

Abstract
Indole-3-carbinol (I3C), an anti-cancer phytochemical derived from cruciferous vegetables, strongly inhibited proliferation and down-regulated protein levels of the melanocyte master regulator micropthalmia-associated transcription factor (MITF-M) in oncogenic BRAF-V600E expressing melanoma cells in culture as well as in vivo in tumor xenografted athymic nude mice. In contrast, wild type BRAF-expressing melanoma cells remained relatively insensitive to I3C anti-proliferative signaling. In BRAF-V600E-expressing melanoma cells, I3C treatment inhibited phosphorylation of MEK and ERK/MAPK, the down stream effectors of BRAF. The I3C anti-proliferative arrest was concomitant with the down-regulation of MITF-M transcripts and promoter activity, loss of endogenous BRN-2 binding to the MITF-M promoter, and was strongly attenuated by expression of exogenous MITF-M. Importantly, in vitro kinase assays using immunoprecipitated BRAF-V600E and wild type BRAF demonstrated that I3C selectively inhibited the enzymatic activity of the oncogenic BRAF-V600E but not of the wild type protein. In silico modeling predicted an I3C interaction site in the BRAF-V600E protomer distinct from where the clinically used BRAF-V600E inhibitor Vemurafenib binds to BRAF-V600E. Consistent with this prediction, combinations of I3C and Vemurafenib more potently inhibited melanoma cell proliferation and reduced MITF-M levels in BRAF-V600E expressing melanoma cells compared to the effects of each compound alone. Thus, our results demonstrate that oncogenic BRAF-V600E is a new cellular target of I3C that implicate this indolecarbinol compound as a potential candidate for novel single or combination therapies for melanoma.
Keywords: indole-3-carbinol, MITF-M, human melanoma, anti-proiferative signaling, MITF-M promoter activity, BRN2, I3C, Vemurafenib combinations
Introduction
Human melanoma, the most aggressive form of malignant skin cancer [1], can be categorized by distinct mutational profiles that determine their corresponding cellular phenotypes, proliferative capabilities, and therapeutic options. Well-established driver mutations of melanoma include expression of oncogenic forms of BRAF and NRAS and loss of tumor suppressor proteins such as PTEN, TP53, and p16INK4a [2]. Notably, in more than 60% of melanoma patients, a point mutation occurs within the BRAF gene, with approximately 90% of these mutations being T1799A that substitutes valine for glutamate at residue 600 in the kinase domain forming the constitutively active Ser/Thr protein kinase BRAF-V600E. In human melanomas, a key down stream target of oncogenic BRAF signaling, which triggers constitutive hyperactivation of MEK and ERK/MAPK, is the enhanced expression of microphthalmia-associated transcription factor isoform M [MITF-M], the master regulator of melanocyte and melanoma biology [3]. MITF-M is a “lineage survival” oncogene that is required for both tissue-specific tumorigenesis and progression [4] and elevated MITF-M levels correlate with decreased overall patient survival [5]. Furthermore, one mechanism of acquired resistance to BRAF inhibitors involves the amplification and restoration of transcriptional activity of MITF-M [6]. Hence, the levels of MITF-M can be considered a critical factor that determines the efficacy of a given melanoma targeted therapy and probability of relapse of the disease.
Indole-3-carbinol (I3C), a natural indolecarbinol compound derived from hydrolysis of glucobrassicin produced in cruciferous vegetables of the Brassica genus such as broccoli, Brussels sprouts, and cauliflower, is a promising anti-cancer molecule because of its anti-proliferative effects in a wide range of human cancers with negligible toxicity and minimal side effects [7–10]. I3C activates several distinct and complementary anti-proliferative signaling cascades in human cancer cells [11–16], and is currently in clinical trials for treatment and prevention of breast and prostrate cancer, respectively [17]. In Phases I and II, clinical trials adult oral doses of I3C as high as 800 mg/d has been shown to be well tolerated and lacking significant toxicity in humans [18]. Additionally, I3C has been shown to be effective in promoting regression of precancerous cervical lesions [19], vulvar epidermal neoplasia [20], and recurrent respiratory papillomatosis [21] and chemoprevention of breast cancer [22]. In pre-clinical studies, a dose of 100–200 μM I3C has been reported to be optimal in causing an antitumorigenic effect in hepatocellular carcinoma [23] hepatic stellate cells [24] and breast cancer cells [25,26]. We originally established in different subtypes of human breast cancer cells that I3C induces its anti-proliferative response by the direct inhibition of elastase enzymatic activity and subsequent regulation of CD40-directed cell signaling cascades [27–29]. Thus, an essential concept that emerged from our studies is that the presence of specific I3C target proteins expressed in human cancer cells mediates the efficacy by which I3C selectively inhibits distinct oncogenic proliferative signaling cascades [27–30].
In human melanoma and squamous cell carcinoma, I3C treatment has been shown to increase sensitivity to UV induced apoptosis and enhance cytotoxic responses, respectively [31,32]. Also, ectopic application of I3C directly inhibits skin tumor formation in mouse models [33]. However, relatively little mechanistic information has been uncovered concerning the effects of I3C on skin cancers. We observed that human melanoma cells with distinct mutational profiles are sensitive to different extents to the anti-proliferative effects of I3C [30], suggesting that the ability of I3C to trigger its anti-cancer signaling is linked to its interaction with specific melanoma target proteins expressed in each cell type. In this regard, we have recently shown that I3C directly binds to the NEDD4-1 ubiquitin ligase and induces the stabilization of the wild type PTEN tumor suppressor protein [30]. Enhanced levels of PTEN trigger the loss of activated Akt cell survival signaling; however, this effect is limited to the subset of melanoma cells expressing wild type PTEN [30]. In the present study, we demonstrate that I3C also directly inhibits oncogenic BRAF-V600E kinase activity with no corresponding effect on the wild type BRAF protein. This selective interaction accounts for the loss of down stream BRAF-V600E signaling, reduced MITF-M gene expression, and elevated sensitivity of oncogenic BRAF expressing melanoma cells to the anti-proliferative effects of I3C. Furthermore, combinations of I3C and Vemurafenib, a clinically employed oncogenic BRAF inhibitor, cooperatively down-regulates MITF-M expression and inhibits melanoma cell proliferation, thereby implicating the potential use of I3C-based compounds in the development of new monotherapeutic or combinational therapeutic strategies for human melanoma. Analogous to I3C, other natural phytochemicals, such as Quercetin and Myrecetin, have also been previously reported to have multiple mechanisms of action making these natural compounds unique in their ability to induce a multipronged inhibition of multiple oncogenic signaling cascades [34]. This characteristic of I3C can potentially prevent the development of BRAF inhibitor induced acquired resistance from targeting a single dominant oncogenic pathway such as mutant BRAF signaling in melanoma.
Materials and Methods
Cell Culture
Melanoma cell lines G-361, SK-MEL-2, SK-MEL-24, and RPMI-7951 were purchased from American Type Culture Collection (ATCC) (Manasas, VA), and were authenticated according to the ATCC guidelines. DM738 melanoma cells were acquired from the tissue culture facility at University of California, Berkeley. The G361 melanoma cells were cultured in Modified McCoy's 5A cell media supplemented with 10% fetal bovine serum (Gemini Bio Products, West Sacramento, CA), 2mM l-glutamine, and 2.5 ml of 10,000 U/ml penicillin/streptomycin mixture (Gibco, Life Technologies, Carlsbad, CA). SK-MEL-24 cells were cultured in DMEM with 4.5 g/L glucose supplemented with 10% fetal bovine serum, 2mM l-glutamine, 2.5 ml of 10,000 U/ml penicillin/streptomycin mixture in addition to 1× of MEM Non-Essential Amino Acid (Gibco, Life Technologies, Carlsbad, CA). DM738, RPMI-7951, and SK-MEL-2 melanoma cells were cultured in DMEM containing 4.5 g/L glucose, 114 mg/L sodium pyruvate, and 2 mM l-glutamine, supplemented as described above. The cells were incubated in tissue culture dishes (Nalgene Nunc, Penfield, NY) at 37°C with controlled humidity and 5% CO2 air content.
Treatment With Indole-3-Carbinol, Vemurafenib, and UO126
I3C was purchased from Sigma–Aldrich (St. Louis, MO), Vemurafenib (Adooq Bioscience, Irvine, CA), and UO126 (Cell Signaling, Danvers, MA). To study the effect of I3C, cells were treated with or without 200 μM I3C every 24 h for 72 h and harvested at 24, 48, and 72 h for western blot and flow cytometric analysis. Cells were treated with Vemurafenib and UO126 for 48 h before harvesting them for western blots. For the experiments on the combination of I3C and Vemurafenib treatment, cells were treated with the compounds for 24 h. I3C, Vemurafenib, and UO126 were dissolved in 99.9% HPLC grade DMSO (Sigma–Aldrich, Milwaukee, WI) and the final dilution was performed in the media aliquots used for treatment.
