

The precise arrangement of the N-terminal actin- and tropomyosin-binding sites within leiomodin-2 is determined. A novel model is given of the roles of leiomodin-2 at thin filament pointed ends.
Abstract
Leiomodin is a potent actin nucleator related to tropomodulin, a capping protein localized at the pointed end of the thin filaments. Mutations in leiomodin-3 are associated with lethal nemaline myopathy in humans, and leiomodin-2–knockout mice present with dilated cardiomyopathy. The arrangement of the N-terminal actin- and tropomyosin-binding sites in leiomodin is contradictory and functionally not well understood. Using one-dimensional nuclear magnetic resonance and the pointed-end actin polymerization assay, we find that leiomodin-2, a major cardiac isoform, has an N-terminal actin-binding site located within residues 43–90. Moreover, for the first time, we obtain evidence that there are additional interactions with actin within residues 124–201. Here we establish that leiomodin interacts with only one tropomyosin molecule, and this is the only site of interaction between leiomodin and tropomyosin. Introduction of mutations in both actin- and tropomyosin-binding sites of leiomodin affected its localization at the pointed ends of the thin filaments in cardiomyocytes. On the basis of our new findings, we propose a model in which leiomodin regulates actin polymerization dynamics in myocytes by acting as a leaky cap at thin filament pointed ends.
INTRODUCTION
Filamentous actin is a major component of thin filaments in muscle cells. Leiomodin (Lmod) is an actin-binding protein and the only known homologue of tropomodulin (Tmod), a capping protein found at the slow-growing (pointed) end of the actin filament (Conley et al., 2001). In contrast to the weak actin-nucleating ability of Tmod (Fowler et al., 2003; Fischer et al., 2006; Yamashiro et al., 2014), Lmod is a potent actin nucleator in vitro (Chereau et al., 2008; Boczkowska et al., 2015; Yuen et al., 2015). Whereas Tmod inhibits polymerization and depolymerization of actin filament pointed ends (Weber et al., 1994), Lmod elongates thin filaments at their pointed ends (Tsukada et al., 2010; Pappas et al., 2015). Together with Tmod, Lmod modulates the length of the filaments in a tropomyosin (Tpm)-dependent manner (Chereau et al., 2008; Tsukada et al., 2010). In our model, Lmod competes with Tmod for binding to actin filaments at the pointed ends, which antagonizes Tmod’s actin-capping function, resulting in elongation of the filaments (Tsukada et al., 2010).
There are three Lmod isoforms with different expression patterns. Lmod1 is predominantly expressed in tissues containing smooth muscles (Conley et al., 2001) and is also found in extraocular muscle (voluntary striated muscle; Conley, 2001). Lmod2 and Lmod3 are found mainly in cardiac and skeletal muscle (Conley et al., 2001; Nanda and Miano, 2012; Yuen et al., 2015). Lmod2 expression is higher in cardiac muscle than in skeletal muscle (Yuen et al., 2015). Lmod2 is highly expressed in mature cultured cardiomyocytes and therefore was suggested to play an important role in maintenance and repair of thin filaments in cardiac muscle rather than contribute to early premyofibril assembly (Skwarek-Maruszewska et al., 2010). Recently it was found that Lmod2 knockout results in severe dilated cardiomyopathy in mice and their death 3 wk after birth (Pappas et al., 2015). Several mutations in Lmod3 result in severely disrupted sarcomeric organization in skeletal muscle and are linked to lethal nemaline myopathy in humans (Yuen et al., 2015).
The Lmod2 isoform is the best characterized and least divergent from Tmod proteins (Conley et al., 2001; Yuen et al., 2015). Lmod2 shares ∼50% sequence identity with the first 100 N-terminal residues of Tmod1. This region in Tmod1 contains a Tpm-binding site (residues 1–38) and an actin-capping site (residues 48–92; Greenfield et al., 2005; Kostyukova et al., 2005, 2006). Based on sequence homology, the Tpm-binding site and the first actin-binding site in Lmod2 are located at residues 5–42 and 43–95, respectively. The presence of the Tpm-binding site in Lmod2 was confirmed by circular dichroism (Kostyukova, 2007). However, recent isothermal titration calorimetry data suggest that the actin-binding site does not exist in Lmod1 and Lmod2 and that this site is only conserved in the Tmods (Boczkowska et al., 2015).
The region composed of ∼100 residues between the putative N-terminal actin-binding site in Lmod2 and the leucine-rich repeat (LRR) domain has very low homology not only between Lmod2 and the Tmods but also between Lmod isoforms. In the Tmods, this region contains a second Tpm-binding site. Attempts to localize the second Tpm-binding site in Lmod2 based on the secondary structure prediction were unsuccessful (Kostyukova, 2007). Despite this, it was shown by blot overlay that bacterially expressed Lmod1 lacking the first 30 N-terminal amino acids (and therefore the first Tpm-binding site) is able to bind striated α-tropomyosin, which is expressed in cardiac and skeletal muscles (Conley, 2001). Therefore it is still not known whether Lmod has a second Tpm-binding site.
In this study, we analyze the Tpm- and actin-binding properties of the N-terminal region of Lmod2 using several in vitro and cell culture approaches. We provide additional evidence that Lmod2 contains only one Tpm-binding site, which is necessary for Lmod2’s pointed-end localization and thin filament elongation in cardiomyocytes. We find the presence of an N-terminal actin-binding site that influences Lmod2’s assembly and dynamics at the pointed end. We provide evidence of additional actin-binding interactions within residues 124–201 of Lmod2. On the basis of our findings, we propose a model in which Tmod1 functions as a tight cap and Lmod2 acts as a leaky cap at the pointed end of the thin filament. In this model, Lmod2 allows actin to polymerize at the pointed end, although at a lower, and consequently more easily regulated, rate than in the absence of these proteins.
RESULTS
Lmod2 fragments 1–94 and 1–201 differentially inhibit actin polymerization
Previously we demonstrated that when the WH2 domain is removed, truncated Lmod2 (Lmod2 [1-514]) inhibits pointed-end actin polymerization, similar to Tmod1 (Tsukada et al., 2010). The difference is that in saturation, Lmod2 [1-514] inhibits 75–80% of what is possible to achieve with Tmod1. The ability to measure potent inhibitory activity of truncated Lmod2 was exploited in pointed-end polymerization assays to study the actin-binding ability of Lmod2 N-terminal fragments Lmod2 [1-94] and Lmod2 [1-201]. They were chosen based on sequence alignment with fragments Tmod1 [1-92] and Tmod1 [1-159] (Figure 1), which were previously used to study Tpm- and actin-binding sites within Tmod1 (Greenfield et al., 2005; Kostyukova et al., 2005, 2006). One would predict no difference in the ability of Lmod2 [1-94] and Lmod2 [1-201] to inhibit actin polymerization, provided that a second Tpm-binding site (Tpm2) is not present in Lmod2 and the arrangement of the first Tpm-binding site (Tpm1) and first actin-binding site (A1) in Lmod2 is the same as in Tmod1.
FIGURE 1:

(A) Domain structure of Tmod1 and Lmod2 based on biochemical and structural analysis of Tmod1 and sequence homology of Lmod2 to Tmod1. A1, A2, and WH2 are actin-binding sites; Tpm1 and Tpm2 are Tpm-binding sites; LRR is the leucine-rich repeat domain; and P is the proline-rich domain. Truncated fragments are indicated by lines. (B) Amino acid sequence alignment of N-terminal fragments for Lmod2 and Tmod1 made using MultAlin. Identical amino acids in the alignment are shaded. The prediction of the secondary structure in Lmod2 was made using Jpred. α-Helical regions are shown in bold italic. Arrowheads and vertical dashed lines show the end of Lmod2 fragment 1–94 and Tmod1 fragment 1–92. Arrows indicate the residues mutated in Lmod2. Lmod2 residues 45–51 are shown in bold underlined.
We prepared actin seeds (short actin filaments capped at their barbed ends by gelsolin) and studied polymerization of G-actin on the seeds in the presence of Lmod2 [1-94] and Lmod2 [1-201]. When striated muscle α-tropomyosin, or Tpm1.1 according to a recently introduced nomenclature (Geeves et al., 2015), was added to the seeds at a saturating concentration, both Lmod2 [1-94] and Lmod2 [1-201] inhibited pointed-end polymerization (Figure 2). Surprisingly, we observed that Lmod2 [1-201] had a stronger inhibitory effect than Lmod2 [1-94], suggesting that the arrangement of binding sites in Lmod2’s N-terminal region differs from what had been proposed (Chereau et al., 2008; Boczkowska et al., 2015).
FIGURE 2:

Both Lmod2 [1-94] and Lmod2 [1-201] inhibit pointed-end polymerization. Effect of 0.2 μM Lmod2 fragments in the absence (A) and presence (B) of a saturating concentration of Tpm1.1 (1 μM) on the polymerization of 1.1 μM G-actin at the pointed end of gelsolin-capped actin filaments. Control, actin alone in the absence of Tpm1.1 and Lmod2 fragments. (C) Dependence of inhibition of actin polymerization on the concentration of Lmod2 fragments in the presence of 1 μM Tpm1.1. (D) The inhibition of actin polymerization by 0.2 μM Lmod2 fragments calculated as Rexp/Rcontrol. Initial rates (R) were determined as the first derivatives at time 0 after fitting. Asterisks indicate statistically significant groups determined using one-way ANOVA with Tukey–Kramer post hoc test. All values are mean ± SD (n = 3–6); *p < 0.05, **p < 0.01, ***p < 0.001.
Of interest, in the absence of Tpm1.1, Lmod2 [1-201] caused slight but statistically significant decrease of actin polymerization, whereas Lmod2 [1-94] had no effect (Figure 2). This indicates that residues 95–201 may provide previously unrecognized additional interactions with actin, and these interactions are not Tpm dependent.
Nucleation ability of Lmod2 [1-94] and Lmod2 [1-201] was tested (Supplemental Figure S1). As expected, both Lmod2 [1-94] and Lmod2 [1-201], which lack LRR and WH2 domains, had no concentration-dependent nucleating activity, which indicates that they do not bind more than one G-actin molecule.
