

Proteolytic processing is a major posttranslational regulatory mechanism. In the stress-responsive slt pathway, both the transcription factor SltA and the signaling protein SltB undergo proteolysis. SltB catalyzes both processes, producing a functional form of SltA to mediate the responses to cation stress and alkalinity in Aspergillus nidulans.
Abstract
Tolerance of Aspergillus nidulans to alkalinity and elevated cation concentrations requires both SltA and SltB. Transcription factor SltA and the putative pseudokinase/protease signaling protein SltB comprise a regulatory pathway specific to filamentous fungi. In vivo, SltB is proteolytically cleaved into its two principal domains. Mutational analysis defines a chymotrypsin-like serine protease domain that mediates SltB autoproteolysis and proteolytic cleavage of SltA. The pseudokinase domain might modulate the protease activity of SltB. Three forms of the SltA transcription factor coexist in cells: a full-length, 78-kDa version and a processed, 32-kDa form, which is found in phosphorylated and unphosphorylated states. The SltA32kDa version mediates transcriptional regulation of sltB and, putatively, genes required for tolerance to cation stress and alkalinity. The full-length form, SltA78kDa, apparently has no transcriptional function. In the absence of SltB, only the primary product of SltA is detectable, and its level equals that of SltA78kDa. Mutations in sltB selected as suppressors of null vps alleles and resulting in cation/alkalinity sensitivity either reduced or eliminated SltA proteolysis. There is no evidence for cation or alkalinity regulation of SltB cleavage, but activation of sltB expression requires SltA. This work identifies the molecular mechanisms governing the Slt pathway.
INTRODUCTION
The biological activity of many proteins, including transcription factors (TFs), is regulated by posttranslational modifications (PTMs). There are diverse PTM systems, which are widely varied and can be reversible or irreversible in response to extracellular signals (Benayoun and Veitia, 2009). The well-known phosphoryl modification systems involving protein kinases and phosphatases are examples of reversible PTMs. Among irreversible examples are PTMs involving a controlled proteolytic activity.
In Aspergillus nidulans, tolerance to alkaline ambient pH requires the activities of at least three zinc-finger transcription factors: PacC, CrzA, and SltA (Tilburn et al., 1995; Spielvogel et al., 2008). In addition, CrzA and SltA mediate tolerance to high concentrations of certain cations (Spielvogel et al., 2008; Findon et al., 2010). We previously demonstrated that the activities of the PacC and CrzA transcription factors are regulated by PTMs. The calcineurin-responsive signaling to CrzA (Crz1p in Saccharomyces cerevisiae; Cyert, 2003) is based on a phosphoryl modification mechanism that determines both subcellular localization and functionality. GskA, an orthologue of human glycogen synthase kinase-3β (Hernández-Ortiz and Espeso, 2013), and CkiA, a casein kinase I homologue (Apostolaki et al., 2012), are protein kinases that phosphorylate CrzA. The reverse reaction is mediated by the calcium-dependent protein phosphatase calcineurin (CN), which dephosphorylates CrzA in response to the presence of calcium or alkalinity (Hernández-Ortiz and Espeso, 2013). Nuclear import of CrzA, Crz1p, and NFATs, a family of unrelated mammalian nuclear factors, requires the activity of CN, and consequently their nuclear accumulation has been linked to a reduction in phosphorylation levels (Crabtree and Olson, 2002; Cyert, 2003; Hernández-Ortiz and Espeso, 2013). Both calcium and alkaline pH have a limited, transient effect due to the activation of CN regulatory mechanisms, which prevent deleterious effects associated with its hyperactivation (reviewed in Espeso, 2016). In consequence, phosphorylation levels of these TFs increase and nuclear export mechanisms become active, restricting their presence in the nucleus and, hence, their function as modulators of transcription.
Unlike phosphorylation, proteolysis is not reversible. Proteolysis can remove regulatory proteins or segments of a protein to enable or prevent its biological function and, in addition, result in changes of subcellular localization by removing targeting sequences (reviewed in Ehrmann and Clausen, 2004). Relevant animal examples of signaling cascades in which proteases play a decisive role are the regulation of sterol metabolism through regulated-intramembrane proteolysis of sterol regulatory elements–binding proteins (reviewed in Brown et al., 2000) and cellular differentiation mediated by hedgehog signaling to Drosophila Cubitus interruptus or its vertebrate homologues, the glioblastoma-associated proteins Gli2 and Gli3 (Aza-Blanc et al., 1997; Pan et al., 2006).
In the fungal kingdom, PacC (Rim101 in yeast-like fungi) is a well-characterized example of a TF regulated by a proteolytic mechanism (Orejas et al., 1995). PacC is synthesized as a primary full-length form of 72 kDa, PacC72, and it is subjected to proteolysis in response to extracellular alkaline pH. Two consecutive proteolytic steps are required for PacC activation. The primary step is pH dependent, mediated by the putative calpain protease PalB, to yield a 53-kDa form, PacC53 (Orejas et al., 1995; Diez et al., 2002; Peñas et al., 2007). PacC53 is a transient intermediate, committed to proteolysis by the proteasome, generating a 27-kDa form of PacC (Diez et al., 2002; Hervas-Aguilar et al., 2007). The truncated form PacC27, containing the DNA-binding region, is required for correct regulation of acid- and alkaline-expressed genes in response to alkaline pH (Diez et al., 2002; Espeso and Arst, 2000; Espeso and Peñalva, 1996).
O’Neil et al. (2002) identified sltA (stzA) in A. nidulans, which encodes a transcription factor with three zinc fingers of the C2H2 class. Further studies identified SltA orthologues in species belonging to the Pezizomycotina subphylum (Chilton et al., 2008; Mellado et al., 2015), but the only characterized homologue is Ace1 of Hypocrea jecorina, a repressor of cellulase and xylanase genes (Saloheimo et al., 2000). Owing to the high conservation in amino acid sequence of the zinc finger domains, both SltA and Ace1 recognize the same target DNA sequence, 5′-AGGCA-3′ (Saloheimo et al., 2000; Spielvogel et al., 2008).
Initial characterization of the A. nidulans sltA1 mutant showed that SltA was required for tolerance to a variety of abiotic stresses, such as elevated concentrations of sodium chloride, arginine, exposure to ultraviolet light, and presence of the alkylating agent N-methyl-N′-nitro-N-nitrosoguanidine (Clement et al., 1996; O’Neil et al., 2002). Additional studies of null and other mutant sltA alleles have shown the role of SltA in tolerance to high extracellular concentrations of various cations (sodium, potassium, lithium, cesium, magnesium) and alkalinity. In addition, sltA mutants are hypervacuolated, with increased number, size, and distribution of vacuoles, and have defects in morphological development and sterigmatocystin production (Spielvogel et al., 2008; Findon et al., 2010; Shantappa et al., 2013). Most of these phenotypes are attenuated when Ca2+ is added to culture medium, suggesting a defect in calcium homeostasis. In fact, double mutants lacking SltA and the protein kinase HalA, homologous to the Hal4p and Hal5p saline-response kinases of S. cerevisiae, are auxotrophic for calcium due to cytoplasmic Ca2+ depletion (Findon et al., 2010).
It is easy to select sltA mutations as suppressors that rescue the near-lethal phenotypes caused by certain vacuolar-protein-sorting deletion (vpsΔ) alleles (Calcagno-Pizarelli et al., 2011). This technique also allows selection of mutations in sltB, a second gene of the sltA pathway (Mellado et al., 2015). The sltB gene encodes a protein of 1272 amino acids with two putative functional domains: a pseudokinase (Psk) and chymotrypsin-like protease. SltB homologues have a similar restricted phylogenetic distribution to SltA. Null sltB and null sltA mutations have identical phenotypes and are nonadditive in double mutants, suggesting that SltB might participate in the activation of SltA. Of interest, sltB is positively regulated by SltA (Mellado et al., 2015).
Here we demonstrate that activation of SltA requires the proteolytic removal of the N-terminal domain and that the functional C-terminal moiety is subject to phosphorylation. We further show that SltB is a serine protease required for SltA processing proteolysis and that SltB is autoproteolyzed as a prerequisite for SltA proteolysis. We propose models for regulation of SltA via SltB activity for this specific cation/alkaline pH tolerance pathway of filamentous fungi.
RESULTS
Cleavage of SltB separates its pseudokinase and protease domains
To understand SltB function, we constructed strains expressing tagged versions of this signaling protein (Figure 1). Using our standard tagging techniques (see Materials and Methods), we obtained a strain expressing an SltB–green fluorescent protein (GFP) chimera. Because GFP tagging might impair SltB function, we tested this strain, MAD3625, for tolerance to elevated sodium concentrations (1 M NaCl) and alkalinity (100 mM Na2HPO4, pH 8). Supplemental Figure S1 shows that colony growth of MAD3625 was comparable to that of the wild-type (WT) strain MAD1427 and clearly different from the sensitive phenotype of a null sltB strain (MAD3669), indicating that the SltB-GFP fusion is functional. Next we obtained a total protein extract from MAD3625 and determined the expression level and electrophoretic mobility of the SltB-GFP chimera. Using an antibody against GFP, we detected a prominent band with an estimated mass of 100 kDa (Figure 1, right). Its mobility was high compared with that expected from the calculated 161-kDa mass of the SltB-GFP fusion (Figure 1, left). To confirm the absence of artifacts resulting from the GFP tag, we constructed MAD3734, which expresses an SltB-Myc3 (having three copies of the Myc tag) chimera (Figure 1, left). This fusion protein is also functional because no sensitivities were observed in a strain expressing SltB-Myc3 compared with WT strain (MAD1427; Supplemental Figure S1). In Western blot analyses, using anti-myc antibody (α-Myc), we detected a 70-kDa band for the SltB-Myc3 fusion (Figure 1, right), again showing that the mobility of the major band is higher than that expected for the chimera. The difference in mobility between SltB-GFP and SltB-Myc3 fusions reflects the difference in size of the tags. We reasoned that the unexpectedly high chimera mobilities might indicate proteolytic cleavage of the SltB protein, in which case, the bands would correspond to the C-terminal protease domain of SltB. Consequently we tagged SltB at both ends to confirm its cleavage by detection of at least two chimera fragments. We analyzed extracts of strain MAD4899, which expresses the GFP-SltB-Myc3 fusion protein in Western blots (Figure 1). Despite the fact that the GFP-SltB-Myc3 fusion is not a functional protein (Supplemental Figure S1), using anti-myc antibody, we again detected a 70-kDa band as in extracts from the strain expressing SltB-Myc3. Anti-GFP antibody revealed a band with an estimated mass of 125 kDa. The two chimera bands were present in similar amounts when the same membrane was probed with a mixture of antibodies (anti-GFP and anti-Myc), and the sum of the predicted masses conformed reasonably well to the expected mass of a full-length GFP-SltB-Myc3 chimera, ∼179 kDa. These data clearly support a working model in which SltB undergoes a cleavage process resulting in the separation of the predicted pseudokinase and protease domains (Figure 1).
FIGURE 1:

Evidence for proteolytic cleavage of signaling protein SltB. Top left, schematic representation of SltB fusions to GFP and/or Myc3 tags. Estimated molecular masses (Mw) are indicated for SltB, tags, and full-length chimeras. Top right, Western blot analysis of total protein extracts from strains expressing SltB fusions using specific antibodies against GFP (α-GFP) and/or Myc (α-Myc) tags. Bottom, antibodies against the two tags of the double-tagged chimera are used in separate Western blots. Estimated sizes of the detected bands are indicated, with interpretations of the bands shown to the left. The strains used are indicated MADnnnn (see Supplemental Table S1 for the genotypes of strains).
