

Abstract
Purpose:
Germ-line testing for panels of cancer genes using next-generation sequencing is becoming more common in clinical care. We report our experience as a clinical laboratory testing both well-established, high-risk cancer genes (e.g., BRCA1/2, MLH1, MSH2) as well as more recently identified cancer genes (e.g., PALB2, BRIP1), many of which have increased but less well-defined penetrance.
Genet Med 18 8, 823–832.
Methods:
Clinical genetic testing was performed on over 10,000 consecutive cases referred for evaluation of germ-line cancer genes, and results were analyzed for frequency of pathogenic or likely pathogenic variants, and were stratified by testing panel, gene, and clinical history.
Genet Med 18 8, 823–832.
Results:
Overall, a molecular diagnosis was made in 9.0% of patients tested, with the highest yield in the Lynch syndrome/colorectal cancer panel. In patients with breast, ovarian, or colon/stomach cancer, positive yields were 9.7, 13.4, and 14.8%, respectively. Approximately half of the pathogenic variants identified in patients with breast or ovarian cancer were in genes other than BRCA1/2.
Genet Med 18 8, 823–832.
Conclusion:
The high frequency of positive results in a wide range of cancer genes, including those of high penetrance and with clinical care guidelines, underscores both the genetic heterogeneity of hereditary cancer and the usefulness of multigene panels over genetic tests of one or two genes.
Genet Med 18 8, 823–832.
Keywords: BRCA1/2, hereditary breast and ovarian cancer, hereditary cancer panels, Lynch syndrome, next generation sequencing
Introduction
Next-generation sequencing (NGS) has reduced the cost of sequencing, increased the capacity to analyze many genes in parallel, and enabled significant progress in our understanding of the genetic etiology and architecture of human disease. While hereditary breast and ovarian cancer has been a common indication for germ-line genetic evaluation since BRCA1 and BRCA2 were identified, many additional genes have subsequently been implicated in hereditary cancer.
Commercial laboratories began offering clinical NGS panels for inherited cancer within the past 3 years. Large panels can increase the yield of genetic testing for diseases with genetic heterogeneity or variable and overlapping phenotypes, and they have been used routinely for other indications, including hereditary cardiac and neurological diseases. One drawback to testing a larger number of genes, especially those that are less well characterized, is the potential for higher numbers of variants of uncertain clinical significance (VUSs).1,2 For many patients and providers, however, the possibility of finding an explanation for a family's cancer history often outweighs the potential burden of results with uncertain significance. In addition, risk estimates for some less well-established genes are derived from studies with potential ascertainment bias and small numbers of patients.3 Despite the limitations of large panels, they present an opportunity to identify genetic contributions to cancer in families and to offer more tailored management to patients and family members. Such information can be used to reduce cancer risk, morbidity, and mortality.
Here we report our experience from the first 10,030 consecutive patients referred for hereditary cancer panel testing at a single clinical diagnostic laboratory, providing data on the frequency of pathogenic genetic variants by gene, gene panel, and cancer history.
Materials and Methods
We reviewed the genetic test results and clinical data from a consecutive series of 10,030 patients referred for evaluation by an NGS hereditary cancer panel between August 2013 and October 2014. Consent was obtained from all patients to have their information used in anonymized studies. Personal and family histories of cancer were provided by the referring clinician. Patients were categorized as having breast, colon, stomach, ovarian, endometrial, or pancreatic cancer; other cancer types were not singled out for analysis. We stratified patients with breast and ovarian cancer according to reported previous BRCA1/2 genetic testing; for most analyses we focused on those without previous testing. Individuals with colon or stomach cancer were combined because of the small number of patients with stomach cancer.
Eight multigene cancer panels comprising combinations of 29 genes were included in this analysis (Table 1). Genes included in the panels had published evidence of association with hereditary cancer in at least two independent publications by two different groups. The genes were broadly grouped into three risk categories based on penetrance data available in 2012, when the tests were developed:
High risk: APC, BMPR1A, BRCA1, BRCA2, CDH1, CDKN2A, EPCAM, MLH1, MSH2, MSH6, MUTYH, PMS2, PTEN, SMAD4, STK11, TP53, VHL
Moderate risk: ATM, CHEK2, PALB2
Increased but less well-defined risk: AXIN2, BARD1, BRIP1, CDK4, FANCC, NBN, RAD51C, RAD51D, XRCC2
Table 1. Next-generation sequencing cancer panels.

Testing included NGS and exon-level array comparative genomic hybridization–based or multiplex ligation-dependent probe amplification (MLPA)–based deletion/duplication analysis of all exons and adjacent noncoding regions (see “Technical methods” below for details). Genetic variants were classified as pathogenic, likely pathogenic, VUS, likely benign, or benign/polymorphism, following the 2007 guidelines from the American College of Medical Genetic and Genomics.4 The overall classification of any test was based on the most severe variant reported. Reports with pathogenic or likely pathogenic variants were considered positive for many analyses in this study, and those with only benign or likely benign variants were considered negative. Because MUTYH-associated polyposis is an autosomal-recessive syndrome, only patients with two positive variants in MUTYH were counted in yield figures. All variants identified are submitted to ClinVar for public access.5
A subset of variants underwent reclassification since originally being reported; the variant classifications used in this analysis represent the current classifications. Providers were notified of changes in variant classifications.
