

Abstract
Processing bodies (PBs) are conserved cytoplasmic aggregations of translationally repressed mRNAs assembled with mRNA decay factors. The aggregation of mRNA-protein (mRNP) complexes into PBs involves interactions between low-complexity regions of protein components of the mRNPs. In Saccharomyces cerevisiae, the carboxy (C)-terminal Q/N-rich domain of the Lsm4 subunit of the Lsm1-7 complex plays an important role in PB formation, but the C-terminal domain of Lsm4 in most eukaryotes is an RGG domain rather than Q/N rich. Here we show that the Lsm4 RGG domain promotes PB accumulation in human cells and that symmetric dimethylation of arginines within the RGG domain stimulates this process. A mutant Lsm4 protein lacking the RGG domain failed to rescue PB formation in cells depleted of endogenous Lsm4, although this mutant protein retained the ability to assemble with Lsm1-7, associate with decapping factors, and promote mRNA decay and translational repression. Mutation of the symmetrically dimethylated arginines within the RGG domain impaired the ability of Lsm4 to promote PB accumulation. Depletion of PRMT5, the primary protein arginine methyltransferase responsible for symmetric arginine dimethylation, including Lsm4, resulted in loss of PBs. We also uncovered the histone acetyltransferase 1 (HAT1)-RBBP7 lysine acetylase complex as an interaction partner of the Lsm4 RGG domain but found no evidence of a role for this complex in PB metabolism. Together, our findings suggest a stimulatory role for posttranslational modifications in PB accumulation and raise the possibility that mRNP dynamics are posttranslationally regulated.
INTRODUCTION
Posttranscriptional gene regulation is critical for maintaining proper cellular function. The combination of proteins interacting with mRNAs making up the messenger ribonucleoproteins (mRNPs) determines the state of the mRNP; whether it is actively translated, targeted for mRNA decay, or stored in a translationally repressed state. These alternative fates of mRNPs can be regulated globally or in an mRNP-specific manner in response to intracellular or extracellular cues (1, 2).
Eukaryotic cytoplasmic mRNAs are bound on the 5′ cap by the translation initiation complex eIF4F and on the 3′ poly(A) tail by poly(A)-binding protein (PABP), which act synergistically to stimulate translation (3). A major pathway of mRNA turnover in eukaryotes initiates by mRNA deadenylation (4, 5). This is followed by decapping by the Dcp2-decapping complex and ultimately 5′-to-3′ exonucleolytic decay by Xrn1 (6). The Lsm1-7 complex associates with its cofactor Pat1 (known as PatL1 in humans) and the 3′ ends of deadenylated mRNAs and promotes decapping by a mechanism that is not fully understood but involves the association of Lsm1-7–Pat1 with decapping enhancers (7–11).
mRNAs that are targeted for deadenylation-initiated mRNA decay can accumulate in the cytoplasm in RNP granules known as processing bodies (PBs) (12). These granules are highly dynamic and sensitive to the level of mRNP intermediates undergoing deadenylation-initiated mRNA decay in the cell. PBs require RNA to assemble (13), and manipulations that prevent the accumulation of mRNPs targeted for deadenylation-initiated decay, such as trapping of mRNAs with ribosomes with translation elongation inhibitors (14, 15) or depletion of factors acting early in the pathway, result in loss of visible PBs (16). Contrasting with this, depletion of factors acting late in the pathway leads to the accumulation of decay intermediate mRNPs, which in general results in an increased size and/or number of PBs in cells (17).
In Saccharomyces cerevisiae, mRNP aggregation into PBs is known to involve the glutamine-asparagine (Q/N)-rich C-terminal domain of the Lsm4 subunit of the Lsm1-7 complex (18, 19). In human cells, depletion of Lsm4 also leads to a loss of PBs (20), but the C terminus of human Lsm4, as in most metazoans, is divergent from that of S. cerevisiae Lsm4 and consists of an arginine-glycine (R/G)-rich RGG domain rather than a Q/N-rich region. Recent studies have implicated low-complexity polypeptide regions of proteins, including R/G-rich regions, in protein polymerization and aggregation (21). The arginines of the human Lsm4 RGG domain have been shown to be symmetrically dimethylated (22), raising the intriguing question of whether posttranslational modifications (PTMs) play a role in PB formation. Here we show that the RGG domain of human Lsm4 stimulates PB formation. While the RGG domain does not appear to play a role in the formation of the Lsm1-7 complex, association with mRNA-decapping factors, translational repression, or mRNA decay, our findings indicate that the arginines of the RGG domain and their dimethylation promote PB formation. This is evidenced by the inhibition of PB accumulation when the dimethylated arginines of Lsm4 are mutated, as well as when symmetric arginine dimethylation is inhibited by depletion of the arginine methyltransferase PRMT5. Interestingly, we have also discovered a novel interaction of the Lsm4 RGG domain with histone acetyltransferase 1 (HAT1) and RBBP7, components of a cytoplasmic lysine acetyltransferase complex, leading to the possibility of a PTM network involved in mRNP regulation.