SltA is posttranslationally modified by proteolysis and phosphorylation
SltB has a small degree of similarity to the endoprotease Ssy5p of S. cerevisiae (25% in a 68–amino acid [aa] overlap in the protease region; Mellado et al., 2015). In view of our previous results and the known role of Ssy5p in the proteolytic activation of TFs Stp1p and Stp2p in yeast (reviewed in Ljungdahl, 2009), we analyzed the electrophoretic mobility of tagged versions of the SltA transcription factor. It was not possible to obtain fusions of SltA to GFP at either the N- or C-terminus. Transformants were unstable, giving rise to sectors lacking a functional sltA gene (unpublished data). Therefore we constructed a strain expressing an SltA-HA3 fusion with three copies of the hemagglutinin (HA) tag. This SltA chimera was functional, as judged by tolerance to alkalinity and elevated concentrations of cations (strain MAD3652; Supplemental Figure S1). MAD3652 was used to construct MAD3751, expressing both SltB-GFP and SltA-HA3 fusions, which also displayed a WT-like phenotype (Supplemental Figure S1).
Detection of the SltA-HA3 fusion in total protein extracts of MAD3652 grown under standard Aspergillus minimal medium (AMM) conditions, using anti-HA, identified three bands in Western blots (Figure 2A). The low-mobility band had a predicted mass of 100 kDa, which was comparable to the calculated mass of 83 kDa for the full-length SltA-HA3 protein (Figure 2A, diagrams). The other two bands have mobility close to that expected for a 45-kDa form, and we postulated that these truncated forms might comprise the C-terminus containing the zinc-finger DNA-binding domain plus a few additional residues N-terminal to it, approximately the region between amino acids 400 and 698.
FIGURE 2:

Three forms of the SltA-HA3 fusion are detectable. (A) Total protein extract from cells of strain MAD3652 expressing the SltA-HA3 chimera was subjected to immunodetection using anti-HA primary antibody (α-HA). Three bands were detectable and estimated, and theoretical molecular masses (Mw) are indicated in kilodaltons. Right, interpretation of fragment sizes. (B) Phosphatase assay of protein extracts of MAD3652 expressing SltA-HA3 using λ-protein phosphatase (λPP). The absence of phosphatase and the presence of the specific phosphatase inhibitor orthovanadate (o-van) were used as controls. Loss of the lower-mobility band in proximity to the 45-kDa Mw marker and increase in intensity of the higher-mobility band at the same proximity demonstrate the presence of two forms of SltA32kDa, of which the lower-mobility band is phosphorylated. Asterisk indicates a nonspecific band detected with the anti-HA antibody when protein extracts were obtained with the A50 extraction procedure. (C) SltA proteolytic cleavage requires SltB. Comparison of immunodetection of SltA-HA3 bands in total protein extracts from strains having (sltB+) or lacking (sltBΔ) SltB. Only the SltA78kDa band is detected in the null sltB background. Levels of actin were used as loading controls.
A phosphatase assay clarified the number of truncated forms. Lambda-phosphatase treatment of protein extracts of a strain expressing SltA-HA3 demonstrated that the less mobile of the two high-mobility bands in the 45-kDa range corresponds to a phosphorylated derivative of the lower SltA band (Figure 2B).
Thus we conclude that SltA undergoes proteolysis, resulting in at least three forms of the SltA transcription factor in the cell: a full-length version designated SltA78kDa, a truncated version designated SltA32kDa, and a phosphorylated form of SltA32kDa. We have not detected a phosphorylated form of SltA78kDa.
Proteolysis of SltA requires SltB activity
On the basis of precedents in A. nidulans and S. cerevisiae, we hypothesized that proteolysis of SltA might depend on SltB activity. We constructed MAD3693, which expresses the SltA-HA3 fusion in the absence of SltB (Supplemental Table S1). Only the full-length SltA78kDa form of SltA is detectable in total protein extracts of MAD3693 (Figure 2C). This result and the analysis of sltB mutations (see later discussion) indicate that SltA proteolysis constitutes the signaling role of SltB.
It is worth noting that a null sltB strain expressing the SltA-HA3 protein, MAD3693, has the same sensitivity to high sodium concentrations or alkalinity as the reference null sltB strain (MAD3669; Supplemental Figure S1). Given the identical phenotypes of null sltA and sltB single mutants (Calcagno-Pizarelli et al., 2011; Mellado et al., 2015) and the presence of the SltA78kDa form in a null sltB background, we conclude that the full-length version of SltA must have little or no role in tolerance to alkaline pH and high cation concentrations.
SltA32kDa is the functional form of SltA in the cell
Because SltA78kDa appears not to have a physiological role in alkaline pH and cation tolerance, we constructed a strain expressing a truncated version of SltA resembling the proteolyzed form. We estimated that the SltA32kDa form should contain the C-terminal 300 amino acids (estimated molecular weight of 32 kDa). At position 400 of SltA protein, there is a methionine, and we took advantage of Met400 to construct a transformation cassette including the thiamine-repressible thiA promoter (thiAp; Calcagno-Pizarelli et al., 2011) and the sltA genomic fragment from codon 400 through the C-terminal Leu-698 codon (Supplemental Figure S2A). This cassette also encodes an in-frame triple-HA tag and homologous flanking regions for gene replacement. The thiamine-repressible thiA promoter was previously used to express sltA conditionally (Calcagno-Pizarelli et al., 2011), and we used it here to prevent any adverse effects of expressing a truncated version of sltA from the endogenous promoter. In fact, attempts to express truncated versions of SltA starting at amino acid 200 (Met) or 331 (Arg) resulted in poorly growing transformants, and additional mutations in the sltA constructs were found, preventing expression or functionality of SltA (Supplemental Figure S2).
A strain expressing the SltA400–698-HA3 form displayed an sltA null phenotype when thiamine was present in the medium (Figure 3A). This sltA null phenotype is also visible in the strain expressing the full-length SltA fused to HA3 expressed from thiAp (Figure 3A). Consistent with the null phenotype, SltA-HA3 fusion proteins were not detected in mycelia grown in the presence of thiamine (unpublished data). In cells grown in minimal medium without thiamine supplementation, the SltA fusions were expressed, and the strain expressing SltA400–698-HA3 is fully tolerant to 1 M NaCl and partially tolerant to 0.3 M LiCl and alkalinity. Partial tolerance to lithium ion seems to be characteristic of SltA fusions to HA3 (Figure 3A; compare rows 3–6 with WT in row 1 and the null sltA strain in row 2), but the reduced tolerance to alkalinity is specific to the SltA400–698-HA3 protein. In Western blots, we detected two bands for the SltA400–698-HA3 protein with almost identical electrophoretic mobilities to those of the SltA32kDa HA3-tagged form (Figure 3B). The lower-mobility band of the SltA400–698-HA3 fusion corresponded to a phosphorylated form, as shown for SltA32kDa (Figures 3C and 2B). These results clearly show that the SltA400–698-HA3 resembles the SltA32kDa HA3-tagged form and that phosphorylation of this form occurs in the absence of the first 399 residues of SltA.
FIGURE 3:

Investigation of the role of SltA32kDa forms. (A) Conditional expression of SltA chimeras driven by the thiamine-repressible thiAp promoter. Addition of 100 μM thiamine to AMM prevents expression of thiAp. When SltA fusions were expressed through thiAp as sole source of this transcription factor, the presence of thiamine led to a null sltA phenotype for sensitivity to high concentrations of sodium or lithium and to alkalinity but a nearly WT phenotype in the absence of vitamin B1. (B) Western blots detect the three forms of SltA in strain MAD4294 (thiAp::sltA::ha3) in the absence of thiamine. Two bands having almost identical mobilities to the SltA32kDa bands are detectable when SltA400–698-HA3 is expressed. The absence of SltB has no effect on the presence or mobility of SltA400–698-HA3 bands. (C) Dephosphorylation assay of extracts from strain MAD4661 expressing the SltA400–698-HA3 fusion. Treatment with λ-protein phosphatase (λPP) results in loss of the low-mobility band and increases the intensity of the high-mobility band for the SltA400–698-HA3 fusion, demonstrating phosphorylation of this chimera (indicated on the left); c, contains total crude protein extracts obtained using the alkaline lysis procedure, resulting in a higher level of detectable SltA400–698-HA3 forms. (D) Western blots showing the presence of the 100-kDa truncated SltB-GFP form when the SltA400–698-HA3 fusion was expressed. Actin was used as loading control. The SltA-HA3 forms in those protein extracts are shown below.
If the SltA32kDa form is the final product of SltB signaling, then functionality of this form should be independent of SltB function. Data in Figure 3A verify this hypothesis. In the absence of thiamine, sltA400–698-ha3 suppresses sltBΔ (Figure 3A; compare rows 4, 6, and 7). An additional conclusion can be drawn from the electrophoretic mobilities of the SltA400–698-HA3 forms (Figure 3, C and D): phosphorylation of SltA is independent of SltB activity. Thus the SltB Psk domain must not play any role in the phosphorylation of SltA32kDa. We also determined that pseudokinase PskA, having similarity to SltB (Mellado et al., 2015), is required neither for SltA phosphorylation nor for its proteolysis (unpublished data), in agreement with its absence having an slt+ phenotype (Mellado et al., 2015). Finally, proteolysis of SltB was also demonstrated in an SltA400–698 background. A strain expressing SltB-GFP was constructed, and the cleaved SltB protease domain of SltB was detected in protein extracts of both SltA-HA3 and SltA400–698-HA3 strains (Figure 3D).
Mutagenesis confirms that SltB contains a chymotrypsin-like serine-protease domain
Previous in silico searches identified a putative chymotrypsin-like domain in the C-terminal moiety of SltB with low similarity to a protease domain present in the fungal family of Ssy5-like endoproteases (Mellado et al., 2015). Consequently we predicted a catalytic triad composed of Ser-1142 and nearby Asp and His residues. Figure 4A shows the position and boundaries of this protease domain and the conservation of sequences (see Logos) containing the putative catalytic triad of His, Asp, and Ser residues. After a similar modeling of this domain in Ssy5p (Poulsen et al., 2006), we generated a refined model of the region between coordinates 900 and 1192 of SltB (Figure 4B). Figure 4B, left, shows, in a rainbow display (from N-terminus in blue to C-terminus in red), a possible three-dimensional structure of the protease domain of SltB, which was generated using as templates several chymotrypsin-like serine proteases from prokaryotes (see Materials and Methods). Seven of 20 crystal structures were used to generate the model by the Phyre2 server; 58% of the model, corresponding to amino acids 1001–1180, has 98% confidence. These in silico results strongly support the hypothesis that SltB is a serine protease based on the presence of the highly conserved Ser-1142. The proximity of Asp-1036 to Ser-1142 predicts its role in the active site, and the strong conservation of His-1008 and His-1033 makes them the likeliest candidates for the third residue of the triad (Figure 4B, right).
