

Abstract
Chemical ligation has been used to alter motions in specific regions of dihydrofolate reductase from E. coli and to investigate the effects of localized motional changes on enzyme catalysis. Two isotopic hybrids were prepared; one with the mobile N‐terminal segment containing heavy isotopes (2H, 13C, 15N) and the remainder of the protein with natural isotopic abundance, and the other one with only the C‐terminal segment isotopically labeled. Kinetic investigations indicated that isotopic substitution of the N‐terminal segment affected only a physical step of catalysis, whereas the enzyme chemistry was affected by protein motions from the C‐terminal segment. QM/MM studies support the idea that dynamic effects on catalysis mostly originate from the C‐terminal segment. The use of isotope hybrids provides insights into the microscopic mechanism of dynamic coupling, which is difficult to obtain with other studies, and helps define the dynamic networks of intramolecular interactions central to enzyme catalysis.
Keywords: chemical ligation, enzyme catalysis, isotope effects, microscopic mechanisms, protein dynamics
The use of protein isotope labeling in enzyme kinetic studies is perhaps the most direct approach to probe protein motional coupling to catalysis.1 When replacing non‐exchangeable atoms in an enzyme with their heavy counterparts (15N, 13C, and 2H), protein fluctuations ranging from fs vibrations to ms structural changes are slowed. However, the electrostatic properties of the enzyme remain unperturbed as predicted by the Born–Oppenheimer theory.1a,b Hence, protein motional coupling to enzyme catalysis can be revealed as a reactivity difference between an isotopically labeled “heavy” enzyme and the natural‐abundance “light” enzyme. Analyses of different enzymology models by protein isotope labeling have shown that protein environmental vibrations couple to the chemical coordinate.1a–h,j,k However, the role of dynamic effects in enzyme catalysis still remains unclear. It has been suggested that “promoting vibrations” enhance enzyme‐catalyzed reactions by coupling to the transition state.2 Some of these studies have also proposed that dynamic coupling might have been selected during evolution, and maximizes the survival of the host. However, others have strongly questioned the existence of such a beneficial coupling and argued that the ability of an enzyme to sample an ideal electrostatic configuration is the key to catalytic efficiency.3 This argument originates from the fact that the microscopic mechanism involved in dynamic coupling has not been solved.
Dihydrofolate reductase from Escherichia coli (EcDHFR) has developed into a classical model in protein dynamic studies.1f–h, 4 This enzyme catalyzes the formation of tetrahydrofolate (H4F) by transfer of the pro‐R hydride from the C4 position of NADPH and a solvent proton to the C6 and N5 positions of dihydrofolate (H2F). In EcDHFR, the M20 loop (residues 9–24) forms part of the active site, and its different conformations are key to progression through the catalytic cycle (Figure 1).5 On binding of substrate and cofactor, the M20 loop closes over the active site by forming hydrogen bonds with the FG loop (residues 116–132)5b to create an optimal electrostatic environment for hydride transfer.5b, 6 Once the products have been formed, the M20 loop releases the nicotinamide ring of the oxidized cofactor and occludes parts of the active site by forming alternative hydrogen bonds with the GH loop (residues 142–149),5b triggering exchange of NADP+ and NADPH. Finally, the M20 loop resumes the closed conformation when the product H4F is released.5b
Figure 1.
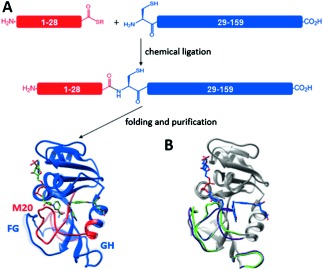
A) Synthetic scheme for EcDHFR by chemical ligation. The N‐terminal thioester peptide (red) and C‐terminal cysteine peptide (blue) are composed of residues 1–28 and residues 29–159, respectively. The M20, FG, and GH loops are indicated. B) Cartoon representations of the X‐ray crystal structures of EcDHFR in the closed (PDB No. 1RX2) and occluded (PDB No. 1RX4) conformations.5b The M20, FG, and GH loops are highlighted in green (closed) and purple (occluded). The ligands NADP+ and folate in the closed complex are shown.
