

Abstract
A 33-year-old white woman arrived at the hospital to undergo a hysterectomy due to uterine fibroids. Blood smear review identified macrothrombocytopenia and Döhle body–like cytoplasmic leukocyte inclusions. Genetic testing identified a mutation in exon 39 of the myosin heavy chain gene (MHY9; OMIM 160775), which confirmed the diagnosis of May-Hegglin anomaly. May-Hegglin anomaly is one of a spectrum of MYH9 disorders that also includes Sebastian, Epstein, and Fechtner syndromes. Herein, we describe the clinical and laboratory presentation of a patient with May-Hegglin anomaly and provide an update on the molecular findings and a discussion of the genotypic-phenotypic correlations in this potentially underdiagnosed disorder.
Keywords: macrothrombocytopenia, May-Hegglin, MYH9-related disease, Döhle body, granulocyte inclusions, giant platelets, blood smear
A 33-year-old white woman who had had 1 pregnancy but no live births (G1 P0) arrived at Virginia Mason Medical Center in Seattle, WA with severe pelvic pain, menorrhagia, and uterine fibroids; for these reasons, she consented to undergo laparoscopic hysterectomy. Her medical history included chronic stable thrombocytopenia, which first had been identified after a miscarriage 6 years previously. The platelet count of the patient varied from 48,000 × 109 per L to 61,000 × 109 per L. Her medical record indicated a history of occasional mild gingival bleeding, easy bruising, and occasional hematomas but no history of major hemorrhage and no requirement for transfusions. At the age of 3 years, the patient had undergone a successful appendectomy, with no requirement for transfusion or excessive bleeding. A computed tomography (CT) scan confirmed a 5.1-cm right subserosal fundic mass consistent with uterine fibroids. An incidental small angiomyolipoma was also identified in the right kidney.
The patient’s preoperative management included a complete blood count (CBC). White blood count (WBC), red blood count (RBC), and indices were within normal limits. Platelet count was 61,000 × 109 per L (reference range, 150 to 400).
Laboratory Findings
Blood Smear Review
A blood smear review was triggered by thrombocytopenia with an increased mean platelet volume (MPV) of 14.0 fL (reference range, 7.4 to 10.7). Hematology technologist and pathologist (S.M.R) slide review further revealed Döhle body–like spindle- and lancet-shaped, light-blue cytoplasmic inclusions in nearly every neutrophil, some eosinophils, and a few monocytes. Large and giant platelets were also noted (Image 1 and Image 2).
Image 1.
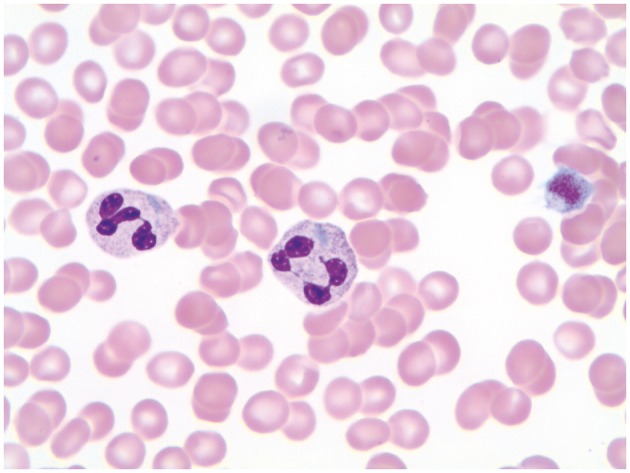
Neutrophils with lancet shaped, intracytoplasmic Dohle-body like inclusions and associated giant platelet (Giemsa stain, original magnification x1000).
Image 2.

Neutrophil with lancet shaped, intracytoplasmic Dohle-body like inclusions and associated giant platelet (Giemsa stain, original magnification x1000).
Chemistry, Urinalysis, and Coagulation
The results of standard chemistry panels were within normal limits, with a creatinine value of 0.8 mg per dL (reference range, 0.57 to 1.11) (estimated glomerular filtration rate [eGFR] > 60 mL/min/1.73 m2). Liver enzyme levels were also within normal limits. The results of a urinalysis were normal, with no evidence of hematuria or proteinuria. Prothrombin time (PT) and partial thromboplastin time (PTT) were also normal.
