

Abstract
An efficient and exceptionally mild intramolecular nickel-catalyzed carbon–oxygen bond-forming reaction between vinyl halides and primary, secondary, and tertiary alcohols has been achieved. Zinc powder was found to be an essential additive for obtaining high catalyst turnover and yields. This operationally simple method allows direct access to cyclic vinyl ethers in high yields in a single step.
Keywords: nickel, C–O bond formation, cross-coupling, vinyl ethers
Nickel-O–C-eon: It’s child’s play
An unprecedented nickel-catalyzed intramolecular C–O bond forming reaction between aliphatic hydroxyl nucleophiles and tethered vinyl halides is disclosed. The reaction conditions are exceptionally mild, operationally simple, and provide access to various cyclic vinyl ethers in a single step. We believe that exploration of this new reactivity of nickel catalysts can provide further insight into the unique properties and opportunities afforded by nickel catalysts.
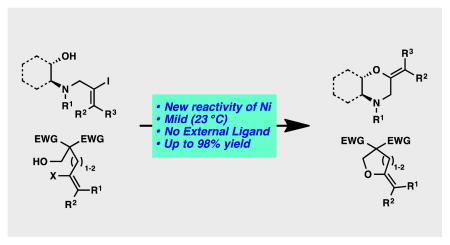
Transition metal-catalyzed cross-coupling reactions have served a powerful tool for efficient carbon–carbon and carbon–heteroatom bond formations over the past several decades.[1] Recently, nickel catalysis has emerged in the synthetic community as an exceptionally useful strategy for cross-coupling.[2] Although tremendous advances in nickel-catalyzed carbon–carbon bond formation have been achieved (e.g., Negishi, Suzuki, Stille, Kumada, Hiyama couplings),[3] nickel-catalyzed carbon–oxygen bond forming processes have proven significantly more challenging. The rationale behind this is that reductive elimination of Ni(II) alkoxide complexes is often cited as being significantly challenging even at elevated temperatures.[4] To circumvent this challenge, stoichiometric oxidation of Ni(II) to the less stable Ni(III) analogs has been required. Additionally, reductive elimination of Ni(II) alkoxides is reported to be endothermic via computational analysis. This is in contrast to that of Pd(II) alkoxides, which are exothermic.[5] In 1997, Hartwig and co-workers developed the first nickel-catalyzed cross-coupling between electron-deficient aryl halides and pre-formed sodium alkoxides or sodium siloxides.[6] In 2014, the Ranu group reported a copper-assisted nickel-catalyzed coupling of phenol derivatives and vinyl halides.[7] However, both of these reactions require high temperatures, and the scope of the nucleophiles is limited to pre-formed alkoxides or phenols. Most recently, MacMillan and co-workers developed the nickel-catalyzed intermolecular cross-coupling of aryl bromides and aliphatic alcohols in the presence of light and a photoredox catalyst.[8] Importantly, MacMillan and co-workers could did observe their desired C–O coupling products in the absence of either the photocatalyst or light. Although Pd- or Cu-catalyzed C–O bond forming reactions have been significantly developed, most of these reactions require high temperature that can limit their utility in the synthesis of multifunctional complex molecules. Moreover, the vast majority of these examples are for aryl ether synthesis, not enol ether synthesis.[9, 10, 11] To our knowledge, a mild and efficient nickel-catalyzed intramolecular cross-coupling cyclization between aliphatic hydroxyl nucleophiles and tethered vinyl halides is unprecedented.
In the course of an alkaloid synthesis effort, we attempted a nickel-catalyzed reductive Heck reaction of vinyl iodide 1 with the aim of producing tricycle 2 (Scheme 1, red arrows).[12] Surprisingly, instead of the desired intramolecular C–C bond-forming reaction, a C–O bond-forming cyclization between the vinyl iodide and the free hydroxyl group furnished morpholine derivative 3 (Scheme 1, blue arrows). Given the lack of precedent in the literature for such a transformation with nickel catalysis, we set out to explore the generality of this reaction. Herein, we describe the first nickel-catalyzed cycloetherification of aliphatic alcohols with pendant vinyl halides.