Certain residues within EcDHFR have been suggested to dynamically couple to the reaction coordinate. Point mutations of these residues reduced the hydride‐transfer rate constant significantly,4d–f and their thermal motions were shown to correlate with one another.4j, 7 The hydride‐transfer rate constant for EcDHFR was mildly altered upon protein isotope labeling, thus providing evidence for the coupling of protein motions to the reaction coordinate.1g These motions, however, do not modulate the barrier to hydrogen transfer, as indicated by computational analysis; instead, they mildly enhance the frequency of barrier recrossing, reducing the frequency of successful transfers from reactants to products.1g Further protein isotope labeling studies of DHFRs from the hyperthermophile Thermotoga maritima (TmDHFR)1i and the thermophile Geobacillus stearothermophilus (BsDHFR)1j also showed the non‐beneficial side of dynamic effects and its minimization under physiological conditions. However, dynamic coupling becomes enhanced in certain scenarios.1g,j,k For instance, the catalytically compromised variant EcDHFR‐N23PP/S148A, which is less able to adopt a favorable configuration for hydride transfer,3b revealed enhanced dynamic coupling to the reaction on the fs–ps timescale.1h Similarly, BsDHFR shows an increase in dynamic coupling at low temperature.1j Furthermore, dynamic coupling appears to exist in many enzymes under or near physiological conditions, but the nature of this has not been clearly revealed.1a–e,g,h,j,k To explain this paradoxical phenomenon, the microscopic mechanism that causes dynamic coupling needs to be solved. Hence, the region of the enzyme whose motions couple to the transition state must be identified.
Native chemical ligation allows the construction of functional proteins by trans thioesterification reactions between unprotected peptides (Figure 1).8 Here, we report the first use of this technique to localize dynamic effects in hydrogen transfer reactions. EcDHFR derivatives that were isotopically labeled in the N‐terminal (residues 1–28; NT‐EcDHFR) and C‐terminal (residues 29–159; CT‐EcDHFR) segments were constructed. NT‐EcDHFR carries heavy isotopes (15N, 13C, and non‐exchangeable 2H) in the βA strand and the active‐site M20 loop, whereas CT‐EcDHFR possesses heavy isotopes only in the C‐terminal portions of the enzyme including the FG and GH loops. Investigation of “heavy” NT‐EcDHFR and CT‐EcDHFR located the regions of the enzyme responsible for dynamic coupling on the ms and fs–ps timescales.
Ala 29 was changed to a cysteine residue to facilitate the chemical ligation, and the effect of this mutation was first analyzed. The 1H–15N HSQC NMR spectroscopic and kinetic characterization of wild‐type EcDHFR and its A29C variant showed that the structural and catalytic properties were not affected by the point mutation (Table 1; see also the Supporting Information, Figure S1 and Table S1). Hence, chemical ligation was used to synthesize EcDHFR‐A29C isotopic hybrids. An N‐terminal thioester peptide comprising residues 1–28 was generated in an intein‐mediated approach, whereas the C‐terminal cysteine peptide (residues 29–159) was produced with a hexahistidine tag using methods from a published semisynthesis of EcDHFR.9 Ligation of the purified peptides and subsequent refolding yielded “light” (isotopes of natural abundance) ligated EcDHFR. “Heavy” peptides were prepared in minimal medium containing isotopically labeled ingredients. Ligation of the heavy N‐terminal peptide with the natural‐abundance C‐terminal peptide yielded NT‐EcDHFR, whereas CT‐EcDHFR was obtained through chemical ligation of the natural‐abundance N‐terminal peptide with the isotopically substituted C‐terminal peptide. Mass spectrometry showed that the purified EcDHFR derivatives were of the expected masses (Figures S2–S4). After purification in buffers made of 1H2O, NT‐EcDHFR and CT‐EcDHFR showed molecular mass increases of 1.8 % and 8.9 %, respectively, indicating that >99.8 % of the 14N, 12C, and non‐exchangeable 1H atoms had been replaced by their heavier isotopes in both proteins.
Table 1.