Discovery of an actin-binding site within the N-terminal region of Lmod2
The ability of N-terminal fragments of Lmod2 to inhibit actin polymerization at the pointed end suggests that there is at least one actin-binding site within the N-terminal region of Lmod2. In contrast, it was suggested that Lmod2 lacks an N-terminal actin-binding site (Boczkowska et al., 2015). To resolve this contradiction, we used nuclear magnetic resonance (NMR) to study interactions between G-actin and Lmod2 fragments. NMR is a well-established method to detect micromolar to millimolar binding between proteins and ligands using differential resonance peak perturbation (Williamson, 2013). To establish the presence of an actin-binding site in the N-terminal region of Lmod2, we investigated the effects of G-actin on NMR spectra of the Lmod2 fragments. The presence of G-actin added to a peptide creates equilibrium between the free and bound peptide, affecting its spectrum. Using an excess molar peptide/actin ratio (e.g., 10–30/1) allows us to see mainly signals from the peptide, which are affected by actin only if binding exists.
To map the putative N-terminal actin-binding site, we divided the N-terminal region of Lmod2 into several overlapping peptides. Residues shown to bind Tpm (Kostyukova, 2007) were excluded. Each fragment was designed to include 10 overlapping residues with adjacent fragments to minimize the chance of disruption of a possible actin-binding site.
The fragments Lmod2 [45-94], Lmod2 [85-123], Lmod2 [114-150], Lmod2 [140-180], and Lmod2 [172-210] were subcloned and expressed. We successfully purified all of the fragments with the exception of Lmod2 [114-150]. Owing to low expression and high content of Glu and Asp (54.1% Glu and 21.6% Asp), we were unable to obtain Lmod2 [114-150] in amounts necessary for NMR experiments. However, it is unlikely that this fragment contains sites of specific interactions, due to its low sequence complexity.
One-dimensional (1D) proton NMR spectra of the fragments Lmod2 [45-94], Lmod2 [85-123], Lmod2 [140-180], and Lmod2 [172-210] were recorded in the presence and absence of G-actin. Adding G-actin resulted in drastic spectral changes in the NMR spectrum of Lmod2 [45-94], indicating the presence of a continuous stretch of interacting residues in the peptide (Figure 3). Figure 3 displays two spectral regions, one corresponding to the aromatic/amide proton resonances (top) and the other to the side chain proton (nonaromatic, noncarboxamide) resonances (bottom). In the presence of actin, many resonance peaks experienced differential broadening/shifts. The peak broadening led to a marked loss of peak resolution and height (e.g., see groups of peaks at ∼0.7, 0.95, 1.3, 1.55, 6.65, 7.1, 7.4, 7.75, and 7.85 ppm). The marked differential broadening of both amide and side chain proton peaks is typical of specific interactions between proteins and their peptide ligands with Kd values in the micromolar range (Ni et al., 1990, 1995; Yu et al., 1998; Tolkatchev et al., 2003). This result is consistent with the high degree of homology between Lmod2 [45-94] and the first actin-binding site in Tmod1 (Kostyukova et al., 2005; Figure 1B). Together with the results of the polymerization assays, our data indicate that the N-terminal region of Lmod2 may bind to G-actin with a lower affinity than to F-actin.
FIGURE 3:
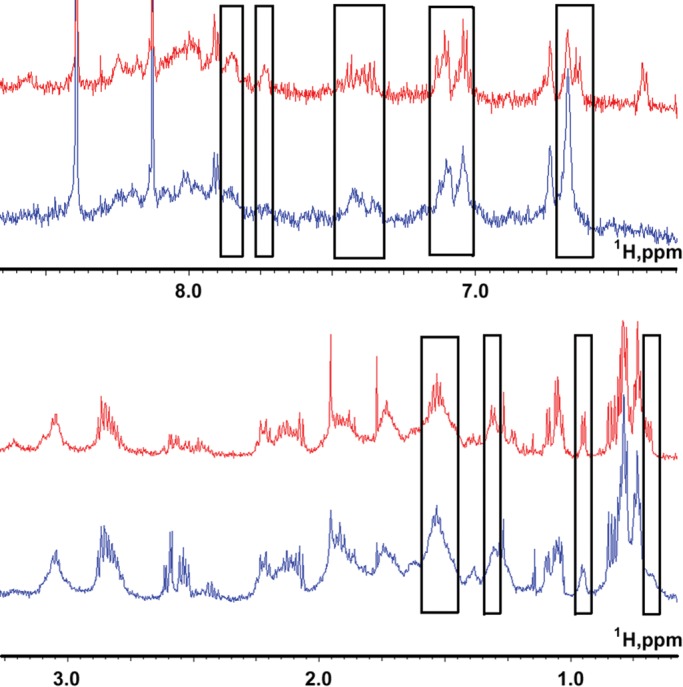
Formation of a complex between the fragment Lmod2 [45-94] and G-actin. NMR proton spectra of 0.2 mM Lmod2 [45-94] in the absence (red) and presence (blue) of 10 μM G-actin in the aromatic/amide (top) and side-chain (bottom) chemical shift regions in 2 mM potassium phosphate, 0.1 mM ATP, 0.1 mM CaCl2, and 0.01% NaN3 at pH 6.8. In the presence of actin, a large number of resonance peaks manifested differential broadening/shifts (regions with broadened peaks are boxed). The NMR spectra were recorded at 25°C.
There were no observable differences in NMR spectra of Lmod2 [85-123] in the presence and absence of G-actin (Supplemental Figure S2). Therefore the fragment Lmod2 [85-123] cannot account for differences in actin-binding abilities between fragments Lmod2 [1-94] and Lmod2 [1-201].
We observed subtle changes in the aromatic/amide chemical shift regions of the spectra of fragments Lmod2 [140-180] and Lmod2 [172-210] (Supplemental Figures S3 and S4), indicating a possibility that some of the amino acid residues of the Lmod2 region [124-201] participate in interactions with actin. The fragment Lmod2 [172-210] contains two residues, N207 and T209, that correspond to residues N179 and T181, respectively, in Tmod1 and are conserved in the Tmod family. The residue N179 in Tmod1 has been shown to interact with actin (Rao et al., 2014), and therefore some of the changes in the NMR spectra of Lmod2 [172-210] may be attributed to interaction of N207 with actin. Other spectral changes (e.g., in Lmod2 [140-180]) are consistent with our observation that removal of residues 95–201 reduces the actin-binding ability of N-terminal fragments of Lmod2 in the absence of Tpm. We propose the existence of additional interactions with actin within residues 124–201 of Lmod2.
Because changes in the spectra of Lmod2 [140-180] and Lmod2 [172-210] were comparatively small, we were concerned that dividing the region represented by residues 140–201 into shorter peptides damaged the additional actin-binding site. We produced a continuous Lmod2 fragment (residues 103–201) that partly overlaps with Lmod2 [85-123]. Because the fragment Lmod2 [103-201] is longer, we did not expect that its proton resonance peaks could be resolved sufficiently well in 1D proton NMR spectra. To improve spectral resolution, we generated 15N-labeled Lmod2 [103-201] and recorded two-dimensional (2D) 15N–heteronuclear single quantum coherence (HSQC) spectra. Unfortunately, even in the labeled sample, about two-thirds of the peaks overlapped. Among the resolvable peaks, we did not observe spectral changes in the presence of actin (Supplemental Figure S5). This region may interact, however, with actin via a few residues located apart from each other, which are difficult to resolve on the 2D 15N-HSQC spectra.
Lmod2 [1-201] binds one Tpm molecule
To complete the mapping of binding sites in the N-terminal region of Lmod2, we studied the Tpm-binding properties of Lmod2 [1-201]. The N-terminal Tpm-binding site in Lmod2 is highly homologous to the first Tpm-binding site in Tmod1 (Kostyukova, 2007). In the Tmod1 [1-92] fragment, which contains only the first Tpm-binding site (Figure 1), the mutation of Leu-27 to Glu (L27E) destroys the formation of the hydrophobic surface in the amphipathic helix and results in loss of Tpm binding (Greenfield et al., 2005). However, the same amino acid substitution did not prevent Tpm from binding to a longer Tmod1 fragment [1-159] and full-length Tmod1 due to the presence of a second Tpm-binding site (Kostyukova et al., 2006). We introduced the homologous mutation into the Lmod2 [1-201] fragment, changing Leu-30 to Glu (L30E; Figure 1B).
Previously we demonstrated that Lmod binds both short and long Tpm isoforms (Kostyukova, 2007). We tested the ability of Lmod2 [1-201] and Lmod2 [1-201(L30E)] to form complexes with αTM1bzip, a chimeric peptide that contains the binding site of short Tpm isoforms (19 N-terminal residues encoded by exon 1b of the Tpm1 gene) for Tmod and Lmod (Kostyukova, 2007), using native gel electrophoresis. This 19-residue sequence is different from that of Tpm1.1, which is encoded by exon 1a of the Tpm1 gene (Geeves et al., 2015). The use of this peptide is preferred in this assay because complex formation with Tpm1.1 cannot be detected in native gel electrophoresis due to its low melting temperature and dissociation during electrophoresis.
Lmod2 [1-201] and Lmod2 [1-201(L30E)] were mixed with αTM1bzip in 1:1 and 1:2 M ratios and loaded onto a native polyacrylamide gel (Figure 4). αTM1bzip is positively charged and does not enter the gel. Binding of αTM1bzip changes the mobility of Lmod2 fragments in a gel, and a shift of the Lmod2 band should be observed when the complex is formed. An additional band appeared when αTM1bzip was mixed with wild type but not mutated Lmod2 [1-201]. Therefore the wild-type fragment Lmod2 [1-201] binds αTM1bzip, whereas the mutant fragment Lmod2 [1-201(L30E)] does not.