Scheme 1.

Ni-Catalyzed C–O Bond Formation
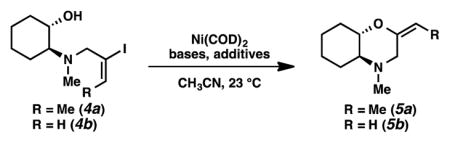
Given this interesting preliminary data, we chose aminocyclohexanols 4a and 4b as simplified substrates for reaction optimization studies (Table 1). Our initial reaction conditions afforded the corresponding morpholines 5a and 5b in 53% and 42% yield, respectively (entries 1 and 2). A wide variety of bases and additives were investigated to improve the yield and catalytic efficiency (entries 3–10). We found triethylamine to be superior to others examined (entries 3–5). The use of a 1:1 mixture of triethylamine and DABCO allowed etherification in 69% yield with reduced catalyst loading (i.e., 20 mol % Ni(COD)2, entry 6). Gratifyingly, the use of 2 equiv of zinc powder as an additive resulted in a significant improvement in yield (84%) with only 5 mol % of Ni(COD)2 (entry 10).[12, 14, 15, 16, 17]
Table 1.
Optimization of Reaction Parameters[a]
| ||||||
|---|---|---|---|---|---|---|
| entry | substrate | conc(M) | Ni mol % | base | additive | yield (%)[b] |
| 1[c] | 4a | 0.02 | 50 | Et3N (10 equiv) | – | 53 |
| 2[c] | 4b | 0.02 | 50 | Et3N (10 equiv) | – | 42 |
| 3[c] | 4a | 0.02 | 35 | Et3N (10 equiv) | – | 42[d] |
| 4[c] | 4a | 0.02 | 35 | Cs2CO3 (3.0 equiv) | – | 30[d] |
| 5[c] | 4a | 0.02 | 35 | K3PO4 (3.0 equiv) | – | 34[d] |
| 6 | 4b | 0.10 | 20 | Et3N (1.0 equiv) | DABCO (1.0 equiv) | 69 |
| 7 | 4b | 0.10 | 20 | Et3N (1.0 equiv) | CsF (1.0 equiv) | 51 |
| 8 | 4a | 0.04 | 20 | DABCO (1.0 equiv) | – | 24 |
| 9 | 4a | 0.04 | 20 | DABCO (2.0 equiv) | – | 50[d] |
| 10 | 4b | 0.15 | 5 | Et3N (1.1 equiv) | Zn (2.0 equiv) | 84 |
Reactions were performed in a N2-filled glove box.
Yield of isolated product.
DMF (0.04 M) was used as a co-solvent.
The reaction proceeded with incomplete conversion of starting material.
With optimized conditions in hand, we investigated the substrate scope of the transformation (Table 2). In addition to simple vinyl iodides (e.g., 4b, R1 = Me, R2 = H, R3 = H),[18] a (Z)-styrenyl iodide (i.e., 4c, R1 = Me, R2 = H, R3 = Ph)[19] furnished vinyl ether 5c in high yield and without loss of olefin stereochemical fidelity. Aminocyclohexanols bearing isopropyl and allyl substituents on nitrogen afforded the corresponding products in reduced yields (5d and 5e). Electronically variable benzyl groups were compatible under the reaction conditions, and even an aryl bromide was well tolerated (5f–5h). Additionally, a silyl ether group remained intact, generating substituted morpholine 5i in good yield. Moreover, we discovered that a cis-aminocyclohexanol derived substrate was competant in the reaction, providing the cis-fused bicyclic product 5j.
Table 2.

|
Reactions were performed in a N2-filled glove box.
Yield of isolated product.
96 h.