Kinetic parameters for the EcDHFR‐catalyzed hydride transfer reactions at 25 °C.
| Enzymes | E A [kcal mol−1] | ΔE A [kcal mol−1] | ΔS ≠ [cal mol−1 K−1] | ΔΔS ≠ [cal mol−1 K−1] | ΔH ≠ [kcal mol−1] | ΔΔH ≠ [kcal mol−1] | ΔG ≠ [kcal mol−1] |
|---|---|---|---|---|---|---|---|
| Light WT EcDHFR[a] | 7.3±0.2 | −26±1 | 6.7±0.3 | 14.4±1.5 | |||
| Light EcDHFR‐A29C | 7.3±0.3 | −25±1 | 6.8±0.3 | 14.3±1.5 | |||
| Heavy WT EcDHFR[a] | 6.0±0.3 | 1.3±0.4 | −30±2 | 4±2 | 5.4±0.6 | 1.3±0.7 | 14.4±2.5 |
| Light, chemically ligated EcDHFR | 8.3±0.3 | −22±1 | 7.8±0.3 | 14.3±1.3 | |||
| NT‐EcDHFR | 8.4±0.2 | −0.1±0.4 | −22±2 | 0±2 | 7.8±0.6 | 0.0±0.7 | 14.3±1.9 |
| CT‐EcDHFR | 7.0±0.4 | 1.3±0.5 | −27±2 | 5±2 | 6.5±0.8 | 1.3±0.9 | 14.3±3.5 |
[a] Data from Ref. 1g. WT=wild type.
The circular dichroism (CD) spectra of ligated EcDHFR, NT‐EcDHFR, and CT‐EcDHFR revealed only minor differences to the spectrum of the wild‐type enzyme, suggesting that the secondary structural organization of EcDHFR is not sensitive to the modifications made for expressed protein ligation or to isotope substitution (Figure S5), an observation that is in agreement with previous CD analyses of completely isotopically substituted EcDHFR.1f,g However, thermal unfolding and binding kinetic investigations had shown that heavy isotope substitution can alter the ground‐state conformational ensemble and ligand interactions of EcDHFR.1f Michaelis constants (K M) of ligated EcDHFR for NADPH and H2F were 7.6±2.4 and 0.4±0.1 μm (Table S2) at pH 7, similar to those of the wild‐type enzyme (4.8±1.0 and 0.7±0.2 μm, respectively).10 The K M values for the isotopic hybrids were also similar to those of wild‐type EcDHFR (Table S1), with the exception of the K M value for NADPH at pH 7.0, which was reduced by a factor of approximately three for NT‐EcDHFR, suggesting a small segment isotope effect on cofactor binding.
The effect of segment isotopic substitution on catalytic turnover was investigated by determining steady‐state turnover rate constants (k cat) at pH 7.0, where product release is mostly rate‐limiting10 (Figure 2, Figure S6, and Table S3). At 20 °C, the k cat value for unlabeled, ligated EcDHFR was 9.1±0.3 s−1, which is in good agreement with that measured for the wild‐type enzyme (8.5±1.2 s−1).10 The turnover rate constant for NT‐EcDHFR (k cat NT) was similar to that of unlabeled ligated EcDHFR (k cat LE) at low temperatures, but began to differ with increasing temperature. At 40 °C, the N‐terminal kinetic isotope effect (KIEcat NT=k cat LE/k cat NT) was 1.16±0.05. The behavior of KIEcat NT was therefore similar to that of the enzyme kinetic isotope effect (KIEcat=k LE/k HE) reported previously for fully labeled wild‐type EcDHFR,[1 g] and consistent with conformational changes in the N‐terminal peptide during turnover.5, 10 In contrast, the maximum C‐terminal kinetic isotope effect (KIEcat CT=k cat LE/k cat CT) was 1.04±0.02 at 40 °C. This is similar to the KIEcat measured for DHFRs that lack significant conformational changes during catalysis.1h,i, 4l, 11
Figure 2.
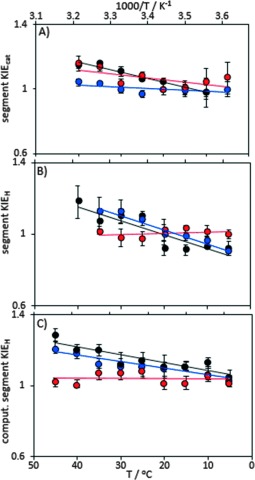
Enzyme and segment kinetic isotope effects measured at pH 7.0 for fully labeled EcDHFR (black),1g NT‐EcDHFR (red), and CT‐EcDHFR (blue) under steady‐state (A) and single‐turnover, pre‐steady‐state (B) conditions. C) Theoretical isotope effects calculated from the recrossing coefficients.