FIGURE 4:
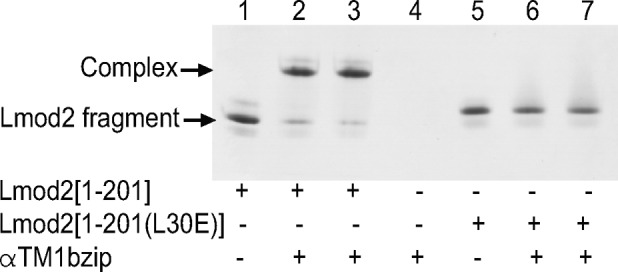
Complex formation between αTM1bzip and Lmod2 [1-201] or Lmod2 [1-201(L30E)] monitored by native (nondenaturing) PAGE. αTM1bzip/Lmod2 molar ratio was 0:1 (lanes 1 and 5), 1:1 (lanes 2 and 6), 2:1 (lanes 3 and 7), and 1:0 (lane 4). Concentration of Lmod2 fragments was 5 μM. αTM1bzip is positively charged and does not enter the gel. Arrows indicate Lmod2 fragments and the complex.
Further, Lmod2 [1-201] was titrated by αTM1bzip (Figure 5). The decrease of free Lmod2, as well as the complex formation in the mixtures, was monitored by scanning and quantifying bands in the native polyacrylamide gel. The position of the complex band did not change at all Tpm/Lmod2 molar ratios. The density of the complex band increased and the density of Lmod2 [1-201] band decreased between 0.125 and 1 molar ratio and remained unchanged at higher molar ratios. These results demonstrate that each molecule of Lmod2 [1-201] binds only one Tpm molecule, and the binding affinity is abolished if Leu30 located in Tpm1 is replaced with Glu, confirming that there is only one Tpm-binding site in Lmod2.
FIGURE 5:
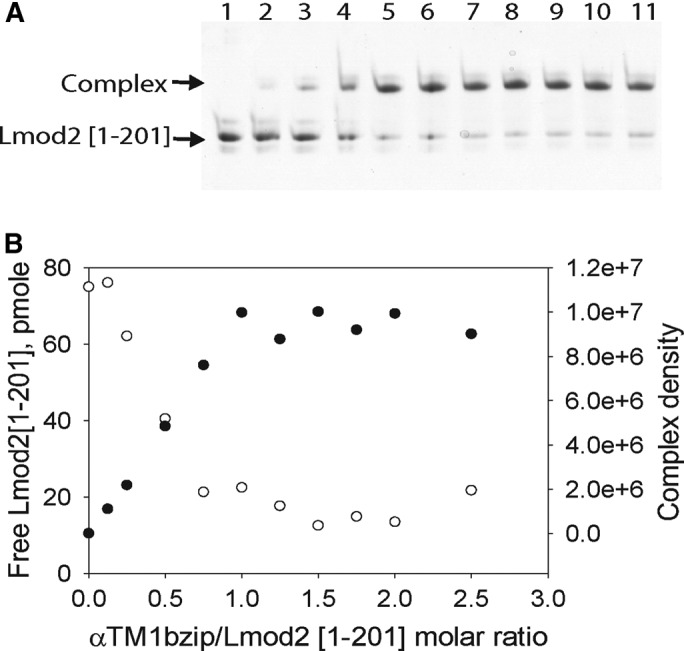
Titration of Lmod2 [1-201] by αTM1bzip. A stock solution of Lmod2 [1-201] was diluted to a final concentration of 5 μM with different concentrations of Tpm peptide in 20 mM Tris-HCl, pH 7.4. The decrease of free Lmod2 [1-201] and the increase of the complex were monitored by scanning and quantifying the Lmod2 [1-201] band in native polyacrylamide gels. (A) 9% native gel. (B) Dependence of the amount of free Lmod2 [1-201] (○) and density of the complex band (●) on the amount of αTM1bZip added. Lanes on the gel correspond to points on the graph. Lanes 1–11 contain αTM1bzip/Lmod2 in ratios of 0, 0.125, 0.25, 0.5, 0.75, 1.0, 1.25, 1.5, 1.75, 2.0, and 2.5, respectively.
To analyze further the locations of the Tpm- and actin-binding sites within the N-terminal region of Lmod2, we performed pyrene-actin polymerization assays in the presence of full-length Tpm1.1 and Lmod2 [1-201] containing mutations in these sites.
Mutations L30E and W73D decrease the actin-binding ability of Lmod2 [1-201] in the presence of Tpm1.1
Using native gel electrophoresis, we did not detect any binding of Lmod2 [1-201(L30E)] with αTM1bzip. Here Lmod2 [1-201(L30E)] was tested in the pyrene-actin polymerization assay in the presence of Tpm1.1. The inhibition of pointed-end actin polymerization by Lmod2 [1-201(L30E)] was approximately fivefold lower than that of Lmod2 [1-201]. The inhibitory activity of Lmod2 [1-201] reached 50% at a concentration ∼50 nM, whereas Lmod2 [1-201(L30E)] reached 50% inhibition at a concentration of ∼500 nM (Figure 6, A and C). Although binding of Tpm is important for the inhibitory effect, the L30E mutation does not eliminate it completely.
FIGURE 6:
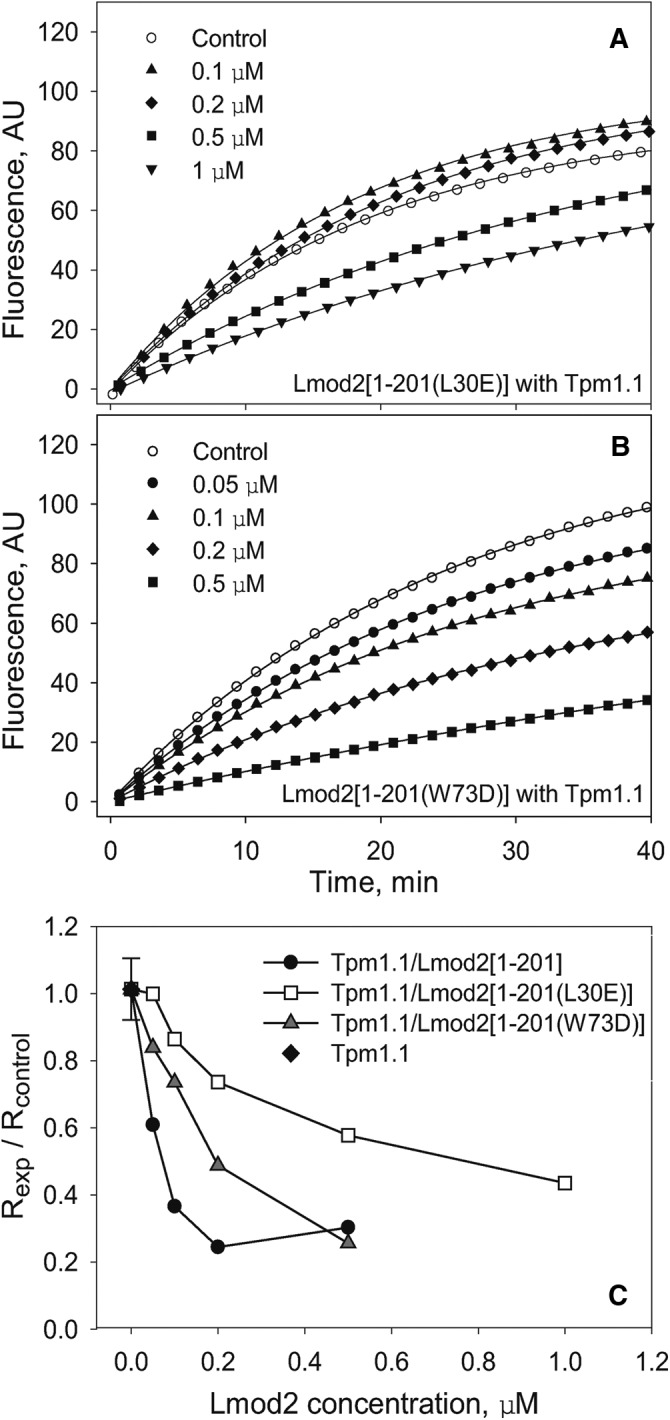
Mutations L30E and W73D reduce the capping ability of the Lmod2 [1-201] truncated fragment. Polymerization of 1.1 μM G-actin at the pointed end of gelsolin-capped actin filaments was measured in the presence of a saturating concentration of Tpm (1 μM) and various concentrations of Lmod2 [1-201(L30E)] (A) and Lmod2 [1-201(W73D)] (B). Control, actin alone in the absence of Tpm1.1 and Lmod2 fragments. (C) Dependence of inhibition of actin polymerization on concentration of Lmod2 [1-201] fragments, wild type, and with the mutations. Initial rates (R) were calculated as the first derivatives at time 0 after fitting. The inhibition of polymerization was calculated as Rexp/Rcontrol.
To confirm further the presence of the functional actin-binding site that we localized to residues 45–94 by NMR, we mutated Trp-73 to Asp (W73D) in Lmod2 [1-201]. The position of Trp-73 in Lmod2 corresponds to Leu-71 of Tmod1 (Figure 1B). Mutating Leu-71 to Asp decreased the actin-capping ability of Tmod (Kostyukova et al., 2005). In pointed-end polymerization experiments, introduction of W73D into Lmod2 [1-201] caused threefold decrease of the ability of the fragment to inhibit actin polymerization (50% inhibition reached at ∼150 nM; Figure 6, B and C), suggesting that Trp73 belongs to the actin-binding site. However, the effect of the W73D mutation is lower than that of the L30E mutation.
In addition, we introduced the W73D mutation into full-length Lmod2. Lmod2 (W73D) was purified and tested in actin nucleation assays. The mutation had almost no effect on the nucleation ability of Lmod2; Lmod2 (W73D) nucleated actin similarly to wild-type Lmod2.
Mutations L30E and W73D affect Lmod2’s assembly and function in neonatal rat cardiomyocytes
To study the functional effects of mutating the first Tpm- and actin-binding sites of Lmod2, we introduced L30E and W73D mutations into GFP-Lmod2. GFP, GFP-Lmod2, GFP-Lmod2 (L30E), and GFP-Lmod2 (W73D) were expressed in neonatal rat cardiomyocytes using adenovirus. Cells with similar expression levels were then analyzed by immunofluorescence microscopy to determine the localization of each construct and thin filament lengths (Figure 7).