FIGURE 4:
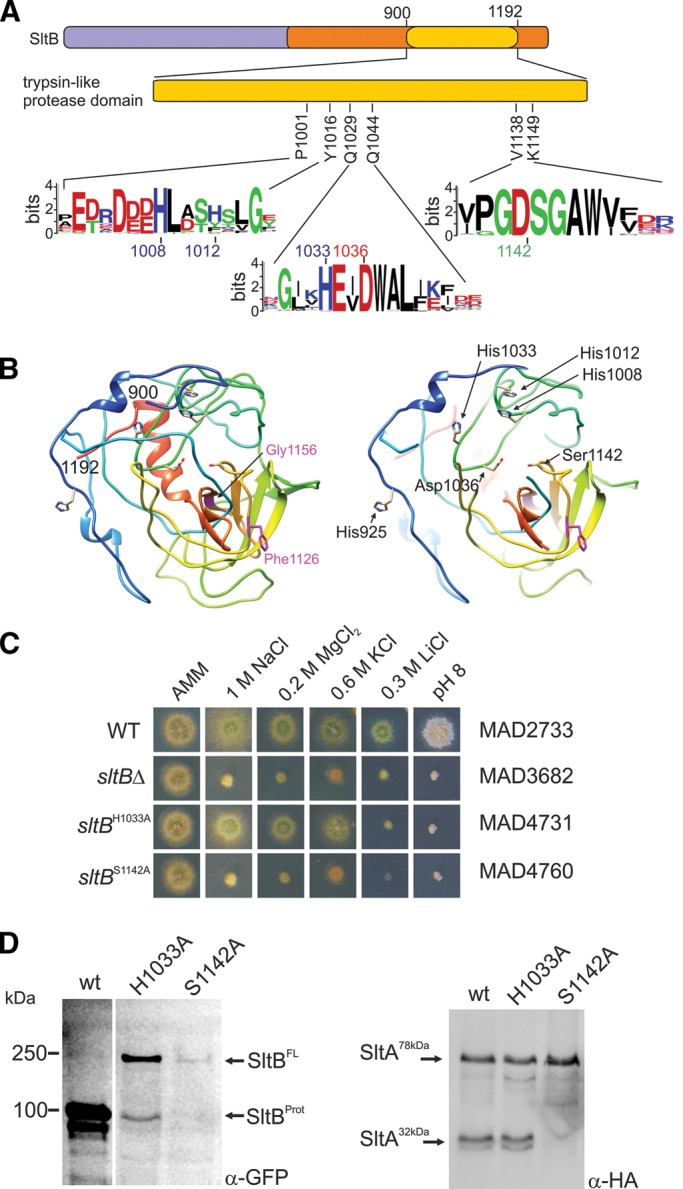
The chymotrypsin-like serine protease domain in SltB. (A) Diagram focusing on the relevant region, showing the conservation of putative key HDS (His/Asp/Ser) triad residues, constituting the catalytic domain of a serine protease. Logos indicate sequence conservation in residues putatively constituting the catalytic triad and flanking regions. (B) Two views of the model generated by the Phyre2 server of the SltB fragment between coordinates 900 and 1192. Left, rainbow representation (see text). Indicated in purple are the positions of Gly-1156 and Phe-1126, in which mutations suppressing null vps alleles have been selected. See Discussion and Mellado et al. (2015) for Gly1156 and Results for the sltB57 mutation deleting F1126. Right, focused (partial) view of the possible catalytic site of the SltB protease. S1142, D1037, and several His residues that might form part of the triad are indicated. Mutations affecting S1142 and H1033 are described in Results. (C) Phenotype analysis of mutant strains expressing chimeras carrying alanine substitutions for either S1142 or H1033. (D) Immunodetection of WT and mutant tagged forms of SltB and the effects of mutations S1142A and H1033A on the proteolysis of SltA-HA3.
To investigate the role of Ser-1142, we constructed a strain carrying an allele of sltB expressing a mutant GFP-tagged SltB in which an alanine replaces Ser-1142 (Figure 4C). This mutant, MAD4760, displayed a null sltB phenotype, exhibiting strong sensitivity to high concentrations of sodium, magnesium, potassium, and lithium ions and alkaline pH (Figure 4C). To determine the basis for the loss-of-function phenotype by the Ser1142Ala substitution, we examined the electrophoretic mobility of this mutant protein. The SltBS1142A-GFP protein was not proteolyzed. The expected 100-kDa band containing the protease domain was not detected and, in contrast, a band corresponding to a mass of nearly 250 kDa was present and identified as the full-length form of SltB (Figure 4D, SltBFL). We then investigated the effect of this mutation on the proteolysis of SltA. Figure 4D, right, shows the absence of SltA32kDa forms in the SltBSer1142Ala mutant background. Thus Ser-1142 is required for SltB protease function, suggesting involvement of the protease domain of SltB in proteolysis of both SltA and SltB. The reduction in levels of mutant SltBS1142A-GFP protein might result from the dependence of sltB transcription on processed SltA (see later discussion).
We then constructed another mutant version of SltB, substituting the highly conserved (and N-proximal to the modeled catalytic triad aspartate-1036) His-1033 by alanine (Figure 4B, right). This mutant, MAD4731, exhibits nearly WT tolerance to high levels of sodium, magnesium, and potassium ions but is strongly sensitive to high concentrations of lithium ions and alkalinity (Figure 4C). Thus sltBH1033A results in partial loss of function. In fact, we detected a reduction in SltBH1033A proteolysis. Although the 100-kDa band corresponding to the SltB protease domain was present, the full-length mutant SltB band was very prominent, in contrast to the extremely low level in the strain expressing the WT SltB-GFP fusion (Figure 4D, left). Consistent with the presence of the proteolyzed SltB form, SltA32kDa was also evident in the sltBH1033A mutant background (Figure 4D, right). Despite the reduction in efficiency of SltB proteolysis, levels of SltA32kDa and SltA78kDa in the sltBH1033A mutant are comparable to those observed in a WT sltB background (Figure 4D).
This mutant analysis indicates that the protease domain of SltB has a dual function, mediating proteolysis of both SltB and SltA. It also demonstrates that Ser-1142 is crucial to protease activity, identifying SltB as a member of the family of serine proteases.
sltB mutations suppressing vpsΔ alleles affect SltB and/or SltA proteolysis
Most mutations in sltB have been isolated as suppressors of the poor, slow growth resulting from certain vpsΔ alleles (Calcagno-Pizarelli et al., 2011; Mellado et al., 2015). We focused on three mutations that affect the Slt pathway to different degrees. sltB53 is the most extreme mutation, with a complete loss-of-function phenotype; sltB56 has a leaky phenotype; and sltB57 has almost no phenotype related to cation and alkaline pH tolerance (Mellado et al., 2015). To investigate the effect of each mutation on SltA signaling, we constructed a collection of sltB mutant strains expressing the tagged SltA-HA3 protein.
sltB53 was selected as suppressing vps20Δ (Calcagno-Pizarelli et al., 2011; Mellado et al., 2015). The SltB53 mutant protein lacks Phe-1126 and results in a null sltB phenotype (Figure 5A; Mellado et al., 2015). In sltB53 strains, SltA is not proteolyzed, and only the SltA78kDa form is detected (Figure 5B). This result established that SltB53 has impaired protease activity on SltA. Because Phe-1126 is in the protease domain, SltB proteolysis was also investigated. A strain expressing an SltB53-GFP fusion protein was constructed (MAD4013). Its phenotype is indistinguishable from that of sltB53 strains (Supplemental Figure S3). Lack of Phe-1126 also prevented SltB proteolysis (Figure 5C), indicating that deletion of Phe-1126 debilitates the protease domain, supporting a model in which the protease domain of SltB is responsible for proteolysis of both SltB and SltA.
FIGURE 5:
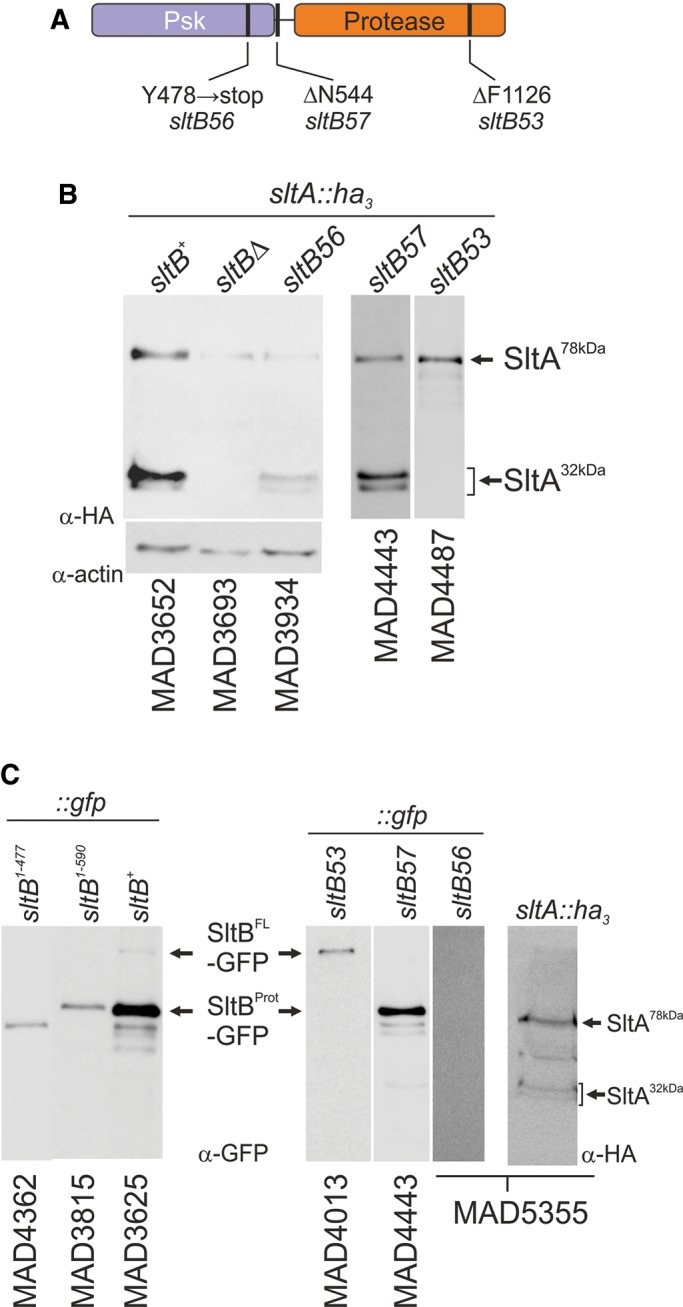
Effects of vpsΔ suppressor mutations sltB53, 56, and 57 on the proteolysis of SltA and SltB. (A) Representation of the SltB protein, indicating the localization of amino acid changes present in mutant alleles sltB53, 56, and 57. (B) SltA-HA3 proteolysis in sltB mutant backgrounds. Actin levels are included as controls in order to show the reduction in SltA protein levels in a sltB56 background. (C) SltB proteolysis in vpsΔ suppressors and constructed truncated forms containing only the Psk domain.
However, the paradoxical finding that the sltB56 mutation truncates SltB upstream of the protease domain while resulting in only a partial-loss-of-function phenotype challenged this working model of Slt regulation. The sltB56 mutant allele was also selected as suppressing a vps20 deletion. sltB56 creates a stop codon, leading to termination of SltB after amino acid 477 (Y478 → stop, TAA; Figure 5A; Mellado et al., 2015). To determine why a mutation leading to loss of nearly two-thirds of the SltB protein nevertheless has a partial-loss-of-function phenotype, we first analyzed its effect on SltA proteolysis. MAD3934 expresses the SltA-HA3 fusion in the sltB56 mutant background. Immunodetection of SltA in protein extracts of MAD3934 showed a reduction in levels of the SltA32kDa forms, but these were nevertheless detectable (Figure 5B). To resolve this paradox, we constructed a strain expressing a truncated version of SltB resembling the presumed sltB56 translation product (SltB1–477) fused to GFP. This strain (MAD4362) has a complete-loss-of-function phenotype (Supplemental Figure S3). To eliminate the possibility that the GFP moiety might be responsible for this null phenotype, we constructed another, similar strain expressing only the SltB fragment (1–477) by inserting a stop codon (TGA) between the SltB and GFP coding sequences. This strain, MAD5268, also has a null sltB phenotype (Supplemental Figure S3). These results suggested that the cause of the leaky cation-sensitive phenotype of sltB56 might be a readthrough of the mutant TAA codon. To test this possibility, we replaced the TAA codon with a TGA stop codon. The resulting sltB mutant strain, MAD5384, has a null sltB phenotype (Supplemental Figure S3). The mutant SltB56 protein was then tagged with GFP in an attempt to visualize the truncated protease domain of SltB or the full- length protein (MAD5355); however, immunodetection experiments failed to reveal any form (Figure 5C). SltA proteolysis was again observed in strain MAD5355 (Figure 5C), and once again, levels of SltA32kDa were reduced. We conclude that readthrough of the ochre stop codon created by sltB56 and the apparent ability of low amounts of SltB to mediate processing of SltA are responsible for the partial-loss-of-function phenotype of sltB56.