These results demonstrate that segmental heavy‐isotope labeling can be used to localize effects of protein motions on enzyme catalysis. The observation that KIEcat NT is temperature‐dependent and accounts for almost the entire KIEcat of EcDHFR whereas KIEcat CT is close to unity indicates that residues 1–28 undergo substantial movement during the catalytic turnover. X‐ray crystallographic studies of EcDHFR previously demonstrated that a substantial movement of the M20 loop (residues 9–24) is required for product release, whereas only a minor rearrangement is seen in the FG loop in the C‐terminal segment and other regions of the enzyme (Figure 1).10 Solution NMR measurements revealed that various regions of the enzyme in the product complex show conformational motions on the same timescale as product release.10 Although a contribution from other regions of the enzyme may be possible, our results confirm that motions of the M20 loop are dominant in limiting k cat. These results therefore show that segment isotope labeling is able to pinpoint regions of the enzyme whose motions on the ms timescale are involved in the rate‐limiting physical step of catalysis.
Having demonstrated the utility of our method in locating motional effects in EcDHFR, we performed pre‐steady‐state stopped‐flow kinetic measurements at pH 7.0 to localize the origin of the fast motions that couple to the chemical step of the catalytic cycle, hydride transfer from NADPH to predominately protonated H2F. The hydride‐transfer rate constants for unlabeled, ligated EcDHFR (k H LE), which carries a hexahistidine tag, are slightly lower than those of the wild‐type, untagged enzyme and its A29C variant (Tables S1 and S4). However, the primary H/D KIE on the reaction was largely unaffected by the presence of the hexahistidine tag (Table S5), which was therefore not removed.12 The hydride‐transfer rate constants for NT‐EcDHFR (k H NT) were identical to those measured for unlabeled, ligated EcDHFR giving a KIEH NT (k H LE/k H NT) value close to unity at all temperatures (Figure S6 and Table S4). In contrast, measurable reactivity differences for CT‐EcDHFR were observed, with an isotope effect KIEH CT (k H LE/k H CT) that increases from 0.90±0.02 at 5 °C to 1.13±0.08 at 35 °C. Both the magnitude and temperature dependence of KIEH CT were similar to those of the enzyme KIE measured for fully labeled wild‐type EcDHFR.1g Indeed, the difference in the activation energies between the light, chemically ligated enzyme and CT‐EcDHFR is statistically identical to that between the light and heavy wild‐type enzymes (Table 1). In contrast, this parameter is not affected by labeling the N‐terminal segment of the chemically ligated enzyme. These observations provide strong evidence for dynamic coupling of the C‐terminal region of EcDHFR to the chemical coordinate at pH 7.0; no evidence for such a coupling to the first 28 amino acid residues was found.
Previously, hydride‐transfer rate constants of isotopically substituted DHFRs were estimated in the framework of transition state theory (TST), which includes a temperature‐dependent transmission coefficient to account for tunneling contributions and dynamic effects:(1), 13
| (1) |
where R is the ideal gas constant, T the temperature, k
B the Boltzmann constant, and h Planck′s constant, 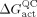 is the quasiclassical activation free energy, κ(T) the tunneling coefficient that accounts for reactive trajectories not reaching the classical threshold energy, and γ(T,ξ) is the recrossing transmission coefficient that includes the trajectories that recross the dividing surface from the product back to the reactant. Several lines of evidence indicated that deuterium substitution does not alter the electrostatic properties of the active site and consequently the activation free energy,1a,b,g and that protein isotope labeling does not alter the tunneling coefficient despite the smaller size of a fully deuterated protein.1g,h,j However, the recrossing trajectories were changed upon protein isotope labeling, thus giving a subtle difference in the hydride‐transfer rate constant:(2), 1g,h,j
is the quasiclassical activation free energy, κ(T) the tunneling coefficient that accounts for reactive trajectories not reaching the classical threshold energy, and γ(T,ξ) is the recrossing transmission coefficient that includes the trajectories that recross the dividing surface from the product back to the reactant. Several lines of evidence indicated that deuterium substitution does not alter the electrostatic properties of the active site and consequently the activation free energy,1a,b,g and that protein isotope labeling does not alter the tunneling coefficient despite the smaller size of a fully deuterated protein.1g,h,j However, the recrossing trajectories were changed upon protein isotope labeling, thus giving a subtle difference in the hydride‐transfer rate constant:(2), 1g,h,j
