channels would prevent the activation-dependent localized diffusion trap. Collectively larger oligomers of Orai1, nonselective permeation, and higher mobility could also explain the apparent expanded size of Orai1 puncta previously observed in TIRF images of α-SNAP–depleted cells (Miao et al., 2013).
STORM imaging of Stim-Orai clusters did not reveal any ultrastructural defects in α-SNAP–depleted cells. However, in WT cells, coassociation of α-SNAP with Stim1 and Orai1 could be observed during early as well as late stages of Stim1-Orai1 clustering, suggesting its continuous engagement during this process. The STORM data also indicate that mEos-Orai1 localizations are consistently clustered toward the interior of small, circular structures demarcated by Stim1-myc localizations. The reason their relative localizations are correlated but do not perfectly overlap could be purely technical or related to the precise arrangement of Orai1 and Stim1. For instance, previous STORM studies (Dani et al., 2010; Suleiman et al., 2013; Szymborska et al., 2013) demonstrated distinct nonoverlapping localizations from N- or C-terminal domains of proteins that are highly oriented within multiprotein complexes. Similarly, the Orai1 N-terminal and Stim1 C-terminal labeling used here could indicate that the CRAC complex is oriented with the Orai1 N-terminal ends clustered together and lining the pore. Stim1 C-terminus is known to activate Orai1 channels by direct binding to its C- as well as N-terminus. A lack of substantial overlap between Stim1-C and Orai1-N localizations could reflect transient interactions. However, these interpretations are complicated by the fact that Stim1 and α-SNAP were labeled using primary and secondary antibodies, which could offset protein localizations somewhat, in comparison to Orai1 localizations obtained using mEos fluorescent protein fusion. Future experiments designed to label Orai1, Stim1, and α-SNAP at various domains using comparable labeling methods along with simultaneous validation of whether the position and choice of fusion tag alters the native distribution of the protein within the complex will enable distinction between these possibilities.
Stim1 is believed to interact with Orai1 at a ratio of ∼1:2 for full activation of SOCE (Hoover and Lewis, 2011; Li et al., 2011; McNally and Prakriya, 2012). According to this ratio, 4-, 8- or 12-mers of Stim1 may be required for full activation of Orai1 dimer, tetramer, and hexamer assemblies. Yet, we could detect only up to eight steps in photobleaching of Stim1 oligomers. However, given the lower likelihood of efficiently detecting higher step numbers, it is possible that even larger oligomers of Stim1 exist but could not be discerned with discrete steps. Therefore our data do not conclusively establish Stim1 stoichiometry or address the calcium selectivity of dimeric or tetrameric versus hexameric CRAC oligomers. Future electrophysiological studies using single-channel recordings may be able to distinguish quantitatively among the functions as well as selectivities of different Orai1 assemblies. Regardless of their oligomeric state, our data demonstrate dynamic on-site assembly of Orai1 multimers and show their dependence on α-SNAP for optimal function as well as calcium selectivity.
The findings presented here also suggest that the correct intracellular ratio of α-SNAP:Orai1:Stim1 is required to ensure full functionality as well as calcium selectivity of Orai1 assemblies. Transient expression of selective components would therefore yield a range of Orai1 stoichiometries with progressive levels of calcium selectivity and function. In vivo, this dynamic process could be rate limiting and may, in part, explain the slow activation of CRAC current. Although the present study could not address precisely how α-SNAP orchestrates functional assembly of Orai1 multimers in the plasma membrane, future structure–function analysis of their interactions may illuminate the precise mode of action of α-SNAP in Orai1 assembly and calcium selectivity in ER-PM junctions with further precision.
MATERIALS AND METHODS
Plasmids and molecular cloning
Human Orai1 was subcloned at the C-terminal end of mEos3.2 to generate mEos3.2-Orai1, using the pEGFP-C1 vector backbone. Stim1-myc and Stim1-mEos3.2 were generated by cloning human Stim1 in the pcDNA 4/TO/myc-His and at the N-terminal end of mEos3.2 plasmid. CFP-α-SNAP was generated by subcloning in the pECFP-C1 vector (Clontech, Mountain View, CA).
siRNA
Double-stranded siRNAs against mouse and human α-SNAP were custom synthesized and purified by high-performance liquid chromatography (Sigma-Genosys, St. Louis, MO) using the following 5′-3′ sequences: mNAPA, CGCCAAAGACUACUUCUUCAA; hNAPA, CCGGGAATGCAAGTTGATGAA; and scrambled (SCR), AACAAGAUGAAGAGCACCAA. siRNAs were labeled with Cy5 fluorescent dye at 1:1 M labeling ratio using the Label IT Tracker Nucleic Acid Localization kit (Mirus Bio, Madison, WI) and purified according to manufacturer’s instructions.