Technical methods
Genomic DNA was isolated from whole blood using a QIAsymphony DNA kit, and from oral rinse using a QIAsymphony DSP Virus/Pathogen Midi Kit (Qiagen, Valencia, CA). Genomic DNA was enriched for the complete coding region and splice-site junctions of the genes of interest using custom SureSelect targeted capture (Agilent, Santa Clara, CA). NGS and deletion/duplication analysis were performed for all coding regions as well as a portion of the 5′ untranslated region, 3′ untranslated region, and intronic regions for all the genes on each panel, with the exception of EPCAM, for which only deletion/duplication analysis was performed. The entire promoter region of the PTEN gene was also analyzed using the same methodology as for the coding regions. The products were sequenced on Illumina MiSeq or HiSeq instrument with paired-end reads (Illumina Inc., San Diego, CA). DNA sequence was mapped to a masked version of the published human genome build UCSC hg19/GRCh37 reference sequence using BWA-Mem version 0.7.8.6,7 Local realignment around insertion/deletion sites and regions with poor mapping quality was performed using the Genome Analysis Toolkit version 1.6 IndelRealigner.8 Variant calls were generated by the union of SAMtools version 0.1.18,9 Genome Analysis Toolkit UnifiedGenotyper,8 and a GeneDx-developed heuristic caller. Capillary sequencing (Applied Biosystems/Life Technologies, Grand Island, NY) on a newly extracted DNA sample was used to confirm all variants with clinical or uncertain significance and to fill in sequence for regions with fewer than 15 reads by NGS. Any potential variant position with coverage of fewer than 50 reads was reviewed by analysts and analyzed by capillary sequencing if suspect. Of cases analyzed, 75% required no capillary sequencing for additional coverage, 3.2% required capillary sequencing of one amplicon for additional coverage, 17.4% required two amplicons, and 4.4% required more than two amplicons. Long-range polymerase chain reaction was used to distinguish variants in PMS2 from those in the PMS2 pseudogene, PMS2CL. Deletion/duplication analysis was performed via custom-designed exon-targeted array comparative genomic hybridization (Agilent) or, for STK11, MLPA (MRC-Holland, Amsterdam, The Netherlands). Confirmation of copy-number changes detected on array comparative genomic hybridization was performed by MLPA or repeat microarray analysis, and those originally detected by MLPA were confirmed by repeat MLPA or quantitative polymerase chain reaction using the Universal Probe Library (Roche, Indianapolis, IN).
Variant classification
We utilized the 2007 American College of Medical Genetics and Genomics framework as a guideline for variant classification.4 Every variant was analyzed with a four-level comprehensive review process by master's- and PhD-level analysts, board-certified genetic counselors, and PhD-level Fellow of the American College of Genetics and Genomics clinical molecular geneticists. Specific tools and resources include the Exome Sequencing Project,10 SIFT,11 PolyPhen2,12 MutationTaster,13 splice site prediction model BDGP,14 NetGene2,15 Softberry,16 Human Gene Mutation Database,17 and locus-specific databases including the Breast Cancer Information Core18 and InSiGHT.19
Evidence supporting benign classification of a variant included, but was not limited to, silent variants without evidence of splicing defects; published functional assessment with results similar to wild type; lack of cosegregation in large, published pedigrees; population frequencies that are too high to be consistent with a rare inherited cancer syndrome; and a lack of evolutionary conservation. Evidence supporting pathogenicity of a variant included, but was not limited to, truncating variants predicted to cause nonsense-mediated messenger RNA decay; published functional assessment demonstrating a clear effect on function; cosegregation with disease in large, published pedigrees; absence of the variant in population databases; evolutionary conservation across species; and location of a variant in a functional domain. Sufficient published information was required for a missense or in-frame variant to be classified as pathogenic. Variants classified as having uncertain significance were often missense for which there was a lack of published information to inform a pathogenic or benign classification, or, alternatively, conflicting data regarding clinical effect. VUSs often were not observed in population databases or were not frequent enough to be considered polymorphisms.
Lines of evidence used to inform a classification held different weights and were generally characterized into stand-alone, strong, and moderate categories. For example, since loss of function is an established mechanism of all the genes currently on the panels, variants creating a null allele fulfilled a stand-alone criterion for pathogenicity. Similarly, an allele frequency of at least 1% among sufficiently large groups within population databases was often sufficient for a benign classification. Well-established functional studies served as strong criteria for classification in many cases, and in silico models were weighted as moderate lines of evidence. Likely pathogenic variants had a high probability of being pathogenic. Variants reached the threshold for likely pathogenic but not pathogenic if, for example, functional studies revealed reduced but not absent activity or the variant was present at a low frequency among healthy controls. The literature about a particular variant is re-reviewed if the variant is observed again to ensure robust, up-to-date classifications. Information from new cases, such as the establishment of phase for two variants in the same gene, or establishing that a variant is de novo, may influence the classification of a variant.