MTT Proliferation Assay
Melanoma cell lines were seeded on a 48-well plate in triplicates and upon 80–90% confluency were either treated with different concentrations of I3C, Vemurafenib, combinations of the two or DMSO for 24 h. Subsequently, inhibition of proliferation was assessed using the Dojindo Cell counting Kit-8 as per the protocol in the user's manual. Briefly, 50 μl of theCCK-8 solution was added to each well along with 450 μl media and incubated for 2–3 h. The absorbance was read at 450 nm and percent inhibition was calculated for each condition standardizing DMSO to zero.
Flow Cytometry Analysis of DNA Content
Melanoma cells were either treated with the indicated concentrations of I3C in triplicates cell cultures for 48 h, or treated with 200 μM I3C in triplicate cultures for a 72 h time course. The DNA content of propidium iodide stained nuclei from harvested cells were determined by flow cytometry. Briefly, cells were hypotonically lysed in 300 ml of DNA staining solution (0.5 mg/ml propidium iodide, 0.1% sodium citrate, and 0.05% Triton-X 100). Emitted fluorescence from the nuclear of wavelengths more than 585 nm was measured with a Coulter Elite instrument with laser output adjusted to deliver 15 mW at 488 nm. Ten thousand nuclei were analyzed from each sample at a rate of 300–500 nuclei/s. The percentage of cells within the G1, S, and G2/M phases of the cell cycle were determined by analysis with the multicycle computer program provided by Phoenix Flow Systems in the Cancer Research Laboratory Microchemical Facility of the University of California, Berkeley.
Western Blot Analysis
Western blot analyses of samples electrophoretically fractionated on 8–10% acrylamide gels were carried out as previously described [29,30]. ECL lightening reagents were used to visualize the primary antibody bound protein bands in nitrocellulose membranes and the results captured on ECL Autoradiography Film (GE Healthcare, Piscataway, NJ). The western blots employed the following primary antibodies: MITF-M (Thermo-Scientific, Waltham, MA), HSP 90 (BD Bio-sciences, San Jose, CA), CDK2, CDK4, BRAF, ERK-p, ERK1/2 (Santacruz, Dallas, TX), P21, Cyclin D1, MEK-p, MEK1/2 (Cell Signaling).
RT-PCR Analysis
Total RNA was extracted from harvested cells using the RNeasy extraction kit for mammalian cells (obtained from Qiagen, Hercules, CA) and spectro-photometrically quantified by absorbance at 260 nm. Reverse transcription (RT) reactions were carried out using RT-MMLV reverse transcriptase (Invitrogen, Eugene, OR) and the cDNA was used for PCR reactions using 10 pM of the following primers: MITF-M forward 5′-CCG TCT CTC ACT GGA TTG GT-3′, MITF-M reverse 5′-TAC TTG GTG GGG TTT TCG AG-3′ GAPDH forward, TGAACGGGAAGCT-CACTGG, and reverse, TCCACCACCCTGTTGCTG TA. PCR conditions were as follows: 30 s at 94°C, 30 s at 55°C, and 30 s at 72°C for 28 cycles. PCR products were electrophoretically fractionated in 1.5% agarose gels containing 0.01% Gel Red (Biotium, Hayward, CA) for DNA staining along with 1 kb plus DNA ladder and further visualized by a UV transilluminator.
Luciferase Reporter Assay
G361 melanoma cells were cultured to approximately 80% confluency in six well adherent culture plates in triplicates for each experimental condition for 24 h. The cells were then transiently transfected with either a wild-type pGL2-pMITF −333/+120-luciferase reporter plasmid or a similar plasmid with the BRN2 binding site mutated at −48, pGL2-pMITF −333/+120-ΔBRN2-luciferase; (ΔBRN2) generated using the following primers: BRN2 Forward, 5′-TACATGCATAACTAGCGAGCTTAGGTTATTATAAGC-3′, BRN2 reverse, 5′-GCTTATATTAACCTAAGCTCGCTAGT-TATGCATGTA-3′. PCR conditions used were as follows: 30 s hotstart at 95°C, 30 s at 95°C, 1 min at 55°C, 8 min at 68°C for 16 cycles. Mutagenesis was performed using QuickChange II kit (Alligent, Stirling, NJ) per the manufacturer's instructions. PCR products were extracted and purified using QIAquick Gel Extraction Kit (Qiagen). Sequence was confirmed by automated DNA sequencing (University of California Berkeley Sequencing Facility). SuperFect Transfection Reagent (Qiagen) was used for the transfections as per instructions in the user manual. The next day, cells were either treated with 200 μM I3C or an equal volume of the DMSO vehicle control for both wild-type plasmid transfected as well as mutant plasmid transfected cells. Twenty-four hours later, luciferase assays were performed by harvesting cells in 1× Promega Lysis Buffer, and then the cell extracts were incubated for 15 min at room temperature, and another 15 min on ice. The cell extracts were then pelleted by a spin at 15,000 rpm for 1 min at 4°C. Twenty microliters of supernatant from each sample was combined with 100 μl luciferase substrate (Promega) and emitted fluorescence was measured using luminometer, Lumat LB 9507 (EG&G Berthold) and the relative light units (RLU) were recorded. The RLU/μg for each sample was calculated by dividing the data obtained with their specific protein concentration determined by a Bradford assay using 1× BioRad Protein Assay reagent and measuring O.D at 595 nm.
Chromatin Immunoprecipitation
Chromatin immunoprecipitation was performed using G361 melanoma cells treated with or without 200 μM I3C for 72 h. Proteins were cross-linked to DNA by the addition of formaldehyde at 2.5% of the final concentration for 5 min at room temperature to cultured cells. Fixation was quenched with glycine for a final concentration of 125 mmol/L for 5 min. Harvested cells were sonicated and the chromatin was immunoprecipitated overnight and 15 μl of Brn2 antibodies (Santacruz Biotechnology) was added to the cell lysates to immunoprecipitated the chromatin-bound Brn2 protein. A fragment of the MITF-M promoter (−170 +120) containing the BRN2 binding site (−53 to −27) was amplified using the primers-MITF-M Forward: 5′-CGT CACTTA AAA AGG TAC CTT TAT ATT TAT G-3′ MITF-M Reverse: 5′-TGT TTT AGC TAG CAC CAA TCC AGT GAG AGA CGG-3′ by cycling 36 times (95°C, 30 s/52°C, 30 s/72°C, 30 s) with a 72°C, 10 min extension. The PCR products were electrophoretically fractionated on a 1.5% agarose gel and visualized using a transilluminator.
Immunoprecipitation Kinase Assay
BRAF immunoprecipitations were carried out either using G-361 cells treated with DMSO, I3C, or Vemurafenib for 72 h or from untreated G-361 and SK-MEL-2 cells. BRAF protein was pulled down from the cell lysates using anti-BRAF antibodies bound to protein G-coupled Sepharose beads, while IgG was used as a negative control. Post-immunoprecipitation supernatants were probed with anti BRAF antibody by western blots to determine pull-down efficiency. In only the “in vitro treated” samples, DMSO, I3C, or Vemurafenib was added to the immunoprecipitated wild type BRAF or mutant BRAF prior to the kinase assay, and reactions were incubated at 30°C for 2 h. Kinase assay was subsequently performed with the immunoprecipitated wild type or oncogenic BRAF from the pre-treated as well as the “in vitro treated” samples by incubation with recombinant inactive MEK(K97R) (Millipore, Darmstadt, Germany) supplemented with ATP/Mg+2 in kinase reaction buffer at 30°C for 30 min. The reactions were terminated by adding 4× SDS sample buffer and boiling the mixture at 100°C for 5 min. The samples were then electrophoresed, immunoblotted, and probed for relative protein levels of phosphorylated MEK and total MEK to assess the kinase activity of the immunoprecipitated BRAF.
In Silico Modeling
The structure of the BRAF-V600E protein was obtained from the Protein Data Bank with accession number 3OG7. The PRODRG server was used to produce the topology files for modeling the I3C structure. The BRAF-V600E and ligand (I3C) structures were subsequently loaded into the Hex Protein Docking program. Prior to docking the structures, all water molecules and hetero molecules were manually removed by editing each PDB file. Shape and electrostatics were used as restrictive parameters to model binding between the receptor and ligand. Modeling results were visualized using PyMol (The PyMol Molecular Graphics System, version 1.7.4 Schrodinger, LLC).
