

Abstract
Small changes in DNA sequence can often have major biological effects. Here the rates and yields of guanine photo‐oxidation by Λ‐[Ru(TAP)2(dppz)]2+ have been compared in 5′‐{CCGGATCCGG}2 and 5′‐{CCGGTACCGG}2 using pico/nanosecond transient visible and time‐resolved IR (TRIR) spectroscopy. The inefficiency of electron transfer in the TA sequence is consistent with the 5′‐TA‐3′ versus 5′‐AT‐3′ binding preference predicted by X‐ray crystallography. The TRIR spectra also reveal the differences in binding sites in the two oligonucleotides.
Keywords: DNA, electron transfer, photooxidation, ruthenium, time‐resolved spectroscopy
There is continued interest in DNA photo‐oxidation by intercalating compounds due to its possible role in photo‐therapeutic applications1 and in the details to be learned about the fundamental processes of photo‐induced electron transfer (ET) in biological systems.2 However, accurate descriptions of the processes influencing ET require knowledge of the intercalator’s location in a DNA sequence that may contain multiple binding sites. Attempts to address this issue have been made by covalently tethering the intercalator to an oligodeoxynucleotide (ODN) strand,3 although even in this case the precise orientation of the intercalating ligand is uncertain.
One interesting class of DNA‐interacting compounds is ruthenium polypyridyls, as their photophysical properties, photochemical reactivity and DNA binding can be readily controlled by variation of the chelating ligands,2c and this area has also been the subject of recent insightful computational studies.4 A significant development is the access to detailed information on the binding modes of non‐covalently bound [RuL2(dppz)]2+ (dppz=dipyrido[3,2‐a:2′,3′‐c]phenazine) intercalators provided by crystal structures of Λ‐[Ru(TAP)2(dppz)]2+ (Λ‐1, Figure 1 a),5a Λ and Δ‐[Ru(phen)2(dppz)]2+,5b,c and Δ‐[Ru(bpy)2(dppz)]2+ 5d in ODNs (TAP=1,4,5,8‐tetraazaphenanthrene, phen=1,10‐phenanthroline, bpy=2,2′‐bipyridyl). Nevertheless, it can be unclear whether these crystal structures reflect what occurs under more dilute conditions in solution, where the majority of experiments on ligand–DNA interactions are performed.
Figure 1.
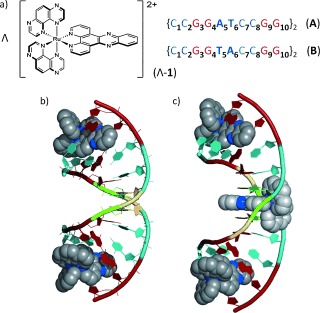
a) Structures of Λ‐[Ru(TAP)2(dppz)]2+ and ODNs used in this study. b,c) Crystal structures of Λ‐[Ru(phen)2(dppz)]2+ bound to b) ODN A and c) ODN B from Niyazi et al.,5b showing binding at central T5A6:T5A6 step in {CCGGTACCGG}2 but not {CCGGATCCGG}2. Intercalated complexes only are shown. Color code : Guanine, red; cytosine, cyan; adenine, green; thymine, yellow; nitrogen, blue.
A striking revelation from crystallography has been from the comparative study of Λ‐[Ru(phen)2(dppz)]2+ bound to 5′‐{CCGGATCCGG}2 (A) and 5′‐{CCGGTACCGG}2 (B) (Figure 1 b,c and Supporting Information (SI), Figures S1 and S2) where dppz intercalation occurs at the central 5′‐TA‐3′ but not 5′‐AT‐3′ sites.5b This observation is significant because binding at the TA step would remove the intercalator from the readily oxidized guanine bases in these sequences.6 It is therefore intriguing to study these ODNs with the structurally similar [Ru(TAP)2(dppz)]2+ (Λ‐1), which, unlike the phen analog, is known to efficiently photo‐oxidize guanine.7 In particular, we aimed to demonstrate whether a TA versus AT selectivity could be identified in solution and how this may help our understanding of ET in these systems.