Platelet Aggregation Studies
We sent the blood specimens to the Bloodworks Northwest hemostasis laboratory in Seattle, Washington, for platelet aggregation studies. The results of these studies were normal, with the exception of a slightly decreased aggregation to low-dose adenosine diphosphate (ADP; possibly an artifact of the low platelet count), including normal response to ristocetin and other agonists.
Molecular Findings
We also sent blood specimens to the BloodCenter of Wisconsin, Inc. in Milwaukee, WI for MYH9 molecular studies. Bidirectional polymerase chain reaction (PCR) amplification and DNA sequence analysis were performed on MYH9 exons 2, 11, 17, 22, 25-27, 31-33, 35, and 38-41; this technique can identify 99% of reported MYH9 mutations. A pathogenic heterozygous sequence variant in exon 39 (c.5521G > A, p.E1841K) was detected, which changes the glutamate at codon 1841 to lysine.
Based on thrombocytopenia with increased MPV and large and giant platelets and neutrophilic Döhle body–like cytoplasmic inclusions, we diagnosed the patient with May-Hegglin anomaly. The molecular findings confirmed this diagnosis.
Discussion
Thrombocytopenia with large or giant platelets, or macrothrombocytopenia, often triggers a blood smear review. This review can confirm the macrothrombocytopenia and also can investigate for an underlying more serious pathologic manifestation, such as a myelodysplastic process or other significant abnormalities. However, most commonly, no additional blood smear abnormalities are observed. Neutrophilic cytoplasmic Döhle bodies are a relatively nonspecific blood smear finding that can be observed in reactive inflammatory and infectious processes. However, when observed in the presence of macrothrombocytopenia, the identification of Döhle-like bodies should raise the possibility of May-Hegglin anomaly or one of the other MYH9 disorders. Another important aspect of this finding is that it can help exclude other causes of acquired macrothrombocytopenia, such as immune thrombocytopenic purpura, or inherited macrothrombocytopenia, such as Bernard-Soulier syndrome, grey platelet syndrome, or von Willebrand disease type IIB.
May-Hegglin anomaly (MHA), Sebastian syndrome (SS), Fechtner syndrome (FS), and Epstein syndrome (ES) are related disorders that are characterized by thrombocytopenia with giant platelets and mutations in the MYH9 gene. Platelet counts range from less than 30,000 as many as 100,000; patients manifest a bleeding tendency with symptoms such as bruising, epistaxis, and menorrhagia and display chronic stable macrothrombocytopenia.1 Severe deep internal bleeding is not usually observed, but rare cases of postpartum, life-threatening or intracranial hemorrhage have been reported.2 Also, patients may be misdiagnosed as having immune thrombocytopenic purpura, prompting treatment with corticosteroids and/or even splenectomy.3-5 Each of the aforementioned syndromes are characterized by macrothrombocytopenia; however, late- or early-onset nephropathy leading to renal failure (FS, ES), sensorineural hearing loss (FS, ES), and cataracts variably may be observed (FS). Some patients with SS may develop high-tone sensorineural hearing loss and cataracts later in life.1 Large leukocyte cytoplasmic inclusion bodies are observed in MHA, whereas small leukocyte cytoplasmic inclusion bodies are observed in FS and SS. Inclusions cannot be observed via standard light microscopy in ES1,6 (Table 1).
Table 1.
Clinical and Laboratory Manifestations in MYH-9 Related Disordersa
| Variable | Macrothromboytopenia | Large Inclusions | Small Inclusions | Nephritis | Hearing Loss | Cataracts |
|---|---|---|---|---|---|---|
| MHA | + | + | – | – | – | – |
| SS | + | – | + | – | Late onset | Late onset |
| FS | + | – | + | + | + | + |
| ES | + | – | – | + | + | – |
MHA, May-Hegglin anomaly; SS, Sebastian syndrome; FS, Fechtner syndrome; ES, Epstein syndrome.
aModified from reference 1.