| (2) |
Accordingly, the segment isotope effects were calculated by evaluating the recrossing transmission coefficients of light and heavy EcDHFR, NT‐EcDHFR, and CT‐EcDHFR from 278 to 318 K (Figure 1, Tables S6 and S7). Computational segmental KIEH values were estimated from Eq. (2) and follow the same trend as the experimental values obtained under pre‐steady‐state conditions (Figure 2). Isotope labeling of the C‐terminal fragment caused a greater difference in the recrossing coefficients and consequently a larger isotope effect at high temperature, whereas contributions from the N‐terminal fragment were small. However, theoretical estimations always yield a normal enzyme KIEH, whereas the experimental values show an inverse effect at temperatures below 20 °C. It may be that heavy‐isotope substitution also modifies the zero‐point energies of protein vibrational modes in the reactant state and transition state differently, altering the value of 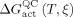 . This could result in a tighter interaction of the isotopically labeled enzyme with the transition state and a slightly higher reactivity at lower temperature. A previous study of pentaerythritol tetranitrate reductase also showed that the heavy enzyme is more reactive under certain conditions.1c However, effects on zero‐point energies are not directly related to dynamic coupling, and thus our approximation for the protein isotope labeling effect remains valid within the TST model with dynamic corrections.1h
. This could result in a tighter interaction of the isotopically labeled enzyme with the transition state and a slightly higher reactivity at lower temperature. A previous study of pentaerythritol tetranitrate reductase also showed that the heavy enzyme is more reactive under certain conditions.1c However, effects on zero‐point energies are not directly related to dynamic coupling, and thus our approximation for the protein isotope labeling effect remains valid within the TST model with dynamic corrections.1h
Residues implicated in a proposed “hydride‐transfer‐driving” dynamic network are located mainly in the M20 loop (residues 9–23), and interact with residues dispersed throughout the enzyme (e.g., Met 43, Thr 113, and Asp 122).4g–l If motions of this network couple to the reaction coordinate, then a measureable KIEH NT would be expected. However, dynamic coupling to the chemical step at pH 7.0 does not involve the first 28 amino acids of EcDHFR, even though this region shows the largest thermal motions; it originates exclusively from the C‐terminal region. The movements involving the M20 loop therefore generate a reaction‐ready active‐site configuration, from which hydride transfer can take place,1f,h, 5b, 6 but do not significantly couple to the reaction coordinate. This is the first line of experimental evidence that clearly shows the region of EcDHFR that is coupled to the hydride transfer event. Instead of “promoting motions”, the observed dynamic effects are likely due to “residual” motions caused by an incomplete electrostatic preorganization of the enzyme, which must bind the Michaelis complex and reorganize to reach the transition state. The enzymatic reorganization therefore results from the unavoidable balance between catalysis and substrate binding. Accordingly, dynamic effects are tied to the efficiency of electrostatic preorganization, which acts to maintain a low reorganization energy in enzyme chemistry.
Measurement of the segment isotope effect is a practical method to investigate dynamic effects on enzyme catalysis with spatial resolution. It can identify regions of an enzyme that couple to the reaction coordinate and can distinguish regions of the enzyme involved in millisecond conformational motions from those whose fast dynamics couple to barrier crossing. Millisecond conformational motions of the M20 loop play a crucial role in the physical step of product release, whereas fast vibrations from the C‐terminal segment affect the chemical step itself. When complemented with computational studies,3–, 4c,g,m,o, 14 measurements of the segment isotope effect provide insights that are not easily obtained with other methods. This may help design inhibitors of motions crucial for the catalytic cycle. Most importantly, the current study demonstrates the applicability of segment isotope labeling to studies of a wide variety of protein models in the field of protein dynamics and beyond.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
miscellaneous_information
This work was supported by Cardiff University through grants BB/J005266/1 and BB/L020394/1 (to R.K.A.) from the UK′s Biotechnology and Biological Sciences Research Council (BBSRC), by FEDER and Ministerio de Economía y Competitividad funds (project CTQ2012‐36253‐C03), the Generalitat Valenciana (ACOMP/2014/277 and PrometeoII/2014/022), and by the Universitat Jaume I (Project P1⋅1B2011‐23).