To determine whether exchanging the TA:TA step for AT:AT would affect the behavior in solution the ET was monitored using transient spectroscopy.8 Parallel experiments were performed using 1) transient visible absorption (TrA) to track the formation and removal of the reduced Ru species formed by ET,7c,d and 2) time‐resolved IR (TRIR) to probe the effect on the DNA by monitoring the nucleobase vibrations.9 Experiments were performed in D2O at 0.8:1 Ru:duplex ratios (400 μm Ru, 500 μm duplex), where on average only one metal complex is available per duplex.
Picosecond‐TrA measurements show that 400 nm laser excitation of 1 in the presence of A results in removal of the ground state (450 nm) and formation of a broad transient feature (λ max=600 nm) (SI Figure S3a), assigned to the [RuIII(TAP)(TAP.−)(dppz)]2+* metal‐to‐TAP‐ligand charge transfer (MLCT) excited state.7c,d Subsequently the absorption at 600 nm decreases while simultaneously that at 515 nm increases with a rate constant of 1/(480±60) ps−1 (Figure 2 a and SI Figure S3a). This is comparable to that observed for Λ‐1 bound to other G‐rich ODNs,7e and is ascribed to ET from guanine and formation of the reduced metal complex [RuII(TAP)(TAP.−)(dppz)]+. The subsequent reverse ET was monitored by TrA experiments on the ns timescale (Figure 2 b) where the decay of the reduced species fitted to a rate constant of 1/(17±3) ns−1.
Figure 2.
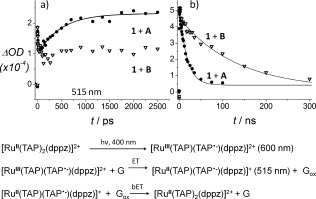
Kinetics of Λ‐[Ru(TAP)2(dppz)]2+ in ODN A (filled circles) and ODN B (open triangles) from TrA spectra on a) ps and b) ns timescales. Measured at 515 nm (except 1 + B in panel (b); 600 nm) λ exc=400 nm.
Strongly contrasting behavior was observed when the experiment was repeated with B where the central step is TA:TA. The TrA spectra recorded from ps to ns shows the [RuIII(TAP)(TAP.−)(dppz)]2+ excited state and there was no evidence for significant formation of the reduced species (Figure 2 a and SI Figure S3b). The excited state decayed with a lifetime of 120±15 ns (Figure 2 b), which was significantly longer than that observed with ODN A. However, it may be noted that this lifetime is shorter than that for the excited state of [Ru(TAP)2(dppz)]2+ either unbound (1080 ns) or bound to a duplex that does not contain G (e.g. poly{dA‐dT}2; τ=1580 ns).7b This implies that excited‐state quenching occurs, but with a much lower rate than in A.
Experiments were then performed using TRIR in order to observe directly the effect on DNA (Figure 3 and SI Figure S4). In the FTIR spectra, the region above 1600 cm−1 is dominated by C (1650 cm−1) and G (1680 cm−1) carbonyl vibrations, while Λ‐1 has no significant bands in this region.7d In the presence of ODN A, bleaching of the CG bands is observed at early times (<20 ps), before ET has occurred (Figure 3 a). This may be due to the intimate association of the photoexcited complex with the nucleobases in the intercalation site.7c, 10 (note that the ODN is not directly excited at 400 nm).
Figure 3.
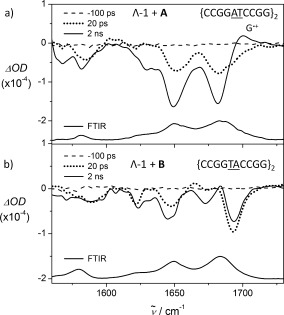
TRIR spectra of Λ‐[Ru(TAP)2(dppz)]2+ bound to a) {CCGGATCCGG}2 and b) {CCGGTACCGG}2 at selected delays. [Ru]=400 μm, [ODN]=500 μm in 50 mm phosphate‐buffered D2O. λ exc=400 nm (100 fs, 1 μJ). A baseline adjustment has been applied to the spectra. ODN FTIRs are also shown.
Subsequently the bleaches increase in magnitude and a new transient feature emerges at 1700 cm−1, which has been assigned as the G radical cation,11 confirming the formation of photo‐oxidized G. On the nanosecond timescale this feature decays and the bleaches recover (SI Figure S5), consistent with the reverse ET recorded by ns‐TrA for the re‐oxidation of reduced Λ‐1.