To our delight, we found that the nickel-catalyzed intramolecular etherification reactions also proceeded with linear aminoalcohol substrates to generate monocyclic morpholine derivatives in moderate to high yields (Table 3). The steric environment of the alcohol fragment did not hinder the performance, as substrates containing primary, secondary, or highly congested tertiary hydroxyl nucleophiles furnished the corresponding cyclic vinyl ethers in excellent yields (7a–7c). Finally, a benzyl-substituted linear aminoalcohol substrate was transformed to the desired vinyl ether in modest yield (7d).
Table 3.

|
Reactions were performed in a N2-filled glove box.
Yield of isolated product.
We were pleased to discover that additional acyclic substrates undergo the nickel-catalyzed carbon–oxygen cross-coupling to furnish alkylidene tetrahydrofurans and dihydropyrans (Table 4). Intramolecular etherification of vinyl iodide 8a furnished the cyclic ether 9a in good yield in only 1 h (entry 1). Although less reactive, a vinyl bromide also fared well in the reaction, affording the corresponding product in 52% yield after 12 h (entry 2). Unfortunately, attempts to employ a vinyl chloride as the coupling partner led predominantly to recovery of starting material (entry 3). Mono-methyl and mono-phenyl substituted vinyl iodides (8d and 8e)[20] afforded the desired ethers (9d and 9e) in good yields with retention of olefin stereochemistry (entries 4 and 5). Gratifyingly, tetrasubstituted vinyl bromide 8f[21] furnished the corresponding tetrahydrofuran product 9f in excellent yield (entry 6). Di-tert-butyl and dibenzyl malonates (8g and 8h) were tolerated under the standard reaction conditions to afford the desired ethers (9g and 9h) in good yields (entries 7 and 8). Additionally, carbon–oxygen bond formations were achieved with nitrile- and amide-containing substituents in 61% and 70% yield, respectively (entries 9 and 10). Formation of a six-membered cyclic vinyl ether (9k) from substrate 8k was found to be challenging with only low levels of conversion (entry 11). Interestingly, cycloetherification of 8l did indeed produce a pyran derivative in good yield, but only isomerized product 9l was isolated (entry 12).[22]
Table 4.
Synthesis of Substituted Tetrahydrofuran and Dihydropyran Rings[a]

| ||||
|---|---|---|---|---|
| entry | substrate | product | time (h) | yield (%)[b] |
| 1 |
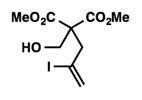 8a |
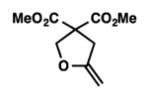 9a |
1 | 87 |
| 2 |
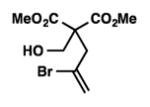 8b |
9b |
12 | 52 |
| 3 |
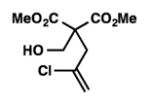 8c |
9c |
60 | –[c] |
| 4 |
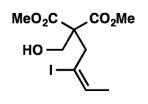 8d |
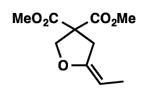 9d |
2 | 87 |
| 5 |
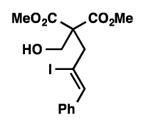 8e |
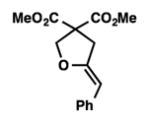 9e |
12 | 91 |
| 6 |
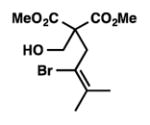 8f |
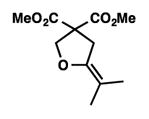 9f |
48 | 98 |
| 7 |
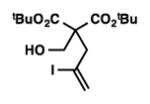 8g |
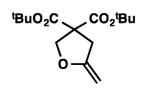 9g |
1 | 87 |
| 8 |
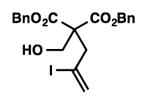 8h |
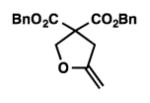 9h |
1 | 66 |
| 9 |
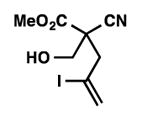 8i |
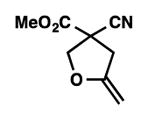 9i |
12 | 61 |
| 10 |
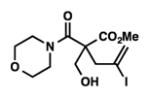 8j |
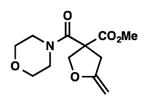 9j |
1 | 70 |
| 11 |
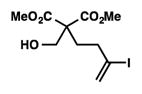 8k |
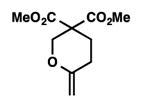 9k |
72 | –[c] |
| 12 |
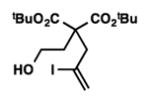 8l |
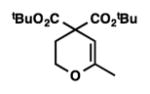 9l |
12 | 76 |
Reactions were performed in a N2-filled glove box.