Mice and MEFs
Orai1+/− mice were generated as described previously (Vig et al., 2008) and backcrossed onto C57BL/6 background for five generations. Stim1+/− mice were a gift from Yoshihiro Baba and Tomohiro Kurosaki (Osaka University, Osaka, Japan; Baba et al., 2008). To generate Orai1−/− and Stim1−/− MEF cells, timed mating of Orai1+/− and Stim1+/− (heterozygous) mice were set up, and pregnant females were killed 12.5 d postcoitus. Embryos were detached from the uterus and amniotic sac, washed with Hanks balanced salt solution (HBSS), and placed in separate 10-cm dishes. To extract MEFs, embryos were decapitated, genotyped, and minced in trypsin/EDTA solution for 20 min. The cells obtained after dissociating the embryonic tissue were washed and cultured in 10-cm dishes. After two or three passages, cells from homozygous Orai1−/− and Stim1−/− embryos were transfected with SV-40 T-antigen plasmid using an Amaxa kit (Lonza, Mapleton, IL) for immortalization and passaged for additional three times to generate stable Orai1−/− and Stim1−/− MEF lines. WT MEFs were generated from C57BL/6 mice using a similar approach.
Single-cell calcium imaging
Cells were loaded with 1 μM Fura-2 AM (Life Technologies) in Ringer’s buffer at 37°C for 30 min in the dark, washed, stimulated with 1 μM TG (Sigma-Aldrich, St. Louis, MO), and imaged as previously described (Miao et al., 2013). Approximately 20–30 cells were analyzed per group in each experiment. Fura-2 AM was calibrated with a Fura-2 Calcium Imaging Calibration Kit (Life Technologies, Carlsbad, CA) according to manufacturer’s instructions.
Single-cell sodium imaging
Cells were loaded with 2.5 μM SBFI-AM (Life Technologies) in HBSS buffer at room temperature for 40 min in the dark, washed, stimulated with 1 μM TG, and imaged in HBSS buffer. SBFI was alternatively excited at 340 and 380 nm, and images were collected at 510-nm emission wavelength. Approximately 20–30 cells were analyzed per group in each experiment. SBFI was calibrated as described previously (Negulescu and Machen, 1990; Donoso et al., 1992).
Cell culture and transfection
Single-molecule bleaching and STORM experiments were performed using cells adhered to carbon-coated, 18-mm round #1.0 coverglasses (Marienfeld-Superior, Germany). Coverslips were cleaned by sequential bath sonication in concentrated nitric acid, water, 1 M KOH, and distilled water, followed by blow-drying using pressurized filtered air. Cleaned coverslips were sputter coated with a thin layer of carbon and glow discharged to promote cell attachment (Stap et al., 2000) and reduce deposition of extracellular fluorescent spots on the coverslip surface. MEF cells were cultured on the carbon-coated surface by placing coverslips inside a 12-well plate. Single-molecule live imaging was performed by plating cells onto #1.0 glass coverslip bottom dishes (MatTek, Ashland, MA) that were similarly carbon sputter coated.
For single-molecule bleaching experiments, MEF cells were initially cultured in six-well plates and transfected with SCR or α-SNAP siRNA, 10 ng/well, using Trans-IT-TKO reagent (Mirus Bio). At 36 h after siRNA treatment, cells were replated onto carbon-coated coverslips placed inside a 12-well plate and transfected with plasmids mEos-Orai1, Stim1-myc, and pcDNA3.1 empty vector at a 1:1:2 weight ratio and 0.8 μg of total plasmid DNA per well using the Viafect transfection reagent (Promega, Madison, WI). The total time of siRNA treatment was 48 h, of which Orai1 and Stim1 transient protein expression was performed for the last 12 h before imaging. Calcium store depletion with 1 μM TG was performed before imaging as described previously (Miao et al., 2013).
Imaging setup
Images were acquired on a custom-built single-molecule imaging rig similar to that described previously (Dani et al., 2010). Briefly, the imaging rig was constructed on an active dampening vibration isolation table (Newport Corporation, Irvine, CA) and around a Nikon TiE inverted microscope. Lasers, 642 nm (Vortran Laser, Sacramento, CA), 561 and 488 nm (Sapphire; Coherent, Santa Clara, CA), and 405 nm (Cube, Coherent), were shuttered individually via mechanical shutters as well as an acousto-optical tunable filter (Crystal Technologies, Palo Alto, CA). Lasers were combined, expanded, collimated, and focused at the back focal plane of the microscope. The diameter of the laser beam was cut using a field diaphragm to just cover a 256 × 256–pixel area of the charge-coupled device (CCD) camera. A mechanical translation stage enabled objective-type total internal reflection (TIR) illumination via a 100×/1.4 numerical aperture (NA) objective (UPlanSApo; Olympus, Japan). The Nikon perfect focus system was used to lock focus at the coverslip surface during imaging. A quad-band dichroic mirror, zt/zet-405/488/561/640rpc (Chroma Technology, Bellows Falls, VT), was used to excite with laser light, and fluorescence collected by the same objective was filtered using emission filters et525/50 m for mEos-green, et600/50 m for mEos-red, and et705/72 m for Alexa 647 placed in a filter slider. A motorized stage (Marzhauser, Wetzlar, Germany) was used to record and save positions of cells. Images were acquired on a back-illuminated electron-multiplying CCD camera (DU-897, iXon+; Andor Technology, Belfast, United Kingdom) using custom software.