Again, strongly contrasting behavior was observed when Λ‐1 was bound to ODN B (Figure 3 b). Notably the structure of the bleaches differs to those observed in ODN A, implying that the environment of the complex is different. Also there is no further bleaching of the GC bands over the 100 s of picoseconds timescale as seen with ODN A and no evidence for formation of the G radical cation (G.+). Interestingly the spectrum in ODN B more closely resembles that found for the complex bound to poly{dAdT}2 (SI Figures S6 and S7), suggesting that the profile with ODN B is diagnostic of binding primarily to an adenine–thymine rich site. Based on these results, assignments may be proposed for the bleaches at 1621 cm−1 (A,νring), 1647 cm−1 (T,νring), 1672 cm−1 (T,νC4=O4), and 1694 cm−1 (T,νC2=O2).12
The above results demonstrate the sensitivity of the ET to the reversal of a single base‐pair step in the duplex. The behavior when 1 is bound to ODN A is similar to that found with G‐rich ODNs such as {G5C5}2.7d This suggests that the complex is bound close to G,13, 14 allowing efficient formation of the reduced species and subsequent reverse ET. In support of this hypothesis the TRIR signal obtained at 20 ps (i.e. before ET occurs) shows strong bleach‐bands associated with G and C. While the complex intercalates at the C1C2:G9G10 step in the crystal structure,5b it is possible that the C2G3:C8G9 site is also occupied in solution (Figure 4 a,b).
Figure 4.
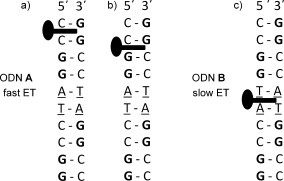
Proposed favored intercalation sites for a single bound Λ‐[Ru(TAP)2(dppz)]2+ in solution and how they may result in different ET behavior. a) ODN A; the G9G10:C1C2 (GG) step predicted by crystallography;5b b) ODN A at the 5′‐pyrimidine:purine‐3′ C8G9:C2G3 (CG) site; and c) the T5A6:T5A6 site in ODN B.
By contrast to what is observed with ODN A, the TRIR spectrum of Λ‐1 bound to B is fully consistent with the binding site being preferentially at the TA:TA step (Figure 4 c). Steady‐state UV/visible and luminescence experiments are also consistent with this model, with slightly stronger binding, and less emission quenching, when the complex is bound to ODN B compared to ODN A (while the structure of the native ODNs are similar, SI Figures S8–S10 and Table S1). Furthermore the yield of ET products is low, showing that transfer of an electron to the metal complex excited state only proceeds efficiently when it is inserted beside a G–C basepair. This observation may be contrasted with studies on tethered intercalators, where it was found that the ET yield, though not necessarily the rate, was affected by distance.3b This may suggest the involvement of different mechanisms in our case, such as proton‐coupled ET.7b,c Our experiments are consistent with the reactive excited state being a triplet ML(TAP)CT species, although a role for triplet ππ* state, as suggested by recent computational studies4 (and experiments on related systems)15 cannot be excluded.
In summary, transient visible and IR spectroscopy shows significant differences in guanine photo‐oxidation dynamics for Λ‐[Ru(TAP)2(dppz)]2+ bound to either {CCGGATCCGG}2 or {CCGGTACCGG}2. These results confirm the prediction of a 5′‐TA‐3′ vs 5′‐AT‐3′ binding preference for RuII dppz complexes, and show the sensitivity of the electron transfer to a separation of one step between donor and acceptor. This is further evidence of the importance of neighboring bases in the photo‐oxidation of guanine in DNA14, 16 Importantly we also report that the bleach bands for the DNA obtained by TRIR allow the identification of the binding site in solution. This observation should have more general applicability to the study of DNA intercalators and could offer an alternative to NMR studies, which are often rather difficult to interpret.17 It is hoped that the present study and investigations on different ODN sequences will shed further light on the structural factors governing binding and photo‐oxidation by intercalators, in both solution and crystal states.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
miscellaneous_information
The work was supported by a Royal Irish Academy/Royal Society International Exchange Scheme award (to C.J.C., J.M.K. and T.G.) and by BBSRC grants BB/K019279/1 and BB/M004635/1 (to C.J.C., M.T. and J.P.H.), by Science Foundation Ireland grants 10/IN.1/B2999 and 13/IA/1865 (T.G.), the Irish Research Council (F.E.P.), the College of Science, UCD (S.J.Q.) and by the STFC (access to the CLF‐App13230047).