The third mutation investigated was sltB57 (ΔΝ544), selected as a suppressor of vps32Δ but having no effect on tolerance of cations or alkalinity (Mellado et al., 2015). In agreement, proteolysis of SltB and SltA proceeds normally in a strain expressing SltB57-GFP and SltA-HA3 (MAD4443; Figure 5, B and C). We postulate that residue Asn-544, located in the central region of SltB, occurs in a region specifically involved in suppression of vpsΔ mutations.
Functional analysis of the protease domain and evidence for intramolecular autoproteolytic processing of SltB
To determine whether the protease domain of SltB, when expressed alone, can mediate proteolysis of SltA, we constructed a strain expressing residues 692–1272 of SltB fused to GFP under the control of the endogenous sltB promoter, MAD3711. The resulting sltB mutant strain has a null sltB phenotype, indicating that the SltB692–1272 fragment alone is nonfunctional (unpublished data). This truncated fusion is detectable in Western blots, and its electrophoretic mobility approximates that of the proteolyzed fragment of a full-length SltB fused to GFP (Figure 6A). This result suggested, first, that cleavage occurs close to residue 692 and, second, that the Psk domain is required for SltB functionality.
FIGURE 6:

Analysis of mutations in the protease domain of SltB. (A) Construction of strains expressing the fragment between codons 692 and 1272, which encodes the protease domain of SltB. SM indicates selectable marker. Right, immunodetection of Myc3- and GFP-tagged versions of SltB692–1272. Bottom, immunodetection of SltA-HA3 forms in a strain (MAD3750) expressing SltB692–1272. (B) Scheme illustrating the SltB forms expressed by the constructed diploids. (C) Immunodetection of SltB forms in haploids and diploids. Coexpression of SltB53-GFP and SltB692–1272-Myc3 does not result in proteolysis of SltB53. Coexpression of SltB53-GFP and the wild-type SltB results in low levels of the SltB53Prot-GFP form (right, longer exposure). In the experiment on the left, a mixture of anti-GFP and anti-Myc primary antibodies was used to visualize both types of SltB-tagged proteins simultaneously.
To investigate the proteolytic processing of SltB, we used the proteolysis-recalcitrant SltB53 protein and determined whether the SltB692–1272 (Figure 6A) fragment or the full-length SltB protein can act in-trans to mediate SltB53 proteolysis. We constructed diploid strains expressing the SltB53-GFP fusion together with either SltB (WT) or the SltB692–1272 fragment fused to the Myc3 tag (Figure 6B). Using the two independent SltB53-GFP/ SltB692–1272 diploids (MAD4510 and MAD4511), we found that only the bands corresponding to each fusion protein were present, and the ∼100-kDa SltBProt (expected from a SltB692–1272-mediated proteolysis of SltB53-GFP) was not detectable. In the case of SltB53-GFP/SltBwt diploids (MAD4519 and MAD4520), we detected higher levels of SltB53-GFP than those detected in the haploid SltB53-GFP strain (MAD4013; Figure 6C), and by increasing protein loading and exposure of Western blots, we were able to detect very low levels of the ∼100-kDa SltBProt from SltB53-GFP processing that were not detectable in protein extracts of the haploid expressing SltB53-GFP. It is worth noting that sltB53 is recessive and the diploids MAD4519 and MAD4520 have a WT phenotype (unpublished data), suggesting that processing of SltA is normal. In contrast, SltB53-GFP/SltB692–1272 diploids MAD4510 and MAD4511 have a null sltB phenotype. Therefore the SltB692–1272 fragment containing the serine protease domain of SltB is not functional in-trans for either SltA or SltB53. These data support a model for intramolecular autoproteolysis of SltB, and the diploids show that any trans autoproteolytic activity of SltB must be very limited. Alternatively, the SltB53 mutant protein might be in an irreversibly inaccessible conformation for autoproteolytic processing. However, the SltB53 protein apparently does not interfere with processing of WT SltA or SltB. The extremely low levels of SltBS1142A protein precluded its use in these experiments.
Intracellular localization of SltB
S. cerevisiae Ssy5p is a peripherally associated plasma membrane protein (Forsberg and Ljungdahl, 2001). To determine the localization of SltB in A. nidulans, we exploited our collection of SltB-GFP fusion strains. SltB-GFP is homogeneously distributed throughout the cytoplasm but is excluded from nuclei (Figure 7A; see also Supplemental Figure S4 for 4′,6-diamidino-2-phenylindole dihydrochloride [DAPI] staining of nuclei in fixed cells). No relationship with internal or plasma membranes was observed. Given that the SltB-GFP fusion is mostly proteolyzed to the 100-kDa form containing the protease domain, the observed fluorescence might result mainly from this form. The fluorescence of SltB692–1272-GFP, mimicking the proteolyzed form, is similarly distributed throughout the cytoplasm with exclusion from nuclei (Figure 7B and Supplemental Figure S4B). To determine the localization of a full-length SltB, we used MAD4013 expressing SltB53-GFP. Consistent with the lower SltB53 protein levels seen in Western blots (Figure 5C), less fluorescence was seen in cells expressing SltB53-GFP than in those expressing SltB-GFP, but fluorescence distribution was similar to that seen with SltB-GFP and SltB692–1272-GFP (Figure 7C and Supplemental Figure S4C). We additionally used the doubly tagged GFP-SltB-Myc3 fusion, our only SltB chimeric protein carrying the GFP tag at the N-terminus. Fluorescence distribution in this case was also similar to that of the other forms (Figure 7D and Supplemental Figure S4D). We conclude that the full-length form of SltB and both fragments resulting from proteolysis maintain a strongly preferential cytoplasmic localization. Thus SltB must preferentially function in the cytoplasm.
FIGURE 7:
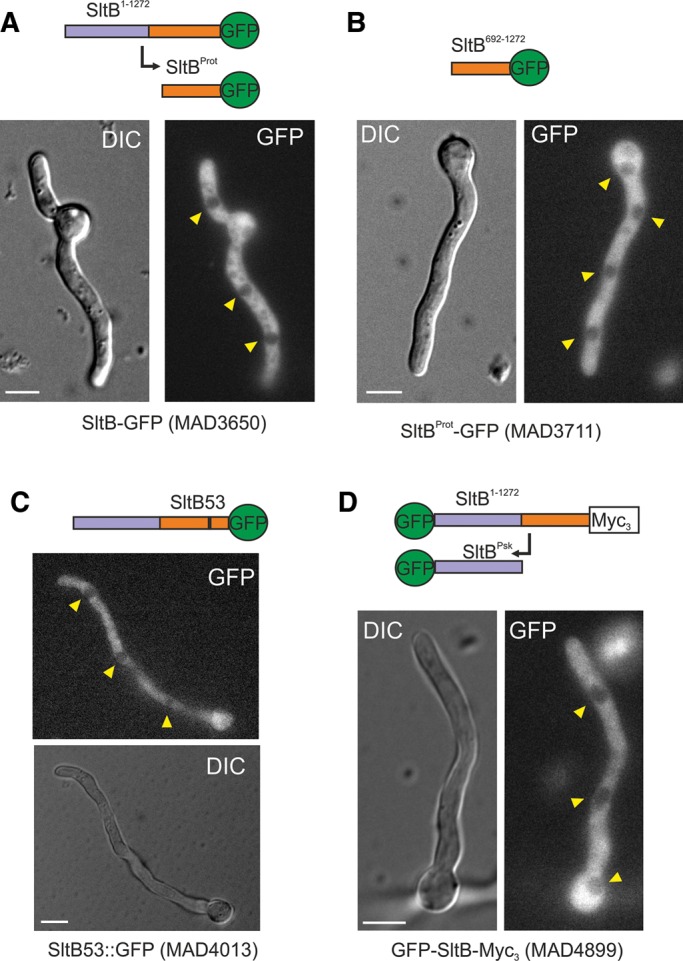
Intracellular localization of SltB. Various GFP-tagged forms of SltB were analyzed for localization. A representative germling is shown for each strain: (A) MAD3650, (B) MAD3711, (C) MAD4013, and (D) MAD4899. In all cases, the fluorescence is confined to the cytoplasm and excluded from nuclei. Nuclei are indicated by yellow arrowheads. A schematic representation of the predicted fusion proteins in each cell is shown on top of each pair of differential interference contrast (Normarski optics) and GFP-labeled images. Scale bars, 5 μm.
Intracellular localization of SltA
The instability of transformants potentially expressing SltA-GFP fusions (see earlier discussion) precluded their use in determining the intracellular localization of SltA. Because null alleles of sltA allow full viability, fusions of GFP to SltA would appear to be deleterious. Therefore we used indirect immunofluorescence to determine the subcellular localization of SltA-HA3 fusions under different growth conditions. In cells of strain MAD3652, full-length SltA-HA3 accumulated throughout the cytoplasm but was barely detectable in nuclei (Figure 8A, red arrowheads in Alexa Fluor 488 image), suggesting a possible differential localization of the 78- and 32-kDa SltA forms. Consequently we determined the subcellular localization of the primary 78-kDa form of SltA using the null sltB strain MAD3693. Figure 8B shows fluorescence accumulations along the cytoplasm but no nuclear SltA-HA3 in the absence of SltB. As a precaution to validate our indirect immunofluorescence detection of SltA-HA3, we analyzed the localization of HypA, a component of the TRAPPII complex (Pinar et al., 2015), tagged with HA3. HypA-HA3 showed a similar localization to that reported for the HypA-GFP fusion (Pinar et al., 2015)—accumulation at the Spitzenkörper (asterisk) and in vesicles (Figure 8C, blue arrowhead). Next we studied the localization of the functional, truncated SltA400-HA3 protein (strain MAD4661), since it mimics both phosphorylated and unphosphorylated 32-kDa forms of SltA-HA3. Figure 8D shows that the SltA400-HA3 protein was detected mainly in nuclei. Despite the fact that SltA400-HA3 and 32-kDa truncated forms of SltA-HA3 are detected to a similar extent in Western blots (Figure 3, B and D), differences in detection of SltA400-HA3 and SltA-HA3 proteins in nuclei were observable. Our overall results indicate that SltA78kDa is cytoplasmic, and SltA32kDa forms are mainly nuclear. However, it is possible that the 78-kDa form of SltA might regulate nuclear accumulation of the 32-kDa forms. Of note, the subcellular distribution of these proteins did not change in cells subjected to alkaline or cation (1 M NaCl) stress (unpublished data), suggesting that subcellular localization is not crucial to the regulation of SltA function.