Single-molecule photobleaching
Resting or store-depleted cells were fixed with 2% PFA for 10 min at room temperature, followed by three or four washes of 1× phosphate-buffered saline (PBS). Transfected MEF cells were first screened using the mEos green fluorescence to identify cells expressing weak levels of mEos-Orai1 at the PM. Cells were further screened for intracellular presence of Cy5-labeled SCR or α-SNAP siRNA. For each cell, mEos-Orai1 molecules were photoconverted to the red form using the 405-nm laser at an intensity of 7 W/cm2 (measured at the sample) for 6 s. Photobleaching of the mEos red form was initiated by turning on the 561-nm laser at 2 W/cm2 with simultaneous image acquisition at 10-Hz frequency and was stopped when the intensity of all molecules reached background.
Photobleaching movies were processed to subtract background, enhance contrast, and automatically identify fluorescent spots, using the following steps. All steps were performed in Matlab, unless mentioned otherwise.
Noise removal and spot identification. In the first stage, each movie was processed to remove noise in the temporal and spatial scales to unambiguously detect all fluorescent spots in the cell. First, a Kalman filter was applied in ImageJ (http://rsb.info.nih.gov/ij/plugins/kalman.html) to maintain transient signal changes due to bleaching while reducing camera noise across the time series. To further remove spatial blurring and increase contrast between the spot signals and background, we averaged the first 10 frames of the movie and filtered this averaged image with a Laplacian of Gaussian (LoG) kernel filter to generate an image mask (Figure 1A). This image mask was used to locate spot positions and filter spots in subsequent steps.
- Filter spots. We removed spots using the following criteria:
- Size. We removed spots of <2 × 2 pixels and >4 × 4 pixels. Smaller spots are likely due to nonspecific fluorescence on the coverslip surface, and larger spots could arise from an overlap of multiple nearby spots. Consistent with this, we did not observe discrete bleaching steps from most spot sizes of 5 × 5 and larger pixel dimensions.
- Shape and distance. We further performed a two-dimensional elliptical Gaussian fitting using a 3 × 3–pixel region of interest (ROI) to extract center positions, x- and y-widths (Wx and Wy, respectively), and peak intensity for each spot. We removed spots if their centroid distance was <4 pixels and elliptical spots since these could arise from a merger of two neighboring spots. Elliptical shapes were defined as follows:
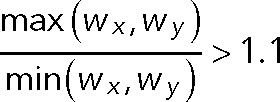
-
Intensity. We removed extremely weak spots whose intensity was <10% of background as a low-intensity threshold.Application of these filters typically resulted in the elimination of 10–20% of spots.
Step detection: To automatically detect steps during the bleaching process, we adopted the progressive step detection algorithm as described by McGuire et al. (2012) and combined it with nonparametric Bayesian inference (Hines et al., 2015) to estimate the existing noise level in the bleaching time series and identify the number of discrete bleaching states. For each bleaching trace, we averaged the observed step sizes and used half of the average step size to set as a noise level threshold for automated step level detection. Each fluctuation in the fluorescence signal was compared with noise level to determine whether it was due to bleaching of the molecule or noise in the signal. Subsequently, through iterated scanning, annealing, and reevaluation phases, all fluctuations from noise were averaged and bleaching events found. Automating these steps avoids subjective bias in selecting spots as well as determining step counts.
Correction of step overcounting/undercounting. Finally, we manually examined the data to identify cases of overcounting or undercounting of steps. Because a small fraction of mEos molecules can spontaneously photoreactivate after bleaching and the intensity after photoreactivation would be larger than the averaged step size, the automated algorithm could falsely assign such a change in fluorescence state as a bleaching step. We identified such cases, and as long as the spot intensity had reached the background CCD noise, we regarded the molecule as completely bleached. Undercounting of steps could be identified when two molecules bleach simultaneously. These could be identified as a large step that was roughly twice the size of the average step value for that trace and was regarded as two steps instead of one.
- Data representation and stoichiometry modeling. Examples of spots exhibiting step-photobleaching fluorescence traces are shown in Figure 1B, where fluorescence values for each spot are integrated from a 3 × 3–pixel ROI. Bleaching steps from multiple cells in each group were pooled and plotted as a histogram in Origin software. The probability of observing k steps each subunit follows a binomial distribution:
where n is the number of homogeneous subunits in one complex (e.g., n = 2 for dimers and n = 6 for hexamers) and p denotes the fraction of active-state fluorescent proteins. In case of a dynamic process in which Orai1 molecules can reconstitute into multiple coexisting subunit configurations, we considered other possibilities to model the distribution of bleaching steps. For instance, in case of a combination of dimer, tetramer, and hexamer, the probability of observing k steps is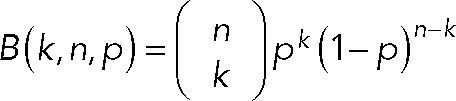
where a, b, and c are fractions of dimer, tetramer, and hexamer, respectively. Excluding all dark state subunits, the predicted distribution is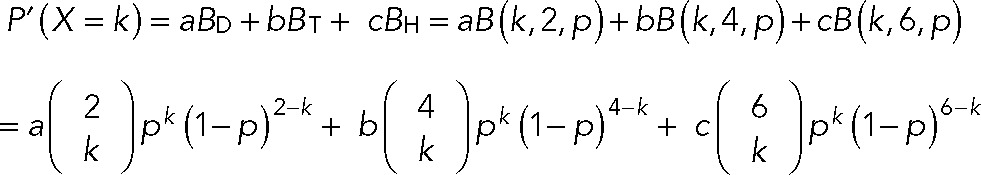
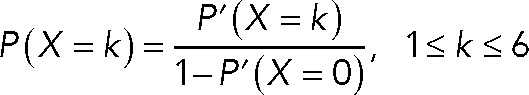
For each model comprising pure dimers, tetramers, hexamers, and combinations thereof, we initially iterated the value of p and the relative fraction of each molecule to calculate the chi- square error between the experimentally observed distribution and the expected distribution from each model. A model was found to fit best when the chi-square error between the observed and expected distribution was minimum. In all cases, we found that models fited best at p around 0.7.