FIGURE 7:
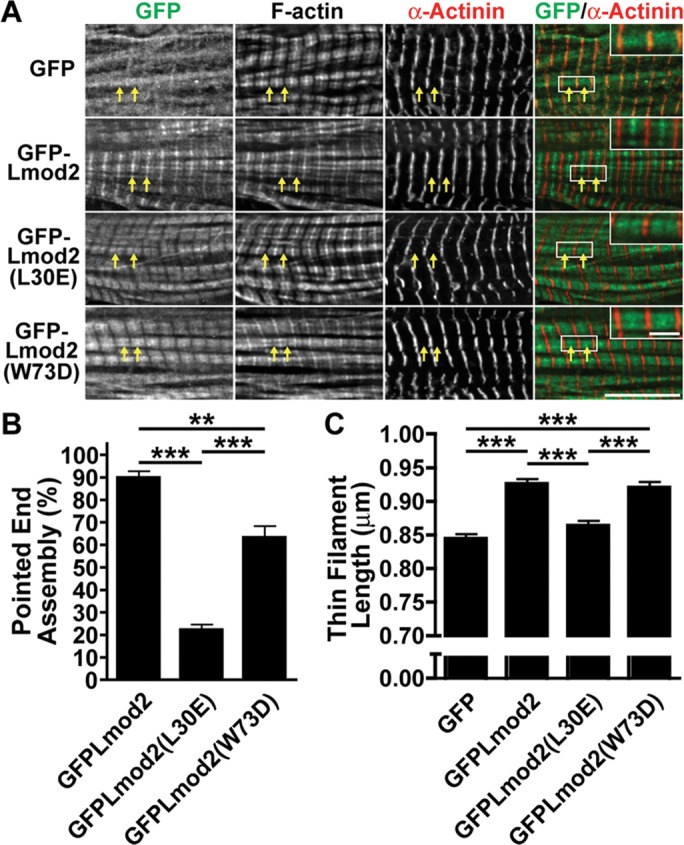
The N-terminal TM- and actin-binding domains of Lmod2 contribute to its localization and thin filament elongation function. (A) Representative images of rat neonatal cardiomyocytes expressing GFP alone, GFP-Lmod2, GFP-Lmod2 (L30E), or GFP-Lmod2 (W73D) stained with phalloidin to label F-actin and for sarcomeric α-actinin to label the Z-disks. Arrows mark pointed-end assembly. Scale bar, 10 μm. Insets in GFP/α-actinin merged images represent twofold magnification to highlight pointed-end assembly of each construct. Scale bar, 2 μm. (B) Pointed-end assembly of cytosol-extracted rat neonatal cardiomyocytes transduced with GFP, GFP-Lmod2, GFP-Lmod2 (L30E), and GFP-Lmod2 (W73D). Graph shows the percentage of cardiomyocytes demonstrating pointed-end assembly (mean ± SEM); 300 total counts from ∼100 cells/culture, three cultures; **p < 0.01, ***p < 0.001, one-way ANOVA. (C) Thin filament lengths of rat neonatal cardiomyocytes transduced with GFP, GFP-Lmod2, GFP-Lmod2 (L30E), and GFP-Lmod2 (W73D). All values are mean ± SEM; 54–66 total measurements from ∼10 cells/culture, 3-4 cultures; ***p < 0.001, one-way ANOVA.
GFP-Lmod2 and GFP-Lmod2 (W73D) both localize to the pointed end and along the length of the thin filaments but are absent from the Z-disk (Figure 7A). However, GFP-Lmod2 (W73D) does not visibly assemble as well at the pointed end as does wild type (Figure 7A). GFP-Lmod2 (L30E) also assembles along the filament but displays very little pointed-end localization (Figure 7A). Note that assembly of GFP-Lmod2 along the length of the thin filament was previously observed and is likely nonspecific or of low affinity (Tsukada et al., 2010; Boczkowska et al., 2015). Therefore, to aid in quantifying the location of the GFP-Lmods, the cytosol of cardiomyocytes was extracted before fixation, which removes nonspecific sarcomere/cytoskeleton associations. GFP-Lmod2 assembles at the pointed end of the thin filament in 90 ± 2.9% of cardiomyocytes analyzed. Introduction of the W73D mutation or the L30E mutation significantly reduces assembly at the pointed end of the thin filaments to 63 ± 5.2% or 22 ± 2.4% of cardiomyocytes analyzed, respectively (Figure 7B).
The average thin filament length increases upon Lmod2 expression (Figure 7C), as we previously reported (Tsukada et al., 2010). The W73D mutation does not significantly alter the ability of Lmod2 to elongate thin filaments (GFP-Lmod2 [W73D], 0.92 ± 0.009 μm; GFP-Lmod2, 0.93 ± 0.007 μm). However, the presence of the L30E mutation abolishes Lmod2-induced thin filament elongation (GFP-Lmod2 [L30E], 0.87 ± 0.007 μm; GFP, 0.84 ± 0.008 μm; Figure 7C).
To study the effect of the W73D mutation on exchange of Lmod2 molecules, we performed fluorescence recovery after photobleaching (FRAP) experiments. Rat neonatal cardiomyocytes transduced with either GFP-Lmod2 or GFP-Lmod2 (W73D) were photobleached in a rectangular area spanning nearly four sarcomeres and fluorescence recovery monitored at the pointed end for ∼1 min after photobleach (Supplemental Figure S6). Recovery was fitted using a single-exponential association equation for each cell. The mutant has a slight increase in the mean rate of recovery and a shorter mean half-time (Figure 8), suggesting that it exchanges more quickly than the wild type. A faster exchange is characteristic of a weaker affinity (Sprague et al., 2004). The wild-type and mutant Lmod2 have the same mobile fraction (i.e., the same fraction of exchangeable molecules).
FIGURE 8:
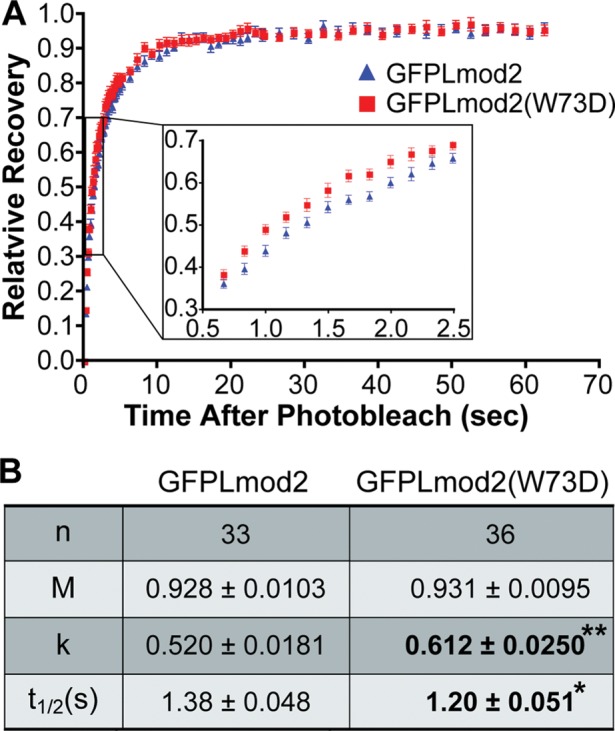
Fluorescence recovery after photobleaching of GFP-Lmod2 and GFP-Lmod2 (W73D). (A) Mean relative recovery after photobleaching over time for GFP-Lmod2 (blue triangles) and GFP-Lmod2 (W73D) (red squares). (B) Recovery data fit using nonlinear regression curves with the single-exponential association equation R = M[1 − exp(−kt)] for each individual cell, where R is the relative recovery of fluorescence at time t. Mean mobile fraction (M), rate (k), and half-time of recovery (t1/2) are indicated ± SEM. Bold indicates values that are statistically significantly different between GFPLmod2 and GFPLmod2 (W73D). n = 34–36 from three independent cultures; *p < 0.05, Student’s t test.
DISCUSSION
The role of Lmod2 in sarcomeres of cardiac muscle is under debate. There are data demonstrating its ability to act as a potent actin nucleator (Chereau et al., 2008; Tsukada et al., 2010; Boczkowska et al., 2015) and an elongation factor for thin filaments (Tsukada et al., 2010). Moreover, the location and roles of Lmod2’s TM- and actin-binding sites are being constantly revised (Chereau et al., 2008; Tsukada et al., 2010; Boczkowska et al., 2015). Assignment of binding sites is the fundamental basis for understanding Lmod2’s function in myocytes, and the aim of this study was to determine the arrangement of binding sites within the N-terminal region of Lmod2.
Recent work on the actin-binding properties of Lmod2 suggested that Lmod2 has no N-terminal actin-binding site (Boczkowska et al., 2015). In the present study, we discovered the presence of an actin-binding site immediately following the only Tpm-binding site in the N-terminal region of Lmod2 (residues 7–41; Colpan et al., 2016). Our NMR data show numerous interactions between actin and the Lmod2 fragment encompassing residues 45–94 and no interaction between actin and the Lmod2 fragment encompassing residues 85–123. Because, due to introduced charges of amine and carboxyl groups, terminal residues of an interacting fragment may lose interactions otherwise present in the entire protein, we can safely assign the actin-binding site to be within residues 43–90. Neighboring residues beyond this region are not likely to participate in actin binding. These data are corroborated by results of the pyrene-actin assays demonstrating that the W73D mutation decreases the ability of Lmod2 to inhibit actin polymerization and therefore binding at the pointed end of the actin filament.
Boczkowska et al. (2015) detected no interaction between actin and an Lmod2 peptide that included residues 52–103; taken together with our data, this indicates that Lmod2 residues 45–51 are critical for binding to actin. Residues 45–51 (NLPVGQR, underlined in Figure 1B) in Lmod2 correspond to residues 43–49 (LLPAGLR) in Tmod1 (Figure 1B), residues MLPAGFR in Tmod2 (accession number AF177169.1), residues LLPAGFR in Tmod3 (accession number AF177171.1), and residues LLPAGLR in Tmod4 (accession number AF177173.2), indicating that residues *LPAG*R are conserved in different members of tropomodulin family and important for the common function of the isoforms in this region. It is possible that excluding these residues in the peptides used in isothermal titration calorimetry experiments resulted in the decrease of actin binding for the Lmod2 peptide below the sensitivity of the technique (Boczkowska et al., 2015).