MATERIALS AND METHODS
Plasmid constructs.
The full coding sequence (CDS) of human Lsm4 was inserted into pcDNA5/FRT/TO (Invitrogen) encoding an N-terminal FLAG epitope and mutated at nucleotide 248 to CGTCGCGAAA (mutations are in bold) by site-directed mutagenesis (SDM) to confer small interfering RNA (siRNA) resistance. The ΔRGG construct was created by cleaving the pcDNA5/FRT/TO-Lsm4 plasmid with NotI, followed by religation; NotI cleaves at the end of the Lsm domain of the Lsm4 CDS and in the plasmid polylinker downstream of the Lsm4 CDS, leaving the first 270 nucleotides of the Lsm4 CDS. AGG and KGG mutant proteins were generated by inserting DNA oligonucleotides between the two NotI sites in the pcDNA5/FRT/TO-Lsm4 plasmid, with the DNA oligonucleotides encoding the final 49 amino acids of Lsm4 with all of the arginines mutated to alanines (AGG) or lysines (KGG). The CDS of human Hat1 was amplified from cDNA reverse transcribed with Superscript II (Invitrogen) from total RNA of HeLa cells isolated with TRIzol (Thermo Fisher) and inserted into pcDNA5/FRT/TO-FLAG and pcDNA3-FLAG (23) and mutated by SDM to create E187Q and W119A mutant proteins. Lsm4 and mutant forms, as well as the NMS2 sequence from pcFLAG-NMS2 (24), were cloned into pcDNA5/FRT/TO with a Gibson Assembly kit (New England BioLabs). The firefly and Renilla luciferase genes were subcloned from pGL2 and pRL (Promega) into pcDNA3 and pcDNA3-3xMS2, a plasmid containing six MS2 RNA stem-loops prior to the bovine growth hormone poly(A) site. The pcTET2 β-GAP plasmid has been previously described (25). pcTET2 β-c-fos was created by inserting the c-fos AU-rich element (ARE) following the β-globin CDS of plasmid pcTET2-βwtβ as described previously (26). Sequences are available upon request.
Cell culture.
HeLa or human embryonic kidney (HEK) 293 Flp-In T-REx (Thermo Fisher) cells were grown in Dulbecco's modified Eagle's medium (DMEM; Gibco) with 10% heat-inactivated fetal bovine serum (FBS). Stable cell lines were generated from Flp-In T-REx 293 cells according to the manufacturer's recommendations (Thermo Fisher). Briefly, cells were transfected with 1 ng of pcDNA5/FRT/TO containing the wild-type (WT) or mutant Lsm4, Lsm4-NMS2, or Hat1 CDS and 9 ng of pOG44 (Thermo Fisher) with Transit 293 transfection reagent (Mirus). At 48 h after transfection, cells were split to ∼10% confluence and grown in DMEM–10% FBS with 100 μg/ml hygromycin and 15 μg/ml blasticidin until visible colonies formed. Colonies were selected and tested by Western blot (WB) assays for protein expression by titration with tetracycline to achieve expression at a nearly endogenous level.
Antibodies.
Antibodies used for WB and immunofluorescence (IF) assays were obtained from the following sources and used at the concentrations indicated. Rabbit anti-Dcp1a (27) (IF assays, 1:100), anti-Edc3 (28) (WB assays, 1:1,000), and anti-Hedls (28) (WB assays, 1:1,000) antibodies were used as previously described. Human IC-6 serum (IF assays, 1:9,000), which recognizes the Edc4 and lamin proteins, was graciously provided by Ed Chan and Donald Bloch (29). Mouse anti-Hat1 (sc-376268; IF assays, 1:100), mouse anti-HuR (sc-5261; WB assays, 1:1,000), and goat anti-Hat1 (sc-8752; WB assays, 1:1,000) sera were obtained from Santa Cruz Biotechnology. Rabbit anti-Rbbp7 (ab109285; WB assays, 1:1,000) and anti-PABP (ab21060; WB assays, 1:1,000) sera were obtained from Abcam. Rabbit anti-DDX6 (A300-461A; WB assays, 1:1,000) serum was obtained from Bethyl Laboratories. Rabbit anti-Lsm4 (PA5-25731; WB assays, 1:500) serum was obtained from Thermo Fisher. Rabbit polyclonal antisera (Cocalico Biologicals Inc.) were raised against PatL1 (amino acids 1 to 240) and fused to an N-terminal glutathione S-transferase tag (WB assays, 1:1,000).
siRNAs and depletions.