Single-particle diffusion
Single-particle tracking (SPT) was performed in live HEK293 cells cultured on coverslip bottom dishes. Cells were transiently cotransfected with mEos-Orai1 along with Stim1-myc or empty vector, and imaging was performed at 12–16 h posttransfection. Sparse single molecules of photoconverted mEos-Orai1 were imaged using continuous TIR illumination of the 561-nm laser at 200 W/cm2 plus the 405-nm laser at 0.3 W/cm2 (Supplemental Video S2). Image acquisition was performed at 50-Hz frequency and stopped at 6 min after addition of TG. This SPT movie was processed to subtract uneven illumination background by using the rolling ball algorithm in ImageJ. Subpixel localizations of single particles were extracted using the comet detection algorithm and u-track software (Jaqaman et al., 2008; Jaqaman and Danuser, 2009). Particle localization in adjacent frames was tracked and trajectories linked using u-track and the following empirically determined criterion: to account for molecule blinking, gap length was set at six frames, search radius to capture displacement was set with an upper bound of 3 pixels, and lower bound was taken as equal to the localization uncertainty of 30 nm. Only trajectories >20 points were taken into consideration for analysis. Diffusion coefficients were obtained using the moment scaling spectrum algorithm (Ewers et al., 2005) implemented in u-track. The cumulative frequency distribution was plotted for all trajectories, as well as the mean square displacement as a function of time lapse.
STORM/PALM
Two- and three-channel STORM images were acquired on the same imaging setup. Coverslips with cells expressing mEos-Orai1 and Stim1-myc were fixed after store depletion, washed, permeabilized with 3% bovine serum albumin plus 0.1% NP-40 in PBS, and immunolabeled with the 9E10 anti-myc antibody (Developmental Studies Hybridoma Bank, Iowa City, IA). Primary antibody was detected using a donkey anti-mouse immunoglobulin G (IgG) secondary antibody (Jackson ImmunoResearch, West Grove, PA) labeled with the reporter and activator fluorescent dyes Alexa 647 and Cy2. A coverslip was inverted into a slide with an imaging buffer of 100 mM Tris-HCl, pH 8.0, and 150 mM NaCl and containing an oxygen-scavenging system comprising glucose, glucose oxidase, catalase, and the reducing agent 2-mercaptoethylamine (Bates et al., 2007). After removal of excess buffer, edges of the coverslip were sealed with nail polish before imaging. Sparse single- molecule images were acquired at 60-Hz frequency by alternating pairs of imaging plus activator laser sequence: the 642-nm laser at 560 W/cm2 plus weak 488-nm laser for the dye-labeled antibody and the 561-nm laser at 250 W/cm2 plus weak 405-nm laser for mEos. The amount of 488- or 405-nm activation laser was adjusted to ensure sparse single-molecule events in each camera frame. Alexa 647 and mEos-red fluorescence emissions were filtered using an et561/640m dual-bandpass emission filter (Chroma). Raw image stacks were fitted with the DAOSTORM algorithm (Holden et al., 2011; Babcock et al., 2012) to determine the centroid positions of fluorescent intensity peaks. These STORM localizations were rendered as images using custom software.
For three-channel STORM, cells transfected with mEos-Orai1, Stim1-myc, and CFP–α-SNAP were immunolabeled with rabbit anti-GFP and mouse anti-myc primary antibodies, followed by donkey anti-rabbit and donkey anti-mouse IgG-specific secondary antibodies (Jackson ImmunoResearch). Secondary reagents were custom conjugated with the fluorescent dye pairs Cy2–Alexa 647 and Alexa 405–Alexa 647. Sequential image acquisition was performed to first image the Alexa 647 antibodies as a two-channel STORM sequence similar to one previously described (Dani et al., 2010), followed by imaging of the mEos fluorescent protein using the 561- and 405-nm lasers. To compensate for drift during image acquisition, we used the redundancy cross-correlation drift correction algorithm in Matlab (Wang et al., 2014). For antibody channels, drift in all frames was corrected to align with the last frame, whereas localizations from all mEos frames were aligned to their first frame.