Contributor Information
Dr. Páraic M. Keane, Email: keanepa@tcd.ie
Prof. Christine J. Cardin, Email: c.j.cardin@rdg.ac.uk
Prof. John M. Kelly, Email: jmkelly@tcd.ie
References
- 1.
- 1a. Puckett C. A., Barton J. K., J. Am. Chem. Soc. 2007, 129, 46–47; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1b. Elmes R. B. P., Orange K. N., Cloonan S. M., Williams D. C., Gunnlaugsson T., J. Am. Chem. Soc. 2011, 133, 15862–15865; [DOI] [PubMed] [Google Scholar]
- 1c. Gill M. R., Thomas J. A., Chem. Soc. Rev. 2012, 41, 3179–3192; [DOI] [PubMed] [Google Scholar]
- 1d. Smith N. A., Sadler P. J., Phil. Trans. R. Soc. A 2013, 371, 20120519; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1e. Wachter E., Heidary D. K., Howerton B. S., Parkin S., Glazer E. C., Chem. Commun. 2012, 48, 9649–9651; [DOI] [PubMed] [Google Scholar]
- 1f. Marcélis L., Moucheron C., Kirsch‐De Mesmaeker A., Phil. Trans. R. Soc. A 2013, 371, 20120131; [DOI] [PubMed] [Google Scholar]
- 1g. Palmer A. M., Burya S. J., Gallucci J. C., Turro C., ChemMedChem 2014, 9, 1260–1265. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Smith J. A., George M. W., Kelly J. M., Coord. Chem. Rev. 2011, 255, 2666–2675; [Google Scholar]
- 2b. Barton J. K., Olmon E. C., Sontz P. A., Coord. Chem. Rev. 2011, 255, 619–634; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2c. Vos J. G., Kelly J. M., Dalton Trans. 2006, 4869–4883. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Le Gac S., Foucart M., Gerbaux P., Defrancq E., Moucheron C., Kirsch‐De Mesmaeker A., Dalton Trans. 2010, 39, 9672–9683; [DOI] [PubMed] [Google Scholar]
- 3b. Wan C., Fiebig T., Kelley S. O., Treadway C. R., Barton J. K., Zewail A. H., Proc. Natl. Acad. Sci. USA 1999, 96, 6014–6019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.
- 4a. Véry T., Ambrosek D., Otsuka M., Gourlaouen C., Assfeld X., Monari A., Daniel C., Chem. Eur. J. 2014, 20, 12901–12909; [DOI] [PubMed] [Google Scholar]
- 4b. Daniel C., Coord. Chem. Rev. 2015, 282, 19–32. [Google Scholar]
- 5a. Hall J. P., O’Sullivan K., Naseer A., Smith J. A., Kelly J. M., Cardin C. J., Proc. Natl. Acad. Sci. USA 2011, 108, 17610–17614; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5b. Niyazi H., Hall J. P., O’Sullivan K., Winter G., Sorensen T., Kelly J. M., Cardin C. J., Nat. Chem. 2012, 4, 621–628; [DOI] [PubMed] [Google Scholar]
- 5c. Hall J. P., Cook D., Morte S. R., McIntyre P., Buchner K., Beer H., Cardin D. J., Brazier J. A., Winter G., Kelly J. M., et al., J. Am. Chem. Soc. 2013, 135, 12652–12659; [DOI] [PubMed] [Google Scholar]
- 5d. Song H., Kaiser J. T., Barton J. K., Nat. Chem. 2012, 4, 615–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.The crystal structure of Λ‐[Ru(phen)2(dppz)]2+ bound to ODNs A and B show that the centrally intercalated complex is likely to be approx. 2.8 Å further away from guanine than a complex bound at the terminal GG step; see also SI Figures S1 and S2.