For all depletions, cells were treated with siRNAs by using siLentFect (Bio-Rad) according to the manufacturer's protocol. For Lsm4 and PRMT5 depletions, cells were treated 96 and 48 h prior to harvesting at a 20 or 30 nM final siRNA concentration, respectively. Hat1-depleted cells were transfected once at a 20 nM final siRNA concentration 72 h prior to harvesting. All siRNAs were from GE Dharmacon and had UU overhangs with the following sequences: siLsm4, AGGAGGAGGUGGUGGCCAA; siPRMT5-1, GGCCAUCUAUAAAUGUCUG (30); siPRMT5-2, ACCGCUAUUGCACCUUGG (31); HAT1, GUUUAGAGUUUAUGAGCAU; RBBP7, AGAGAAGAAGUUGCUUAA.
Coimmunoprecipitation (co-IP) and immunoprecipitation (IP), followed by liquid chromatography-tandem mass spectrometry (LC-MS/MS) assays.
Ten-centimeter plates seeded with cells stably expressing Lsm4 were treated with tetracycline (WT, 7.5 ng/ml; ΔRGG mutant form, 100 ng/ml; AGG mutant form, 7 ng/ml; KGG mutant form, 7 ng/ml) to express exogenous Lsm4 and treated with siRNA to deplete the endogenous Lsm4 (see above). Cells were washed with phosphate-buffered saline (PBS; 136 mM NaCl, 2.6 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4 [pH 7.4]), lysed on the plate with 1 ml of isotonic lysis buffer (ILB; 10 mM Tris-HCl [pH 7.5], 150 nM NaCl, 2 mM EDTA, 0.1% Triton X-100, 1 mM phenylmethylsulfonyl fluoride, 2 μM aprotinin, 2 μM leupeptin, 1 ng/ml FLAG peptide [Sigma]), and incubated for 10 min on ice with 50 μg/ml RNase A. Cell debris was pelleted by centrifugation at 21,000 × g for 10 min at 4°C, and the supernatant was nutated with 50 μl of anti-FLAG M2–agarose beads (Sigma) for 4 h to overnight, depending on the experiment. The beads were washed eight times with NET-2 (50 mM Tris-HCl [pH 7.5], 150 mM NaCl, 0.05% Triton X-100) and resuspended in SDS loading buffer (100 mM Tris-HCl [pH 6.8], 4% SDS, 20% glycerol, 0.1% bromophenol blue, 200 mM dithiothreitol). For IP–LC-MS/MS assays, cells were treated as described above, with the following modifications. Two 15-cm plates of cells were used and lysed at 2.5 ml/plate with ILB containing 2.5 ng/ml FLAG peptide. Clarified lysates were incubated with 125 μl of anti-FLAG M2–agarose per plate and washed six times with NET-2. Beads were eluted three times with 125 μl of NET-2 containing 300 ng/ml FLAG peptide. Eluents were combined and precipitated by the addition of trichloroacetic acid (TCA) to 20% and incubation on ice for 1 h or overnight at −20°C. Protein was collected by centrifugation at 21,000 × g for 30 min at 4°C, washed with ice-cold 10% TCA, washed three times with ice-cold acetone, and centrifuged at 21,000 × g for 10 min at 4°C after each washing. LC-MS/MS was carried out as previously described (32).
Luciferase assays.
Cells stably expressing Lsm4-NMS2 were seeded into 3.5-cm dishes with 2 ml of DMEM–10% FBS and treated with tetracycline to induce nearly endogenous levels of Lsm4 (WT, 8 ng/ml; ΔRGG mutant form, 11 ng/ml; AGG mutant form, 10 ng/ml; KGG mutant form, 17 ng/ml). HEK 293 Flp-In T-Rex parental cells were also seeded into 3.5-cm dishes for transient transfection with pcDNA3-NMS2 and pcDNA3-RGG-NMS2 (ΔRGG mutant form only). At 24 h later, cells were transfected with 0.25 μg of pc-FLuc-3xMS2, 0.05 μg of pc-RLuc, 0.04 μg of pcDNA3-RGG-NMS2 (ΔRGG mutant form only), 0.04 μg of pcDNA3-NMS2 (none), and pcDNA3 to a total of 2.0 μg with TransIT HeLa (Mirus) according to the manufacturer's recommendations. Cells were lysed with 0.5 ml of passive lysis buffer (Promega) at room temperature with gentle rocking for 20 min. A 10-μl sample of lysate was analyzed for luciferase activity with Dual-Luciferase reporter assay (Promega) reagents and a NOVOstar plate reader.