Tumor Xenografts and Immunofluorescence
Approximately, 1 million G361 melanoma cells in a total volume of 0.1 ml of Matrigel were injected subcutaneously into each lateral flank of NIH III athymic nude mice. The resulting tumors were allowed to grow to a mean starting volume of 146 ± 10 mm3. The animals were then randomized into two groups; a vehicle control group that was treated with DMSO and an I3C treated group that were injected daily with 200 mg/kg body weight of I3C. The resulting tumor volumes were measured every other day for 4 wk using calipers. The tumor volumes were calculated using the standard formula: (width2 × length)/2, and changes in tumor volumes were calculated using the formula: 100 + {(Tf – Ti)/Ti × 100}, where Tf is the final mean tumor volume and Ti is the initial mean tumor volume. At terminal sacrifice, tumor xenografts were harvested, and a portion of each tumor was fixed in 4% paraformaldehyde for 1 h at room temperature, followed by a PBS wash and subsequently immersed in 3% sucrose overnight at 4°C. The resulting tissues were embedded in optimal cutting temperature medium and 10 μm thin cryostat sections were taken for immunofluorescence studies.
Results
I3C Inhibits Cellular and In Vivo Proliferation and Down-Regulates MITF-M in BRAF-V600E Expressing Melanoma Cells
Because MITF-M plays a central role in the regulation of melanoma cell proliferation and differentiation [35], we initially examined whether I3C disrupts the in vivo production of this transcription factor in human melanoma cell-derived tumor xenografts. G-361 melanoma cells, which express oncogenic BRAFV600E and are sensitive to the anti-proliferative effects of I3C [30], were subcutaneously injected into NIH III athymic mice to generate xenografted tumors. The mice were injected with either I3C (200 mg/kg body weight) or DMSO vehicle control throughout a 4-wk course. I3C strongly inhibited tumor growth that was noticeable within the first week of injections and sustained over the entire time course (Figure 1A). Immunofluorescence analysis of tumor xenograft sections demonstrated that concomitant with the in vivo inhibition of tumor growth, I3C injected animals showed a strong down-regulation of MITF-M protein relative to vehicle treated control animals (Figure 1B, left panels). Similar down regulation of MITF-M protein was observed by immunofluorescence analysis of cultured G-361 melanoma cells treated for 48h with or without 200 μM I3C (Figure 1B, right panels), which is the optimal I3C concentration for the anti-proliferative effect in cultured human cancer cells [36]. This result indicated that the I3C down regulation of MITF-M levels observed in vivo is a direct effect in the melanoma cells, rather than an indirect consequence of other tissues conceivably producing a factor in response to I3C that acts on the melanoma-derived tumor xenografts.
Figure 1.
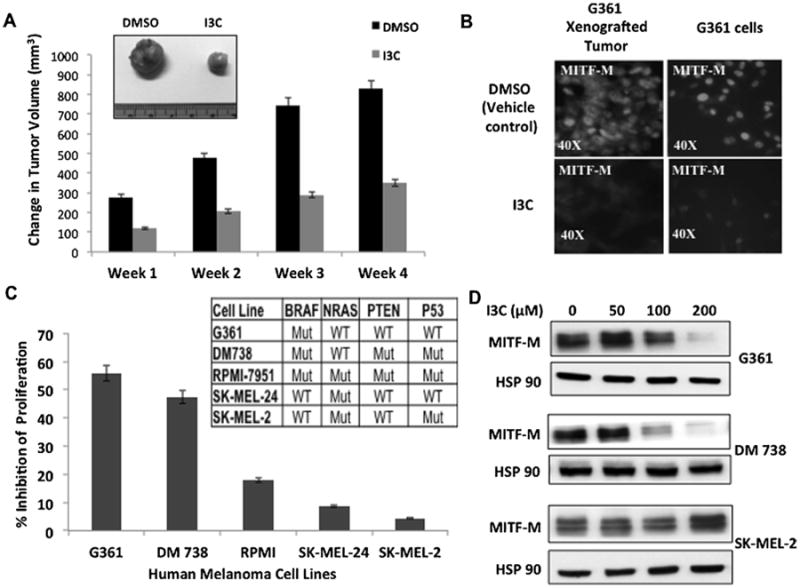
Effects of I3C on in vivo growth of melanoma tumor xenografts, production of MITF-M, and melanoma cell proliferation. (A) Athymic mice with G-361 cell-derived tumor xenografts were injected subcutaneously with either I3C or with DMSO vehicle control, and resulting tumor volumes were calculated as described in the Supporting Information. The micrograph insert shows tumors harvested at the end of week 4. (B) At terminal sacrifice, tumor sections were analyzed for MITF-M expression by immunofluorescence using primary antibodies to MITF-M (left panel). Cultured G361cells treated with 200 μM I3C for 72 h were similarly probed for MITF-M levels (right panel). (C) Human melanoma cell lines with distinct genotypes were treated with or without 200 μM I3C for 48 h and the effects on cell proliferation measured using a CCK-8 assay relative to the vehicle control. (D) The levels of MITF-M protein were determined in melanoma cells treated with the indicated concentrations of I3C for 48 h by western blots.
The potential effects of I3C on melanoma cell proliferation and production of MITF-M was tested in five different human melanoma cell lines with distinct mutation profiles, including G-361 cells. Each of the cell lines was treated for 72 h with or without 200 μM I3C, and cell proliferation determined using a CCK-8 proliferation assay. Most sensitive to the I3C anti-proliferative effects were G-361, DM738 and RPMI-7951 cells, which harbor the BRAF-V600E oncogenic mutation, although each cell line differs with respect to their NRAS, PTEN, and P53 genotypes (Figure 1C). Western blot analysis of melanoma cells treated with varying concentrations of I3C for 48 h showed that I3C strongly down-regulated MITF-M levels in G-361 and DM738 cells at approximately the same concentration range (Figure 1D, upper and middle set of panels). Consistent with cell cycle regulators such as CDK2, CDK4, and cyclin D1 being transcriptional target genes of MITF-M [37], the I3C inhibition of MITF-M production dose dependently and temporally correlated with the G1 cell cycle arrest and down-regulated levels of cell cycle genes in BRAF-V600E expressing melanoma cells (see Supplementary Material). I3C has been reported to arrest cell cycle progression in other cancer cell types [38], although we propose that in melanoma cells the loss of MITF-M helps to mediate this response. In contrast, wild type BRAF expressing melanoma cell lines, SK-MEL-24 and SK-MEL-2, were significantly less sensitive to the anti-proliferative effects of I3C (Figure 1C), SK-MEL-2 expressed high levels of MITF-M protein at all concentrations of I3C (Figure 1D, lower set of panels) and did not down regulate MITF-M targets or display a G1 cell cycle arrest in the presence of I3C (see Supplementary Material).
Expression of Exogenous MITF-M Rescues the I3C Down-Regulation of MITF-M Target Genes CDK2 and CDK4 and Attenuates the I3C Proliferative Arrest of Melanoma Cells
Exogenous MITF-M was expressed in G-361 melanoma cells to functionally test whether the I3C induced down regulation of MITF-M is required to mediate the I3C anti-proliferative response. As shown in Figure 2A, cells transfected with an MITF-M expression vector (pCMV-MITF) displayed high levels of MITF-M protein in the presence or absence of I3C, whereas MITF-M protein levels were strongly down regulated by I3C in empty expression vector (pCMV) transfected cells or in untransfected cells. Western blots further revealed that the I3C down-regulation of two MITF-M target genes, CDK2 and CDK4, was rescued in cells expressing exogenous MITF-M protein (Figure 2A). Consistent with a critical role of MITF-M down-regulation for the I3C anti-proliferative effects, expression of exogenous MITF-M protein strongly attenuated this process in cells treated with I3C for 24 h relative to vehicle control treated cells expressing exogenous MITF-M (Figure 2B). Empty vector transfected or untransfected melanoma cells showed only a 60% proliferative arrest because of the submaximal incubation time of 24 h employed to ensure maximal expression of exogenous MITF-M.
Figure 2.