FRET microscopy
FRET microscopy experiments were performed using live HEK 293 cells transiently transfected with Orai1-CFP or CFP-Orai1 along with Stim1-YFP plasmids. Images were acquired on a Nikon A1R+ laser scanning confocal microscope using a 60× Plan Apo 1.4 NA objective lens. CFP and YFP were sequentially excited using 441- and 514-nm lasers, and images were filtered using 400–457/514 dichroic mirror and 540/30 and 585/65 emission filters and recorded on GaAsP photomultiplier tube detectors. Images were scanned at a rate of 800 ms/frame. After background subtraction, fluorescence pixel intensity values were extracted from ER-PM junctional regions marked by Orai1-Stim1 punctae in each cell using a 3 × 3–pixel ROI. Enhanced FRET emission was calculated as previously described (Xia and Liu, 2001):
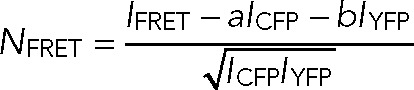 |
where IFRET is the CFP excitation-YFP emission intensity, ICFP and IYFP are the respective fluorescence intensities of CFP and YFP, and a and b are cross-talk constants obtained by imaging single-color CFP and YFP samples in the same experiment.
Electron microscopy
For ultrastructural analysis, HEK 293 cells treated with SCR or α-SNAP RNAi were fixed in 2% paraformaldehyde/2.5% glutaraldehyde in 100 mM cacodylate buffer, pH 7.2, for 1 h at room temperature. Samples were washed in cacodylate buffer, postfixed in 1% osmium tetroxide (Polysciences) for 1 h, and rinsed extensively in distilled H2O before en bloc staining with 1% aqueous uranyl acetate (Ted Pella, Redding, CA) for 1 h. After several rinses in distilled H2O, samples were dehydrated in a graded series of ethanol and embedded in Eponate 12 resin (Ted Pella). Sections of 95 nm were cut with a Leica Ultracut UCT ultramicrotome (Leica Microsystems, Bannockburn, IL), stained with uranyl acetate and lead citrate, and viewed on a JEOL 1200 EX transmission electron microscope (JEOL USA, Peabody, MA) equipped with an AMT 8-megapixel digital camera (Advanced Microscopy Techniques, Woburn, MA).
Membrane protein cross-linking
Cross-linking experiments were performed using the stably transfected T-REx cell line (Miao et al., 2013), in which flag-Orai1 and Stim1-myc expression was induced by overnight treatment with 1 μg/ml doxycycline. Cells were harvested from the culture plate and centrifuged, and the cell pellet was freeze-thawed twice by alternating it on dry ice and warm water bath for few seconds. Cell pellet was suspended in 50 mM phosphate buffer and 150 mM KCl, pH 8.0, containing protease inhibitors leupeptin, aprotinin, and pepstatin, and cell membranes were disrupted by bath sonication for 1 min, followed by 10 strokes through a 2-ml Dounce homogenizer. Cell debris and nuclei were removed by centrifugation at 2500 × g for 10 min at 4°C. Supernatant containing the membrane fraction was ultracentrifuged at 105,000 × g for 1 h at 4°C. Membrane pellet was suspended in the same buffer by passage through two to four strokes of a 0.5-ml Dounce homogenizer. Proteins were cross-linked for 30 min at room temperature by adding freshly reconstituted BS3 cross-linker at a final concentration of 900 μM. Unreacted cross-linker was quenched by addition of 20 mM Tris-HCl, pH 8.0. Cross-linked proteins were dissolved by adding Laemmli sample buffer containing 100 mM dithiothreitol, incubated at 37°C for 15 min, and resolved by electrophoresis on a 4–15% gradient SDS–polyacrylamide gel. Proteins were transferred by Western blotting to nitrocellulose membrane, and flag-Orai1 protein bands were detected using indirect chemiluminescence with rabbit anti-flag primary, donkey anti-rabbit, horseradish peroxidase–labeled secondary antibody.
Viability assay
MEF cells were treated with SCR or α-SNAP siRNA for the same duration as described in all experiments, suspended in annexin V binding buffer (Biolegend, San Diego, CA) containing annexin V-APC and PI for 15 min, and analyzed using FACSCalibur or LSR Fortessa analyzers and Flow Jo software (BD Biosciences, San Jose, CA).
Estimation of mitochondrial content
For gross morphology, SCR or α-SNAP siRNA–treated MEFs were incubated with 50 nM MitoTracker Green (Molecular Probes, Eugene, OR) diluted in DMEM without fetal bovine serum for 30 min at 37°C, washed with warm HBSS, and imaged on an Olympus IX71 wide-field fluorescence microscope using a 60× UPlanFL 1.25 NA objective lens and a Photometrics CoolSNAP HQ2 CCD camera. For total mitochondrial content, cells were stained with MitoTracker Green as described earlier, washed, and analyzed using FACSCalibur or LSR Fortessa analyzers and Flow Jo software.
Statistical analysis
Statistical significance represented as p values was calculated using Student’s t test.
Supplementary Material
Acknowledgments
We thank Pingyong Xu (Institute of Biophysics, Chinese Academy of Sciences, Beijing, China) for kindly providing the mEos3.2 expression plasmid, Tao Xu for the Orai1-SS construct, Tomohiro Kurosaki and Yoshihiro Baba for Stim1-knockout mice, Michael Cahalan and Joe Dynes for their advice on the manuscript, Wandy Beatty (Washington University School of Medicine) for help with electron microscopy, and the Washington University Center for Cellular Imaging for access to the A1R confocal microscope. Funding was provided by National Institutes of Health Grants NIAID-AI108636, ACS-RSG 14-040-01-CSM, and CCFA SRF 3798 to M.V. and NIMH-R21MH099798 to A.D.