Yield of isolated product.
Low conversion.
In conclusion, a highly efficient, mild, and operationally simple nickel-catalyzed intramolecular carbon–oxygen bond-forming reaction between vinyl halides and aliphatic alcohols has been developed. We discovered that zinc powder plays an important role in improving catalyst turnover and isolated yields. The reaction is tolerant of many functional groups, affording various cyclic vinyl ethers in good to excellent yields. This work further expands the capability of nickel catalysis in the context of small molecule chemical synthesis. Additional studies are ongoing to expand the scope of the reaction, to understand the mechanism, and to deploy the cyclization in the context of a complex molecular target.[23] These efforts will be reported in due course.
Supplementary Material
Acknowledgments
The authors wish to thank the NIH-NIGMS (R01GM080269), Amgen, the Gordon and Betty Moore Foundation, the Caltech Center for Catalysis and Chemical Synthesis, and Caltech for financial support. S.-J. H. thanks the Fulbright program (Foreign Student Program, No. 15111120) and the Ilju Foundation of Education & Culture (Pre-doctoral Research Fellowship) for financial support. R.D. is grateful for support as a JSPS research fellow. Dr. Mona Shahgholi (Caltech) and Naseem Torian (Caltech) are acknowledged for high-resolution mass spectrometry assistance. Dr. Scott C. Virgil (Caltech) is thanked for helpful discussions.
Footnotes
Supporting information for this article is given via a link at the end of the document.((Please delete this text if not appropriate))
References
- 1.Selected recent reviews: Han FS. Chem Soc Rev. 2013;42:5270–5298. doi: 10.1039/c3cs35521g.Jana R, Pathak TP, Sigman MS. Chem Rev. 2011;111:1417–1492. doi: 10.1021/cr100327p.Hartwig JF. Nature. 2008;455:314–322. doi: 10.1038/nature07369.Terao J, Kambe N. Bull Chem Soc Jpn. 2006;79:663–672.
- 2.a) Tasker SZ, Standley EA, Jamison TF. Nature. 2014;509:299–309. doi: 10.1038/nature13274. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ananikov VP. ACS Catal. 2015;5:1964–1971. [Google Scholar]
- 3.Selected recent reviews: Netherton MR, Fu GC. Adv Synth Catal. 2004;346:1525–1532.Rosen BM, Quasdorf KW, Wilson DA, Zhang N, Resmerita AM, Garg NK, Percec V. Chem Rev. 2011;111:1346–1416. doi: 10.1021/cr100259t.Hu X. Chem Sci. 2011;2:1867–1886.Standley EA, Tasker SZ, Jensen KL, Jamison TF. Acc Chem Res. 2015;48:1503–1514. doi: 10.1021/acs.accounts.5b00064.
- 4.For fundamental studies involving stoichiometric nickel-alkoxides, see: Matsunaga PT, Hillhouse GL, Rheingold AL. J Am Chem Soc. 1993;115:2075–2077.Han R, Hillhouse GL. J Am Chem Soc. 1997;119:8135–8136.