FIGURE 8:
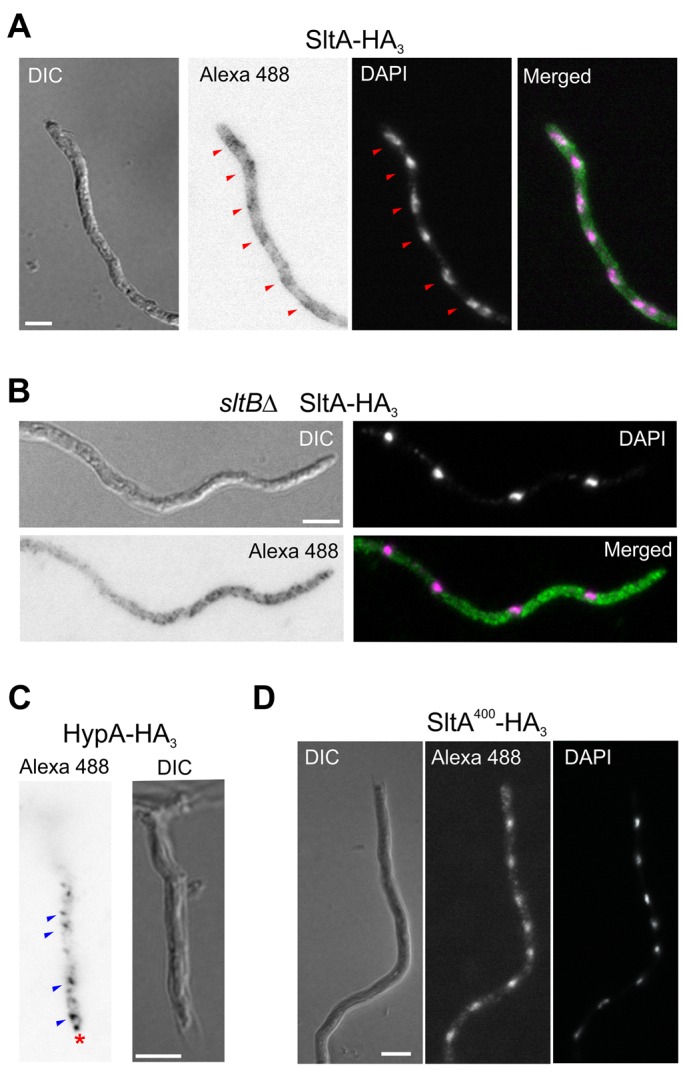
Intracellular localization of SltA. HA3 fusions were detected using a secondary antibody labeled with Alexa Fluor 488 (Alexa 488). (A) Immunofluorescence detection of SltA-HA3 expressed by strain MAD3652. (B) Detection of SltA400-HA3 expressed by null sltB strain MAD3693 under the regulation of the thiamine-repressible promoter in the absence of thiamine. (C) Detection of HypA-HA3 fusion as a quality control for immunodetection procedure. HypA-HA3 (MAD4784) displayed a similar localization to HypA-GFP, as described in Pinar et al. (2015). The red asterisk indicates the Spitzenkörper, and blue arrowheads indicate the intracellular vesicles, probably the Golgi. (D) Detection of SltA400-HA3 expressed by strain MAD4661 under the regulation of the thiamine-repressible promoter in the absence of thiamine. Bar, 5 μm.
The effects of cation stress and alkaline pH on SltA and SltB proteolysis
Because SltA and SltB are both required for tolerance to cation stress and alkaline pH, we inquired whether these conditions affect proteolysis. The foregoing experiments were all done in nonstressing conditions. Strains expressing SltB-Myc3 (MAD3734) and SltA-HA3 (MAD3652) were cultivated in alkalinized AMM or AMM containing elevated concentrations of sodium, potassium, or magnesium ions, conditions in which the Slt activities are required. After 16 h of incubation at 37°C in these media, the distribution of SltA forms was analyzed. Figure 9A shows that the three forms of SltA are observed in similar ratios to those found in AMM. SltB proteolysis was also observed in all cases, with a similar pattern to that found in nonstressing conditions, in which the processed SltB form is mainly detected (Figure 9B). We then examined the effect of stress applied for short times. Mycelia grown in AMM were subjected to addition of an excess of sodium and to alkalization of the medium. The pattern of SltA bands at 15 and 30 min after the stress showed only a relatively minor modification. In the case of excess sodium, we observed an increase in the levels of the phosphorylated SltA32kDa form and, after alkalization, higher levels of the SltA78kDa form (Figure 9C). In any case, the phosphorylation status of SltA does not seem to be markedly regulated under any of the conditions examined, despite the predicted role of SltA in transcribing stress response genes under such conditions. There was no change in the pattern or mobility of SltB forms, which mainly consist of the SltBProt form (Figure 9D). In contrast to other regulatory systems dependent on proteolysis, such as the pH regulatory system mediated by PacC or the S. cerevisiae amino acid–sensing system mediated by Spt1p and Spt2p, there is no known growth condition in which SltA and/or SltB proteolysis is severely or completely curtailed. Because of the dependence of SltA proteolysis upon SltB activity and SltB activity on autoproteolysis, it appears that proteolysis of SltB is also a not markedly regulated process in this pathway. The identity of kinases and phosphatases participating in SltA signaling remains to be determined. However, the calcium-dependent phosphatase calcineurin does not participate in the PTM process of SltA, although its absence reduces levels of all SltA forms (Supplemental Figure S5).
FIGURE 9:
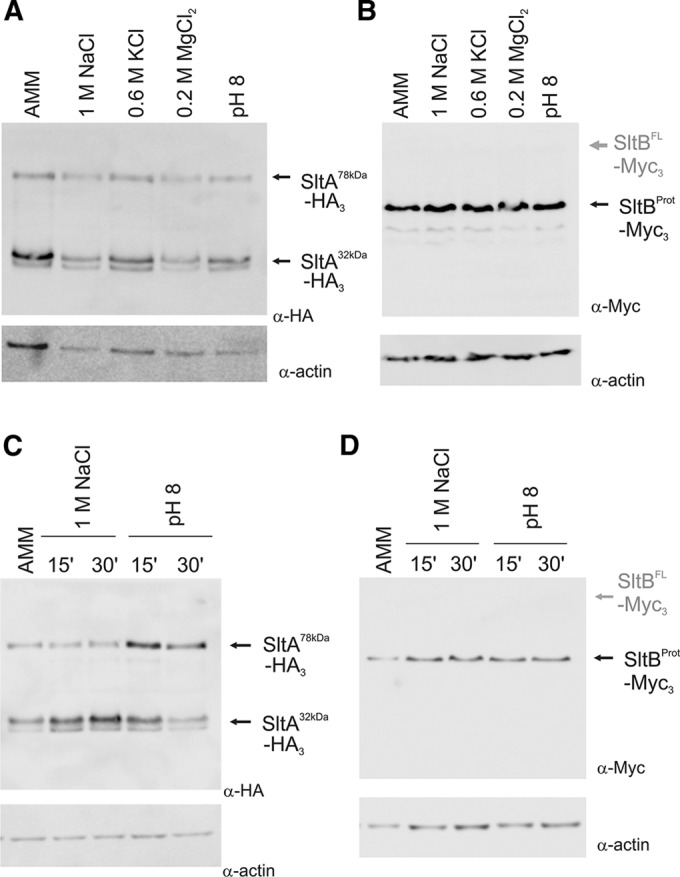
Cation or alkaline pH stress does not appreciably affect cleavage of either SltA or SltB. (A) Immunodetection experiment showing the pattern of SltA-HA3 bands in protein extracts from strain MAD3652 grown under various conditions. (B) Immunodetection of SltB-Myc3 fusion in protein extracts of strain MAD3734. (C) Mycelia grown in nonstressing conditions (AMM) were subjected to an elevated concentration of sodium or to medium alkalinization. Immunodetection of the SltA-HA forms after 15 and 30 min of strain MAD3652 treatment. (D) Immunodetection of SltB-Myc3 fusion in protein extracts from cells of strain MAD3734 grown and treated as in C.
Transcriptional regulation and protein expression levels of SltA and SltB forms
Previous work demonstrated that sltB mRNA levels are low in the absence of the SltA protein (Mellado et al., 2015). To study the levels of the SltB protein and its proteolysis in a null sltA background, we generated a sltAΔ::riboBAf allele by transformation (see Materials and Methods) in strain MAD3625, creating strain MAD3705. Of interest, the SltB-GFP fusion was not immunodetectable in the absence of SltA (Figure 10A, left). This result showed the strong dependence of SltB regulation on SltA activity. Figure 10B shows a Northern blot in which transcript levels of sltB are visualized in different sltA and sltB mutant backgrounds. Loss-of-function mutations in sltA, such as sltA1, resulting in truncation at amino acid 502 (O’Neil et al., 2002), and sltA114, substituting a key residue in the DNA-binding domain (Mellado et al., 2015; Supplemental Figure S2D), reduced the expression levels of sltB similar to those found in sltAΔ strains (Figure 10B; compare lanes 7 and 8 with lanes 3 and 4). Transcription of sltB is also strongly reduced in nonproteolyzed SltB53 (Figure 10B, lanes 2 and 13) and SltBS1142A (Figure 10B, lane 14) backgrounds, in which the SltA78kDa form predominates (Figure 5B). These results, together with the effects of sltA1 and sltA114 mutations, corroborate our previous conclusion that SltA78kDa has little or no role in sltB transcriptional regulation and that one or both nuclear SltA32kDa forms is a positive-acting inducer of sltB expression, most probably via direct binding to any or all of the five putative SltA-target sites previously identified in the promoter region of SltB (Mellado et al., 2015). This hypothesis was confirmed when the thiamine-repressible expression of the truncated SltA400–698 up-regulated expression of sltB to levels comparable to those found in the WT (Figure 10B; compare lanes 5 and 6 for the thiamine-repressible regulation and lane 1 for WT).
FIGURE 10:

SltA-dependent and -independent transcriptional and functional analyses of sltB. (A) Immunodetection of SltB tagged proteins in null and WT sltA backgrounds. Left, full-length SltB-GFP fusion; right, truncated SltB1–590-GFP fusion. (B) Northern blots showing transcript levels of sltB in several sltA and sltB mutant backgrounds. The probe covered the first 1600 base pairs of the sltB coding region. Absence of hybridization with the transcript of the sltB692–1272::gfp construct was used as a negative control to show sltA-independent levels of the sltB transcript. As total RNA loading control, we used methylene blue–stained 18S rRNA. (C) Northern blot containing samples of total RNA of strains expressing alcAp-driven sltA or sltB. The presence of 1% EtOH in AMM as sole carbon source is an inducing, derepressing condition for alcAp, whereas 3% glucose (Glu) is a noninducing, repressing condition. (D) Western blots showing the presence of the proteolyzed form of SltB-Myc3 when alcAp is induced in strains MAD4015 and MAD4016. Here 1x indicates the presence of a single copy of the construct, and nx indicates more than three copies of the construct. Actin levels were used as loading control.
The undetectable levels of SltB forms in the absence of SltA and the predicted lack of transcriptional function for SltA78kDa contrasts with the messenger levels for several truncated versions of sltB constructed in this work. In Figure 10B, right, the steady-state mRNA level is shown for a strain expressing the nonfunctional SltB1–590-HA3 fusion, which is surprisingly high for a mutant incapable of making SltA32kDa. As expected from Northern blots, SltA-independent expression of SltB1–590-HA3 protein was observed (Figure 9A, right). Transcript levels of another nonfunctional sltB chimera, sltB1–536TGA::gfp, are also unexpectedly higher than sltB levels in null sltA, sltB53, or sltBS1142A::gfp backgrounds (Figure 10B; compare lane 12 with lanes 9, 13, and 14). These results suggest that detection of truncated nonfunctional chimeras such as SltB1–590-HA3 or -GFP (Figure 5C), SltB1–477-GFP (Figure 5C), and SltB692–1272-GFP or -Myc3 (Figure 6A) is possible because of SltA-independent, high levels of their respective mutant mRNAs. It is possible that these truncation mutant mRNAs are significantly more stable than WT sltB mRNA and that their high steady-state levels reflect this stability.
To investigate the effect of SltA-independent expression of SltB, we constructed strains expressing sltB in an ethanol-dependent manner from the alcohol dehydrogenase I promoter, alcAp. The Northern blot in Figure 10C shows the conditional expression of WT sltB when induced by ethanol (EtOH) or repressed by glucose (Glu) in sltA-null and sltA+ strains. The Western blot in Figure 10D shows that the level of the SltBProt-Myc3 form is similar when expressed from alcAp and EtOH induced to that when expressed from the endogenous promoter. This result also demonstrates that SltB-Myc3 proteolysis does not require SltA function. Despite these similar levels of proteolyzed SltB protein, this strain, MAD4015, does not display tolerance to cation or alkaline pH stress (Supplemental Figure S6). Even the presence of extra copies (nx) of the alcAp::sltB::myc3 construct, which results in greater-than-physiological levels of proteolyzed SltB protein (Figure 10D), did not compensate the lack of SltA. Strains expressing sltA solely from the alcAp (Northern blot in Figure 10C) are cation and alkalinity tolerant in EtOH-inducing conditions but not in glucose-repressing conditions (unpublished data). Thus the combined results of this section establish that the proteolytically processed SltA32kDa form is the ultimate mediator of cation and alkalinity tolerance via the Slt pathway.