MHY9 Gene
The MHY9 gene is located on chromosome 22q11.2 and is composed of 40 coding exons and 1960 amino acid residues.5,7 This gene encodes for the heavy chain of nonmuscular myosin IIA (NMM-IIA), a class-II myosin belonging to the myosin superfamily. This cytoskeletal protein plays a role in cellular motility, phagocytosis, and maintenance of cell shape. Exon 1 is not translated. Exons 2 through 19 code for the motor or “head” domain. Exon 20 codes for the “neck” domain, and exons 21 through 40 code for the coiled “tail” domain. Finally, exon 41 for the nonhelical C-terminal sequence. NMM-IIA is expressed in muscle but also in nonmuscle cells, including granulocytes, platelets, and podocytes, as well as in distal tubules of the kidneys and in the cochlea and lens.7,8
Inclusions and Immunofluoresence
A hallmark attribute of MYH9 disorders is the formation of Döhle body–like inclusions in leukocytes. These inclusions represent an aggregation of abnormal and wild-type nonmuscle myosin IIA protein, MYH9 messenger RNA (mRNA), and clusters of ribosomes forming ribonucleotide-protein complexes in the cytoplasm.9 Utilization of immunofluorescence has enabled these inclusions to be categorized into three types: I, II, and III. The morphological characteristics and localization of these inclusions yield some insight into the type of MYH9 mutation. Type I inclusions are large (0.5μm to 2μm) and arise in the cytoplasm as double or single aggregates. These granules stain intensely and range from oval to spindle shaped. Type II granular inclusions are small (≤ 1 μm), numerous (as many as 20), and are dispersed throughout the cytoplasm in oval or circle shapes. Type III NMM-IIA aggregates give the cell a speckled appearance and generally cannot be observed via light microscopy.7,9 Also, the degree of expression of these inclusions amongst different types of granulocytes suggests that regulatory mechanisms are different for each cell type. Inclusions routinely are observed in neutrophils but are observed only occasionally in monocytes.10,11 The specific mutation seems to associate with the type of inclusion. For example, exon 39 and exon 41 mutations yield type I inclusions, exon 27 and exon 31 mutations are usually associated with type II inclusions, and exon 2 and exon 17 mutations may be associated with type II or type III inclusions.6,7,9,11
MYH9 Genotype-Phenotype Correlation
Most MYH9 disorders are inherited in an autosomal dominant fashion, although as many as 35% may occur due to sporadic mutations.5 As of the year 2013, a total of 44 MYH9 alterations involving only 35 of 1960 MYH9 amino acids were implicated in the disease. Further, alterations of only 6 residues account for 80% of cases, including Ser96, Arg702, Arg1165, Asp1424, Glu1841, and Arg1933.5 A more recent study12 updates the number of alterations to 83, but these alterations still involve the most common exons and residues. These mutations are usually of the missense type. However, deletions and nonsense and frameshift mutations have been documented. The nonsense and frameshift mutations that predict a truncated protein all involve the terminal exon 41.13 It is believed colloquially that gene mutations in the “head” region (exons 2-19) result in more serious manifestations compared with the “neck” and “tail” regions (exons 20-41). However, although the specific mutation gives some insight into the clinical presentation of the disease, individuals with the same mutation may express a variation in phenotype14 (Table 2).
Table 2.
Genotype-Phenotype Correlationa
| Domain | Exon | Mutation | Nephropathy | Deafness | Cataracts | Inclusion Type | Bleeding Risk |
|---|---|---|---|---|---|---|---|
| Motor Domain | Exon 2 | S96i | Low to intermediate risk | Before age 60 years in all patients | Low risk | Type 2 or 3 | Higher |
| Exon 17 | R702 | Before age 40 in all patients: higher risk of progression to ESRD | Before age 40 years in all patients | Low risk | Type 2 or 3 | ||
| Tail Domain | Exon 27 | R1165 | Low risk | Before age 60 years in all patients | Low risk | Type 2 or 3 | Lower |
| Exon 31 | D1424H | High risk | Intermediate risk | Probably higher risk | Type 1 or 2 | ||
| D1424N | Very low risk | Low risk | Low risk | ||||
| Exon 39 | E1841K | Low risk | Low to intermediate risk | Low risk | Type 1 | ||
| Exon 41 | NHT deletion | Very low risk | Low risk | Very low risk | Type 1 |
aModified from references 12 and 13.