Our data also indicate that there are additional interactions with actin within residues 124–201. Based on the NMR data, actin binding in the region is not localized to a long, continuous stretch of residues but is instead mediated by interactions with a few individual, and most likely separated in sequence, residues. It is possible that these residues form transient yet biologically significant contacts with actin. The presence of these interactions is also supported by the increased ability of Lmod2 [1-201] to inhibit actin polymerization at the pointed end compared with the shorter Lmod2 fragment [1-94] both with and without Tpm1.1 (Figure 2). Whereas in the presence of Tpm1.1, maximal possible inhibition of actin polymerization by Lmod2 [1-201] was reached at 0.2 μM (Figure 6C), inhibition by Lmod2 [1-94] was weaker at the same concentration (Figure 2D). Moreover, even in the absence of Tpm1.1, Lmod2 [1-201] was able to decrease actin polymerization.
Although the mutation W73D decreased the ability of Lmod2 [1-201] to inhibit actin polymerization, it did not affect the actin nucleation activity of full-length Lmod2. However, recently it was shown that the double mutation Y72D/W73D caused a slight but visible decrease in nucleation ability (Chen et al., 2015). These observations agree with our mapping of an actin-binding site in the N-terminal region. In addition, removal of the whole N-terminal region up to the beginning of the LRR domain caused an even larger decrease of Lmod2’s nucleating ability, although much less than the removal of the WH2 domain (Chereau et al., 2008; Boczkowska et al., 2015). Together all of the data indicate that interaction of the N-terminal region of Lmod2 with actin is less important for actin nucleation but has an essential role in the previously demonstrated ability of Lmod2 to inhibit actin polymerization (Tsukada et al., 2010).
In cardiomyocytes in culture, assembly of Lmod2 (W73D) is reduced compared with wild-type Lmod2. Furthermore, in FRAP experiments, the W73D mutant displays a slight increase in rate of recovery at the pointed ends and a resulting decrease in half-time of recovery compared with the wild-type protein. The rate of recovery after photobleaching is indicative of either dissociation rate of a molecular complex (off-rate) or binding affinity (the ratio between association [on-rate] and off-rate [ Sprague et al., 2004]). Together these data suggest that the W73D mutation results in a decrease in the pointed-end binding affinity, consistent with the results of the in vitro actin polymerization experiments. However, it is not clear why, with an overall reduction in binding affinity/assembly, the W73D mutation is still able to elongate thin filaments as well as wild type. It is possible that due to the relatively high level of expression of the constructs in the cells, we are unable to detect a difference in activity between the W73D mutant and wild type. This is potentially supported by actin polymerization assays that show a fairly narrow range in concentration in which the W73D mutation and wild type show a difference in inhibition. Alternatively, maintenance of thin filament length may depend not only on the binding of Lmod2 but also on the rate of filament elongation when Lmod2 is bound. Depending on the balance between on-rate, off-rate, and rate of elongation, Lmod2 mutations may influence thin filament maintenance in a way that does not directly correlate with Lmod2 binding affinity. Thus, the W73D mutation could increase the elongation rate of bound filaments, possibly explaining why the mutant elongates thin filaments even though it does not assemble as well.
Based on our data, Lmod2 has only one Tpm-binding site and binds to one Tpm per molecule. Titration of Lmod2 [1-201] by αTM1bZip showed that no more than one Tpm peptide was bound by this Lmod2 fragment. The mutation L30E, which is predicted to disrupt the formation of an amphipathic helix within this site, resulted in loss of Tpm-binding ability and approximately fivefold decrease in actin-capping ability of Lmod2 [1-201]. Consistently, our cell data showed that the mutation L30E in Lmod2 led to a drastic decrease in assembly at the pointed end and the loss of Lmod2’s ability to increase thin filament lengths in cardiomyocytes. These results support that pointed-end assembly is necessary for Lmod2’s ability to increase thin filament length. It also suggests that the Tpm-binding site is necessary for Lmod2’s pointed-end assembly, which is similar to that seen for Tmod. Of interest, the comparable mutation in Tmod1 (L27E) reduces pointed-end assembly by only 25% (Tsukada et al., 2011), but the mutation L30E in Lmod2 reduces pointed end assembly in 68% of cardiomyocytes analyzed. This further supports the notion that, unlike Tmod1, Lmod2 does not have a second Tpm-binding site.
We propose a model in which Tmod1 functions as a tight cap at the pointed end and Lmod2 acts as a leaky cap. Lmod2 allows actin to polymerize at the pointed end, although at a lower, and therefore more easily controlled, rate than in the absence of these proteins. This is substantiated by our observation that only <80% inhibition can be achieved in actin polymerization assays with Lmod2 [1-201] (Figure 6) and Lmod2 [1-514] (Tsukada et al., 2010). According to this model, the Tpm-binding site is the major determinant of binding at the pointed end and competition with Tmod1, whereas the N-terminal actin-binding site ensures only limited inhibition at the end, allowing elongation of the filament.
MATERIALS AND METHODS
Plasmid construction
The constructs Lmod2 [1-94], Lmod2 [1-201], Lmod2 [1-201(L30E)], and Lmod2 [1-201(W73D)] were made using the previously described N-terminal histidine (His)-tagged chicken Lmod2 (cLmod2) construct (Tsukada et al., 2010). Reverse primers were designed to truncate the full-length cLmod2 and add a single alanine (GCA) to the C-terminus of Lmod2 (for efficient dithiothreitol [DTT]-induced cleavage). The product was inserted between NcoI and SapI within pTYB3 (New England BioLabs, Ipswich, MA), which fuses an intein-tag to the C-terminus of Lmod2. For expression in cardiomyocytes, mouse Lmod2 was cloned from cDNA generated from left ventricle tissue and the coding sequence inserted into pEGFP-C2 (Clontech, Mountain View, CA) using XhoI and HindIII restriction sites. Mutations (L30E and W73D) were introduced using a QuikChange Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA). All sequences were confirmed by DNA sequencing.
Synthetic DNA coding Gallus gallus Lmod2 (NP_001186644) and optimized for Escherichia coli expression was obtained from GenScript (Piscataway, NJ). DNA regions coding for amino acid residues 43–94, 85–123, 140–180, 172–210, and 103x201 were subcloned into the pET-21b(+) vector (EMD Millipore, Billerica, MA) between EcoRI and XhoI sites as MFH-fusion proteins (Tolkatchev et al., 2010). An enterokinase cleavage site (5′-GACGATGATGACAAG-3′, coding for DDDDK) was inserted between the coding regions for Lmod2 [43-94] and MFH. An alanine residue was also inserted at the beginning of Lmod2 [43-94] coding sequence to facilitate efficient enterokinase cleavage. For Lmod2 [85-123], Lmod2 [140-180], Lmod2 [172-210], and Lmod2 [103-201] fusion constructs, a cyanogen bromide cleavage site (5′-ATG-3′ coding for one methionine residue) was inserted between the Lmod2 coding regions and the MFH tag protein. The cleavage sites enabled the removal of the MFH tag from Lmod2 peptides. Primers for subcloning were purchased from Integrated DNA Technology (Coralville, IA). Subcloning enzymes and buffers were purchased from New England Biolabs (Ipswich, MA). DNA sequencing was confirmed by GENEWIZ (South Plainfield, NJ).
MultAlin (multalin.toulouse.inra.fr/multalin/; Corpet, 1988) and Jpred (www.compbio.dundee.ac.uk/www-jpred/; Drozdetskiy et al., 2015) were used for amino acid sequences alignment and secondary structure prediction, respectively.
Protein expression and purification
Lmod2 [1–94], Lmod2 [1-201], Lmod2 [1-201(L30E)], and Lmod2 [1-201(W73D)] were expressed and purified as described (Chereau et al., 2008). After purification, proteins were exchanged into 20 mM Tris-HCl, pH 7.4, for biochemical analysis.
Lmod2 [43-94], Lmod2 [85-123], Lmod2 [140-180], and Lmod2 [172-210] fusion proteins were expressed in regular Luria–Bertani (LB) medium. Lmod2 [114-150] was expressed in ZYP medium (Studier, 2005). Lmod2 [103-201] was expressed in M9 minimal medium with 28 mM (15NH4)2SO4 as the only nitrogen source. All cell pellets, except for those expressing the Lmod2 [140-180] construct, were solubilized in 8 M urea, 300 mM NaCl, 10 mM imidazole, and 50 mM sodium phosphate buffer at pH 8.0, followed by sonication at 50% amplitude for 5–6 min and centrifugation at 17,000 rpm for 30 min at 4°C. For the Lmod2 [140-180] construct, cell pellets were resuspended in a buffer containing 300 mM NaCl, 10 mM imidazole, 50 mM sodium phosphate at pH 6.9, 0.7 mM phenylmethylsulfonyl fluoride (PMSF), and one cOmplete Mini, EDTA-free tablet. The cell suspension was sonicated at 50% amplitude for 5–6 min, followed by centrifugation at 17,000 rpm for 30 min at 4°C. For all constructs, the supernatant was purified with His60 Ni Superflow (Clontech) and the fusion proteins were eluted with 250 mM imidazole. Lmod2 [85-123], Lmod2 [114-150], Lmod2 [140-180], Lmod2 [172-210], and Lmod2 [103-201] fusions were cleaved with CNBr at a 500:1 molar ratio and in 0.3 M HCl. Several trials showed that Lmod2 [85-123] and Lmod2 [140-180] were cleaved by CNBr more efficiently in 6 M GuHCl than in 8 M urea. After CNBr cleavage, each reaction was loaded onto reversed-phase chromatography (Sep-Pak C18; Waters, Milford, MA). All constructs were eluted with 30% acetonitrile and 0.1% trifluoroacetic acid (TFA), which were then frozen at −80°C and lyophilized overnight.