Indirect IF assays.
HEK 293 T-REx cells stably expressing Lsm4 or Hat1 were seeded into 3.5-cm wells with 2 ml of DMEM–10% FBS containing a 12-mm coverslip pretreated with poly-d-lysine (Corning BioCoat), treated with siRNA and tetracycline to deplete the endogenous Lsm4, and rescued with exogenous Lsm4 as described above (for Hat1 overexpression, there was no siRNA treatment). Treated cells were fixed in 3 to 4% formaldehyde in PBS for 15 min at room temperature, permeabilized in 0.5% Triton X-100 in PBS containing 1% goat serum (GS; Life Technologies) for 15 min, and incubated with primary antibodies at the concentrations listed above in PBS–1% GS for 1 h. Cells were then washed two times for 5 min each time with PBS–1% GS, treated with secondary antibodies in PBS–1% GS for 1 h (anti-rabbit antibody–Texas Red, 1:1,000; Invitrogen), anti-mouse antibody–Alexa Fluor 488 (1:1,000; Thermo Fisher), and anti-human antibody–fluorescein isothiocyanate (1:500; Jackson ImmunoResearch), treated with 4′,6-diamidino-2-phenylindole (DAPI; 5 μg/ml in PBS–1% GS; Sigma) for 2 min, washed three times with PBS–1% GS, and washed once with distilled water. Coverslips were briefly air dried and mounted on slides. Images are from a Zeiss AX10 or 200m microscope with a 60× objective. Mutant Hat1 was overexpressed in HeLa cells grown in 3.5-cm wells transfected with 2 μg of pcDNA3-Hat1 E187Q or W199A vector with TransIT HeLa according to the manufacturer's recommendations (Mirus). At 24 h after transfection, cells were trypsinized and transferred to eight-well chamber slides (Thermo Scientific) for 24 h and treated as described above. PBs were counted with CellProfiler (33), and the project file is available upon request.
mRNA decay assays.
For ARE-mRNA decay assays, HeLa Tet-Off cells (Clontech) were grown in 3.5-cm wells with DMEM–10% FBS containing 50 ng/ml tetracycline. At 48 h prior to time point 0, cells were transfected with 0.6 μg of pcTET2-β-c-fos, 0.1 μg of pcDNA3-β-GAP, and 1.3 μg of pcDNA3 with TransIT HeLa (Mirus). At 6.5 h prior to time point 0, reporter mRNA expression was induced by changing the medium to fresh medium lacking tetracycline. At 30 min prior to time point 0, reporter expression was halted by changing to fresh medium containing 1 μg/ml tetracycline. Cells were then harvested in 1 ml of TRIzol (Thermo Fisher) at the time points indicated. For histone mRNA decay assays, HEK 293 T-REx cells stably expressing exogenous Lsm4 were grown in 3.5-cm wells. At 30 min prior to time point 0, cells were changed into fresh medium containing 5 mM hydroxyurea. Cells were then harvested in 1 ml of TRIzol at the time points indicated. Total RNA was prepared according to the manufacturer's recommendations and resolved in 1.1% agarose-formaldehyde gels, followed by Northern blotting, visualization on a Typhoon Trio (Amersham Biosciences), and quantification with ImageJ.
RESULTS
The RGG domain of human Lsm4 is required for PB formation.