Our patient had a heterozygous mutation in exon 39, which induced a glutamate-to-lysine change at amino acid 1841 (E1841K). The literature varies in its accounting of which exon is linked with the E1841K mutation. Historically, the E1841 mutation has been ascribed to exon 38, although more recently, it has been referred to as exon 39, depending on whether or not the first noncoding exon is counted.15 E1841K locates to the “tail” portion of the protein. As one might guess based on the literature, our patient has a history of macrothrombocytopenia and menorrhagia, along with the finding of large type I inclusions in granulocytes that is diagnostic of May-Hegglin anomaly. She has not shown any signs or symptoms of renal disease, hearing impairment, or cataracts up to this point. One study investigated MYH9-related disorders (MYH9-RD) with specific respect to genotype-phenotype correlation. E1841K was considered to confer a low to intermediate risk of clinical manifestations because 43% of patients with that change manifested some degree of sensorineural hearing loss; renal disease and cataracts each affected approximately 12% of patients.13 In contrast, patients with alterations involving exon 17 (amino acid position 702) are at high risk of developing renal disease leading to renal failure, which pathologically manifests as focal segmental glomerulosclerosis.4,16 The specific amino-acid alteration is important for nephropathy risk determination for exon 31: D1424H is associated with a high risk of renal disease, whereas D1424N is associated with a very low risk. For patients who develop renal disease, renin-angiotensin system pharmacologic blockade is thought to potentially forestall progression to renal failure.17
Clinical follow-up consisting of renal function studies, ophthalmologic examination, and sensorineural hearing studies is recommended. Our patient received desmopressin before her hysterectomy, as is currently recommended, and did not experience any harmful or unusual bleeding during or after the procedure. However, she experienced an arrhythmic event, during which she had to be resuscitated. We believe that this happened due to an extreme vasovagal response to abdominal insufflation and was unrelated to her MHA. As indicated previously herein, patients with MYH9-RD generally do not experience significant bleeding events during an operation. Nevertheless, as in our patient, desmopressin may be indicated before a surgical procedure to potentiate platelet function. The use of eltrombopag, a thrombopoietin receptor analogue, should be used with caution in patients with MYH9-RD because the thrombocytopenia in these disorders is present due to ineffective platelet maturation while pro-platelet formation is still intact. In fact, rare patients with MYH9-RD have been reported to have postsurgical thromboses.18
In summary, MHA is an inherited disorder manifested by bleeding tendency, macrothrombocytopenia, and Döhle body–like inclusions. In rare instances, patients with this disorder may experience hearing loss, cataracts, and renal insufficiency or renal failure. The combination of macrothrombocytopenia and Döhle body–like inclusions on blood-smear review offers laboratory professionals a unique opportunity to diagnose this uncommon but possibly underrecognized inherited disorder. Clinical suspicion of MYH9-RD is further enhanced if the patient has a family history of minor bleeding tendency and has thrombocytopenia that is resistant to corticosteroid therapy. If MYH9-RD is suspected, a blood smear should be examined for Dohle body–like cytoplasmic inclusions within 2 to 4 hours because after that timeframe, inclusions dissipate and are more difficult to identify.7,18 A manual platelet count should be performed because automated instruments may underestimate the true platelet count, as large or giant platelets may be mistaken for RBC's or neutrophils. If there is a strong clinical suspicion for an MYH9-RD, NMM-IIA immunofluorescence studies should be performed in cases in which the blood-smear result is negative because that type of test is more sensitive and specific than alternative tests. If the results of blood smear or NMM-IIA immunofluorescence studies are positive, molecular studies of the MYH9 gene should be performed for confirmation of diagnosis and for evaluation of risk of extrahematopoietic manifestations.