Results
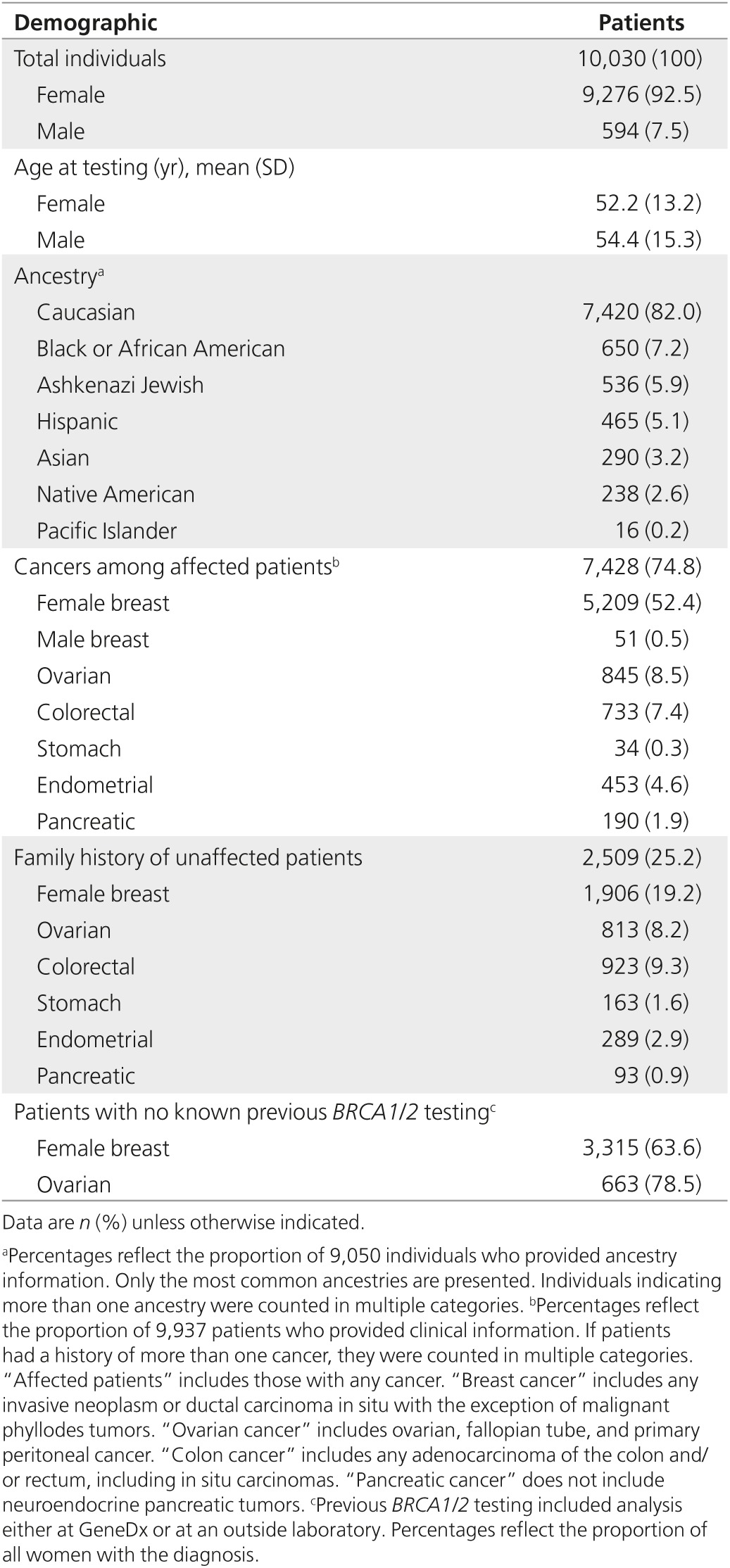
During the first 15 months in which we offered cancer panels, 10,030 patients underwent analysis with a NGS hereditary cancer panel. Sixteen patients were tested with two different panels for a total of 10,046 panels. Patient characteristics including age at genetic testing, reported ancestry, and cancer diagnosis are summarized in Table 2. Over half of the individuals referred for testing were women with breast cancer (n = 5,209). Of those, 3,315 (63.6%) had not, to our knowledge, had previous BRCA1/2 testing. Unaffected individuals comprised 25.2% of the study population.
Table 2. Demographics of individuals tested with a next-generation sequencing hereditary cancer panel.
Frequency of pathogenic and likely pathogenic variants
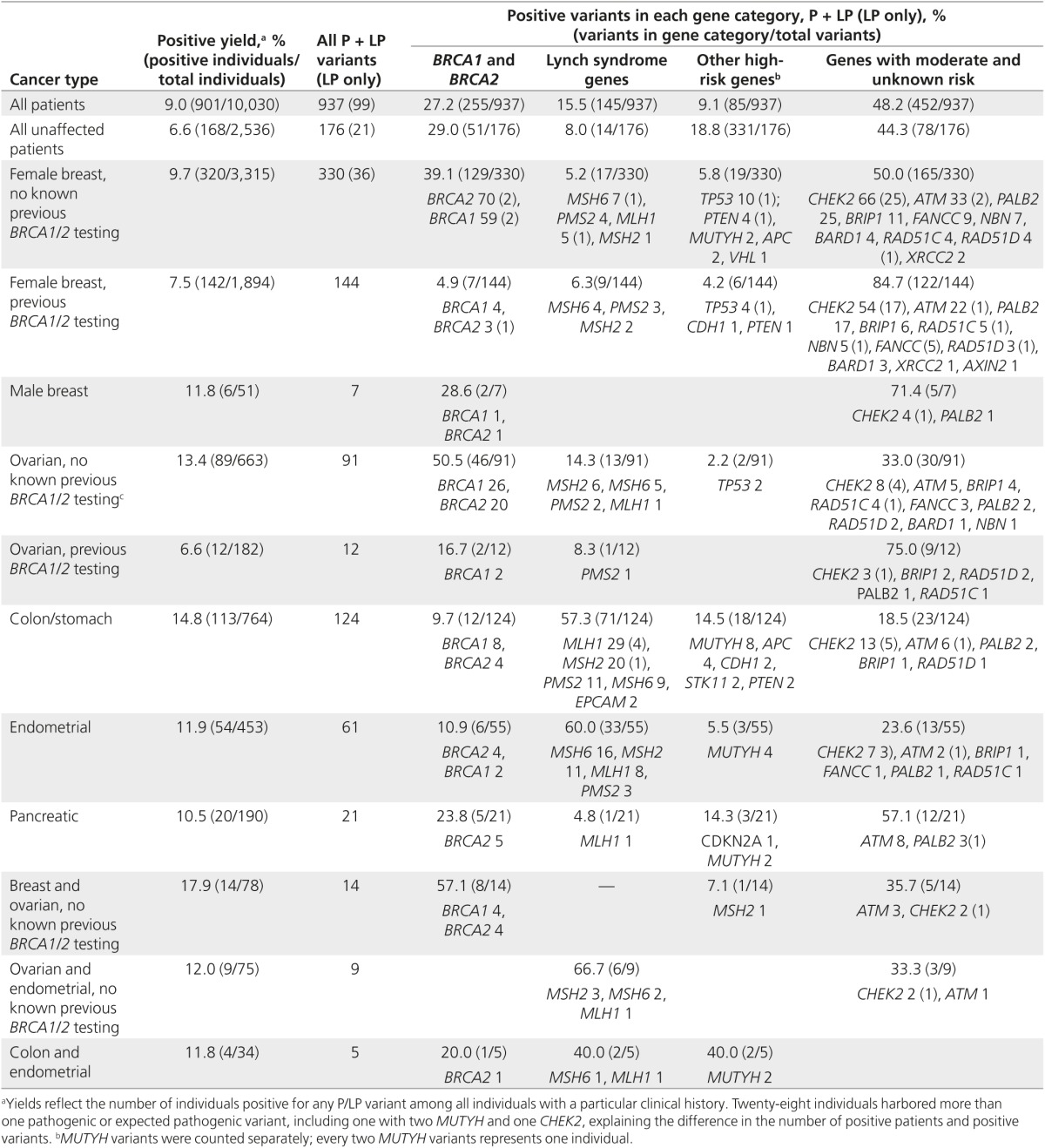
Overall, 9.0% (901/10,030) of patients in this series were found to carry at least one pathogenic or likely pathogenic variant, totaling 937 variants (Supplementary Table S1 online). Positive results were split relatively evenly between well-established genes—including BRCA1/2, Lynch syndrome, and other high-risk genes (51.8%)—and more recently described genes with moderate or unknown risk (48.2%) (Table 3). Likely pathogenic variants comprised 10.6% (99/937) of all positive results, with CHEK2 accounting for the majority of all likely pathogenic variants (68.7%; 68/99). Missense variants accounted for 18.4% (172/937) of variants, whereas the remainder included splicing, gross rearrangements, frameshifts, nonsense, and regulatory variants (Supplementary Table S2 online). Large deletions and duplications across all panels accounted for 7.0% (66/937) of positive results, composing 0.7% of the entire testing population. The majority of large rearrangements were in BRCA1 (n = 15) and MSH2 (n = 10).
Table 3. Yields by cancer type and genes with pathogenic (P) and likely pathogenic (LP) variants among patients with specific clinical histories.
The yields for positive results, stratified by panel and affected status, are shown in Figure 1. The Lynch syndrome/colorectal cancer panel, containing MLH1, MSH2, MSH6, PMS2, EPCAM, APC, and MUTYH, had the highest yield (13.7% overall, 17.6% among affected individuals), whereas the breast cancer high-risk panel containing BRCA1, BRCA2, CDH1, PTEN, STK11, and TP53 had the lowest yield (3.8% overall, 4.2% among affected individuals). On every panel, the positive yield was higher among affected than unaffected individuals.