DISCUSSION
A novel regulatory system involving the zinc-finger transcription factor SltA and the heretofore enigmatic SltB mediates tolerance of high levels of cations and alkaline pH in the filamentous fungus A. nidulans. Using tagged proteins and mutations, we demonstrate that both Slt proteins undergo regulated proteolytic processing. C-terminally tagged SltA exists in two forms—the 78-kDa translation product and a 32-kDa form also containing the DNA-binding region. The 78-kDa version is nonfunctional, unable to mediate transcriptional regulation of genes required for cation and alkalinity tolerance, including sltB. Functionality resides in the truncated 32-kDa SltA, which can activate sltB expression. Thus proteolytic processing of SltA is the main activation mechanism.
Cleavage of SltA requires both the putative pseudokinase and chymotrypsin-like protease domains of SltB. Using protein modeling, we identified SltB Ser-1142 as a key residue of the putative endoprotease catalytic center, confirmed by a Ser1142Ala mutant and defining SltB as a serine protease. Analyses of mutant phenotypes and tagged versions of SltB demonstrate autoproteolysis of SltB, separating Psk and endoprotease domains.
Prediction of the cleavage sites for chymotrypsin- or trypsin-like serine proteases is difficult because specificity is low and Y/F/W or K/R residues, respectively, are preferred at position P1 of target sequences (Di Cera, 2009). Because the electrophoretic mobility of SltA400–698 is close to that of the 32-kDa form, we predict that the SltB-mediated SltA cleavage site is located near Met-400. The K384R385 doublet is highly conserved among SltA homologues and seems a likely target for a trypsin-like activity, and highly conserved Y375 is a putative target for a chymotrypsin-like activity. The sltB53 mutation deleting F1126 prevents both SltB and SltA processing; this deletion in the endoprotease domain might disrupt functionality of the catalytic triad. Based on structural data from other serine proteases (Di Cera, 2009), absence of F1126 might perturb the structure of the predicted β-barrel, typical of this family of proteases, and subsequently collapse the putative oxyanion hole formed by Ser-1142 and the conserved Gly-1140 in SltB. The SltB692–1272 fragment has the same mobility as the truncated version of the SltB protease domain, and thus cleavage must be close to Met-692. Among SltB homologues, strong sequence conservation is observed after SltB residue 700 (Mellado et al., 2015); K703 and K707 are highly conserved, as is W722, presenting likely targets for trypsin- and chymotrypsin-type activities. Similarly to other serine proteases (Page and Di Cera, 2008), SltB is expressed as a zymogen. The apparent requirement for the Psk domain ensures specificity, and there is no evidence that SltB cleaves any proteins other than SltA and itself. There are numerous precedents for proteases having prodomains that interact with the protease after autoproteolysis to modulate activity (reviewed in Di Cera, 2009; Page and Di Cera, 2008). The SltB Psk domain (>600 aa) might constitute a prodomain and, if so, is, to our knowledge (and compared with Ssy5, 381 aa; Poulsen et al., 2006), the largest described to date.
The precise role of the Psk domain remains enigmatic, but our results clearly show that the protease domain alone is insufficient to cleave SltA into the 32-kDa form. Mellado et al. (2015) hypothesized a scaffolding role for the Psk domain, based on predicted activities for this class of nonfunctional kinases; as a prodomain, it might contribute to proper folding of the catalytic domain. Figure 11 accommodates available data and illustrates two of the possible proteolytic scenarios. SltB and SltA are synthesized as 140- and 78-kDa proteins, respectively. In scenario A, SltB is first autoproteolyzed, and the Psk and protease domains continue to associate but in a different configuration, constituting an inactive but primed protease. Then, SltA interacts with and activates the SltB complex and is cleaved by it. In scenario B, the SltB and SltA translation products interact, activating the protease domain, leading to cleavage first of SltB and second to cleavage of SltA. Both A and B lead to the active, 32-kDa form of SltA. Model B envisages a 1:1 stoichiometry between SltA and SltB or at least between the active form of SltA and SltB. Because SltB autoprocessing does not require SltA and SltB is barely detected in its full-length form in all conditions tested, suggesting a preferentially cleaved status for SltB, we consider model A more plausible. In addition, detection of the full-length form of SltA in all conditions and the leaky phenotypes of sltB56 and sltBH1033A mutants suggest a purely catalytic role for SltB.
FIGURE 11:

Working models for SltA/SltB proteolytic processes. Two sequential proteolytic steps ordered in two different ways. Branch A begins with SltB proteolysis, followed by association with SltA to mediate its proteolysis. Branch B presupposes the association of SltB and SltA before cleavage of SltB, with the processing of SltA following. In both cases, the final product of this pathway is the functional SltA32kDa product, which mediates transcriptional regulation in response to cation stress and alkaline pH, one of the targets being the sltB gene. The remaining complex might be inactive and subject to degradation/elimination. The Psk domain of SltB is depicted as a blue hexagon and the protease domain as a pair of scissors. The gray circle represents the DNA-binding domain of SltA, and the red bar is the target sequence for the SltB protease.
Another important issue is how the protease activity of SltB is modulated, since SltA and SltB proteolyses are constitutively detected. SltB Psk prodomain and SltA might play a role in regulating activity and inactivation of the catalytic domain. Because a truncated version of SltB mimicking the protease domain (starting at residue 692) is nonfunctional, disassembly of the SltA/SltB complex after SltA cleavage yields an inactive SltB protease, at least with regard to SltA processing. The role of the N-terminal region of SltA needs clarification at two levels: how it mediates SltA activity and whether it modulates SltB activity. In the SPS system, an N-terminal region of Stp1/2 TFs is central to regulating localization and stability. In addition, the stability of the Ssy5 prodomain (a proteasome substrate upon ubiquitination) is key to maintaining Ssy5 activity (Pfirrmann et al., 2010). Of interest, a recent systematic study of the ubiquitome of A. nidulans reveals that SltA and SltB are ubiquitinated (Chu et al., 2015). Remarkably, K257, 690, and 693 are found to be ubiquitinated in SltB, and the proximity of these PTMs to the target site of proteolysis suggests an alternative mechanism regulating SltB cleavage. In SltA, five lysines were found to be ubiquitinated—K43, 220, 259, 405, and 452—of which three are located in the N-terminal region, K405 close to the cleavage site and the predicted nuclear localization signal, and K452 in the DNA-binding region. SltA ubiquitination might regulate its stability, processing, cellular localization, and DNA-binding activity.
Finally, the possibility that SltB has additional functions cannot be dismissed. Of note, mutations such as sltB57 (ΔN544; Mellado et al., 2015) do not detectably affect SltA and SltB proteolyses or tolerance of cations but are able to suppress deletion of trafficking vpsΔ mutations. sltA mutations having only minor effects on cation or alkalinity tolerance but able to suppress vpsΔ mutations are also known (Calcagno-Pizarelli et al., 2011). Alternative roles of proteases are challenging topics, based on the increasing number of defined pseudoproteases (inactive proteases; reviewed in Reynolds and Fischer, 2015), as is the presence in the cell of inactive but primed proteases (Di Cera, 2009) as important players in cell processes. SltB may well serve as a model to understand these mechanisms in a genetically amenable organism. Future work will aim to identify further components of the cation/alkalinity tolerance system and elucidate its role in intracellular trafficking and calcium homeostasis. Among these elements are the phosphorylation system acting on SltA and its role in transcriptional regulation and the structural genes mediating cation homeostasis. In fact, calcineurin has no role in regulating SltA phosphorylation, but it might participate in regulating SltA levels. Deciphering the molecular mechanisms of Slt pathway will enable an understanding of how filamentous fungi prosper under abiotic stress in the environment and hosts.
MATERIALS AND METHODS
Strains and growth conditions
For standard DNA cloning in plasmids or its propagation, we used the Escherichia coli strains DH1 (endA1 recA1 gyrA96 thi-1 glnV44 relA1 hsdR17(rK- mK+) λ-) and DH5a (F- endA1 glnV44 thi-1 recA1 relA1 gyrA96 deoR nupG Φ80dlacZΔM15 Δ(lacZYA-argF)U169, hsdR17(rK- mK+), λ–). Genotypes of A. nidulans strains used in this work are listed in Supplemental Table S1, and nomenclature of genes is described in Clutterbuck (1993). A. nidulans strains were constructed either by transformation with plasmids or DNA cassettes following the protocol described in Tilburn et al. (1983) or by crossing and selecting recombinants. Diploid strains were made by mixing protoplasts of each strain and regenerating fused protoplasts on selective minimal medium containing 1 M sucrose as osmotic stabilizer. Aspergillus complete medium (ACM) and AMM were as described in Cove (1966), and liquid media were like the solid ones but without the addition of 1% agar. Solid ACM was mainly used to produce conidia for propagation and maintenance of strains. Solid AMM, supplemented for auxotrophies, was routinely used to characterize the phenotypes of mutations in slt genes as described in Mellado et al. (2015). The sensitivity of mutant slt strains to salt/cations and alkalinity was assessed by taking as reference growth on solid AMM containing 1 M NaCl. Sensitivity of slt mutants to alkalinity was assayed by adjusting media to pH 8.0 with 100 mM Na2HPO4. The conditional expression of slt genes driven by the alcA promoter in liquid or solid medium was induced by adding 1% (vol/vol) EtOH as main carbon source and repressed by adding 3% (wt/vol) glucose. Expression driven by thiA promoter was down-regulated by adding 100 μM thiamine. Phenotypes on solid media were scored after incubation of strains for 48 h at 37°C.
Construction of transformation cassettes for epitope and fluorescent protein tagging of Slt proteins
Chimeric genes expressing complete or fragments of SltA or SltB proteins fused to epitopes were constructed by gene replacement technique using DNA cassettes obtained following the fusion-PCR and selection protocols described in Markina-Iñarrairaegui et al. (2011) and Nayak et al. (2006). Supplemental Table S2 lists the specific oligonucleotides used to generate each of the transformation cassettes. Aspergillus fumigatus (Af) pyrGAf, riboBAf, and pyroAAf genes were used as prototrophic selection markers (for details, see Markina-Iñarrairaegui et al., 2011; Mellado et al., 2015). Point mutations in sltB to create the H1033A and S1142A substitutions were introduced by PCR using mutant oligonucleotides (Supplemental Table S2) and a plasmid containing the genomic sltB sequence as template. Plasmids containing each of the mutagenized sltB codons were sequenced to ensure the absence of additional mutations and subsequently used as templates for generation of PCR fragments, which were then used a templates for the generation of transformation DNA cassettes following the fusion-PCR protocol. To construct the double tagged GFP-SltB-Myc3 strain, the null sltB::riboBAf strain (MAD3669) was transformed with a 5′UTR::gfp::sltB::myc3::pyrGAf::3′UTR DNA cassette generated by the fusion-PCR protocol. The 5′ and 3′ untranslated regions (UTRs) correspond to sltB flanking sequences required for homologous recombination. Transformants carrying the replacement sltB allele were selected by growth on regeneration plates containing pyrimidine-less selective medium but supplemented with riboflavin.