Glossary
Abbreviations
- G1 P0
1 pregnancy but no live births
- CT
computed tomography
- CBC
complete blood count
- WBC
white blood cell
- RBC
red blood cell
- eGFR
estimated glomerular filtration rate
- PT
prothrombin time
- PTT
partial thromboplastin time
- ADP
adenosine diphosphate
- PCR
polymerase chain reaction
- MHA
May-Hegglin anomaly
- SS
Sebastian syndrome
- FS
Fechtner syndrome
- ES
Epstein syndrome
- NMM-IIA
nonmuscular myosin IIA
- mRNA
messenger RNA
- E1841K
glutamate-to-lysine change at amino acid 1841
- MYH9-RD
MYH9-related disorders
References
- 1.Althaus K, Greinacher A. MYH9-related platelet disorders. Semin Thromb Hemost. 2009;35:189-203. [DOI] [PubMed] [Google Scholar]
- 2.Min SY, Ahn HJ, Park WS, Kim JW. Successful renal transplantation in MYH9-related disorder with severe macrothrombocytpenia: first report in Korea. Transplant Proc. 2014;46:654-656. [DOI] [PubMed] [Google Scholar]
- 3.Skiver BM, Patel SB, Bose P. It is not always immune thrombocytopenia: a case of MYH9-related platelet disorder caused by a novel mutation. Eur J Haematol. 2013;91:191-192. [DOI] [PubMed] [Google Scholar]
- 4.Sirachainan N, Komwilaisak P, Kitamura K, et al. The first two cases of MYH9 disorders in Thailand: an international collaborative study. Ann Hematol. 2015;94:707-709. [DOI] [PubMed] [Google Scholar]
- 5.De Rocco D, Zieger B, Platokouki H, et al. MYH9-related disease: five novel mutations expanding the spectrum of causative mutations and confirming genotype/phenotype correlations. Eur J Med Genet. 2013;56(1):7-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kunishima S, Matsushita T, Kojima T, et al. Immunofluorescence analysis of neutrophil nonmuscle myosin heavy chain A in myh9 disorders: association of subcellular localization with MYH9 mutations. Lab Invest. 2003;83(1):115-122. [DOI] [PubMed] [Google Scholar]
- 7.Sun X, Wang Z, Yang H, et al. Clinical, pathological, and genetic analysis of ten patients with MYH9-related disease. Acta Haematol. 2013;129:106-113. [DOI] [PubMed] [Google Scholar]
- 8.Baluini C, Pecci A, Savoia A. Recent advances in the understanding and management of MYH9-related inherited thrombocytopenias. Br J Haematol. 2011;154:161-174. [DOI] [PubMed] [Google Scholar]
- 9.Kunishima S, Saito H. Advances in the understanding of MYH-9 disorders. Curr Opin Hematol. 2010;17:405-410. [DOI] [PubMed] [Google Scholar]
- 10.Poopak B, Rezvani H, DiFeo A, et al. The first report of homozygous May-Hegglin anomaly E1841K mutation. Eur J Haematol. 2011;86:357. [DOI] [PubMed] [Google Scholar]
- 11.Kunishima S, Hamaguchi M, Saito H. Differential expression of wild-type and mutant NMMHC-IIA polypeptides in blood cells suggests cell specific regulation mechanisms in MYH9 disorders. Blood 2008;111(6):3015-3023. [DOI] [PubMed] [Google Scholar]
- 12.Saposnik B, Binard S, Fenneteau O, et al. Mutation spectrum and genotype-phenotype correlations in a large French cohort of MYH9-related disorders. Mol Genet Genomic Med. 2014;2(4):297-312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pecci A, Panza E, Pujo-Moix N, et al. Position of nonmuscle myosin heavy chain IIA (NMMHC-IIA) mutations predicts the natural history of myh9-related disease. Hum Mutat. 2008;29(3):409-417. [DOI] [PubMed] [Google Scholar]
- 14.Landi D, Lockhart E, Miller S, et al. Report of a young girl with MYH9 mutation and review of the literature. J Pediatr Hematol Oncol. 2012;34(7):538-540. [DOI] [PubMed] [Google Scholar]
- 15.Savoia A, Pecci A. MYH9-related disorders. Gene Reviews. Genetics Home Reference online MYH-9. https://ghr.nlm.nih.gov/condition/myh9-related-disorder. Updated July 2015. Accessed May 8, 2016. [Google Scholar]
- 16.Bostrom MA, Freedman BI. The spectrum of MYH9-associated nephropathy. Clin J Am Soc Nephrol. 2010;5:1107-1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pecci A, Granata A, Fiore CE, et al. Renin-angiotensin system blockade is effective in reducing proteinuria of patients with progressive nephropathy caused by MYH9 mutations (Fechtner-Epstein syndrome). Nephrol Dial Transplant. 2008;23:2690-2692. [DOI] [PubMed] [Google Scholar]
- 18.Althaus K, Greinacher A. MYH9-related platelet disorders: Strategies for management and diagnosis. Transfus Med Hemother. 2010;37:260-267. [DOI] [PMC free article] [PubMed] [Google Scholar]