Figure 1.

Frequency of positive results. A positive result includes at least one pathogenic or likely pathogenic variant. “All patients” includes the entire testing population regardless of cancer history. “Affected” includes individuals reporting any type of cancer. Clinical information was not provided for 93 patients (94 panel tests); thus the sum of those affected and those unaffected does not equal the total number tested. HR, high risk.
Frequency of VUSs
While VUS as the overall test classification was not associated with affection status, it was associated with the number of genes on a panel. The highest VUS frequency was observed on the largest panel, the comprehensive panel with 29 genes, with the lowest rate on the high-risk breast cancer panel with only six genes (Figure 2). VUS frequencies varied by reported ancestry. Looking at the subset of individuals reporting only one ancestry, those reporting Hispanic or Caucasian ancestry had the lowest rates across all panels (20.4 and 22.7%, respectively), whereas those reporting Asian or African-American ancestry were more likely to have a VUS as the highest classification on the report (37.3 and 39.7%).
Figure 2.

Frequency of variant of uncertain significance (VUS) results. This represents the proportion of patients with a VUS as the overall test result. “All patients” includes the entire testing population regardless of cancer history. “Affected” includes individuals reporting any type of cancer. Clinical information was not provided for 93 patients (94 panel tests); thus the sum of those affected and those unaffected does not equal the total number tested. HR, high risk.
Yield by cancer diagnosis
We also examined positive yields among individuals with the same type of cancer regardless of the panel ordered (Table 3). Individuals with colon/stomach cancer had the highest yield (14.8%; 113/764) of positive results. The majority of those positive results were in well-established colon cancer genes: MLH1, MSH2, MSH6, PMS2, EPCAM, MUTYH, APC, PTEN, and STK11. However, 28.2% (35/124) were observed in genes considered nonclassical for gastrointestinal cancers: BRCA1, BRCA2, CHEK2, ATM, PALB2, BRIP1, and RAD51D. BRCA1/2 alone accounted for 9.7% (12/124) of positive variants identified in individuals diagnosed with colon cancer.