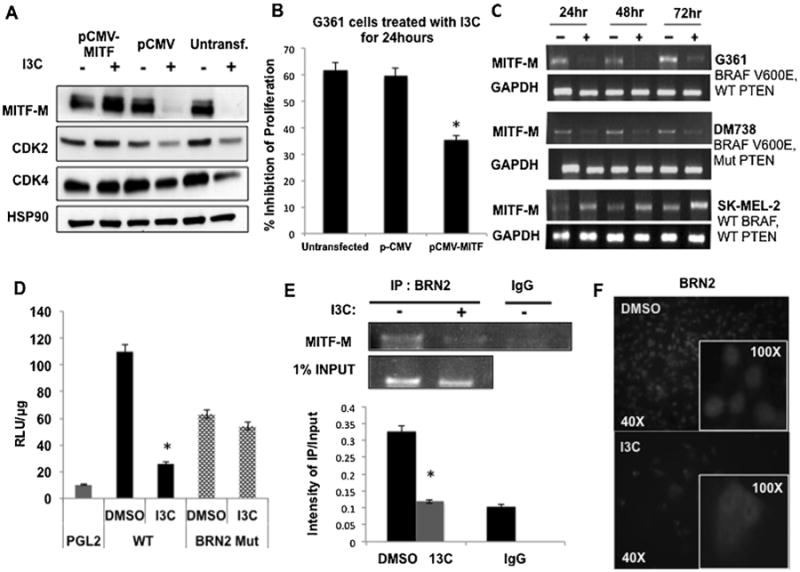
Role of MITF-M in I3C anti-proliferative response and I3C regulation of MITF-M gene expression. (A) G-361 cells were either transfected with pCMV-MITF expression vector, or pCMV empty vector control or left untransfected, and each set of cells were treated with or without 200 μM I3C for a submaximal time of 24 h. Levels of MITF-M, CDK2, CDK4, and HSP90 protein were determined by western blots. (B) Cell proliferation was measured using a CCK-8 assay, and results show the mean of three independent experiments ±SEM (*P<0.01) (C) MITF-M transcript expression in G-361 cells treated with or without 200 μM I3C was determined by RT-PCR analysis in comparison to the GAPDH control. (D) Cells were transfected with reporter plasmids containing either a wild type MITF-M promoter (WT), a BRN2 consensus site mutant (BRN2 Mut) or the PGL2 empty control vector. Luciferase specific activity was measured in cells treated with or without 200 μM I3C for 24 h. The bar graph shows the results of three independent experiments in triplicate ± SEM (*P<0.01). (E) ChIP assay was performed on G361 cells treated with or without 200 μM I3C for 48 h using BRN2 antibodies (IP:BRN2) or the control IgG with one percent input as the loading control. The bar graphs quantify the densitometry results from three independent experiments ± SEM (*P<0.01). (F) G-361 cells were treated with or without 200 μM I3C for 48 h and BRN2 localization was examined by immunofluorescence using anti-BRN2 antibodies. The insets show magnified portions of the larger fields.
I3C Disrupts BRN2 Interactions With the MITF-M Promoter, Inhibiting Promoter Activity, and Gene Expression
I3C effects on MITF-M transcript levels were examined in BRAF-V600E expressing G-361 and DM738 cells as well as wild type BRAF-expressing SK-MEL-2 cells treated for a 72 h time course with or without 200 μM I3C. RT-PCR analysis of total RNA at each time point revealed that I3C strongly down regulated MITF-M transcript levels in both the BRAF-V600E expressing melanoma cell lines, accounting for the loss of MITF-M protein (Figure 2C, upper and middle panels). G-361 cells displayed a greater loss of MITF-M transcripts compared to DM738 cells. In contrast, in wild type BRAF-expressing SK-MEL-2 cells, I3C induced a modest increase in MITF-M transcript levels (Figire 2C, lower panels). Consistent with the loss of MITF-M transcripts, transient transfection of a −333/+120 MITF-M promoter-luciferase reporter plasmid (WT) revealed that I3C strongly down regulated MITF-M promoter activity (Figure 2D). This MITF-M promoter fragment contains a consensus site for the melanoma specific transcription regulator BRN2 (N-Oct3) at −50 to −36, which functions downstream of the oncogenic BRAF signaling pathway to maintains high levels of melanoma MITF-M [3]. Mutation of the BRN2 consensus site (BRN2 Mut) prevented the I3C down regulation of MITF-M promoter activity (Figure 2D), directly implicating involvement of BRN2 in the I3C inhibition of MITF-M promoter activity.
Chromatin immunoprecipitation (ChIP) assay demonstrated that I3C treatment disrupted binding of endogenous BRN2 to the MITF-M promoter. G-361 cells were treated with or without I3C for 48 h, and the genomic fragments cross-linked to protein were immunoprecipitated with anti-BRN2 antibodies or with an IgG control antibody. As shown in Figure 2E, PCR analysis using primers specific to the BRN2 binding site in the MITF-M promoter revealed that I3C significantly down regulated endogenous BRN-2 interactions with this promoter. One percent input was used as a loading control. Immunofluorescence of G-361 melanoma cells treated with or without 200 μM I3C for 48 h revealed that I3C disrupted the nuclear localization of BRN2 and thereby preventing accessibility of this transcription factor to the MITF-M promoter. As shown in Figure 2F (upper panel), in vehicle control DMSO treated cells, BRN2 is highly concentrated in the nucleus, whereas, treatment with I3C disrupted the nuclear localization of BRN2 with this protein remaining diffusely localized throughout the cytoplasm (Figure 2F, lower panel).
I3C Disrupts the Oncogenic BRAF Signaling Pathway
To assess the potential effects of I3C on the immediate down stream effectors of BRAF, signaling namely MEK and ERK/MAPK, melanoma cells were treated with or without I3C over a 72 h time course and protein levels of each signal transduction component was examined by western blots. In the BRAF-V600E expressing G-361 and DM738 cells, I3C strongly down regulated the phosphorylated forms of MEK and ERK/MAPK, which are the active forms of both protein kinases, with no corresponding changes in the level of total MEK or ERK/MAPK protein, respectively (Figure 3A, left and middle panels). The total levels of BRAF-V600E protein and the control protein Hsp90 remained unchanged. In contrast, I3C had no effect on the levels of phosphorylated MEK or phosphorylated ERK/MAPK in wild type BRAF expressing SK-MEL-2 cells (Figure 3A, right panel), which likely accounts for the inability of I3C to down-regulate MITF-M expression in this melanoma cell line (Figure 1D). The in vivo effect of I3C on oncogenic BRAF signaling was analyzed by immuno-fluorescence on sections from G-361 cell-derived tumor xenografts. Consistent with the cellular observations, I3C strongly down regulated the level of phosphorylated ERK/MAPK with no effect on the total levels of ERK/MAPK protein in the tumor xenografts (Figure 3B).
Figure 3.
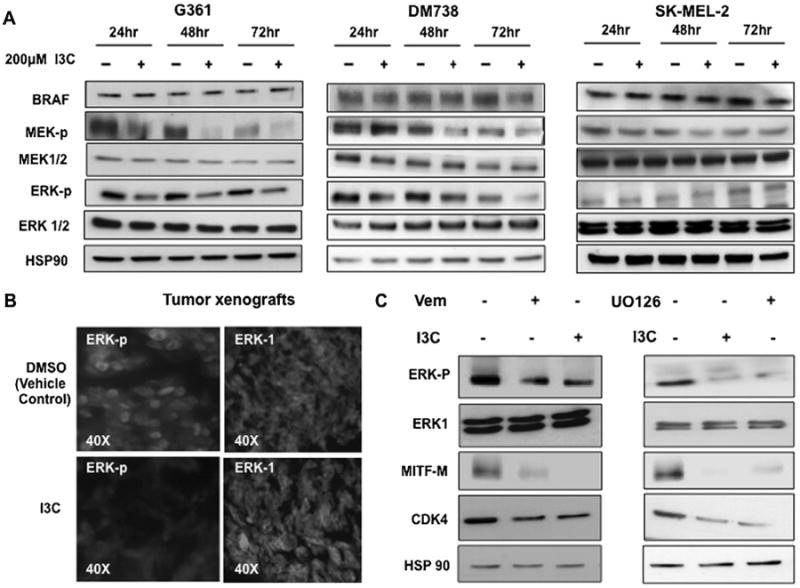
Effect of I3C on BRAF signalling in cells and in vivo tumors (A) BRAF-V600E expressing G-361 and DM-738 cells as well as wild type BRAF expressing SK-MEL-2 cells were treated with or without 200 μM I3C for 24, 48, and 72 h. Western blots were performed on total cell extracts and probed with the indicated antibodies. (B) Tumor xenograft sections from I3C treated and untreated animals were analyzed for ERK-p and total ERK-1 protein by immunofluorescence. (C) G-361 cells were treated with or without 10 μM Vemurafenib BRAF inhibitor, 10 μM U0126 MEK inhibitor, or 200 μM I3C for 48 h. Western blots were probed for the indicated proteins and the results are representative of three independent experiments.