- 5.Macgregor SA, Neave GW, Smith C. Faraday Discuss. 2003;124:111–127. doi: 10.1039/b212309f. [DOI] [PubMed] [Google Scholar]
- 6.Mann G, Hartwig JF. J Org Chem. 1997;62:5413–5418. [Google Scholar]
- 7.Kundu D, Maity P, Ranu BC. Org Lett. 2014;16:1040–1043. doi: 10.1021/ol500134p. [DOI] [PubMed] [Google Scholar]
- 8.Terrett JA, Cuthbertson JD, Shurtleff VW, MacMillan DWC. Nature. 2015;524:330–334. doi: 10.1038/nature14875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.For examples of intramolecular C–O cyclization using non-nickel catalysis, see: Palucki M, Wolfe JP, Buchwald SL. J Am Chem Soc. 1996;118:10333–10334.Torraca KE, Kuwabe SI, Buchwald SL. J Am Chem Soc. 2000;122:12907–12908. doi: 10.1021/ja012046d.Kuwabe S-i, Torraca KE, Buchwald SL. J Am Chem Soc. 2001;123:12202–12206. doi: 10.1021/ja012046d.Sun C, Fang Y, Li S, Zhang Y, Zhao Q, Zhu S, Li C. Org Lett. 2009;11:4084–4087. doi: 10.1021/ol9015578.Fang Y, Li C. J Am Chem Soc. 2007;129:8092–8093. doi: 10.1021/ja072793w.Fang Y, Li C. Chem Commun. 2005:3574–3576. doi: 10.1039/b505006e.
- 10.For examples of intermolecular C–O cyclization using non-nickel catalysis, see: Mann G, Incarvito C, Rheingold AL, Hartwig JF. J Am Chem Soc. 1999;121:3224–3225.Aranyos A, Old DW, Kiyomori A, Wolfe JP, Sadighi JP, Buchwald SL. J Am Chem Soc. 1999;121:4369–4378.Burgos C, Barder TE, Huang X, Buchwald SL. Angew Chem. 2006;118:4427–4432. doi: 10.1002/anie.200601253.Angew Chem Int Ed. 2006;45:4321–4326. doi: 10.1002/anie.200601253.Parrish CA, Buchwald SL. J Org Chem. 2001;66:2498–2500. doi: 10.1021/jo001426z.Shelby Q, Kataoka N, Mann G, Hartwig J. J Am Chem Soc. 2000;122:10718–10719.Wu X, Fors BP, Buchwald SL. Angew Chem. 2011;123:10117–10121. doi: 10.1002/anie.201104361.Angew Chem Int Ed. 2011;50:9943–9947. doi: 10.1002/anie.201104361.Surasani R, Kalita D, Rao AVD, Chandrasekhar KB. Beilstein J Org Chem. 2012;8:2004–2018. doi: 10.3762/bjoc.8.227.Maiti D, Buchwald SL. J Org Chem. 2010;75:1791–1794. doi: 10.1021/jo9026935.Niu J, Guo P, Kang J, Li Z, Xu J, Hu S. J Org Chem. 2009;74:5075–5078. doi: 10.1021/jo900600m.Nordmann G, Buchwald SL. J Am Chem Soc. 2003;125:4978–4979. doi: 10.1021/ja034809y.Shade RE, Hyde AM, Olsen JC, Merlic CA. J Am Chem Soc. 2010;132:1202–1203. doi: 10.1021/ja907982w.
- 11.For an example of a base-mediated intramolecular synthesis of an alkylidene morpholine by C–O bond formation, see: Bottini AT, Mullikin JA, Morris CJ. J Org Chem. 1964;29:373–379.