Focusing on breast cancer, 9.7% (320/3,315) of female breast cancer patients without prior BRCA1/2 testing were found to carry a pathogenic or likely pathogenic variant. BRCA1 and BRCA2 (with 70 and 59 variants, respectively) accounted for 39.1% of positive findings, meaning the majority of positive results identified in women with breast cancer were in genes other than BRCA1/2. Other high-risk genes, including TP53, PTEN, and CDH1, accounted for 5.8% (19/330) of positive results. Furthermore, 5.2% (17/330) of positive variants were in the Lynch syndrome genes.
Moderate and less well-defined risk genes accounted for 50.0% (165/330) of all positive results among women with breast cancer. The moderate-risk genes—CHEK2, ATM, and PALB2—had positive yields of 2.0% (n = 66), 1.0% (n = 33), and 0.8% (n = 25), respectively, among the 3,315 women with breast cancer and no previous testing. Two specific disease-related CHEK2 variants, c.1100delC and p.Ile157Thr, were identified at high frequencies of 0.8 and 0.5%, respectively, in women with breast cancer.
Pathogenic variants were identified in 11.8% (6/51) of male patients with breast cancer and were found in BRCA1, BRCA2, CHEK2, and PALB2; one man was positive for both a BRCA1 and CHEK2 variant. Two of the six men with pathogenic variants (one CHEK2, one PALB2) reported previous negative BRCA1/2 testing.
Of women with ovarian cancer without reported previous BRCA1/2 testing, 13.4% (89/663) were positive. BRCA1 and BRCA2 together accounted for 50.5% of all positive findings, Lynch syndrome genes for 14.3%, and moderate or less well-defined risk genes for 33.0%.
Nearly all women who had undergone prior BRCA1/2 testing were negative for pathogenic variants in those genes. However, seven women with breast or ovarian cancer and previous BRCA1/2 testing were found to have a positive BRCA1/2 finding on an NGS panel in our laboratory. Four of these women knew of their positive status before testing at our laboratory but chose to pursue additional panel testing because of bilineal familial risk or the presence of potentially heritable cancers other than breast and ovarian in the family. The other three had undergone BRCA1/2 testing that would not have detected the variant they carried; previous testing included Ashkenazi Jewish founder testing, sequencing only, and sequencing plus testing for common BRCA1 rearrangements.
Of the 453 women with endometrial cancer, the positive yield was 11.9% (n = 54); 7.3% (n = 33) of these were within a Lynch gene, most commonly MSH6. CHEK2 also had a significant number of positives (n = 7), with an overall frequency of 1.5%. Likewise, six positive results were identified in BRCA1/2, or 10.9% (6/55) of all positive variants identified. Among 190 pancreatic cancer patients, the positive yield was 10.5% (n = 20), and positive results were most commonly identified in ATM (40.0%; 8/20), BRCA2 (25.0%; 5/20), and PALB2 (15.0%; 3/20).
Multiple positive findings
Of 901 positive patients, 28 (3.1%) had more than one positive finding, reflecting 0.3% (28/10,030) of the total testing population (Supplementary Table S3 online). Five had positive results in two highly penetrant genes, 12 had one positive result in a high-risk gene and one in a gene with moderate or unknown risk, and 11 had two positive findings in genes with moderate or unknown risk. Twenty individuals had two pathogenic variants, seven had one pathogenic and one likely pathogenic, and one had two likely pathogenic variants (MSH6 and CHEK2). Twelve women with breast cancer had multiple positive findings: one with findings in two high-risk genes (BRCA2/VHL), seven with a result in one high-risk gene (e.g., BRCA1/PALB2, BRCA2/CHEK2, biallelic MUTYH/CHEK2), and four with positive results in genes of moderate or unknown risk (all CHEK2/ATM or compound heterozygous or homozygous for CHEK2). Of the 28 individuals with multiple positive findings, 6 (21.4%) reported multiple primary cancers.
Findings in TP53 and CDH1
Six of the 18 patients (33%) with positive findings in TP53 did not meet classic Li-Fraumeni syndrome,20 Li-Fraumeni-like,21 2009 Chompret,22,23 or National Comprehensive Cancer Network guideline criteria for TP53 testing,24 resulting in a frequency of 0.06% (6/9,605) unanticipated positive results. Four patients in our cohort had a positive CDH1 result, two of whom did not meet International Gastric Cancer Linkage Consortium testing criteria,25 resulting in a frequency of 0.02% (2/8,708) positive CDH1 results in individuals who do not meet clinical criteria among those tested for the gene.
Discussion
Before the advent of NGS, patients who underwent germ-line genetic testing for hereditary cancer were typically tested for a limited number of genes that were strongly associated with a single hereditary cancer syndrome, such as BRCA1/2 or the Lynch genes. In the current era of NGS there is an opportunity to provide more information on a large number of genes, allowing for more accurate risk stratification and tailored cancer care. We provide data for over 10,000 patients who have undergone germ-line cancer genetic testing to begin to evaluate the overall yield of gene panels by cancer type and to identify the specific genes with the highest yields.
In this study population, the Lynch syndrome/colorectal cancer panel had the highest yield among all panels despite the relatively small number of genes (13.7% among all patients, 18.7% among affected patients). This high yield is likely a result of the well-established association of all genes on this panel with colorectal cancer, as well as the specific clinical history or tumor characteristics (microsatellite instability and/or immunohistochemistry) that prompted providers to order this more focused panel.