Abbreviations used:
- CRAC
calcium release–activated channel
- ER
endoplasmic reticulum
- FRET
fluorescence resonance energy transfer
- MEF
mouse embryonic fibroblast
- PM
plasma membrane
- α-SNAP
α–soluble N-ethylmaleimide–sensitive factor attachment protein
- SOCE
store-operated calcium entry
- TG
thapsigargin.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E16-03-0163) on June 22, 2016.
REFERENCES
- Baba Y, Nishida K, Fujii Y, Hirano T, Hikida M, Kurosaki T. Essential function for the calcium sensor STIM1 in mast cell activation and anaphylactic responses. Nat Immunol. 2008;9:81–88. doi: 10.1038/ni1546. [DOI] [PubMed] [Google Scholar]
- Babcock H, Sigal YM, Zhuang X. A high-density 3D localization algorithm for stochastic optical reconstruction microscopy. Opt Nanoscopy. 2012;1 doi: 10.1186/2192-2853-1-6. doi: 10.1186/2192-2853-1-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates M, Huang B, Dempsey GT, Zhuang X. Multicolor super-resolution imaging with photo-switchable fluorescent probes. Science. 2007;317:1749–1753. doi: 10.1126/science.1146598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colledge M, Froehner SC. To muster a cluster: anchoring neurotransmitter receptors at synapses. Proc Natl Acad Sci USA. 1998;95:3341–3343. doi: 10.1073/pnas.95.7.3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dani A, Huang B, Bergan J, Dulac C, Zhuang X. Superresolution imaging of chemical synapses in the brain. Neuron. 2010;68:843–856. doi: 10.1016/j.neuron.2010.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demuro A, Penna A, Safrina O, Yeromin AV, Amcheslavsky A, Cahalan MD, Parker I. Subunit stoichiometry of human Orai1 and Orai3 channels in closed and open states. Proc Natl Acad Sci USA. 2011;108:17832–17837. doi: 10.1073/pnas.1114814108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donoso P, Mill JG, O’Neill SC, Eisner DA. Fluorescence measurements of cytoplasmic and mitochondrial sodium concentration in rat ventricular myocytes. J Physiol. 1992;448:493–509. doi: 10.1113/jphysiol.1992.sp019053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorwart MR, Wray R, Brautigam CA, Jiang Y, Blount P. S. aureus MscL is a pentamer in vivo but of variable stoichiometries in vitro: implications for detergent-solubilized membrane proteins. PLoS Biol. 2010;8:e1000555. doi: 10.1371/journal.pbio.1000555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durisic N, Godin AG, Wever CM, Heyes CD, Lakadamyali M, Dent JA. Stoichiometry of the human glycine receptor revealed by direct subunit counting. J Neurosci. 2012;32:12915–12920. doi: 10.1523/JNEUROSCI.2050-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durisic N, Laparra-Cuervo L, Sandoval-Alvarez A, Borbely JS, Lakadamyali M. Single-molecule evaluation of fluorescent protein photoactivation efficiency using an in vivo nanotemplate. Nat Methods. 2014;11:156–162. doi: 10.1038/nmeth.2784. [DOI] [PubMed] [Google Scholar]
- Ewers H, Smith AE, Sbalzarini IF, Lilie H, Koumoutsakos P, Helenius A. Single-particle tracking of murine polyoma virus-like particles on live cells and artificial membranes. Proc Natl Acad Sci USA. 2005;102:15110–15115. doi: 10.1073/pnas.0504407102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Parajo MF, Koopman M, van Dijk EM, Subramaniam V, van Hulst NF. The nature of fluorescence emission in the red fluorescent protein DsRed, revealed by single-molecule detection. Proc Natl Acad Sci USA. 2001;98:14392–14397. doi: 10.1073/pnas.251525598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green WN. Ion channel assembly: creating structures that function. J Gen Physiol. 1999;113:163–170. doi: 10.1085/jgp.113.2.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurnett CA, Campbell KP. Transmembrane auxiliary subunits of voltage-dependent ion channels. J Biol Chem. 1996;271:27975–27978. doi: 10.1074/jbc.271.45.27975. [DOI] [PubMed] [Google Scholar]
- Hastie P, Ulbrich MH, Wang HL, Arant RJ, Lau AG, Zhang Z, Isacoff EY, Chen L. AMPA receptor/TARP stoichiometry visualized by single-molecule subunit counting. Proc Natl Acad Sci USA. 2013;110:5163–5168. doi: 10.1073/pnas.1218765110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hines KE, Bankston JR, Aldrich RW. Analyzing single-molecule time series via nonparametric Bayesian inference. Biophys J. 2015;108:540–556. doi: 10.1016/j.bpj.2014.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holden SJ, Uphoff S, Kapanidis AN. DAOSTORM: an algorithm for high- density super-resolution microscopy. Nat Methods. 2011;8:279–280. doi: 10.1038/nmeth0411-279. [DOI] [PubMed] [Google Scholar]