After Ni–nitriloacetic acid purification from cell lysates, Lmod2 [43-94] fusion protein was additionally purified with reverse-phased chromatography to remove imidazole and other salts before being cleaved by enterokinase at pH 6.8. The plasmid pET-15b-EK_C122S_His5 (entry number 49048) for expression of recombinant enterokinase was obtained from the nonprofit plasmid depository Addgene. Enterokinase was purified as described (Skala et al., 2013). Enterokinase cleavage reaction was controlled to reach ∼60–80% cleavage efficiency overnight in order to minimize the degree of nonspecific cleavage. To stop the proteolytic reaction, 1 mM PMSF was added to the overnight reaction mixture, followed by purification with reversed-phase chromatography. Mass spectrometry analysis showed that enterokinase cleaved the fusion after Arg-44, resulting in an Lmod2 [45-94] fragment. During purification with reversed-phase chromatography, the Lmod2 [45-94] fragment was eluted with 40% acetonitrile and 0.1% TFA. The purified product was frozen and lyophilized overnight.
For all Lmod2 constructs, lyophilized samples were purified by high-performance liquid chromatography (HPLC) on a Waters Vydac 218TP reversed-phase C18 column, using a linear (0–50%) gradient of acetonitrile in the presence of 0.1% TFA at a constant flow rate of 1 ml/min. Lmod2 [85-123] and Lmod2 [103-201] were further purified by HPLC on a XBridge C18 column using gradients of acetonitrile in 1% NH4HCO3 and desalted on a XBridge C18 column by a 0–50% acetonitrile gradient in 0.1% TFA. All HPLC-purified samples were freeze-dried overnight.
Actin acetone powder was purified according to protocols described in Pardee and Spudich (1982a, b) with modifications. Purification was carried out in the cold room at 4°C. Breast muscle from a freshly killed chicken was immediately embedded in ice. The visible fat and connecting tissue were trimmed, and the muscle was ground in an electric meat grinder, which was rinsed with cold 0.2 M EDTA. Ground muscle was weighed and placed in a 4-l plastic beaker, and myosin was extracted with 2× (volume/muscle weight) extraction buffer (0.15 M potassium phosphate buffer, pH 6.7, 0.3 M KCl, 20 mM EDTA, 5 mM MgCl2, and 3.3 mM ATP) for 10–15 min with stirring. Then, a double volume of cold, deionized water was added to stop extraction. The mixture was filtered, and the pulp was washed in sequence with 5× volume of 0.4% NaHCO3, 0.1 mM CaCl2, 2× volume of 10 mM NaHCO3, 10 mM Na2CO3, 0.1 M CaCl2, and 10× volume of MQ-H2O. A 2× volume of ice-cold acetone was added to the pulp, and the mixture was stirred for 30 min in ice. Pulp was filtered, and this step was repeated five more times; then the acetone powder was dried and stored at −20°C.
G-actin was purified from acetone powder following protocols previously described (Spudich and Watt, 1971; MacLean-Fletcher and Pollard, 1980) with modifications. Briefly, actin was extracted twice from 1 g of acetone powder with 20 and 10 ml of buffer A (2 mM Tris-HCl, pH 8, 0.5 mM DTT, 0.2 mM ATP, 0.2 mM CaCl2, and 0.01% NaN3) during 10 min on ice. The solution was filtered through a Whatman filter and through a 0.2-mm Millipore filter. To polymerize actin, 50 mM KCl and 2 mM MgCl2 were slowly added to the solution. After 2 h, KCl concentration was adjusted to 0.6 M. The mixture was centrifuged at 50,000 rpm (Beckman Ti-70 rotor) at 4°C for 2.5 h. The pellet was washed twice, resuspended in 2 ml of buffer A, and sonicated for 2 min on ice. ATP concentration was adjusted to 1 mM and dialyzed for 2 d against buffer A at 4°C. Dialysis buffer was changed daily. After centrifugation at 100,000 rpm (Beckman TLA-100 rotor) for 2 h at 4°C, G-actin from the supernatant was purified using a gel-filtration Sephacryl S-300 column equilibrated with buffer A.
Pyrene iodoacetamide–labeled G-actin was prepared as described (Kouyama and Mihashi, 1981; Cooper et al., 1983) and stored in liquid nitrogen. Before experiments, pyrene-actin was defrosted at 37°C and centrifuged to remove aggregates.
Tpm1.1 was purified as in Moroz et al. (2013). Recombinant human gelsolin was a generous gift from John H. Hartwig (Brigham and Women’s Hospital and Hematology Division, Boston, MA). N-acetylated Tpm peptide, αTM1bzip, was synthesized by the Tufts University Core Facility (Boston, MA).
Protein and peptide concentration measurements
The purity of G-actin, Tpm1.1, αTM1bZip, Lmod2 [1–94], Lmod2 [1-201], Lmod2 [1-201(L30E)], and Lmod2 [1-201(W73D)] was evaluated using SDS–PAGE. Actin, Tpm1.1, and αTM1bZip concentrations were determined by measuring difference spectra in 6 M guanidine HCl between pH 12.5 and 7.0 using the equation [C] = A294/(2357NY + 830NW), where [C] is molar protein concentration, A294 is absorbance at 294 nm, 2357 and 830 are extinction coefficients for tyrosine and tryptophan, respectively, and NY and NW are numbers of tyrosines and tryptophans in a protein, respectively (Edelhoch, 1967; Fasman, 1989). Tpm concentrations were determined for two-stranded coiled-coil Tpm1.1 and αTM1bZip. The concentrations of Lmod2 [1–94], Lmod2 [1-201], Lmod2 [1-201(L30E)], and Lmod2 [1-201(W73D)] peptides were determined using a BCA Assay Kit (Thermo Fisher Scientific, Waltham, MA).
The identity of all Lmod2 fragments purified for NMR experiments was confirmed by mass spectrometry: 5814.14 (average mass, expected 5813.6) for Lmod2 [45-94], 4487.6 (monoisotopic mass, expected 4487.8) for Lmod2 [85-123], 4545.2 (monoisotopic mass, expected 4546.0) for Lmod2 [140-180], 4187.1 (monoisotopic mass, expected 4189.0) for Lmod2 [172-201], and 11415.7 (average mass, expected 11415.47) for Lmod2 [103-201] (15N-labeled construct). The differences between the expected and detected values were within the error of the experiment. With the exception of the Lmod2 [140-180] fragment, the concentrations of the Lmod2 fragments were determined by measuring difference spectra as described earlier. The concentration of Lmod2 [140-180] fragment was determined using the BCA Assay Kit.
Fluorescence measurements
Actin polymerization was detected as the change in pyrene-actin fluorescence using a PTI fluorometer (excitation, 366 nm; emission, 387 nm; with a 2-nm slit). To measure actin nucleation, a range of concentrations of Lmod2 wild type, truncated, and mutant was added to 1 μM G-actin/0.1 μM pyrene-G-actin or 1.5 μM G-actin/0.15 μM pyrene-G-actin, and the reaction was initiated by adding the 20× polymerization buffer (final concentration, 100 mM KCl, 1 mM MgCl2, 1 mM ethylene glycol tetraacetic acid (EGTA), 25 mM imidazole, pH 7.0). Actin polymerization in the absence of Lmod2 was measured as a control. To measure polymerization of actin at the pointed end, seeds (short filaments capped at the barbed ends with gelsolin) were prepared as described (Kostyukova and Hitchcock-DeGregori, 2004) by polymerization of 3 μM G-actin in the presence of 30 nM gelsolin. Polymerization was monitored by the increase in fluorescence when the seeds were diluted fivefold with 1 μM G-actin (10% pyrene-actin) in the polymerization buffer. The initial concentrations of F- and G-actin after dilution were 0.6 and 1.1 μM, respectively. We added 1 μM Tpm1.1 and Lmod2, wild type or mutant, to the seeds before dilution with G-actin solution. Gelsolin capped seeds were prepared in sets of four, and fluorescence measurements were carried out in parallel in a four-cuvette holder with actin alone as a control in each set. Exponential curves were fitted to the polymerization data using SigmaPlot 12, and initial rates (R) were calculated as the first derivatives at time zero. Statistical analysis was done using one-way analysis of variance (ANOVA) with a Tukey–Kramer post hoc test in SigmaPlot 12.
The fluorescence signal was normalized using the equation
where F is the normalized fluorescence, Ft(sample) is the real fluorescence at time t; Fmin(sample) is the minimal sample fluorescence after fitting, Fmax (control) is the maximal control fluorescence after fitting, and Fmin (control) is the minimal control fluorescence after fitting
Binding experiments
The chimeric Tpm peptide designed for structural and functional studies (Greenfield et al., 2001) contained 19 N-terminal residues of short Tpm isoforms encoded by exon 1b of the Tpm1 gene. To stabilize the coiled-coil structure, the Tpm peptide contained an additional 18 C-terminal residues of the GCN4 leucine zipper domain. The identity of the synthetic peptide was confirmed using mass spectroscopy.
Lmod2-Tpm binding was detected using native gel electrophoresis in nondenaturing 9% polyacrylamide gels that were polymerized in the presence of 10% glycerol without SDS. To prepare complexes for loading onto gels, Lmod2 [1-201] and Lmod2 [1-201(L30E)] were mixed with αTM1bzip in a 1:1 and 1:2 molar ratios. For titration, a stock solution of Lmod2 [1-201] was diluted to a final concentration of 5 μM with different concentrations of αTM1bzip in 20 mM Tris-HCl, pH 7.4. After electrophoresis, gels were stained by Coomassie R-250, scanned, and analyzed using an Image-Scanner III densitometer with Labscan 6.0 and ImageQuantTL 7.0 image analysis software.