Among patients with specific cancers, yields were 9.7, 13.4, and 14.8% in patients with breast, ovarian, and colon/stomach cancer, respectively (the patients with breast or ovarian cancer did not report previous BRCA1/2 testing). Importantly, a significant proportion of the positive results were in genes that are not typically associated with the referring diagnosis. For instance, 5.8% of positive results among women with breast cancer were in highly penetrant genes other than BRCA1/2. The yield in Lynch genes among breast cancer patients was 0.5% (17/3,315), higher than a published upper estimate of the prevalence of Lynch syndrome among the general population (0.2%).26 In addition, more than a quarter of patients with colon cancer tested positive for genes that are not considered to be classic colorectal cancer genes; BRCA1/2 alone accounted for 9.7% of pathogenic variants identified in individuals with colon cancer. Similarly, over 11% of the positive findings among women with endometrial cancer were in BRCA1/2. Finally, a small number of patients whose personal and family histories were not suggestive of Li-Fraumeni syndrome were nonetheless positive for pathogenic variants in the highly penetrant TP53 gene. Together, these findings illustrate the utility of large panels in identifying pathogenic variants in high-risk genes that might not have been considered based only on a patient's personal cancer history. In some of the cases described above, the family history might have prompted broader testing and uncovered the variant identified on the larger panel, but this is not true for all examples.
Genes with moderate or less well-defined risk
Notably, almost half (48.2%) of positive variants identified in our overall testing population were in genes with moderate or undefined risk. While literature supports an association with cancer, the magnitude of the risk and complete cancer spectrum for variants in these genes is unclear. As Easton et al.3 convey, establishing an accurate risk requires studies that are sufficiently large and without ascertainment bias, and not many of these have been performed. Positive results in genes with moderate or unknown risk can present quandaries for providers given the lack of established guidelines for medical management. Furthermore, it often is not clear whether the identified variant in these genes is the sole genetic cause of the cancer in the family.
Pathogenic variants in CHEK2, ATM, and PALB2 are thought to confer a breast cancer risk that is above the 20% lifetime threshold for which breast magnetic resonance imaging is considered.24,27 Thus identifying pathogenic variants in any of these three genes in particular can have clear implications for clinical management.
CHEK2 is well established as a moderate-risk susceptibility gene for breast cancer, with weaker associations reported for colon and other cancers.28,29,30 The high yield in CHEK2 among individuals with all types of cancer may be in part a result of the high population frequency of the common CHEK2 disease-related variants. CHEK2 c.1100delC is present in up to 1% in Northern Europeans, whereas p.Ile157Thr is found in 1.4% in the 1000 Genomes Project and up to 5% of individuals of Finnish or Polish ancestry.31 Even in our population with multiple ancestries, CHEK2 c.1100delC and p.Ile157Thr comprised notable proportions of positive findings, with overall frequencies of 0.8 and 0.5%, respectively. The positive yield in CHEK2 among women with breast cancer (2.0%; 66/3,315) or endometrial cancer (1.5%; 7/453) were consistent with published frequencies.29,32,33
PALB2 is emerging as a gene that plays a major role in inherited breast cancer; recent data suggest a lifetime risk for PALB2 carriers ranging from 33 to 58%, depending on the family history of cancer.34 If these risks are confirmed in subsequent studies, PALB2 would impart the same level of breast cancer risk as BRCA2 and therefore may warrant similar breast cancer medical management recommendations. PALB2 alone accounted for 7.6% (25/330) of all positive findings among female patients with breast cancer without previous testing, which draws attention to its relevance in breast cancer genetic testing. Among the male patients with breast cancer, five of six who tested positive had a variant in CHEK2 or PALB2, both of which have been previously implicated in male breast cancer.35,36 ATM accounted for 10.0% (33/330) of positive findings among female patients with breast cancer, making it a common finding.
Variants of uncertain significance
Although VUSs are not considered clinically actionable, they can create concern for patients and clinicians. The observed VUS frequency was correlated with the number of genes on each panel, related in part to the fact that many of the larger panels have more newly characterized genes. Less well-studied genes, by definition, contribute to higher VUS rates since few missense variants in those genes have been functionally characterized or studied in families. VUS frequencies also varied by reported ancestry and were highest among individuals of African American and Asian ancestry. This is related, in part, to the higher proportion of Caucasians included in reference databases.
During the course of performing NGS cancer panels, the VUS frequencies at our laboratory varied by panel, decreasing modestly by a few percentage points on the breast/ovarian cancer, Lynch syndrome/colorectal cancer, and high/moderate risk panels. Approximately 200 variants (2.6% of all reported variants) have been reclassified since they were originally reported. The majority (~90%) of reports that were revised were done so because of a variant being downgraded from uncertain to likely benign, often because of increasing numbers of observations in the testing population or in recently available large reference data sets.
Multiple positive findings
Previously, once a pathogenic variant was identified in a hereditary cancer gene, additional testing was rarely performed because it was often assumed that only that one pathogenic variant was necessary to account for the cancer. However, additional pathogenic variants in genes that were never previously tested may also contribute to cancer risk in the family. Current estimates suggest that up to 3% of individuals who test positive on a multigene cancer test have more than one pathogenic finding, and our frequency of 3.1% is consistent with these estimates.1,2,30 In our population, a second positive variant would have been missed in 23 of 28 patients if individuals had been tested only for established high-penetrance genes such as BRCA1/2 or the Lynch syndrome genes. Notably, over 20% of individuals with two positive variants reported multiple primary malignancies. Whether individuals with multiple primary tumors are more likely to carry more than one positive variant is a hypothesis that needs to be examined in future studies.