- Hoover PJ, Lewis RS. Stoichiometric requirements for trapping and gating of Ca2+ release-activated Ca2+ (CRAC) channels by stromal interaction molecule 1 (STIM1) Proc Natl Acad Sci USA. 2011;108:13299–13304. doi: 10.1073/pnas.1101664108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou X, Pedi L, Diver MM, Long SB. Crystal structure of the calcium release-activated calcium channel Orai. Science. 2012;338:1308–1313. doi: 10.1126/science.1228757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaqaman K, Danuser G. Computational image analysis of cellular dynamics: a case study based on particle tracking. Cold Spring Harb Protoc. 2009;2009:pdb top65. doi: 10.1101/pdb.top65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaqaman K, Loerke D, Mettlen M, Kuwata H, Grinstein S, Schmid SL, Danuser G. Robust single-particle tracking in live-cell time-lapse sequences. Nat Methods. 2008;5:695–702. doi: 10.1038/nmeth.1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji W, Xu P, Li Z, Lu J, Liu L, Zhan Y, Chen Y, Hille B, Xu T, Chen L. Functional stoichiometry of the unitary calcium-release-activated calcium channel. Proc Natl Acad Sci USA. 2008;105:13668–13673. doi: 10.1073/pnas.0806499105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Liu L, Deng Y, Ji W, Du W, Xu P, Chen L, Xu T. Graded activation of CRAC channel by binding of different numbers of STIM1 to Orai1 subunits. Cell Res. 2011;21:305–315. doi: 10.1038/cr.2010.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madl J, Weghuber J, Fritsch R, Derler I, Fahrner M, Frischauf I, Lackner B, Romanin C, Schutz GJ. Resting state Orai1 diffuses as homotetramer in the plasma membrane of live mammalian cells. J Biol Chem. 2010;285:41135–41142. doi: 10.1074/jbc.M110.177881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuire H, Aurousseau MR, Bowie D, Blunck R. Automating single subunit counting of membrane proteins in mammalian cells. J Biol Chem. 2012;287:35912–35921. doi: 10.1074/jbc.M112.402057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNally BA, Prakriya M. Permeation, selectivity and gating in store-operated CRAC channels. J Physiol. 2012;590:4179–4191. doi: 10.1113/jphysiol.2012.233098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNally BA, Somasundaram A, Yamashita M, Prakriya M. Gated regulation of CRAC channel ion selectivity by STIM1. Nature. 2012;482:241–245. doi: 10.1038/nature10752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao Y, Miner C, Zhang L, Hanson PI, Dani A, Vig M. An essential and NSF independent role for alpha-SNAP in store-operated calcium entry. Elife. 2013;2:e00802. doi: 10.7554/eLife.00802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mignen O, Thompson JL, Shuttleworth TJ. Both Orai1 and Orai3 are essential components of the arachidonate-regulated Ca2+-selective (ARC) channels. J Physiol. 2008a;586:185–195. doi: 10.1113/jphysiol.2007.146258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mignen O, Thompson JL, Shuttleworth TJ. Orai1 subunit stoichiometry of the mammalian CRAC channel pore. J Physiol (Lond) 2008b;586:419–425. doi: 10.1113/jphysiol.2007.147249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajo K, Ulbrich MH, Kubo Y, Isacoff EY. Stoichiometry of the KCNQ1–KCNE1 ion channel complex. Proc Natl Acad Sci USA. 2010;107:18862–18867. doi: 10.1073/pnas.1010354107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naydenov NG, Brown B, Harris G, Dohn MR, Morales VM, Baranwal S, Reynolds AB, Ivanov AI. A membrane fusion protein alphaSNAP is a novel regulator of epithelial apical junctions. PLoS One. 2012a;7:e34320. doi: 10.1371/journal.pone.0034320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naydenov NG, Harris G, Brown B, Schaefer KL, Das SK, Fisher PB, Ivanov AI. Loss of soluble N-ethylmaleimide-sensitive factor attachment protein alpha (alphaSNAP) induces epithelial cell apoptosis via down-regulation of Bcl-2 expression and disruption of the Golgi. J Biol Chem. 2012b;287:5928–5941. doi: 10.1074/jbc.M111.278358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negulescu PA, Machen TE. Intracellular ion activities and membrane transport in parietal cells measured with fluorescent dyes. Methods Enzymol. 1990;192:38–81. doi: 10.1016/0076-6879(90)92062-i. [DOI] [PubMed] [Google Scholar]