The thiamine-repressible promoter thiAp was used to express the SltAMet400::HA3 fusion protein in a thiamine-dependent manner (Calcagno-Pizarelli et al., 2011, and references therein). A 5′UTR::thiAp::sltA400–678::ha3::pyrGAf::3′UTR DNA cassette was constructed by fusion PCR and used to replace the null allele sltAΔ::riboBAf in the recipient strain MAD3919. To express SltB protein conditionally using the ethanol-inducible promoter alcAp, a DNA fragment containing complete CDS of SltB and the fragment coding for Myc3 was cloned into plasmid pALC-pyroA* (Garzia et al., 2009). Oligonucleotide pair SltBmyc-BamHI sense and SltBmyc-XmaI antisense (Supplemental Table S2) were used to amplify the sltB::mycx3 fusion from genomic DNA of strain MAD3734. After digestion with BamHI and XmaI, this fragment was cloned into the corresponding sites at pALC-pyroA* to generate plasmid pALC-sltBmyc-pyroA*. The truncated version of pyroA (pyroA*) present in the plasmid was used as selection marker for directed integration at the mutant pyroA4 locus to reconstruct a functional copy of pyroA. The pALC-sltBmyc-pyroA* plasmid was transformed into recipient strain MAD3816. Pyridoxine prototrophic single-copy or multicopy transformants were selected for further analyses. In all cases, the appropriate gene replacement event or the integration of plasmids, in single or multiple copies, was verified by Southern blotting following standard procedures.
Protein extraction and immunodetection of tagged Slt proteins
For total protein extraction, strains were routinely cultivated in supplemented liquid AMM for 18 h at 37°C. The effect of environmental stress in SltA and SltB proteolysis was studied by adding the stress agent, for example, 1 M NaCl, 0.2 M MgCl2, 0.6 M KCl, or 0.1 M Na2HPO4, to alkalinize pH, with further incubation for the period indicated. To extract proteins, mycelia were collected by filtration and frozen in dry ice. Samples were lyophilized for 16 h and processed by two different protein extraction methods. For samples used exclusively for immunodetection, we followed the alkaline-lysis extraction method described in Hervas-Aguilar and Peñalva (2010). Native Aspergillus total protein extracts, suitable for in vitro λ-phosphatase assays, were obtained using the protocol of protein extraction in A-50 buffer described in Hernández-Ortiz and Espeso (2013). Protein concentrations of extracts from alkaline lysis were estimated by PAGE; for native protein extracts, we used the Bio-Rad (Hercules, CA) protein assay procedure.
For Western blotting, proteins were separated in 10% SDS–polyacrylamide gels and transferred to nitrocellulose membranes using the Trans-blot Turbo transfer pack and Trans Blot Turbo system (Bio-Rad) following the manufacturer’s instructions. GFP-tagged proteins were detected using polyclonal mouse anti-GFP (1:5000; clones 7.1 and 13.1; Sigma-Aldrich Quimica SL, Madrid, Spain); HA3-tagged chimeras were detected using monoclonal rat anti-HA (1:1000; clone 3F10; Sigma-Aldrich), and Myc3-tagged proteins were detected using monoclonal mouse anti–c-Myc (1:10,000; clone 9E10; Sigma-Aldrich). Actin was detected using monoclonal mouse anti-actin (1:5000; clone C4; MP Biomedicals, Santa Ana, CA). Peroxidase-conjugated goat anti-mouse immunoglobulin G (IgG; 1:4000; Jackson ImmunoResearch Laboratories, Suffolk, UK) or goat anti-rat IgG + IgGM (1:4000; Southern Biotech, Birmingham, AL) was used as secondary antibody. Western blots were developed using the ECL kit (GE Heathcare), and images were taken using a Fujifilm Luminescent Image Analyzer LAS-3000 and processed with Multi-Gauge, version 3.0, software (Fujifilm Europe GmbH, Barcelona, Spain).
Gene expression analyses
Wild-type and mutant strains were cultivated in supplemented liquid AMM for 16 h at 37°C. For total RNA extraction, mycelia were harvested by filtration using Miracloth (Calbiochem, Merck-Millipore, Darmstadt, Germany); 100 mg of mycelial samples was frozen and ground in liquid nitrogen, and 1 ml of TRIreagent (Fluka, Sigma-Aldrich Quimica SL, Madrid, Spain) was added and further processed according to the manufacturer’s protocol. Northern blot analyses were carried out following standard protocols as described in Etxebeste et al. (2008). RNA concentrations were measured using a NanoDrop spectrophotometer, and 10 μg of total RNA was loaded per sample and lane in Northern assays. mRNA-DNA hybridization was detected using a PhosphorImager (Fujifilm) as described in Mellado et al. (2015). Images were processed using Multi-Gauge, version 3.0, software.
Microscopy
Samples for epifluorescence microscopy were prepared in watch minimal medium (WMM) as described in Peñalva (2005). Conidiospores were inoculated in WMM with 17.5 mM NaH2PO4, 7.5 mM NaHPO4, 5 mM ammonium (+)-tartrate, and 0.5% glucose and grown for 16–18 h at 25°C. For DAPI (Sigma-Aldrich) nucleic acid staining, cells grown onto coverslips were fixed with a freshly prepared solution of 4% paraformaldehyde in phosphate-buffered saline (PBS) for 15 min at room temperature. Next cells were washed twice with PBS, and nuclei were stained using a solution of 60 μg/ml DAPI in 50% glycerol. After 10 min of incubation, cells were washed twice for 10 min with a 50% glycerol in PBS and mounted onto slides for observation.
For immunodetection of SltA-HA3, we mainly followed the protocol described in Oakley et al. (1990). Conidia were grown on coverslips submerged in supplemented WMM as indicated for 16 h at 25ºC; for thiAp repression, a final concentration of 100 μM thiamine was added. For fixation and partial cell wall digestion to allow entrance of antibodies, we used solutions, enzymes, and conditions indicated in Oakley et al. (1990). All incubations were done at 25ºC. For HA immunodetection, we used monoclonal rat anti-HA (1:500; clone 3F10; Sigma-Aldrich) and, as secondary, a polyclonal goat antibody against rat IgG coupled to Alexa Fluor 488 (5 μg/ml; Molecular Probes, Thermo Fisher Scientific, Waltham, MA).
GFP, Alexa Fluor 488, and DAPI fluorescence was visualized using a Leica DMI-6000b inverted microscope equipped with a 63× Plan Apo 1.4 numerical aperture oil immersion lens (Leica, Wetzlar, Germany), a Leica GFP-specific filter (GFP and Alexa Fluor 488 detection, excitation filter, bandpass 470/40 nm; and emission filter, bandpass 525/50 nm), and the D filter (DAPI detection, excitation filter, bandpass 355–425 nm; and emission filter, long pass 470 nm). Images were recorded with an ORCA-ER digital camera (Hamamatsu Photonics France S.A.R.L., Barcelona, Spain) and processed with MetaMorph (Universal Imaging, Molecular Devices, Sunnyvale, CA) or ImageJ 1.50e (rsb.info.nih.gov/ij/) software.
Bioinformatic tools
Logos were generated at the Weblogo facility at Berkeley (weblogo.berkeley.edu/logo.cgi) using a multiple alignment of sequences from 65 homologues of SltB from fungal species belonging to the Pezizomycotina subphylum. On the y-axis, the entropy for a given position with a maximum of log2 20 = 4.3 bits is shown, indicating the degree of conservation at each position. On the x-axis, the amino acid sequence from the region of interest is shown. The amino acid color scheme is as follows: black, noncharged; red, acid; blue, basic; and green, polar. Jalview 2.9.0b2, open source (Waterhouse et al., 2009), was used for editing multiple sequence alignments.
Models of the SltB fragment between coordinates 900 and 1192 were made using the Phyre2 server (www.sbg.bio.ic.ac.uk/phyre2; Kelley and Sternberg, 2009) with Modeling mode parameter set to intensive. Seven of 20 crystal structures were used to generate the model by the Phyre2 server, and these correspond to proteases of the chymotrypsin- and trypsin-like family 2oua_A protease A from Nocardiopsis alba; 4ic6_A, a plant HtrA protease; 2h5c_A1, an α-lytic protease from Lysobacter enzymogenes; 2qaa_A1 protease B of Streptomyces griseus; 3k6z_A serine protease Rv3671c of Mycobacterium tuberculosis; 2sga_A protease A of Streptomyces griseus; and 2pfe_A protease A of Thermobifida fusca. UCSF Chimera (Pettersen et al., 2004) software was used to modify display and color of generated models.
Supplementary Material
Acknowledgments
We thank Elena Reoyo for technical assistance and Mario Pinar for suggesting the use of HypA-HA3 fusion as a quality control for immunofluorescence detection of SltA-HA3. This work was supported by the Spanish Ministerio de Economia y Competitividad through Grants BFU2009-08701 (partially supported by the Fondo Europeo de Desarrollo Regional) and BFU2012-33142 to E.A.E. L.M. was the holder of an FPI Fellowship associated with Grant BFU2009-08701.
Abbreviations used:
- AMM
Aspergillus minimal medium
- Psk
pseudokinase
- PTM
posttranslational modification
- TF
transcription factor
- vpsΔ
vacuolar-protein-sorting gene deletion.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E16-01-0049) on June 15, 2016.