- 7.
- 7a. Coates C. G., Callaghan P., McGarvey J. J., Kelly J. M., Jacquet L., Kirsch‐De Mesmaeker A., J. Mol. Struct. 2001, 598, 15–25; [Google Scholar]
- 7b. Ortmans I., Elias B., Kelly J. M., Moucheron C., Kirsch‐De Mesmaeker A., Dalton Trans. 2004, 668–676; [DOI] [PubMed] [Google Scholar]
- 7c. Elias B., Creely C., Doorley G. W., Feeney M. M., Moucheron C., Kirsch‐De Mesmaeker A., Dyer J., Grills D. C., George M. W., Matousek P., et al., Chem. Eur. J. 2008, 14, 369–375; [DOI] [PubMed] [Google Scholar]
- 7d. Keane P. M., Poynton F. E., Hall J. P., Clark I. P., Sazanovich I. V., Towrie M., Gunnlaugsson T., Quinn S. J., Cardin C. J., Kelly J. M., J. Phys. Chem. Lett. 2015, 6, 734–738; [DOI] [PubMed] [Google Scholar]
- 7e. Marcélis L., Rebarz M., Lemaur V., Fron E., De Winter J., Moucheron C., Gerbaux P., Beljonne D., Sliwa M., Kirsch‐De Mesmaeker A., J. Phys. Chem. B 2015, 119, 4488–4500. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Greetham G. M., Burgos P., Cao Q., Clark I. P., Codd P. S., Farrow R. C., George M. W., Kogimtzis M., Matousek P., Parker A. W., et al., Appl. Spectrosc. 2010, 64, 1311–1319; [DOI] [PubMed] [Google Scholar]
- 8b. Greetham G. M., Sole D., Clark I. P., Parker A. W., Pollard M. R., Towrie M., Rev. Sci. Instrum. 2012, 83, 103107. [DOI] [PubMed] [Google Scholar]
- 9. Towrie M., Doorley G. W., George M. W., Parker A. W., Quinn S. J., Kelly J. M., Analyst 2009, 134, 1265–1273. [DOI] [PubMed] [Google Scholar]
- 10. Cao Q., Creely C. M., Dyer J., Easun T. L., Grills D. C., McGovern D. A., McMaster J., Pitchford J., Smith J. A., Sun X.‐Z., et al., Photochem. Photobiol. Sci. 2011, 10, 1355–1364. [DOI] [PubMed] [Google Scholar]
- 11. Parker A. W., Lin C. Y., George M. W., Towrie M., Kuimova M. K., J. Phys. Chem. B 2010, 114, 3660–3667. [DOI] [PubMed] [Google Scholar]
- 12. Banyay M., Sarkar M., Gräslund A., Biophys. Chem. 2003, 104, 477–488. [DOI] [PubMed] [Google Scholar]
- 13.In a GG doublet, the 5′ base is expected to be the most reactive, that is, G9 and G3 in this case (see Ref. [14]).
- 14. Saito I., Nakamura T., Nakatani K., Yoshioka Y., Yamaguchi K., Sugiyama H., J. Am. Chem. Soc. 1998, 120, 12686–12687. [Google Scholar]
- 15.
- 15a. Sun Y., Liu Y., Turro C., J. Am. Chem. Soc. 2010, 132, 5594–5595; [DOI] [PubMed] [Google Scholar]
- 15b. Sun Y., Joyce L. E., Dickson N. M., Turro C., Chem. Commun. 2010, 46, 2426–2428. [DOI] [PubMed] [Google Scholar]
- 16. Markus T. Z., Daube S. S., Naaman R., J. Phys. Chem. B 2010, 114, 13897–13903. [DOI] [PubMed] [Google Scholar]
- 17.
- 17a. Greguric A., Greguric I. D., Hambley T. W., Aldrich‐Wright J. R., Grant Collins J., J. Chem. Soc. Dalton Trans. 2002, 849–855; [Google Scholar]
- 17b. Wu L., Reymer A., Persson C., Kazimierczuk K., Brown T., Lincoln P., Nordén B., Billeter M., Chem. Eur. J. 2013, 19, 5401–5410. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
miscellaneous_information