To determine if the C-terminal RGG domain of human Lsm4 plays a role in mRNA regulation, we created stable HEK 293 T-REx cell lines expressing FLAG-tagged WT Lsm4 or Lsm4 with its RGG domain deleted (ΔRGG mutant form) (Fig. 1A) at nearly endogenous levels under the control of a tetracycline-regulated promoter. We generated silent mutations in the exogenous Lsm4 coding region to allow for complementation assays in which endogenous Lsm4 expression could be depleted by an siRNA that specifically targets the endogenous Lsm4 mRNA while leaving exogenous Lsm4 expression unperturbed (Fig. 1B). To determine if the Lsm4 C terminus is important for PB formation, we depleted cells of endogenous Lsm4, expressed WT or ΔRGG mutant Lsm4 at nearly endogenous levels, and visualized PBs by IF assay using the endogenous PB component Dcp1a as a marker (Fig. 1C; quantified in Fig. 1D). As expected, Lsm4 depletion led to loss of Dcp1a foci, which was rescued by the expression of exogenous WT Lsm4. In contrast, ΔRGG mutant Lsm4 failed to rescue Dcp1a localization to visible foci. This observation, supported by the monitoring of other PB markers (shown below), suggests that the C-terminal RGG domain of human Lsm4 plays an important role in PB accumulation.
FIG 1.

The RGG domain of Lsm4 is required for visible PBs. (A) Schematic showing WT and ΔRGG mutant Lsm4 proteins. aa, amino acids. (B) WB assay showing depletion of endogenous Lsm4 and expression of exogenous WT Lsm4 at nearly endogenous levels in stable HEK 293 T-REx cells. The first three lanes (from the left) are loaded with 100, 33, and 11% of the total protein in the last two lanes. (C) HEK 293 T-REx cells treated with siRNA against GFP (control) or Lsm4, induced to express FLAG (FL)-tagged WT or ΔRGG mutant Lsm4, and stained for Dcp1a (red). DAPI staining is blue. (D) Quantification of PB numbers from three independent indirect IF experiments. Error bars represent standard deviations. *, P < 0.05 (Student's paired two-tailed t test).
The RGG domain of Lsm4 is not required for Lsm4 assembly with Lsm1-7 and decapping factors.
A possible explanation for the failure of ΔRGG mutant Lsm4 to support PB accumulation is that it fails to associate with the larger Lsm complex. To determine if this was the case, we performed IP of Lsm4 and ΔRGG mutant Lsm4, followed by LC-MS/MS. IPs were performed in the presence of RNase A to specifically monitor for interactions that are independent of RNA. These assays showed similar levels of association of WT and ΔRGG mutant Lsm4 with Lsm1-7 subunits (Fig. 2A; see Table S1 in the supplemental material; Lsm5 was below the level of detection in either sample). Similarly, IP–LC-MS/MS assays revealed that the C-terminal RGG domain is not required for the association of Lsm4 with decapping enhancers PatL1, DDX6, or Edc3 (Fig. 2B). These observations suggest that the loss of PB accumulation associated with ΔRGG mutant Lsm4 is not a result of defects in Lsm1-7 complex formation or decapping factor association.
FIG 2.

The RGG domain of Lsm4 is required for interaction with HAT1/RBBP7 but is not necessary for association with Lsm1-7 components or decapping factors. The graphs show the numbers of peptides detected per 1,000 total peptides coprecipitating with FLAG-tagged WT or ΔRGG mutant Lsm4 for Lsm1-7 complex members (A), decapping factors (B), or HAT1 complex members (C). None is a negative-control FLAG (FL) IP performed with parental cells containing no FLAG-Lsm4 protein. See Table S1 in the supplemental material for the primary data.
While the RGG domain of Lsm4 was not important for the association of Lsm4 with Lsm1-7 or decapping factors, our LC-MS/MS assays did reveal two novel protein interactions, which showed an RGG-dependent association with Lsm4: HAT1 and RBBP7 (Fig. 2C). This was intriguing, given that HAT1 and RBBP7 are known to form a lysine acetylase complex that functions in both the cytoplasm and the nucleus (34) and the mouse orthologs were previously reported to inhibit the formation of PB-like mRNP granules known as chromatoid bodies in mouse germ cells (35). However, we found no defect in PB accumulation upon the codepletion of HAT1 and RBBP7 or the overexpression of WT or E187Q and W119A catalytic mutant forms of HAT1 (36), suggesting that HAT1 does not play a limiting role in the accumulation of PBs in HeLa cells (data not shown).
Lsm4 represses translation and activates mRNA decay independently of the RGG domain.