REFERENCES
- Apostolaki A, Harispe L, Calcagno-Pizarelli AM, Vangelatos I, Sophianopoulou V, Arst HN, Jr, Peñalva MA, Amillis S, Scazzocchio C. Aspergillus nidulans CkiA is an essential casein kinase I required for delivery of amino acid transporters to the plasma membrane. Mol Microbiol. 2012;84:530–549. doi: 10.1111/j.1365-2958.2012.08042.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aza-Blanc P, Ramirez-Weber FA, Laget MP, Schwartz C, Kornberg TB. Proteolysis that is inhibited by hedgehog targets Cubitus interruptus protein to the nucleus and converts it to a repressor. Cell. 1997;89:1043–1053. doi: 10.1016/s0092-8674(00)80292-5. [DOI] [PubMed] [Google Scholar]
- Benayoun BA, Veitia RA. A post-translational modification code for transcription factors: sorting through a sea of signals. Trends Cell Biol. 2009;19:189–197. doi: 10.1016/j.tcb.2009.02.003. [DOI] [PubMed] [Google Scholar]
- Brown MS, Ye J, Rawson RB, Goldstein JL. Regulated intramembrane proteolysis: a control mechanism conserved from bacteria to humans. Cell. 2000;100:391–398. doi: 10.1016/s0092-8674(00)80675-3. [DOI] [PubMed] [Google Scholar]
- Calcagno-Pizarelli AM, Hervas-Aguilar A, Galindo A, Abenza JF, Peñalva MA, Arst HN., Jr Rescue of Aspergillus nidulans severely debilitating null mutations in ESCRT-0, I, II and III genes by inactivation of a salt-tolerance pathway allows examination of ESCRT gene roles in pH signalling. J Cell Sci. 2011;124:4064–4076. doi: 10.1242/jcs.088344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chilton IJ, Delaney CE, Barham-Morris JB, Fincham DA, Hooley P, Whitehead MP. The Aspergillus nidulans stress response transcription factor StzA is ascomycete-specific and shows species-specific polymorphisms in the C-terminal region. Mycol Res. 2008;112:1435–1446. doi: 10.1016/j.mycres.2008.06.028. [DOI] [PubMed] [Google Scholar]
- Chu XL, Feng MG, Ying SH. Qualitative ubiquitome unveils the potential significances of protein lysine ubiquitination in hyphal growth of Aspergillus nidulans. Curr Genet. 2015;62:191–201. doi: 10.1007/s00294-015-0517-7. [DOI] [PubMed] [Google Scholar]
- Clement DJ, Stanley MS, Attwell NA, Clipson NJ, Fincham DA, Hooley P. Evidence for sltA1 as a salt-sensitive allele of the arginase gene (agaA) in the ascomycete Aspergillus nidulans. Curr Genet. 1996;29:462–467. [PubMed] [Google Scholar]
- Clutterbuck AJ. Aspergillus nidulans. In: O’Brien SJ, editor. Genetic Maps. Locus Maps of Complex Genomes. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1993. pp. 3.71–3.84. [Google Scholar]
- Cove DJ. The induction and repression of nitrate reductase in the fungus Aspergillus nidulans. Biochim Biophys Acta. 1966;113:51–56. doi: 10.1016/s0926-6593(66)80120-0. [DOI] [PubMed] [Google Scholar]
- Crabtree GR, Olson EN. NFAT signaling: choreographing the social lives of cells. Cell. 2002;109(Suppl):S67–S79. doi: 10.1016/s0092-8674(02)00699-2. [DOI] [PubMed] [Google Scholar]
- Cyert MS. Calcineurin signaling in Saccharomyces cerevisiae: how yeast go crazy in response to stress. Biochem Biophys Res Commun. 2003;311:1143–1150. doi: 10.1016/s0006-291x(03)01552-3. [DOI] [PubMed] [Google Scholar]
- Di Cera E. Serine proteases. IUBMB Life. 2009;61:510–515. doi: 10.1002/iub.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diez E, Alvaro J, Espeso EA, Rainbow L, Suarez T, Tilburn J, Arst HN, Jr, Peñalva MA. Activation of the Aspergillus PacC zinc finger transcription factor requires two proteolytic steps. EMBO J. 2002;21:1350–1359. doi: 10.1093/emboj/21.6.1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrmann M, Clausen T. Proteolysis as a regulatory mechanism. Annu Rev Genet. 2004;38:709–724. doi: 10.1146/annurev.genet.38.072902.093416. [DOI] [PubMed] [Google Scholar]
- Espeso EA. The CRaZy calcium cycle. Adv Exp Med Biol. 2016;892:169–186. doi: 10.1007/978-3-319-25304-6_7. [DOI] [PubMed] [Google Scholar]
- Espeso EA, Arst HN., Jr On the mechanism by which alkaline pH prevents expression of an acid-expressed gene. Mol Cell Biol. 2000;20:3355–3363. doi: 10.1128/mcb.20.10.3355-3363.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espeso EA, Peñalva MA. Three binding sites for the Aspergillus nidulans PacC zinc-finger transcription factor are necessary and sufficient for regulation by ambient pH of the isopenicillin N synthase gene promoter. J Biol Chem. 1996;271:28825–28830. doi: 10.1074/jbc.271.46.28825. [DOI] [PubMed] [Google Scholar]
- Etxebeste O, Ni M, Garzia A, Kwon NJ, Fischer R, Yu JH, Espeso EA, Ugalde U. Basic-zipper-type transcription factor FlbB controls asexual development in Aspergillus nidulans. Eukaryot Cell. 2008;7:38–48. doi: 10.1128/EC.00207-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Findon H, Calcagno-Pizarelli AM, Martinez JL, Spielvogel A, Markina-Iñarrairaegui A, Indrakumar T, Ramos J, Peñalva MA, Espeso EA, Arst HN., Jr Analysis of a novel calcium auxotrophy in Aspergillus nidulans. Fungal Genet Biol. 2010;47:647–655. doi: 10.1016/j.fgb.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsberg H, Ljungdahl PO. Genetic and biochemical analysis of the yeast plasma membrane Ssy1p-Ptr3p-Ssy5p sensor of extracellular amino acids. Mol Cell Biol. 2001;21:814–826. doi: 10.1128/MCB.21.3.814-826.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garzia A, Etxebeste O, Herrero-Garcia E, Fischer R, Espeso EA, Ugalde U. Aspergillus nidulans FlbE is an upstream developmental activator of conidiation functionally associated with the putative transcription factor FlbB. Mol Microbiol. 2009;71:172–184. doi: 10.1111/j.1365-2958.2008.06520.x. [DOI] [PubMed] [Google Scholar]
- Hernández-Ortiz P, Espeso EA. Phospho-regulation and nucleocytoplasmic trafficking of CrzA in response to calcium and alkaline-pH stress in Aspergillus nidulans. Mol Microbiol. 2013;89:532–551. doi: 10.1111/mmi.12294. [DOI] [PubMed] [Google Scholar]
- Hervas-Aguilar A, Peñalva MA. Endocytic machinery protein SlaB is dispensable for polarity establishment but necessary for polarity maintenance in hyphal tip cells of Aspergillus nidulans. Eukaryot Cell. 2010;9:1504–1518. doi: 10.1128/EC.00119-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hervas-Aguilar A, Rodriguez JM, Tilburn J, Arst HN, Jr, Peñalva MA. Evidence for the direct involvement of the proteasome in the proteolytic processing of the Aspergillus nidulans zinc finger transcription factor PacC. J Biol Chem. 2007;282:34735–34747. doi: 10.1074/jbc.M706723200. [DOI] [PubMed] [Google Scholar]
- Kelley LA, Sternberg MJ. Protein structure prediction on the Web: a case study using the Phyre server. Nat Protoc. 2009;4:363–371. doi: 10.1038/nprot.2009.2. [DOI] [PubMed] [Google Scholar]
- Ljungdahl PO. Amino-acid-induced signalling via the SPS-sensing pathway in yeast. Biochem Soc Trans. 2009;37:242–247. doi: 10.1042/BST0370242. [DOI] [PubMed] [Google Scholar]
- Markina-Iñarrairaegui A, Etxebeste O, Herrero-Garcia E, Araújo-Bazán L, Fernández-Martinez J, Flores JA, Osmani SA, Espeso EA. Nuclear transporters in a multinucleated organism: functional and localization analyses in Aspergillus nidulans. Mol Biol Cell. 2011;22:3874–3886. doi: 10.1091/mbc.E11-03-0262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellado L, Calcagno-Pizarelli AM, Lockington RA, Cortese MS, Kelly JM, Arst HN, Jr, Espeso EA. A second component of the SltA-dependent cation tolerance pathway in Aspergillus nidulans. Fungal Genet Biol. 2015;82:116–128. doi: 10.1016/j.fgb.2015.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayak T, Szewczyk E, Oakley CE, Osmani A, Ukil L, Murray SL, Hynes MJ, Osmani SA, Oakley BR. A versatile and efficient gene-targeting system for Aspergillus nidulans. Genetics. 2006;172:1557–1566. doi: 10.1534/genetics.105.052563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakley BR, Oakley CE, Yoon Y, Jung MK. Gamma-tubulin is a component of the spindle pole body that is essential for microtubule function in Aspergillus nidulans. Cell. 1990;61:1289–1301. doi: 10.1016/0092-8674(90)90693-9. [DOI] [PubMed] [Google Scholar]
- O’Neil JD, Bugno M, Stanley MS, Barham-Morris JB, Woodcock NA, Clement DJ, Clipson NJW, Whitehead MP, Fincham DA, Hooley P. Cloning of a novel gene encoding a C2H2 zinc finger protein that alleviates sensitivity to abiotic stresses in Aspergillus nidulans. Mycol Res. 2002;106:491–498. [Google Scholar]
- Orejas M, Espeso EA, Tilburn J, Sarkar S, Arst HN, Jr, Peñalva MA. Activation of the Aspergillus PacC transcription factor in response to alkaline ambient pH requires proteolysis of the carboxy-terminal moiety. Genes Dev. 1995;9:1622–1632. doi: 10.1101/gad.9.13.1622. [DOI] [PubMed] [Google Scholar]
- Page MJ, Di Cera E. Serine peptidases: classification, structure and function. Cell Mol Life Sci. 2008;65:1220–1236. doi: 10.1007/s00018-008-7565-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Y, Bai CB, Joyner AL, Wang B. Sonic hedgehog signaling regulates Gli2 transcriptional activity by suppressing its processing and degradation. Mol Cell Biol. 2006;26:3365–3377. doi: 10.1128/MCB.26.9.3365-3377.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peñalva MA. Tracing the endocytic pathway of Aspergillus nidulans with FM4-64. Fungal Genet Biol. 2005;42:963–975. doi: 10.1016/j.fgb.2005.09.004. [DOI] [PubMed] [Google Scholar]
- Peñas MM, Hervas-Aguilar A, Munera-Huertas T, Reoyo E, Peñalva MA, Arst HN, Jr, Tilburn J. Further characterization of the signaling proteolysis step in the Aspergillus nidulans pH signal transduction pathway. Eukaryot Cell. 2007;6:960–970. doi: 10.1128/EC.00047-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Pfirrmann T, Heessen S, Omnus DJ, Andreasson C, Ljungdahl PO. The prodomain of Ssy5 protease controls receptor-activated proteolysis of transcription factor Stp1. Mol Cell Biol. 2010;30:3299–3309. doi: 10.1128/MCB.00323-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinar M, Arst HN, Jr, Pantazopoulou A, Tagua VG, de lR V, Rodriguez-Salarichs J, Diaz JF, Peñalva MA. TRAPPII regulates exocytic Golgi exit by mediating nucleotide exchange on the Ypt31 ortholog RabERAB11. Proc Natl Acad Sci USA. 2015;112:4346–4351. doi: 10.1073/pnas.1419168112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulsen P, Lo LL, Kielland-Brandt MC. Mapping of an internal protease cleavage site in the Ssy5p component of the amino acid sensor of Saccharomyces cerevisiae and functional characterization of the resulting pro- and protease domains by gain-of-function genetics. Eukaryot Cell. 2006;5:601–608. doi: 10.1128/EC.5.3.601-608.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds SL, Fischer K. Pseudoproteases: mechanisms and function. Biochem J. 2015;468:17–24. doi: 10.1042/BJ20141506. [DOI] [PubMed] [Google Scholar]
- Saloheimo A, Aro N, Ilmen M, Penttila M. Isolation of the ace1 gene encoding a Cys(2)-His(2) transcription factor involved in regulation of activity of the cellulase promoter cbh1 of Trichoderma reesei. J Biol Chem. 2000;275:5817–5825. doi: 10.1074/jbc.275.8.5817. [DOI] [PubMed] [Google Scholar]
- Shantappa S, Dhingra S, Hernández-Ortiz P, Espeso EA, Calvo AM. Role of the zinc finger transcription factor SltA in morphogenesis and sterigmatocystin biosynthesis in the fungus Aspergillus nidulans. PLoS One. 2013;8:e68492. doi: 10.1371/journal.pone.0068492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soriani FM, Malavazi I, Silva Ferreira ME, Savoldi M, Zeska Kress MR, Souza Goldman MH, Loss O, Bignell EM, Goldman GH. Functional characterization of the Aspergillus fumigatus CRZ1 homologue, CrzA. Mol Microbiol. 2008;67:1274–1291. doi: 10.1111/j.1365-2958.2008.06122.x. [DOI] [PubMed] [Google Scholar]
- Spielvogel A, Findon H, Arst HN, Jr, Araújo-Bazán L, Hernández-Ortiz P, Stahl U, Meyer V, Espeso EA. Two zinc finger transcription factors, CrzA and SltA, are involved in cation homoeostasis and detoxification in Aspergillus nidulans. Biochem J. 2008;414:419–429. doi: 10.1042/BJ20080344. [DOI] [PubMed] [Google Scholar]
- Tilburn J, Sarkar S, Widdick DA, Espeso EA, Orejas M, Mungroo J, Peñalva MA, Arst HN., Jr The Aspergillus PacC zinc finger transcription factor mediates regulation of both acid- and alkaline-expressed genes by ambient pH. EMBO J. 1995;14:779–790. doi: 10.1002/j.1460-2075.1995.tb07056.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilburn J, Scazzocchio C, Taylor GG, Zabicky-Zissman JH, Lockington RA, Davies RW. Transformation by integration in Aspergillus nidulans. Gene. 1983;26:205–221. doi: 10.1016/0378-1119(83)90191-9. [DOI] [PubMed] [Google Scholar]
- Waterhouse AM, Procter JB, Martin DM, Clamp M, Barton GJ. Jalview Version 2–a multiple sequence alignment editor and analysis workbench. Bioinformatics. 2009;25:1189–1191. doi: 10.1093/bioinformatics/btp033. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.