Unexpected findings in highly penetrant genes
There is concern among providers about identifying a disease-causing variant in a high-penetrance gene in a family that does not have the classically associated phenotype. Two genes of greatest concern are TP53 (Li-Fraumeni syndrome) and CDH1 (hereditary diffuse gastric cancer), both of which are associated with high risks of cancer and management options that include prophylactic surgery and/or extensive, though often inadequate, surveillance.24,25 If a family without a classic phenotype is found to have a positive finding in either of these genes, the providers ordering the testing may question the appropriateness of applying standard management guidelines.
Two individuals in our referral population who were found to carry CDH1 variants (one pathogenic, one likely pathogenic) did not meet CDH1 testing guidelines. However, their phenotypes were not outside of the CDH1 spectrum; one had diffuse gastric cancer and the other had breast cancer with lobular features plus a family history of lobular breast cancer. By contrast, of the six individuals found to carry pathogenic or likely pathogenic TP53 variants who did not meet testing criteria for TP53, three did not have phenotypes characteristic of Li-Fraumeni syndrome. Those with suspicious histories (bilateral breast cancer at age 42; breast cancer at age 54 and sarcoma at age 56; a strong family history of early-onset colon cancer) all carried pathogenic variants. The three remaining individuals—two with pathogenic variants and one with a likely pathogenic TP53 variant—had cancer at older ages and family histories that are not typical of Li-Fraumeni syndrome. While de novo events may explain weaker family histories in some cases, these findings may also demonstrate our evolving understanding of syndrome phenotypes and penetrance associated with CDH1 and TP53. In sum, panel testing identified variants in high-risk genes that might not otherwise have been targeted, although the overall risk for an unexpected finding in either of these two genes was low for any given patient undergoing testing.
Limitations
As a testing laboratory, the clinical information we report is limited to what is provided by the referring clinician. Age at diagnosis, pathology, and complete family history were not available for every case and were not used in this analysis. Furthermore, to our knowledge, individuals included in the analyses represent unrelated probands, but two or more family members might have been referred for a panel test. Finally, the small number of patients with certain cancers, and the fact that not all individuals referred for testing were tested for the same group of genes, suggest the need for replication. However, the large number of patients tested overall provides important data on the frequency of pathogenic/likely pathogenic variants as well as variants of uncertain significance that are likely to be encountered in routine clinical practice.
Conclusion
We report the largest series to date of patients undergoing NGS for hereditary cancer gene panels. Our experience demonstrates that multigene panels have the potential to identify pathogenic variants in genes that would not typically have been tested, both in high-risk genes not associated with a patient's personal cancer history and in genes with moderate or less well-defined penetrance which, before the availability of multigene panels, were not often tested. The high frequency of positive findings in these more recently identified cancer genes underscores their potential contribution to hereditary cancer risk and evolving impact on medical management. Not all patients or providers are comfortable dealing with uncertainty in management, and expansive gene panels including genes with currently unknown risks are not appropriate in all clinical contexts or for all patients. Many patients and providers, however, would like to have the information about these genes so that as more information becomes available they are prepared to act immediately. Our study provides important empirical data to inform clinical decision making when choosing between single genes and NGS cancer panel testing in a variety of clinical scenarios.
Disclosure
The following individuals are employed by GeneDx/BioReference Labs and have salary as the only disclosure: L.R.S., M.L.M., R.N., K.J.V.P., S.M.W., L.Y., E.M.V., J.B., J.K.B., M.L.C., F.G., P.D.M., D.E.P.-A., G.D.P., Z.X., R.T.K., K.S.H. In addition, G.R. and S.B. hold stock of and are employed by GeneDx/BioReference Labs, and W.K.C. holds stock and has a consulting agreement with GeneDx/BioReference Labs.
Acknowledgments
The authors gratefully acknowledge the following individuals for their contributions: Laura Andolina, Windy Berkofsky-Fessler, Oscar Cano, Wanchun Cheng, Kurston Doonanco, Dana Farengo-Clark, Kristen Grill, Brian Henson, Susan Hiraki, Melanie Hussong, Sarah Jackson, Guang Li, Anna McGill, Erica Rinella, Maegan Roberts, Nina Sanapareddy, Sheila Solomon, Alexis Stasiak, Kristin Theobald, Joaquin Villar, Lisa Vincent, Eve Voultsis, Leslie Walsh, Stephanie Warren, Nicole Warren-Mora, Lashonda White, and Rebekah Zimmerman.