In a complementary pharmacological approach, G-361 cells were treated for 48 h with or without either 10 μM Vemurafenib, a clinically used BRAF-V600E specific inhibitor [39], or 10 μM U0126, a selective inhibitor of MEK1/2 activity [40]. A parallel set of cells were treated with 200 μM I3C and the effects compared to each inhibitor. I3C, Vemurafenib, and U0126 each down regulated the levels of phosphorylated ERK/MAPK, MITF-M, and CDK4 protein (Figure 3C). The Vermurafenib effects on MITF-M protein in melanoma cells is the first such observation for this clinically used BRAF inhibitor. Taken together, these results indicate that in melanoma cells, I3C disrupts oncogenic BRAF signaling at or upstream of MEK activation.
I3C Inhibits the Catalytic Activity of Oncogenic BRAF-V600E But Not the Wild Type BRAF
To initially test whether I3C alters BRAF-V600E enzymatic activity in cells, G-361 cells were treated for 48 h with the DMSO vehicle control, 200 μM I3C, 400 μM I3C, or with 10 μM Vemurafenib. The BRAF-V600E protein was pulled down from each of the cell extracts with anti-BRAF antibodies and the kinase activities assayed in vitro as previously described [41] by adding ATP and inactive MEK protein as the BRAF substrate. After 30 min incubation, the levels of phosphorylated MEK protein in the reaction mixture was assessed using western blots to measure of BRAF-V600E activity. Cellular treatment with either I3C or Vemurafenib significantly inhibited BRAF-V600E enzymatic activity as shown by the loss of phosphorylated MEK, whereas in the absence of either compound, immunoprecipitated BRAF-V600E was highly active (Figure 4A, IP:BRAF panels). The levels of BRAF-V600E, MEK, and the HSP90 gel loading control in each of the reaction mixtures remained constant.
Figure 4.
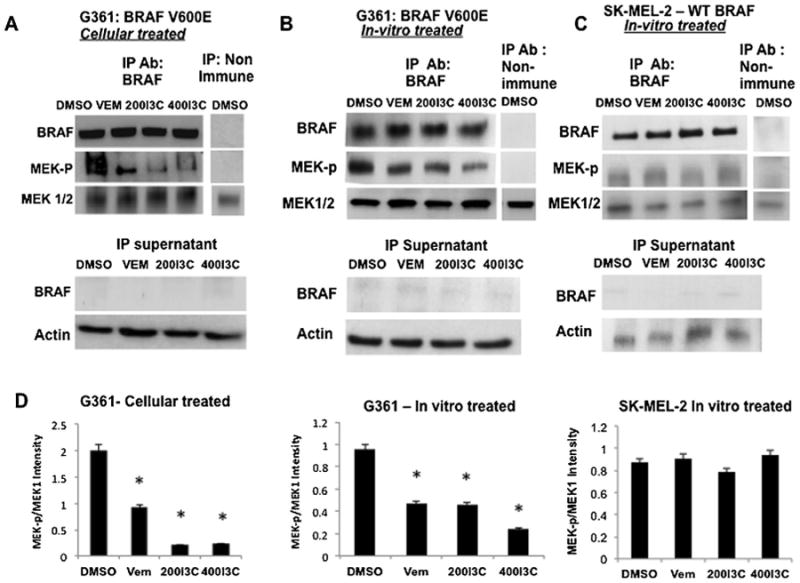
I3C inhibits oncogenic BRAF-V600E enzymatic activity. (A) G-361 cells were treated with 200 μM I3C, 400 μM I3C, 10 μM Vemurafenib, or with DMSO vehicle control for 48 h. BRAF was immunoprecipitated from pretreated cell extracts, and assayed for its intrinsic enzymatic activity in vitro using inactive MEK as the substrate in the presence of ATP. Immunoprecipitation with non-immune IgG was used as a negative control. The level of in vitro generated phospho-MEK was determined by western blot analysis (left panel). The lower panels show the level of BRAF remaining in the cell extracts after immunoprecipitation by western blot analysis. (B) BRAF-V600E was immunoprecipitated from untreated G-361 cells and incubated in vitro with 200 μM I3C, 400 μM I3C, 10 μM Vermurafenib, or the DMSO vehicle control. BRAF-V600E enzymatic activity was assayed using the level of detected in vitro phosphorylation of inactive MEK as described above. (C) Wild type BRAF was immunoprecipitated from SK-MEL-2 melanoma cells and BRAF enzymatic activity post I3C treatment was analyzed as described for (C). (D) I3C regulation of BRAF enzymatic activity in vitro was quantified by densitometry of MEK-P and total MEK protein levels detected by western blots and the ratio of MEK-P:total MEK determined from three independent experiments±SEM (*P<0.01).
The in vitro kinase assay also used to assess the potential direct effects of I3C on BRAF enzymatic activity. BRAFV600E (Figure 4B) or wild type BRAF protein (Figure 4C) were first immunoprecipitated from untreated G-361 or SK-MEL-2 melanoma cells, respectively, and then pre-incubated for 2h with the DMSO vehicle control, 200 μM I3C, 400 μM I3C, or 10 μM Vemurafenib. Subsequently, inactive MEK protein and ATP were added to the in vitro reactions and after30 min the level of BRAF activity was assessed by western blots to determine the level of phosphorylated MEK protein. As shown in Figure 4B (left panels), I3C strongly down regulated BRAF-V600E activity at levels comparable to the well-characterized BRAF-V600E inhibitor Vemurafenib. In contrast, in vitro incubations containing either I3C or Vemurafenib had no effect on wild type BRAF enzymatic activity (Figure 4C, right panels). The relative BRAF activities generated under each condition were quantified by densitometry of the amount of phosphorylated MEK detected in the western blots (Figure 4D). The observed BRAF-V600E kinase activity was dependent on the use of anti-BRAF antibodies in the original immune isolations because no phosphorylated MEK was observed when non-immune antibodies were used for the immunoprecipitations (Figure 4A–C, right panels). Furthermore, the post-pull down supernatant fractions were devoid of any residual BRAF protein showing that the BRAF immunoprecipitations quantitatively brought down all of the BRAF protein in each cell extract (Figure 4A–C, lower panels). Taken together, these results demonstrate that I3C functions in human melanoma cells as a selective inhibitor of BRAF-V600E enzymatic activity.
Combinations of I3C and Vemurafenib Display a Cooperative Anti-Proliferative Effect in Oncogenic BRAF-V600E Expressing Melanoma Cells
Vermurafenib acts as an ATP competitive inhibitor of BRAF-V600E and thereby inhibits downstream ERK pathway activity in a BRAF-V600E specific manner [42]. Based on the known X-ray crystal structure of Vemurafenib in adducts with BRAF protomers (accession number: 3OG7), Figure 5A (right protomer) shows that this small molecule inhibitor is anchored stably at the adenine-binding region of the ATP pocket by forming a hydrogen bond between the backbone NH of Asp594 of one of the protomers and the sulphonamide nitrogen of Vermurafenib [43]. We reasoned that oncogenic BRAF inhibitors that do not compete for the ATP binding pocket of BRAF, could potentially cooperate or synergize with Vermurafenib to further disrupt BRAF-V600E enzymatic activity. The Hex Protein Docking software [44] was employed to initially test this possibility by scanning the crystal structure of oncogenic BRAF-V600E protein surfaces for potential docking sites with I3C. As shown in Figure 5A (left protomer), the computer-aided binding simulations indicate that I3C should make Van der Waals interactions (within 3.5 Å) with two critical catalytic residues Arg 575 and Asp 576 that are located in the HRD motif of the catalytic loop of BRAF-V600E that are in close proximity (less than 10 Å) to the site of the oncogenic mutation at position 600, which also potentially explains I3C's selectivity for oncogenic BRAF.
Figure 5.