- Palty R, Isacoff EY. Cooperative binding of stromal interaction molecule 1 (STIM1) to the N and C termini of calcium release-activated calcium modulator 1 (Orai1) J Biol Chem. 2016;291:334–341. doi: 10.1074/jbc.M115.685289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park CY, Hoover PJ, Mullins FM, Bachhawat P, Covington ED, Raunser S, Walz T, Garcia KC, Dolmetsch RE, Lewis RS. STIM1 clusters and activates CRAC channels via direct binding of a cytosolic domain to Orai1. Cell. 2009;136:876–890. doi: 10.1016/j.cell.2009.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penna A, Demuro A, Yeromin AV, Zhang SL, Safrina O, Parker I, Cahalan MD. The CRAC channel consists of a tetramer formed by Stim-induced dimerization of Orai dimers. Nature. 2008;456:116–120. doi: 10.1038/nature07338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senning EN, Gordon SE. Activity and Ca(2)(+) regulate the mobility of TRPV1 channels in the plasma membrane of sensory neurons. Elife. 2015;4:e03819. doi: 10.7554/eLife.03819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonson PD, Deberg HA, Ge P, Alexander JK, Jeyifous O, Green WN, Selvin PR. Counting bungarotoxin binding sites of nicotinic acetylcholine receptors in mammalian cells with high signal/noise ratios. Biophys J. 2010;99:L81–L83. doi: 10.1016/j.bpj.2010.08.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stap J, Van Marle J, Van Veen HA, Aten JA. Coating of coverslips with glow-discharged carbon promotes cell attachment and spreading probably due to carboxylic groups. Cytometry. 2000;39:295–299. doi: 10.1002/(sici)1097-0320(20000401)39:4<295::aid-cyto7>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- Stathopulos PB, Schindl R, Fahrner M, Zheng L, Gasmi-Seabrook GM, Muik M, Romanin C, Ikura M. STIM1/Orai1 coiled-coil interplay in the regulation of store-operated calcium entry. Nat Commun. 2013;4:2963. doi: 10.1038/ncomms3963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suleiman H, Zhang L, Roth R, Heuser JE, Miner JH, Shaw AS, Dani A. Nanoscale protein architecture of the kidney glomerular basement membrane. Elife. 2013;2:e01149. doi: 10.7554/eLife.01149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szymborska A, de Marco A, Daigle N, Cordes VC, Briggs JA, Ellenberg J. Nuclear pore scaffold structure analyzed by super-resolution microscopy and particle averaging. Science. 2013;341:655–658. doi: 10.1126/science.1240672. [DOI] [PubMed] [Google Scholar]
- Thompson JL, Shuttleworth TJ. How many Orai’s does it take to make a CRAC channel. Sci Rep. 2013;3:1961. doi: 10.1038/srep01961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulbrich MH, Isacoff EY. Subunit counting in membrane-bound proteins. Nat Methods. 2007;4:319–321. doi: 10.1038/NMETH1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vig M, Beck A, Billingsley JM, Lis A, Parvez S, Peinelt C, Koomoa DL, Soboloff J, Gill DL, Fleig A, et al. CRACM1 multimers form the ion-selective pore of the CRAC channel. Curr Biol. 2006a;16:2073–2079. doi: 10.1016/j.cub.2006.08.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vig M, DeHaven WI, Bird GS, Billingsley JM, Wang H, Rao PE, Hutchings AB, Jouvin MH, Putney JW, Kinet JP. Defective mast cell effector functions in mice lacking the CRACM1 pore subunit of store-operated calcium release-activated calcium channels. Nat Immunol. 2008;9:89–96. doi: 10.1038/ni1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vig M, Kinet JP. The long and arduous road to CRAC. Cell Calcium. 2007;42:157–162. doi: 10.1016/j.ceca.2007.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vig M, Peinelt C, Beck A, Koomoa DL, Rabah D, Koblan-Huberson M, Kraft S, Turner H, Fleig A, Penner R, et al. CRACM1 is a plasma membrane protein essential for store-operated Ca2+ entry. Science. 2006b;312:1220–1223. doi: 10.1126/science.1127883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Brautigan DL. alpha-SNAP inhibits AMPK signaling to reduce mitochondrial biogenesis and dephosphorylates Thr172 in AMPKalpha in vitro. Nat Commun. 2013;4:1559. doi: 10.1038/ncomms2565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Schnitzbauer J, Hu Z, Li X, Cheng Y, Huang ZL, Huang B. Localization events-based sample drift correction for localization microscopy with redundant cross-correlation algorithm. Opt Express. 2014;22:15982–15991. doi: 10.1364/OE.22.015982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu MM, Covington ED, Lewis RS. Single-molecule analysis of diffusion and trapping of STIM1 and Orai1 at endoplasmic reticulum-plasma membrane junctions. Mol Biol Cell. 2014;25:3672–3685. doi: 10.1091/mbc.E14-06-1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Z, Liu Y. Reliable and global measurement of fluorescence resonance energy transfer using fluorescence microscopes. Biophys J. 2001;81:2395–2402. doi: 10.1016/S0006-3495(01)75886-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Chang H, Zhang Y, Yu J, Wu L, Ji W, Chen J, Liu B, Lu J, Liu Y, et al. Rational design of true monomeric and bright photoactivatable fluorescent proteins. Nat Methods. 2012;9:727–729. doi: 10.1038/nmeth.2021. [DOI] [PubMed] [Google Scholar]
- Zheng H, Zhou MH, Hu C, Kuo E, Peng X, Hu J, Kuo L, Zhang SL. Differential roles of the C and N termini of Orai1 protein in interacting with stromal interaction molecule 1 (STIM1) for Ca2+ release-activated Ca2+ (CRAC) channel activation. J Biol Chem. 2013;288:11263–11272. doi: 10.1074/jbc.M113.450254. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.