- 12.Teng M, Zi W, Ma D. Angew Chem. 2014;126:1845–1848. [Google Scholar]; Angew Chem Int Ed. 2014;53:1814–1817. doi: 10.1002/anie.201310928. [DOI] [PubMed] [Google Scholar]
- 13.Lebedev SA, Lopatina VS, Petrov ES, Beletskaya IP. J Organomet Chem. 1988;344:253–259. [Google Scholar]
- 14.Importantly, if the Ni(COD)2 catalyst was omitted, no carbon–oxygen bond formation was observed. Addition of other additives such as Mg and CuI decreased the yields (36% and 0% yield, respectively).
-
15.Intramolecular etherification of vinyl iodide 4b proceeded with 5 mol % of NiI2 or NiBr2(dme) to furnish vinyl ether 5b (entries 1 and 2). An increased rate of cycloetherification was observed with 10 mol % of NiBr2(dme) (entries 2 and 3). Unfortunately, attempts to convert vinyl iodide 4b to enol ether 5b outside a N2-filled glove box were unsuccessful (entries 4 and 5).
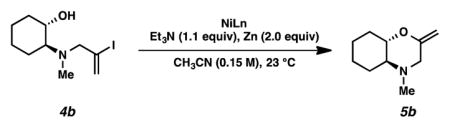
entry NiLn Ni mol % time yield (%)[a] 1[b] Nil2 5 16 h 60 2[b] NiBr2(dme) 5 48 h 53 3[b] NiBr2(dme) 10 16 h 51 4[c] Nil2 5 24 h 0 5[c] NiBr2(dme) 5 24 h 0 [a]Yield of isolated product.
[b]Reactions were performed in a N2-filled glove box.
[c]Reactions were performed under argon outside a N2-filled glove box.
- 16.No significant improvement in yield or catalyst turnover was observed when various ligands (e.g., various NHC, PYBOX, BOX, diamine, BiOX ligands) were employed.
- 17.Since intramolecular etherifications of aminocyclohexanols 4a or 4b proceeded even without Zn powder despite low yields and catalyst turnover (Table 1), we envision that Zn powder likely plays an important role as a scavenger of the forming HI.
- 18.Kurosu M, Lin MH, Kishi Y. J Am Chem Soc. 2004;126:12248–12249. doi: 10.1021/ja045557j. [DOI] [PubMed] [Google Scholar]
- 19.Bowman WR, Bridge CF, Brookes P, Cloonan MO, Leach DC. J Chem Soc Perkin Trans 1. 2002:58–68. [Google Scholar]
- 20.Cao H, Yu J, Wearing XZ, Zhang C, Liu X, Deschamps J, Cook JM. Tetrahedron Lett. 2003;44:8013–8017. [Google Scholar]
- 21.Gatti M, Drinkel E, Wu L, Pusterla I, Gaggia F, Dorta R. J Am Chem Soc. 2010;132:15179–15181. doi: 10.1021/ja108253f. [DOI] [PubMed] [Google Scholar]
-
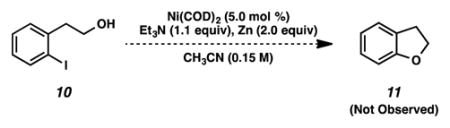
22.Although we attempted to construct 2,3-dihydrobenzofuran 11 from aryl iodide 10 under our standard reaction conditions, only unreacted starting material was recovered.
-
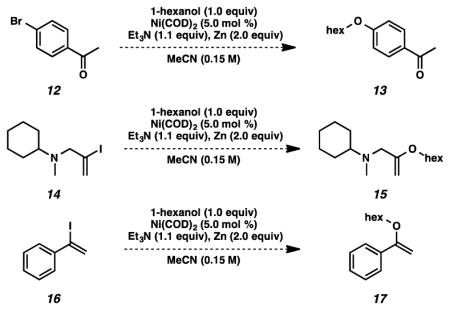
23.An intermolecular etherification of MacMillan’s substrate 12 (ref. 8) with 1-hexanol under our reaction conditions was not successful. Additionally, attempted intermolecular cross-coupling processes of 14 and 16 with 1-hexanol under our standard reaction conditions resulted in no reaction.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.