The inability of ΔRGG mutant Lsm4 to support the accumulation of visible PBs could reflect a role for the RGG domain in translation repression or mRNA decay. We therefore tested the ability of ΔRGG mutant Lsm4 to complement Lsm4 depletion in these processes. The Lsm1-7 complex is limiting for the degradation of mRNAs containing AREs (37) and of replication-dependent histone mRNAs (38). As expected, depletion of Lsm4 caused an increase in the half-life of a β-globin reporter mRNA containing the ARE from c-fos mRNA in the 3′ untranslated region (UTR), which was rescued by the exogenous expression of WT Lsm4 (Fig. 3A). ΔRGG mutant Lsm4 also fully rescued the degradation of the reporter mRNA, suggesting that the Lsm4 RGG domain is not limiting for ARE-containing mRNA degradation. Lsm4 with 5 or 10 C-terminal amino acids deleted was previously observed to impair histone mRNA degradation (38). However, we found that the WT and ΔRGG mutant Lsm4 proteins both completely complement the defect of depleting endogenous Lsm4 in the degradation of histone H2A mRNA induced by hydroxyurea treatment (Fig. 3B). Thus, the Lsm4 RGG domain is not limiting for the degradation of histone H2A mRNA and deletion of the entire RGG domain appears to rescue the defect associated with deleting the C-terminal 5 or 10 amino acids only (38).
FIG 3.
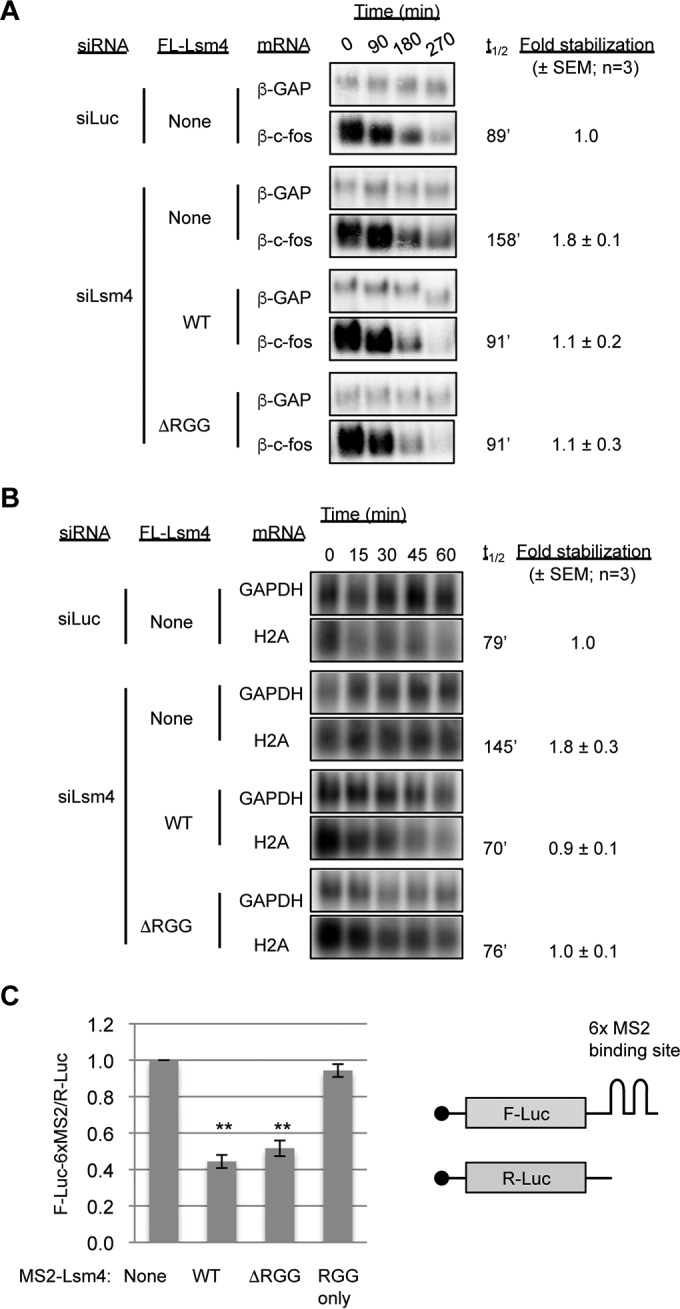
The RGG domain of Lsm4 is not required for mRNA decay or translational repression. (A) Northern blot assays showing the decay of an ARE-containing reporter mRNA (β-c-fos) in HeLa Tet-off cells treated with the siRNAs indicated and transiently expressing the Lsm4 proteins indicated. β-c-fos half-lives (t1/2) were calculated with the constitutively transcribed β-GAP internal control mRNA for normalization. Fold stabilization was calculated relative to the siLuc control condition in three experiments, and the standard errors of the means are indicated. (B) Northern blot assays showing the decay of endogenous H2A mRNA induced by treatment with 5 mM hydroxyurea in HEK 293 T-REx cells treated with the siRNAs indicated and stably expressing the Lsm4 proteins indicated. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) mRNA served as a normalization control. (C) Luciferase luminescence assays of cells transiently transfected with plasmids encoding the MS2 fusion proteins indicated, as well as a firefly luciferase reporter with six MS2 coat protein binding sites in its 3′ UTR (F-Luc-6xMS2) and a Renilla luciferase reporter (R-Luc) as an internal control. F-Luc-6xMS2 was normalized to R-Luc, and all samples were normalized to cells transfected with MS2 and the reporters alone. Error bars represent the standard errors of the means. **, P < 0.01 (Student's paired two-tailed t test).