Contributor Information
Dr. Iñaki Tuñón, Email: ignacio.tunon@uv.es
Prof. Dr. Vincent Moliner, Email: moliner@uji.es
Prof. Dr. Rudolf K. Allemann, Email: allemannrk@cardiff.ac.uk
References
- 1.
- 1a. Silva R. G., Murkin A. S., Schramm V. L., Proc. Natl. Acad. Sci. USA 2011, 108, 18661–18665; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1b. Kipp D. R., Silva R. G., Schramm V. L., J. Am. Chem. Soc. 2011, 133, 19358–19361; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1c. Pudney C. R., Guerriero A., Baxter N. J., Johannissen L. O., Waltho J. P., Hay S., Scrutton N. S., J. Am. Chem. Soc. 2013, 135, 2512–2517; [DOI] [PubMed] [Google Scholar]
- 1d. Toney M. D., Castro J. N., Addington T. A., J. Am. Chem. Soc. 2013, 135, 2509–2511; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1e. Świderek K., Javier Ruiz‐Pernía J., Moliner V., Tuñón I., Curr. Opin. Chem. Biol. 2014, 21, 11–18; [DOI] [PubMed] [Google Scholar]
- 1f. Wang Z., Singh P. N., Czekster C. M., Kohen A., Schramm V. L., J. Am. Chem. Soc. 2014, 136, 8333; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1g. Luk L. Y. P., Ruiz‐Pernia J. J., Dawson W. M., Roca M., Loveridge E. J., Glowacki D. R., Harvey J. N., Mulholland A. J., Tuñón I., Moliner V., Allemann R. K., Proc. Natl. Acad. Sci. USA 2013, 110, 16344–16349; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1h. Ruiz‐Pernia J. J., Luk L. Y. P., García‐Meseguer R., Martí S., Loveridge E. J., Tuñón I., Moliner V., Allemann R. K., J. Am. Chem. Soc. 2013, 135, 18689–18696; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1i. Luk L. Y. P., Loveridge E. J., Allemann R. K., J. Am. Chem. Soc. 2014, 136, 6862; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1j. Luk L. Y. P., Ruiz‐Pernía J. J., Dawson W. M., Loveridge E. J., Tuñón I., Moliner V., Allemann R. K., J. Am. Chem. Soc. 2014, 136, 17317; [DOI] [PubMed] [Google Scholar]
- 1k.R. K. Allemann, E. J. Loveridge, L. Y. P. Luk, Phys. Chem. Chem. Phys. 2015, DOI: 10.1039/C5CP00794A. [DOI] [PubMed]
- 2.
- 2a. Basran J., Sutcliffe M. J., Scrutton N. S., Biochemistry 1999, 38, 3218–3222; [DOI] [PubMed] [Google Scholar]
- 2b. Antoniou D., Caratzoulas S., Kalyanaraman C., Mincer J. S., Schwartz S. D., Eur. J. Biochem. 2002, 269, 3103–3112; [DOI] [PubMed] [Google Scholar]
- 2c. Sutcliffe M. J., Scrutton N. S., Eur. J. Biochem. 2002, 269, 3096–3102; [DOI] [PubMed] [Google Scholar]
- 2d. Knapp M. J., Klinman J. P., Eur. J. Biochem. 2002, 269, 3113–3132; [DOI] [PubMed] [Google Scholar]
- 2e. Klinman J. P., J. Phys. Org. Chem. 2010, 23, 606–612. [Google Scholar]
- 3.
- 3a. Adamczyk A. J., Cao J., Kamerlin S. C. L., Warshel A., Proc. Natl. Acad. Sci. USA 2011, 108, 14115–14120; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3b. Pisliakov A. V., Cao J., Kamerlin S. C. L., Warshel A., Proc. Natl. Acad. Sci. USA 2009, 106, 17359–17364; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3c. Boekelheide N., Salomón‐Ferrer R., Miller T. F., Proc. Natl. Acad. Sci. USA 2011, 108, 16159–16163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.