Cell culture and adenoviral transduction
Cardiomyocytes were isolated from postnatal day 1–3 rat hearts as described (Brand et al., 2010). Isolated cells were plated on 35-mm tissue culture dishes containing 12-mm round glass coverslips thin-coated with Matrigel (1:100; BD Biosciences, San Jose, CA) at ∼4.5 × 105 cells/dish. Cells were maintained in DMEM with 1 g/l glucose (Life Technologies) with the addition of 10% (vol/vol) fetal bovine serum (HyClone, Logan, UT) and 1% penicillin/streptomycin (Cellgro; Mediatech, Manassas, VA). Cardiomyocytes were transduced with adenovirus 2 d after plating at a multiplicity of infection of 1. Expression differences between constructs were further normalized using Western blot analysis (unpublished data). Adenovirus expressing GFP, GFP-Lmod2, GFP-Lmod2 (L30E), and GFP-Lmod2 (W73D) were generated as previously outlined (Bliss et al., 2013).
Immunofluorescence microscopy
At 2–3 d after transduction, cells were incubated in relaxing buffer (150 mM KCl, 5 mM MgCl2, 10 mM 3-(N-morpholino)propanesulfonic acid, pH 7.4, 1 mM EGTA, and 4 mM ATP) for 15 min and fixed with 2% paraformaldehyde in relaxing buffer for 15 min. Transduced myocytes were stained as described (Pappas et al., 2008) with monoclonal anti–sarcomeric α-actinin (1:3000; EA-53; Sigma-Aldrich, St. Louis, MO) and Texas red–phalloidin (1:50; Invitrogen, Carlsbad, CA) to mark Z-disks and F-actin, respectively. Alexa Fluor 350 goat anti-mouse immunoglobulin G (1:200; Invitrogen) was the secondary antibody used. The cells were analyzed, and images were captured using a DeltaVision RT system (Applied Precision, Mississauga, Canada) with an inverted microscope (IX70; Olympus, Tokyo, Japan), a 100×/numerical aperture (NA) 1.3 objective, and a charge-coupled device camera (CoolSNAP HQ; Photometrics, Tucson, AZ) using SoftWoRx 3.5.1 software (Applied Precision). The images were then deconvolved using SoftWoRx and processed using Photoshop CS (Adobe, San Jose, CA). Thin filament and sarcomere lengths were measured using the DDecon plug-in for ImageJ (the precision of the method is 0.01–0.02 μm; Littlefield and Fowler, 2002). Thin filament lengths were analyzed within a sarcomere length range of 2.0–2.2 μm; this sarcomere length was not significantly different between groups. Statistical analysis was done using a one-way ANOVA with Prism 4 software (GraphPad Software, La Jolla, CA). To determine specific interactions of GFP-Lmod2 at the pointed end, cells were extracted in cytoskeleton-stabilization buffer (10 mM 1,4-piperazinediethanesulfonic acid, pH 6.8, 100 mM KCl, 300 mM sucrose, 2.5 mM MgCl2, and 0.5% Triton X-100 plus protease inhibitors) before fixation. Nearly 100 cardiomyocytes per culture (with mature staining for α-actinin) were imaged, and sarcomeric assembly of each construct was analyzed as follows: “pointed-end assembly” refers to clear and well-defined striated staining at the pointed end (>90% of sarcomeres contain GFP striations); “no pointed-end assembly” refers to faint (<90% of sarcomeres contain GFP striations) or no visible striations at the pointed end. The data are presented as the percentage of the total number of GFP-positive mature cardiomyocytes in each group (mean ± SEM). Data are from three independent cultures.
FRAP
A Leica SP5-II spectral confocal with a 63×/NA 1.4 objective and a 488-nm argon laser was used for FRAP experiments. Cells were plated on glass-bottom dishes (MatTek, Ashland, MA) thin-coated with Matrigel (1:100) at ∼1.5 × 105 cells/dish and maintained at 37°C with CO2 for the duration of the experiment. One prebleach image was recorded, followed by photobleaching for 1 s at 100% total laser power. To monitor recovery after photobleaching, images were captured in successive intervals of 0.17 s (for a duration of 5 s), 1 s (for a duration of 20 s), and 2 s (for a duration of 40 s). Analysis of recovery was completed as described previously with minor modifications (Wang et al., 2005). In brief, images were analyzed using Leica Application Suite software, and mean intensities from bleached and nonbleached regions were recorded. After correction for photobleaching caused by image acquisition, intensities were normalized so that prebleach intensity was set to 1 and postbleach intensity was set to 0. Mobile fractions and half-times were determined from nonlinear regression curves fit using the one-exponential association equation R = M[1 − exp(−kt)] with Prism 4 software, where R is the relative recovery at time t, and M is the mobile fraction. Half-time is equal to 0.69/k. Recovery was determined independently at two or three thin filament pointed ends per cell. Approximately 10 cells per treatment were analyzed in each experiment, and three experiments were averaged.
NMR sample preparation
Special care was taken to ensure identical pH for Lmod2 peptide samples in the presence and absence of G-actin. Specifically, purified, lyophilized Lmod2 fragments were dissolved in 1 mM sodium phosphate and pH adjusted to 6.8. Each sample solution was divided into two equal portions and lyophilized to dryness. G-actin was dialyzed against 2 mM potassium phosphate, 0.1 mM ATP, 0.1 mM CaCl2, and 0.01% NaN3, pH 6.8, with one buffer change. For each Lmod2 construct, the dialysis buffer equilibrated with the actin dialysate was used to dissolve the first lyophilized portion, and an equal volume of actin was added to the other lyophilized portion. For NMR measurements, the concentration of the Lmod2 fragments was ∼0.2 mM for the unlabeled fragments and ∼0.1 mM for the 15N-labeled fragment; actin concentration was ∼10 μM.
NMR spectral analysis
The 1D proton and 2D 15N-HSQC NMR spectra were recorded at 25°C on a Varian VNMRS 600 MHz spectrometer (Agilent Technologies, Santa Clara, CA) equipped with a 5-mm triple-resonance probe. The gNhsqc pulse sequence found in the BioPack pulse sequence library (Agilent Technologies, Liberty Lake, CA) was used to record the 2D 15N-HSQC spectra. Extra care was given to the calibration of the water flipback pulse to avoid water saturation and signal loss. For each 2D spectrum, we recorded 2 × 128 increments with 10,000-Hz spectral width in F2 (1H) and 3700-Hz spectral width in F1 (15N).
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health Grant RO1GM081688 and startup funds to A.S.K., a donation from Linda and Jim Lee, and National Institutes of Health Grants R01HL108625 and R01HL123078 to C.C.G. and 1F31HL117520 to S.M.N.
Abbreviations used:
- FRAP
fluorescence recovery after photobleaching
- GFP
green fluorescent protein
- HPLC
high-performance liquid chromatography
- HSQC
heteronuclear single quantum coherence
- Lmod
leiomodin
- NMR
nuclear magnetic resonance
- Tmod
tropomodulin
- Tpm
tropomyosin
- WH2
Wiskott-Aldrich homology 2 domain
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E16-03-0200) on June 15, 2016.
REFERENCES
- Bliss KT, Chu M, Jones-Weinert CM, Gregorio CC. Investigating lasp-2 in cell adhesion: new binding partners and roles in motility. Mol Biol Cell. 2013;24:995–1006. doi: 10.1091/mbc.E12-10-0723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boczkowska M, Rebowski G, Kremneva E, Lappalainen P, Dominguez R. How Leiomodin and Tropomodulin use a common fold for different actin assembly functions. Nat Commun. 2015;6:8314. doi: 10.1038/ncomms9314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand NJ, Lara-Pezzi E, Rosenthal N, Barton PJ. Analysis of cardiac myocyte biology in transgenic mice: a protocol for preparation of neonatal mouse cardiac myocyte cultures. Methods Mol Biol. 2010;633:113–124. doi: 10.1007/978-1-59745-019-5_9. [DOI] [PubMed] [Google Scholar]
- Chen X, Ni F, Kondrashkina E, Ma J, Wang Q, et al. Mechanisms of leiomodin 2-mediated regulation of actin filament in muscle cells. Proc Natl Acad Sci USA. 2015;112:12687–12692. doi: 10.1073/pnas.1512464112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chereau D, Boczkowska M, Skwarek-Maruszewska A, Fujiwara I, Hayes DB, Rebowski G, Lappalainen P, Pollard TD, Dominguez R. Leiomodin is an actin filament nucleator in muscle cells. Science. 2008;320:239–243. doi: 10.1126/science.1155313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colpan M, Tolkatchev D, Grover S, Helms GL, Cort JR, Moroz N, Kostyukova AS. Localization of the binding interface between leiomodin-2 and alpha-tropomyosin. Biochim Biophys Acta. 2016;1864:523–530. doi: 10.1016/j.bbapap.2016.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conley CA. Leiomodin and tropomodulin in smooth muscle. Am J Physiol Cell Physiol. 2001;280:C1645–C1656. doi: 10.1152/ajpcell.2001.280.6.C1645. [DOI] [PubMed] [Google Scholar]
- Conley CA, Fritz-Six KL, Almenar-Queralt A, Fowler VM. Leiomodins: larger members of the tropomodulin (Tmod) gene family. Genomics. 2001;73:127–139. doi: 10.1006/geno.2000.6501. [DOI] [PubMed] [Google Scholar]
- Cooper JA, Walker SB, Pollard TD. Pyrene actin: documentation of the validity of a sensitive assay for actin polymerization. J Muscle Res Cell Motil. 1983;4:253–262. doi: 10.1007/BF00712034. [DOI] [PubMed] [Google Scholar]
- Corpet F. Multiple sequence alignment with hierarchical clustering. Nucleic Acids Res. 1988;16:10881–10890. doi: 10.1093/nar/16.22.10881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drozdetskiy A, Cole C, Procter J, Barton GJ. JPred4: a protein secondary structure prediction server. Nucleic Acids Res. 2015;43:W389–W394. doi: 10.1093/nar/gkv332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edelhoch H. Spectroscopic determination of tryptophan and tyrosine in proteins. Biochemistry. 1967;6:1948–1954. doi: 10.1021/bi00859a010. [DOI] [PubMed] [Google Scholar]