In yeast, several proteins containing RGG domains were shown to interact with eIF4G and repress translation initiation (39). To determine if the Lsm4 RGG domain could be acting in a similar manner, we conducted a tethering assay with MS2 coat protein-Lsm4 fusion proteins and a firefly luciferase reporter mRNA containing a minimal 3′ UTR composed of six MS2 coat protein binding sites. When the MS2-Lsm4 fusion proteins were coexpressed with the luciferase reporter, the WT and ΔRGG mutant Lsm4 proteins were able to repress the luciferase reporter to similar extents, compared to the negative control of expressing the MS2 coat protein alone (Fig. 3C). Expression of an MS2 coat protein fused to the Lsm4 RGG domain alone had no effect on luciferase expression. Collectively, these observations suggest that the RGG domain is neither necessary nor sufficient for mRNA repression by Lsm4.
Arginines of the Lsm4 RGG domain are important for PB accumulation.
R/G-containing domains have been identified as targets for arginine methylation, and the RGG domain of Lsm4 specifically has been identified as a target for symmetric arginine dimethylation (22). To test the importance of the arginines of Lsm4 undergoing symmetric dimethylation in PB accumulation, we created mutant forms of Lsm4 where all of the arginines that were previously demonstrated to be methylated were mutated to either alanines (AGG mutant Lsm4) or lysines (KGG mutant Lsm4). As expected, these mutations prevented the detection of Lsm4 by an antibody specific for symmetric dimethylated arginines that recognizes WT Lsm4 (Sym10) (Fig. 4A). Similar to ΔRGG mutant Lsm4, the AGG and KGG mutant Lsm4 proteins expressed at close-to-endogenous Lsm4 levels supported interaction with Lsm1-7 components and decapping factors, as monitored by co-IP (Fig. 4A and B) and IP–LC-MS/MS (Fig. 4C and D). Moreover, both the AGG and KGG mutant Lsm4 proteins showed impaired association with HAT1 and RBBP7 (Fig. 4D and E), although KGG mutant Lsm4 was only partially impaired for HAT1-RBBP7 association. The AGG and KGG mutant Lsm4 proteins could fully complement the depletion of endogenous Lsm4 in the degradation of the ARE reporter and histone H2A mRNAs tested (Fig. 5A and B) and in the repression of tethered luciferase mRNA (Fig. 5C and D). When monitoring for effects on PBs of complementing endogenous Lsm4 with the mutant Lsm4 proteins expressed at nearly endogenous levels, Lsm4 AGG was unable to support PB accumulation, as monitored with three different PB markers (Fig. 6). KGG mutant Lsm4 also showed an impaired ability to support PB accumulation compared with that of WT Lsm4, although one marker (Dcp1a) indicated partial PB rescue (Fig. 6). Thus, the arginines of the Lsm4 RGG domain that undergo symmetric dimethylation play an important role in PB accumulation.
FIG 4.

Arginines in the Lsm4 RGG domain are required for interaction with Hat1/RBBP7 but dispensable for association with Lsm1-7 and decapping factors. (A, B) WB assay showing Lsm1 and decapping factors coimmunoprecipitating with WT and mutant Lsm4. Input samples are shown on the left. Sym10 is specific for symmetrically dimethylated arginines, and Asym is specific for asymmetrically dimethylated arginines. The asterisk indicates cross-reaction with IgG. (C, D) Graphs showing the numbers of peptides detected per 1,000 total peptides from Lsm factors, decapping factors, and Hat1 complex members coprecipitating with WT or mutant Lsm4, as indicated. (E) WB assay showing HAT1 complex members coimmunoprecipitating with WT and mutant Lsm4.
FIG 5.