Supplementary Material
References
- LaDuca H, Stuenkel AJ, Dolinsky JS, et al. Utilization of multigene panels in hereditary cancer predisposition testing: analysis of more than 2,000 patients. Genet Med 2014;16:830–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tung N, Battelli C, Allen B, et al. Frequency of mutations in individuals with breast cancer referred for BRCA1 and BRCA2 testing using next-generation sequencing with a 25-gene panel. Cancer 2015;121:25–33. [DOI] [PubMed] [Google Scholar]
- Easton DF, Pharoah PD, Antoniou AC, et al. Gene-panel sequencing and the prediction of breast-cancer risk. N Engl J Med 2015;372:2243–2257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards CS, Bale S, Bellissimo DB, et al.; Molecular Subcommittee of the ACMG Laboratory Quality Assurance Committee. ACMG recommendations for standards for interpretation and reporting of sequence variations: revisions 2007. Genet Med 2008;10:294–300. [DOI] [PubMed] [Google Scholar]
- Landrum MJ, Lee JM, Riley GR, et al. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res 2014;42:D980–D985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ, Sugnet CW, Furey TS, et al. The human genome browser at UCSC. Genome Res 2002;12:996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009;25:1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Auwera GA, Carneiro MO, Hartl C, et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics 2013;11:11.10.1–11.10.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, et al.; 1000 Genome Project Data Processing Subgroup. The sequence alignment/map format and SAMtools. Bioinformatics 2009;25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ, Sugnet CW, Furey TS, et al. The human genome browser at UCSC. Genome Res 2002;12:996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc 2009;4:1073–1081. [DOI] [PubMed] [Google Scholar]
- Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods 2010;7:248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods 2014;11:361–362. [DOI] [PubMed] [Google Scholar]
- Celniker SE, Wheeler DA, Kronmiller B, et al. Finishing a whole-genome shotgun: release 3 of the Drosophila melanogaster euchromatic genome sequence. Genome Biol 2002;3:RESEARCH0079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebsgaard SM, Korning PG, Tolstrup N, Engelbrecht J, Rouzé P, Brunak S. Splice site prediction in Arabidopsis thaliana pre-mRNA by combining local and global sequence information. Nucleic Acids Res 1996;24:3439–3452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Softberry. 2014. http://www.softberry.com/. Accessed 27 October 2014.
- Stenson PD, Mort M, Ball EV, Shaw K, Phillips A, Cooper DN. The Human Gene Mutation Database: building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Hum Genet 2014;133:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NHGRI: Breast Cancer Information Core. 2014. https://research.nhgri.nih.gov/projects/bic/Member/index.shtml. Accessed 27 October 2014.
- Thompson BA, Spurdle AB, Plazzer JP, et al.; InSiGHT. Application of a 5-tiered scheme for standardized classification of 2,360 unique mismatch repair gene variants in the InSiGHT locus-specific database. Nat Genet 2014;46:107–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li FP, Fraumeni JF Jr, Mulvihill JJ, et al. A cancer family syndrome in twenty-four kindreds. Cancer Res 1988;48:5358–5362. [PubMed] [Google Scholar]
- Birch JM, Hartley AL, Tricker KJ, et al. Prevalence and diversity of constitutional mutations in the p53 gene among 21 Li-Fraumeni families. Cancer Res 1994;54:1298–1304. [PubMed] [Google Scholar]
- Chompret A, Abel A, Stoppa-Lyonnet D, et al. Sensitivity and predictive value of criteria for p53 germline mutation screening. J Med Genet 2001;38:43–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tinat J, Bougeard G, Baert-Desurmont S, et al. 2009 version of the Chompret criteria for Li Fraumeni syndrome. J Clin Oncol 2009;27:e108–9; author reply e110. [DOI] [PubMed] [Google Scholar]
- National Comprehensive Cancer Network Clinical Practice Guidelines in Oncology. 2014. http://www.nccn.org/professionals/physician_gls/f_guidelines.asp#detection. Accessed 28 October 2014.
- Fitzgerald RC, Hardwick R, Huntsman D, et al.; International Gastric Cancer Linkage Consortium. Hereditary diffuse gastric cancer: updated consensus guidelines for clinical management and directions for future research. J Med Genet 2010;47:436–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Chapelle A. The incidence of Lynch syndrome. Fam Cancer 2005;4:233–237. [DOI] [PubMed] [Google Scholar]
- Saslow D, Boetes C, Burke W, et al.; American Cancer Society Breast Cancer Advisory Group. American Cancer Society guidelines for breast screening with MRI as an adjunct to mammography. CA Cancer J Clin 2007;57:75–89. [DOI] [PubMed] [Google Scholar]
- Cybulski C, Górski B, Huzarski T, et al. CHEK2 is a multiorgan cancer susceptibility gene. Am J Hum Genet 2004;75:1131–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennington KP, Walsh T, Lee M, et al. BRCA1, TP53, and CHEK2 germline mutations in uterine serous carcinoma. Cancer 2013;119:332–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh T, Casadei S, Lee MK, et al. Mutations in 12 genes for inherited ovarian, fallopian tube, and peritoneal carcinoma identified by massively parallel sequencing. Proc Natl Acad Sci USA 2011;108:18032–18037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavtigian SV, Chenevix-Trench G. Growing recognition of the role for rare missense substitutions in breast cancer susceptibility. Biomark Med 2014;8:589–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHEK2 Breast Cancer Case-Control Consortium. CHEK2*1100delC and susceptibility to breast cancer: a collaborative analysis involving 10,860 breast cancer cases and 9,065 controls from 10 studies. Am J Hum Genet 2004;74:1175–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desrichard A, Bidet Y, Uhrhammer N, Bignon YJ. CHEK2 contribution to hereditary breast cancer in non-BRCA families. Breast Cancer Res 2011;13:R119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antoniou AC, Casadei S, Heikkinen T, et al. Breast-cancer risk in families with mutations in PALB2. N Engl J Med 2014;371:497–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adank MA, van Mil SE, Gille JJ, Waisfisz Q, Meijers-Heijboer H. PALB2 analysis in BRCA2-like families. Breast Cancer Res Treat 2011;127:357–362. [DOI] [PubMed] [Google Scholar]
- Wasielewski M, den Bakker MA, van den Ouweland A, et al. CHEK2 1100delC and male breast cancer in the Netherlands. Breast Cancer Res Treat 2009;116:397–400. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.