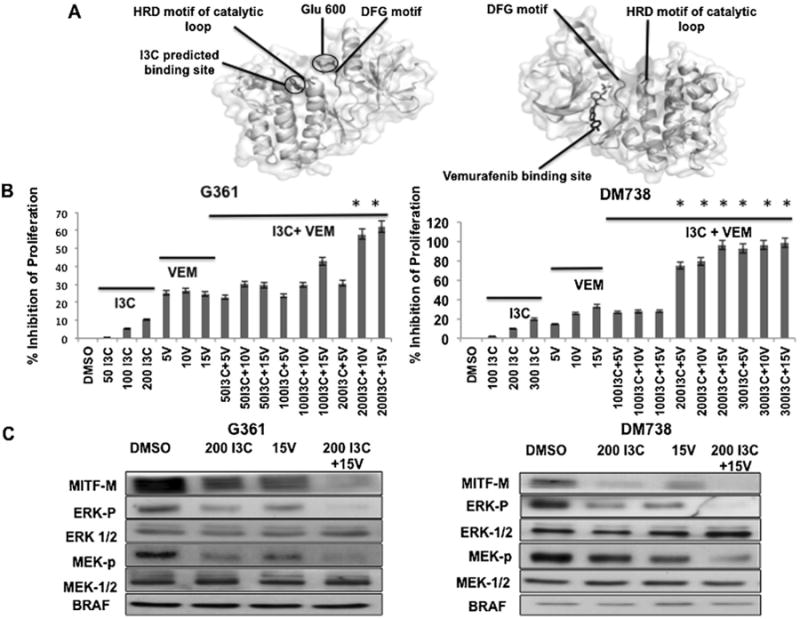
In silico analysis of interactions of I3C and Vermurafenib with BRAF-V600E and combinational effects on melanoma cell proliferation and BRAF-V600E signaling. (A) The crystallographic structures of the BRAF-V600E protomers (accession number: 3OG7) are shown with the known Vemurafenib binding site (right protomer) and the predicted I3C binding site visualized using the PyMol program (left protomer). (B) G-361 or DM-738 cells were treated with the indicated combinations of I3C and Vermurafenib for a suboptimal time of 24 h and inhibition of proliferation was monitored using a CCK-8 assay. The results represent the average of three independent experiments with a mean±SEM (*P< 0.01) shown in the bar graphs. (C) G-361 cells or DM-738 cells were treated with combinations of 200 μM I3C and/or 15 μM Vermurafenib for 24 h and the levels of the indicated proteins determined by western blots on total cell extracts.
The in silico predicted difference between the I3C protomer binding site and the Vemurafenib protomer binding site provided a preliminary indication that I3C and Vemurafenib could potentially induce a stronger anti-proliferative responsein melanoma cells compared to the effects of either compound alone. To directly test this possibility, BRAF-V600E expressing G-361 and DM738 melanoma cells were treated with different concentration combinations of I3C and Vemurafenib for a submaximal response time of 24 h and cell proliferation was determined by CCK-8 assay. As shown in Figure 5B and C, a combination of I3C and Vemurafenib more effectively inhibited the proliferation of both melanoma cell lines compared to the effects of either I3C or Vemurafenib alone. The effects of 24 h treatments with 200 μM I3C and/or 15 μM Vemurafenib on BRAF-V600E cell signaling was examined in G-361 and DM738 melanoma cells. Western blot analysis of total cell extracts revealed that combining I3C and Vemurafenib down-regulated the levels of MITF-M, phosphorylated ERK/MAPK and phosphorylated MEK to a significantly greater extent compared to the individual effects of either I3C or Vemurafenib (Figure 5C and D). The down regulation of MITF-M protein levels mirrored the arrest of melanoma cell proliferation.
Discussion
Our study establishes that I3C directly inhibits the enzymatic activity of oncogenic BRAF, but not of the wild type protein, thereby identifying BRAF-V600E as a new I3C target. The selective targeting of oncogenic BRAF by I3C accounts for BRAF-V600E expressing melanoma cells being highly sensitive to I3C anti-proliferative effects, whereas in contrast wild type BRAF expressing cells are relatively insensitive to I3C. Because BRAF-V600E is a key driver mutation in human melanomas, several BRAF inhibitory compounds have been developed as melanoma targeted therapies, such as Vemurafenib and Dabrafenib to treat nonresectable or metastatic melanoma [45,46]. However, the long-term prognosis of patients treated with these inhibitors is poor because of the development of acquired drug resistance, and an increased risk for cancerous or precancerous non-melanoma skin lesions [47–49]. Among the reported mechanisms of acquired resistance are reactivating mutations in MEK or higher expression levels of ERK/MAPK that can override the inhibition of oncogenic BRAF [50]. Parallel activation of the PI3-kinase/Akt pathway and up-regulating mutations in MITF-M has also been observed in melanoma tumors resistant to oncogenic BRAF inhibitor therapy [51]. In this regard, combinations of the BRAF inhibitor Dabrafenib, the MEK inhibitor Trametinib, and the Akt pathway inhibitor GSK2141795B were shown to delay the emergence of drug resistance in melanoma cells [52]. We previously demonstrated in melanoma cells that I3C interacts with NEDD4-1 ubiquitin ligase and induces higher levels of the wild type PTEN tumor suppressor protein, which leads to the down-regulation of active Akt-1 [30]. Because I3C can disrupt both oncogenic BRAF and Akt-1 signaling in melanoma cells through direct target protein interactions, and also cooperate with Vemurafenib in anti-proliferative signaling, an intriguing clinical possibility is that combining I3C with a BRAF inhibitor will effectively disrupt melanoma proliferation as well as potentially delay or prevent the onset of acquired drug resistance in oncogenic BRAF-expressing melanoma cells.
MITF-M plays a central role in melanoma proliferation and differentiation [3,34,53], and cellular variations in MITF-M levels and/or function correlate with the efficacy of and resistance to anti-melanoma therapeutics [54]. Several studies have shown that MITF-M expression can be regulated transcriptionally in melanocytes and/or melanomas through different transcription factor interactions with the MITF-M promoter including that of CREB, SOX10, LEF, and BRN2 [55]. A key consequence of the I3C inhibition of oncogenic BRAF-V600E activity is the loss of ERK/MAPK signaling and disruption of BRN2 nuclear localization, which in turn prevented accessibility to the MITF-M promoter. As a result, MITF-M promoter activity is down regulated, accounting for reduced MITF-M transcript and protein levels as well as subsequent loss of expressing MITF-M target genes such as CDK2, CDK4, and cyclin D1.
I3C represents a structurally unique small molecule inhibitor of oncogenic BRAF-V600E enzymatic activity. In the BRAF-V600E protein, a salt bridge forms between Glu600 and Lys507 that stabilizes the activation loop in the “DFG-in” conformation. This conformation allows the formation of hydrophobic interactions between the activation domain and the HRD motif in the catalytic loop [56], thus rendering the oncogenic mutant protein constitutively active. Upon ATP binding to BRAF, Asp 576 of the catalytic loop acts as a base catalyst to activate a substrate hydroxyl group and thus increases its nucleophilicity. Intriguingly, our in silico simulations predict that I3C stably interacts with Arg 575 and Asp 576, as well as with several other spatially adjacent amino acid residues, which may conceivably prevent the ability of BRAF-V600E to kinetically drive the phosphorylation reaction and ultimately displaces the HRD motif into positions that are not suited for catalysis. These in silico predictions are substantiated by the in vitro observation that I3C strongly inhibits BRAF-V600E enzymatic activity. Interestingly, the in silico predicted I3C binding site on the catalytic loop is distinct from the known Vemurafenib binding site in the cleft between the N and C lobes of the kinase domain at the adenine-binding region of the ATP pocket of BRAF [43]. The different binding sites within the BRAF-V600E may explain the ability of combinations of I3C and Vermurafenib to cooperatively disrupt BRAF-V600E enzymatic activity and downstream signaling leading to the loss of MITF-M expression.
Our cellular and in vitro observations were further substantiated in vivo in tumor xenografts derived from oncogenic BRAF-expressing melanoma cells in athymic nude mice. Injections with I3C strongly inhibited the growth rate of the tumor xenografts and down-regulated levels of MITF-M protein and phosphorylated ERK/MAPK. These in vivo results implicate I3C and its highly potent derivatives [57] as a new class of potential therapeutic compounds for treatment of BRAF-V600E expressing melanomas. Furthermore, conceivably I3C could be employed topically to treat melanoma, which would circumvent the limited half-life of this natural indolecarbinol in the acidic conditions of the stomach. It is also tempting to consider that I3C-based therapeutics could also potentially be useful to treat other types of human cancers expressing BRAF mutations, such as renal cancer [58], with minimal side effects because this natural indolecarbinol has been reported to be well tolerated in clinical trials with cancer patients [17].
Supplementary Material
Acknowledgments
We greatly appreciate the generous gifts of the MITF expression plasmid from Dr. Vijayasaradhi Setaluri and Ashika Jayanthy (Department of Dermatology, University of Wisconsin, School of Medicine and Public Health), and the wild type MITF promoter-luciferase reporter plasmid was a kind gift from Dr. Richard Marais (Cancer Research UK Centre of Cell and Molecular Biology, London, United Kingdom). We also thank Dr. Peter Walentek for his assistance with the immunofluorescence studies, Kevin Poindexter for sharing the mutant BRN2-pMITF reporter plasmid, as well as Andrew Gabrielson and Carmen Conroy for their help during preliminary stages of the project. This study was supported by National Institutes of Health Public Service grant CA164095 awarded from the National Cancer Institute.
Grant sponsor: National Institutes of Health Public Service; Grant number: CA164095
Abbreviations
- BRN2
brain 2
- I3C
indole-3-carbinol
- MITF-M
microphthalmia-associated transcription factor isoform-M
Footnotes
Supporting Information: Additional supporting information may be found in the online version of this article at the publisher's web-site.