- Fasman GD. Protein conformational prediction. Trends Biochem Sci. 1989;14:295–299. doi: 10.1016/0968-0004(89)90068-6. [DOI] [PubMed] [Google Scholar]
- Fischer RS, Yarmola EG, Weber KL, Speicher KD, Speicher DW, Bubb MR, Fowler VM. Tropomodulin 3 binds to actin monomers. J Biol Chem. 2006;281:36454–36465. doi: 10.1074/jbc.M606315200. [DOI] [PubMed] [Google Scholar]
- Fowler VM, Greenfield NJ, Moyer J. Tropomodulin contains two actin filament pointed end-capping domains. J Biol Chem. 2003;278:40000–40009. doi: 10.1074/jbc.M306895200. [DOI] [PubMed] [Google Scholar]
- Geeves MA, Hitchcock-DeGregori SE, Gunning PW. A systematic nomenclature for mammalian tropomyosin isoforms. J Muscle Res Cell Motil. 2015;36:147–153. doi: 10.1007/s10974-014-9389-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenfield NJ, Huang YJ, Palm T, Swapna GV, Monleon D, Montelione GT, Hitchcock-DeGregori SE, et al. Solution NMR structure and folding dynamics of the N terminus of a rat non-muscle alpha-tropomyosin in an engineered chimeric protein. J Mol Biol. 2001;312:833–847. doi: 10.1006/jmbi.2001.4982. [DOI] [PubMed] [Google Scholar]
- Greenfield NJ, Kostyukova AS, Hitchcock-DeGregori SE. Structure and tropomyosin binding properties of the N-terminal capping domain of tropomodulin 1. Biophys J. 2005;88:372–383. doi: 10.1529/biophysj.104.051128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostyukova AS. Leiomodin/tropomyosin interactions are isoform specific. Arch Biochem Biophys. 2007;465:227–230. doi: 10.1016/j.abb.2007.05.012. [DOI] [PubMed] [Google Scholar]
- Kostyukova AS, Choy A, Rapp BA. Tropomodulin binds two tropomyosins: a novel model for actin filament capping. Biochemistry. 2006;45:12068–12075. doi: 10.1021/bi060899i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostyukova AS, Hitchcock-DeGregori SE. Effect of the structure of the N terminus of tropomyosin on tropomodulin function. J Biol Chem. 2004;279:5066–5071. doi: 10.1074/jbc.M311186200. [DOI] [PubMed] [Google Scholar]
- Kostyukova AS, Rapp BA, Choy A, Greenfield NJ, Hitchcock-DeGregori SE. Structural requirements of tropomodulin for tropomyosin binding and actin filament capping. Biochemistry. 2005;44:4905–4910. doi: 10.1021/bi047468p. [DOI] [PubMed] [Google Scholar]
- Kouyama T, Mihashi K. Fluorimetry study of N-(1-pyrenyl)iodoacetamide-labelled F-actin. Local structural change of actin protomer both on polymerization and on binding of heavy meromyosin. Eur J Biochem. 1981;114:33–38. [PubMed] [Google Scholar]
- Littlefield R, Fowler VM. Measurement of thin filament lengths by distributed deconvolution analysis of fluorescence images. Biophys J. 2002;82:2548–2564. doi: 10.1016/S0006-3495(02)75598-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLean-Fletcher S, Pollard TD. Identification of a factor in conventional muscle actin preparations which inhibits actin filament self-association. Biochem Biophys Res Commun. 1980;96:18–27. doi: 10.1016/0006-291x(80)91175-4. [DOI] [PubMed] [Google Scholar]
- Moroz NA, Novak SM, Azevedo R, Colpan M, Uversky VN, Gregorio CC, Kostyukova AS. Alteration of tropomyosin-binding properties of tropomodulin-1 affects its capping ability and localization in skeletal myocytes. J Biol Chem. 2013;288:4899–4907. doi: 10.1074/jbc.M112.434522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanda V, Miano JM. Leiomodin 1, a new serum response factor-dependent target gene expressed preferentially in differentiated smooth muscle cells. J Biol Chem. 2012;287:2459–2467. doi: 10.1074/jbc.M111.302224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni F, Konishi Y, Scheraga HA. Thrombin-bound conformation of the C-terminal fragments of hirudin determined by transferred nuclear Overhauser effects. Biochemistry. 1990;29:4479–4489. doi: 10.1021/bi00470a030. [DOI] [PubMed] [Google Scholar]
- Ni F, Zhu Y, Scheraga HA. Thrombin-bound structures of designed analogs of human fibrinopeptide A determined by quantitative transferred NOE spectroscopy: a new structural basis for thrombin specificity. J Mol Biol. 1995;252:656–671. doi: 10.1006/jmbi.1995.0527. [DOI] [PubMed] [Google Scholar]
- Pappas CT, Bhattacharya N, Cooper JA, Gregorio CC. Nebulin interacts with CapZ and regulates thin filament architecture within the Z-disc. Mol Biol Cell. 2008;19:1837–1847. doi: 10.1091/mbc.E07-07-0690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pappas CT, Mayfield RM, Henderson C, Jamilpour N, Cover C, Hernandez Z, Hutchinson KR, Chu M, Nam KH, Valdez JM, et al. Knockout of Lmod2 results in shorter thin filaments followed by dilated cardiomyopathy and juvenile lethality. Proc Natl Acad Sci USA. 2015;112:13573–13578. doi: 10.1073/pnas.1508273112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardee JD, Spudich JA. Purification of muscle actin. Methods Enzymol. 1982a;85:164–181. doi: 10.1016/0076-6879(82)85020-9. [DOI] [PubMed] [Google Scholar]
- Pardee JD, Spudich JA. Purification of muscle actin. Methods Cell Biol. 1982b;24:271–289. doi: 10.1016/s0091-679x(08)60661-5. [DOI] [PubMed] [Google Scholar]
- Rao JN, Madasu Y, Dominguez R. Mechanism of actin filament pointed-end capping by tropomodulin. Science. 2014;345:463–467. doi: 10.1126/science.1256159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skala W, Goettig P, Brandstetter H. Do-it-yourself histidine-tagged bovine enterokinase: a handy member of the protein engineer’s toolbox. J Biotechnol. 2013;168:421–425. doi: 10.1016/j.jbiotec.2013.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skwarek-Maruszewska A, Boczkowska M, Zajac AL, Kremneva E, Svitkina T, Dominguez R, Lappalainen P. Different localizations and cellular behaviors of leiomodin and tropomodulin in mature cardiomyocyte sarcomeres. Mol Biol Cell. 2010;21:3352–3361. doi: 10.1091/mbc.E10-02-0109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprague BL, Pego RL, Stavreva DA, McNally JG. Analysis of binding reactions by fluorescence recovery after photobleaching. Biophys J. 2004;86:3473–3495. doi: 10.1529/biophysj.103.026765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spudich JA, Watt S. The regulation of rabbit skeletal muscle contraction. I. Biochemical studies of the interaction of the tropomyosin-troponin complex with actin and the proteolytic fragments of myosin. J Biol Chem. 1971;246:4866–4871. [PubMed] [Google Scholar]
- Studier FW. Protein production by auto-induction in high density shaking cultures. Protein Expr Purif. 2005;41:207–234. doi: 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- Tolkatchev D, Plamondon J, Gingras R, Su Z, Ni F. Recombinant production of intrinsically disordered proteins for biophysical and structural characterization. In: Uversky VN, Longhi S, editors. Instrumental Analysis of Intrinsically Disordered Proteins: Assessing Structure And Conformation. Hoboken, NJ: Wiley; 2010. pp. 653–670. [Google Scholar]
- Tolkatchev D, Xu P, Ni F. Probing the kinetic landscape of transient peptide-protein interactions by use of peptide (15)n NMR relaxation dispersion spectroscopy: binding of an antithrombin peptide to human prothrombin. J Am Chem Soc. 2003;125:12432–12442. doi: 10.1021/ja021238l. [DOI] [PubMed] [Google Scholar]
- Tsukada T, Kotlyanskaya L, Huynh R, Desai B, Novak SM, Kajava AV, Gregorio CC, Kostyukova AS. Identification of residues within tropomodulin-1 responsible for its localization at the pointed ends of the actin filaments in cardiac myocytes. J Biol Chem. 2011;286:2194–2204. doi: 10.1074/jbc.M110.186924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukada T, Pappas CT, Moroz N, Antin PB, Kostyukova AS, Gregorio CC. Leiomodin-2 is an antagonist of tropomodulin-1 at the pointed end of the thin filaments in cardiac muscle. J Cell Sci. 2010;123:3136–3145. doi: 10.1242/jcs.071837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Shaner N, Mittal B, Zhou Q, Chen J, Sanger JM, Sanger JW. Dynamics of Z-band based proteins in developing skeletal muscle cells. Cell Motil Cytoskeleton. 2005;61:34–48. doi: 10.1002/cm.20063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber A, Pennise CR, Babcock GG, Fowler VM. Tropomodulin caps the pointed ends of actin filaments. J Cell Biol. 1994;127:1627–1635. doi: 10.1083/jcb.127.6.1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson MP. Using chemical shift perturbation to characterise ligand binding. Prog Nucl Magn Reson Spectrosc. 2013;73:1–16. doi: 10.1016/j.pnmrs.2013.02.001. [DOI] [PubMed] [Google Scholar]
- Yamashiro S, Gokhin DS, Sui Z, Bergeron SE, Rubenstein PA, Fowler VM. Differential actin-regulatory activities of Tropomodulin1 and Tropomodulin3 with diverse tropomyosin and actin isoforms. J Biol Chem. 2014;289:11616–11629. doi: 10.1074/jbc.M114.555128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen M, Sandaradura SA, Dowling JJ, Kostyukova AS, Moroz N, Quinlan KG, Lehtokari VL, Ravenscroft G, Todd EJ, Ceyhan-Birsoy O, et al. Leiomodin-3 dysfunction results in thin filament disorganization and nemaline myopathy. J Clin Invest. 2015;125:456–457. doi: 10.1172/JCI80057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Vranken W, Goudreau N, de Miguel E, Magny MC, Mort JS, Dupras R, Storer AC, Ni F. An NMR-based identification of peptide fragments mimicking the interactions of the cathepsin B propeptide. FEBS Lett. 1998;429:9–16. doi: 10.1016/s0014-5793(98)00548-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.