- 4a. Pu J., Ma S., Gao J., Truhlar D. G., J. Phys. Chem. B 2005, 109, 8551–8556; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4b. Garcia‐Viloca M., Truhlar D. G., Gao J., Biochemistry 2003, 42, 13558–13575; [DOI] [PubMed] [Google Scholar]
- 4c. Liu H., Warshel A., J. Phys. Chem. B 2007, 111, 7852–7861; [DOI] [PubMed] [Google Scholar]
- 4d. Cameron C. E., Benkovic S. J., Biochemistry 1997, 36, 15792–15800; [DOI] [PubMed] [Google Scholar]
- 4e. Wang L., Tharp S., Selzer T., Benkovic S. J., Kohen A., Biochemistry 2006, 45, 1383–1392; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4f. Singh P., Sen A., Francis K., Kohen A., J. Am. Chem. Soc. 2014, 136, 2575–2582; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4g. Watney L. B., Agarwal P. K., Hammes‐Schiffer S., J. Am. Chem. Soc. 2003, 125, 3745–3750; [DOI] [PubMed] [Google Scholar]
- 4h. Agarwal P. K., Billeter S. R., Rajagopalan P. T. R., Benkovic S. J., Hammes‐Schiffer S., Proc. Natl. Acad. Sci. USA 2002, 99, 2794–2799; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4i. Radkiewicz J. L., C. L. Brooks III , J. Am. Chem. Soc. 2000, 122, 225–231; [Google Scholar]
- 4j. Rod T. H., Radkiewicz J. L., C. L. Brooks III , Proc. Natl. Acad. Sci. USA 2003, 100, 6980–6985; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4k. Stojković V., Perissinotti L. L., Lee J., Benkovic S. J., Kohen A., Chem. Commun. 2010, 46, 8974–8976; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4l. Bhabha G., Lee J., Ekiert D. C., Gam J., Wilson I. A., Dyson H. J., Benkovic S. J., Wright P. E., Science 2011, 332, 234–238; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4m. Fan Y., Cembran A., Ma S., Gao J., Biochemistry 2013, 52, 2036–2049; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4n. Roston D., Kohen A., Doron D., Major D. T., J. Comput. Chem. 2014, 35, 1411; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4o. Arora K., C. L. Brooks III , J. Am. Chem. Soc. 2009, 131, 5642–5647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.
- 5a. Boehr D. D., McElheny D., Dyson H. J., Wright P. E., Science 2006, 313, 1638–1642; [DOI] [PubMed] [Google Scholar]
- 5b. Sawaya M. R., Kraut J., Biochemistry 1997, 36, 586–603. [DOI] [PubMed] [Google Scholar]
- 6. Loveridge E. J., Behiry E. M., Guo J., Allemann R. K., Nat. Chem. 2012, 4, 292–297. [DOI] [PubMed] [Google Scholar]
- 7. Epstein D. M., Benkovic S. J., Wright P. E., Biochemistry 1995, 34, 11037–11048. [DOI] [PubMed] [Google Scholar]
- 8. Dawson P., Muir T., Clark‐Lewis I., Kent S., Science 1994, 266, 776–779. [DOI] [PubMed] [Google Scholar]
- 9. Ottesen J. J., Bar‐Dagan M., Giovani B., Muir T. W., Pept. Sci. 2008, 90, 406–414. [DOI] [PubMed] [Google Scholar]
- 10. Fierke C. A., Johnson K. A., Benkovic S. J., Biochemistry 1987, 26, 4085–4092. [DOI] [PubMed] [Google Scholar]
- 11. Dams T., Bohm G., Auerbach G., Bader G., Schurig H., Jaenicke R., Biol. Chem. 1998, 379, 367–371. [PubMed] [Google Scholar]
- 12. Swanwick R. S., Maglia G., Tey L., Allemann R. K., Biochem. J. 2006, 394, 259–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.
- 13a. Alhambra C., Corchado J., Sanchez M. L., Garcia‐Viloca M., Gao J., Truhlar D. G., J. Phys. Chem. B 2001, 105, 11326–11340; [Google Scholar]
- 13b. Truhlar D. G., Garrett B. C., Klippenstein S. J., J. Phys. Chem. 1996, 100, 12771–12800. [Google Scholar]
- 14. Cui Q., Karplus M., J. Phys. Chem. B 2002, 106, 7927–7947. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
miscellaneous_information