References
- 1.Uzarska M, Czajkowski R, Schwartz RA, Bajek A, Zegarska B, Drewa T. Chemoprevention of skin melanoma: Facts and myths. Melanoma Res. 2013;23:426–433. doi: 10.1097/CMR.0000000000000016. [DOI] [PubMed] [Google Scholar]
- 2.Hodis E, Watson IR, Kryukov GV, et al. A landscape of driver mutations in melanoma. Cell. 2012;150:25–263. doi: 10.1016/j.cell.2012.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wellbrock C, Arozarena I. Microphthalmia-associated transcription factor in melanoma development and MAP-kinase pathway targeted therapy. Pigment Cell Melanoma Res. 2015;28:390–406. doi: 10.1111/pcmr.12370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Garraway LA, Widlund HR, Rubin MA, et al. Integrative genomic analyses identify MITF as a lineage survival oncogene amplified in malignant melanoma. Nature. 2005;436:117–122. doi: 10.1038/nature03664. [DOI] [PubMed] [Google Scholar]
- 5.Ugurel S, Houben R, Schrama D, et al. Microphthalmia-associated transcription factor gene amplification in metastatic melanoma is a prognostic marker for patient survival, but not a predictive marker for chemosensitivity and chemotherapy response. Clin Cancer Res. 2007;13:6344–6350. doi: 10.1158/1078-0432.CCR-06-2682. [DOI] [PubMed] [Google Scholar]
- 6.Van Allen EM, Wagle N, Sucker A, et al. The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Cancer Discov. 2014;4:94–109. doi: 10.1158/2159-8290.CD-13-0617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sarkar FH, Li Y. Harnessing the fruits of nature for the development of multi-targeted cancer therapeutics. Cancer Treat Rev. 2009;35:597–607. doi: 10.1016/j.ctrv.2009.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ahmad A, Sakr WA, Rahman KM. Novel targets for detection of cancer and their modulation by chemopreventative natural compounds. Front Biosci. 2012;4:410–425. doi: 10.2741/e388. [DOI] [PubMed] [Google Scholar]
- 9.Aggarwal BB, Ichikawa H. Molecular targets and anticancer potential of indole-3-carbinol and its derivatives. Cell Cycle. 2005;4:1201–1215. doi: 10.4161/cc.4.9.1993. [DOI] [PubMed] [Google Scholar]
- 10.Ahmad A, Sakr WA, Rahman KM. Anticancer properties of indole compounds: Mechanism of apoptosis induction and role in chemotherapy. Curr Drug Targets. 2010;11:652–666. doi: 10.2174/138945010791170923. [DOI] [PubMed] [Google Scholar]
- 11.Wu TY, Saw CL, Khor TO, Pung D, Boyanapalli SS, Kong AN. In vivo pharmacodynamics of indole-3-carbinol in the inhibition of prostate cancer in transgenic adenocarcinoma of mouse prostate (TRAMP) mice: Involvement of Nrf2 and cell cycle/apoptosis signaling pathways. Mol Carcinog. 2012;51:761–770. doi: 10.1002/mc.20841. [DOI] [PubMed] [Google Scholar]
- 12.Brew CT, Aronchik I, Hsu JC, et al. Indole-3-carbinol activates the ATM signaling pathway independent of DNA damage to stabilize p53 and induce G1 arrest of human mammary epithelial cells. Int J Cancer. 2006;118:857–868. doi: 10.1002/ijc.21445. [DOI] [PubMed] [Google Scholar]
- 13.Tin AS, Park AH, Sundar SN, Firestone GL. Essential role of the cancer stem/progenitor cell marker nucleostemin for indole-3-carbinol anti-proliferative responsiveness in human breast cancer cells. BMC Biol. 2014;12:72. doi: 10.1186/s12915-014-0072-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marconett CN, Sundar SM, Poindexter KM, Stueve TR, Bjeldanes LF, Firestone GL. Indole-3-carbinol triggers AhR-dependent ERalpha protein degradation in breast cancer cells disrupting an ERalpha-GATA3 transcriptional cross-regulatory loop. Mol Biol Cell. 2010;21:1166–1177. doi: 10.1091/mbc.E09-08-0689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marconett CN, Sundar SN, Tseng M, et al. Indole-3-carbinol down-regulation of telomerase gene expression requires the inhibition of estrogen receptor-alpha and Sp1 transcription factor interactions within the hTERT promoter and mediates the G1 cell cycle arrest of human breast cancer cells. Carcinogenesis. 2011;32:1315–1323. doi: 10.1093/carcin/bgr116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brew CT, Aronchik I, Kosco K, McCammon J, Bjeldanes LF, Firestone GL. Indole-3-carbinol inhibits MDA-MB-231 breast cancer cell motility and induces stress fibers and focal adhesion formation by activation of Rho kinase activity. Int J Cancer. 2009;124:2294–2302. doi: 10.1002/ijc.24210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Minich DM, Bland JS. A review of the clinical efficacy and safety of cruciferous vegetable phytochemicals. Nutr Rev. 2007;65:259–267. doi: 10.1301/nr.2007.jun.259-267. [DOI] [PubMed] [Google Scholar]
- 18.Reed GA, Peterson KS, Smith HJ, et al. A phase I study of indole-3-carbinol in women: Tolerability and effects. Cancer Epidemiol Biomarkers Prev. 2005;14:1953–1960. doi: 10.1158/1055-9965.EPI-05-0121. [DOI] [PubMed] [Google Scholar]
- 19.Bell MC, Crowley-Nowick P, Bradlow HL, et al. Placebo-controlled trial of indole-3-carbinol in the treatment of CIN. Gynecol Oncol. 2000;78:123–129. doi: 10.1006/gyno.2000.5847. [DOI] [PubMed] [Google Scholar]
- 20.Naik R, Nixon S, Lopes A, Godfrey K, Hatem MH, Monaghan JM. A randomized phase II trial of indole-3-carbinol in the treatment of vulvar intraepithelial neoplasia. Int J Gynecol Cancer. 2006;16:786–790. doi: 10.1111/j.1525-1438.2006.00386.x. [DOI] [PubMed] [Google Scholar]
- 21.Rosen CA, Bryson PC. Indole-3-carbinol for recurrent respiratory papillomatosis: Long-term results. J Voice. 2004;18:248–253. doi: 10.1016/j.jvoice.2003.05.005. [DOI] [PubMed] [Google Scholar]
- 22.Wong GY, Bradlow L, Sepkovic D, Mehl S, Mailman J, Osborne MP. Dose-ranging study of indole-3-carbinol for breast cancer prevention. J Cell Biochem Suppl. 1997:28–29. 111–116. doi: 10.1002/(sici)1097-4644(1997)28/29+<111::aid-jcb12>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 23.Wang X, He H, Lu Y, Ren W, Teng KY, Chiang C. Indole-3-carbinol inhibits tumorigenicity of hepatocellular carcinoma cells via suppression of microRNA-21 and upregulation of phosphatase and tensin homolog. Biochim Biophys Acta. 2015:244–253. doi: 10.1016/j.bbamcr.2014.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ping J, Li JT, Liao ZX, Shang L, Wang H. Indole-3-carbinol inhibits hepatic stellate cells proliferation of blocking NADPH oxidase/reactive oxygen species/p38 MAPK pathway. Eur J Pharmacol. 2011;650:656–662. doi: 10.1016/j.ejphar.2010.10.057. [DOI] [PubMed] [Google Scholar]
- 25.Cover CM, Hsieh SJ, Tran SH, et al. Indole-3-carbinol inhibits the expression of cyclin-dependent kinase-6 and induces a G1 cell cycle arrest of human breast cancer cells independent of estrogen receptor signaling. J Biol Chem. 1998;273:3838–4387. doi: 10.1074/jbc.273.7.3838. [DOI] [PubMed] [Google Scholar]
- 26.Caruso JA, Campana R, Wei C, Su CH, Hanks AM, Bornmann WG. Indole-3-carbinol and its N-alkoxy derivatives preferentially target ERa-positive breast cancer cells. Cell Cycle. 2014;13:2587–2599. doi: 10.4161/15384101.2015.942210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nguyen HH, Aronchik I, Brar GA, et al. The dietary phytochemical indole-3-carbinol is a natural elastase enzymatic inhibitor that disrupts cyclin E protein processing. Proc Natl Acad Sci USA. 2008;105:19750–19755. doi: 10.1073/pnas.0806581105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Aronchik I, Brar GA, Nguyen DH, Bjeldanes LF, Firestone GL. Direct inhibition of elastase activity by indole-3-carbinol triggers a CD40-TRAF regulatory cascade that disrupts NFkB transcriptional activity in human breast cancer cells. Cancer Res. 2010;70:4961–4971. doi: 10.1158/0008-5472.CAN-09-3349. [DOI] [PubMed] [Google Scholar]
- 29.Aronchik I, Chen T, Durkin KA, et al. Target protein interactions of indole-3-carbinol and the highly potent derivative 1-benzyl-I3C with the C-terminal domain of human elastase uncouples cell cycle arrest from apoptotic signaling. Mol Carcinog. 2012;51:881–894. doi: 10.1002/mc.20857. [DOI] [PubMed] [Google Scholar]
- 30.Aronchik I, Kundu A, Quirit JG, Firestone GL. The antiproliferative response of indole-3-carbinol in human melanoma cells is triggered by an interaction with NEDD 4-1 and disruption of wild-type PTEN degradation. Mol Cancer Res. 2014;12:1621–1634. doi: 10.1158/1541-7786.MCR-14-0018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim DS, Jeong YM, Moon SI, et al. Indole-3-carbinol enhances ultraviolet B-induced apoptosis by sensitizinig human melanoma cells. Cell Mol Life Sci. 2006;63:2661–2668. doi: 10.1007/s00018-006-6306-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dahler AL, Rickwood D, Guminski A, Teakle N, Saunders Indole-3-carbinol induced growth inhibition can be converted to a cytotoxic response in the presence of TPA + Ca2+ in squamous cell carcinoma cell lines. FEBS Lett. 2007;581:3839–3847. doi: 10.1016/j.febslet.2007.07.009. [DOI] [PubMed] [Google Scholar]
- 33.Cope RB, Loehr C, Dashwood R, et al. Ultraviolet radiation-induced non-melanoma skin cancer in the Crl:SKH1:hr-BR hairless mouse: Augmentation of tumor multiplicity by cholorophylin and protection by indole-3-carbinol. Photochem Photobiol Sci. 2006;5:499–507. doi: 10.1039/b515556h. [DOI] [PubMed] [Google Scholar]
- 34.Lee KW, Bode AM, Dong Z. Molecular targets of phytochemicals for cancer prevention. Nat Rev Cancer. 2011;11:211–218. doi: 10.1038/nrc3017. [DOI] [PubMed] [Google Scholar]
- 35.Yajima I, Kumasaka MY, Thang ND, et al. Molecular network associated with MITF in skin melanoma development and progression. J Skin Cancer. 2011;2011:730170. doi: 10.1155/2011/730170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wong GY, Bradlow L, Sepkovic D, et al. Dose-ranging study of indole-3-carbinol for breast cancer prevention. J Cell Biochem Suppl. 1997:28–29. 111–116. doi: 10.1002/(sici)1097-4644(1997)28/29+<111::aid-jcb12>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 37.Koludrovic D, Davidson I. MITF-the Janus transcription factor of melanoma. Future Oncol. 2013;9:235–244. doi: 10.2217/fon.12.177. [DOI] [PubMed] [Google Scholar]
- 38.Aggarwal BB, Ichikawa H. Molecular targets and anticancer potential of indole-3-carbinol and its derivatives. Cell Cycle. 2005;4:1201–1215. doi: 10.4161/cc.4.9.1993. [DOI] [PubMed] [Google Scholar]
- 39.Flaherty KT, Yasothan U, Kirkpatrick P. Vemurafenib. Nat Rev Drug Discov. 2011;10:811–812. doi: 10.1038/nrd3579. [DOI] [PubMed] [Google Scholar]
- 40.Shi R, Lin J, Guo Y, Gong YP. MEK1/2 inhibitor U0126 reverses imatinib resistance through down-regulating activation of Lyn/ERK signaling pathway in imatinib-resistant K562R leukemia cells. Pharmazie. 2014;69:346–352. [PubMed] [Google Scholar]
- 41.Poulikakos PI, Zhang C, Bollag G, et al. RAF inhibitor transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature. 2010;464:427–430. doi: 10.1038/nature08902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Luke JJ, Hodi FS. Vemurafenib and BRAF inhibition: A new class of treatment for metastatic melanoma. Clin Cancer Res. 2012;18:9–14. doi: 10.1158/1078-0432.CCR-11-2197. [DOI] [PubMed] [Google Scholar]
- 43.Tsai J, Lee JT, Wang W, et al. Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proc Natl Acad Sci USA. 2008;105:3041–3046. doi: 10.1073/pnas.0711741105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Macindoe G, Mavridis L, Venkatraman V, Devignes MD, Ritchie HexServer: An FFT-based protein docking server powered by graphics processors. Nucleic Acids Res. 2010;38:445–449. doi: 10.1093/nar/gkq311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim G, McKee AE, Ning YM, et al. FDA approval summary: Vemurafenib for treatment of unresectable or metastatic melanoma with the BRAFV600E mutation. Clin Cancer Res. 2014;20:4994–5000. doi: 10.1158/1078-0432.CCR-14-0776. [DOI] [PubMed] [Google Scholar]
- 46.Ballantyne AD, Garnock-Jones KP. Dabrafenib: First global approval. Drugs. 2013;73:1367–1376. doi: 10.1007/s40265-013-0095-2. [DOI] [PubMed] [Google Scholar]
- 47.Macdonald B, Golitz LE, LoRusso P, et al. Cutaneous adverse effects of targeted therapies: Part II: Inhibitors of intracellular molecular signaling pathways. J Am Acad Dermatol. 2015;72:221–236. doi: 10.1016/j.jaad.2014.07.033. [DOI] [PubMed] [Google Scholar]
- 48.Anforth R, Menzies A, Byth K, et al. Factors influencing the development of cutaneous squamous cell carcinoma in patients on BRAF inhibitor therapy. J Am Acad Dermatol. 2015;72:809–815. doi: 10.1016/j.jaad.2015.01.018. [DOI] [PubMed] [Google Scholar]
- 49.Monsma DJ, Cherba DM, Eugster EE, et al. Melanoma patient derived xenografts acquire distinct vemurafenib resistance mechanisms. Am J Cancer Res. 2015;5:1507–1518. [PMC free article] [PubMed] [Google Scholar]
- 50.Lidsky M, Antoun G, Speicher P, et al. Mitogen-activated protein kinase (MAPK) hyperactivation and enhanced NRAS expression drive acquired vemurafenib resistance in V600E BRAF melanoma cells. J Biol Chem. 2014;289:27714–27726. doi: 10.1074/jbc.M113.532432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shi H, Hugo W, Kong X, et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov. 2014;4:80–93. doi: 10.1158/2159-8290.CD-13-0642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lassen A, Atefi M, Robert L, et al. Effects of AKT inhibitor therapy in response and resistance to BRAF inhibition in melanoma. Mol Cancer. 2014;13:83. doi: 10.1186/1476-4598-13-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kido K, Sumimoto H, Asada S, et al. Simultaneous suppression of MITF and BRAF V600E enhanced inhibition of melanoma cell proliferation. Cancer Sci. 2009;100:1863–1869. doi: 10.1111/j.1349-7006.2009.01266.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cirenajwis H, Ekedahl H, Lauss M, et al. Molecular stratification of metastatic melanoma using gene expression profiling: Prediction of survival outcome and benefit from molecular targeted therapy. Oncotarget. 2015;6:12297–12309. doi: 10.18632/oncotarget.3655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wan PI, Hu Y, He L. Regulation of melanocyte pivotal transcription factor MITF by some other transcription factors. Mol Cell Biochem. 2011;354:241–246. doi: 10.1007/s11010-011-0823-4. [DOI] [PubMed] [Google Scholar]
- 56.Moretti S, De Falco V, Tamburrino A, et al. Insights into the molecular function of the inactivating mutations of B-Raf involving the DFG motif. Biochim Biophys Acta. 2009;1793:1634–1645. doi: 10.1016/j.bbamcr.2009.09.001. [DOI] [PubMed] [Google Scholar]
- 57.Nguyen HH, Lavrenov SN, Sundar SN, et al. 1-Benzyl-indole-3-carbinol is a novel indole-3-carbinol derivative with significantly enhanced potency of anti-proliferative and anti-estrogenic properties in human breast cancer cells. Chem Biol Interact. 2010;186:255–266. doi: 10.1016/j.cbi.2010.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dadone B, Ambrosetti D, Carpentier X, et al. A renal metanephric adenoma showing both a 2p16e24 deletion and BRAF V600E mutation: A synergistic role for a tumor suppressor gene on chromosome 2p and BRAF activation? Cancer Genet. 2013;206:347–352. doi: 10.1016/j.cancergen.2013.09.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.