Arginines in the Lsm4 RGG domain are dispensable for mRNA repression and degradation. (A) Graphs showing histone H2A and β-c-fos mRNA half-lives in the presence of WT and mutant Lsm4 monitored as described in the legend to Fig. 3A and B. Error bars represent the standard errors of the means. (B) WB assay showing nearly endogenous expression of FL-Lsm4 proteins in the stable cell lines used in panel A. (C) Graphs showing luciferase activity from tethering of MS2-Lsm4 WT and mutant proteins monitored as described in the legend to Fig. 3C. Error bars represent the standard errors of the means. **, P < 0.01 (Student's paired two-tailed t test). (D) WB assay showing even expression of FL-MS2-Lsm4 proteins as detected by the FLAG antibody. The asterisk indicates a cross-reacting band.
FIG 6.
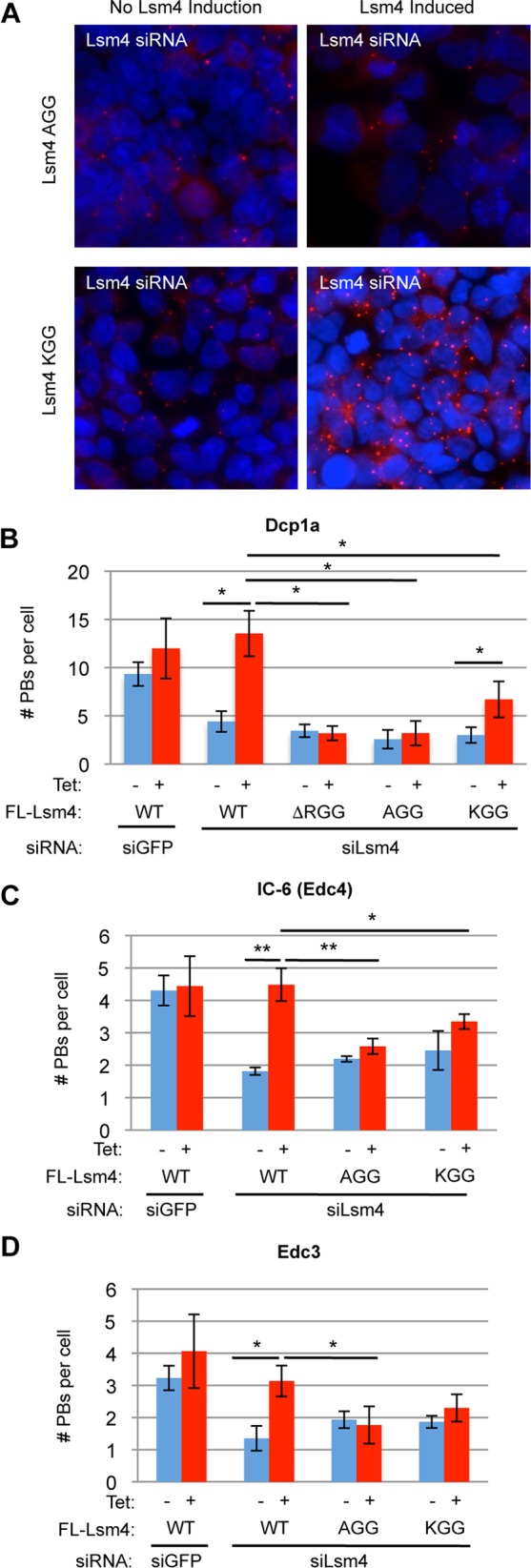
Arginines in the Lsm4 RGG domain are required for PB accumulation. (A) Indirect IF assays for Dcp1a (red) in HEK 293 T-REx cells treated with an siRNA against Lsm4 and induced to express the AGG and KGG mutant Lsm4 proteins. DAPI staining is shown in blue to mark nuclei. (B to D) Quantification of PBs by the PB markers indicated in three independent experiments. Error bars represent standard deviations. **, P < 0.01; *, P < 0.05 (Student's paired two-tailed t test).
PRMT5 depletion disrupts PB accumulation.
To further explore whether symmetric arginine dimethylation plays a role in PB formation, we tested the effect of inhibiting the enzyme responsible for this activity. Indeed, treatment of HeLa cells with the global methylation inhibitor AMI-1 for 1 h resulted in less PB accumulation than in mock-treated cells (data not shown). Symmetric arginine dimethylation, as found in Lsm4, is carried out by the type II PRMTs PRMT5 and PRMT7 (40). A study quantifying the HeLa cell proteome showed PRMT5 to be expressed 20-fold more strongly than PRMT7 (41); we therefore tested the effect of PRMT5 depletion on PB accumulation. Depletion of PRMT5 caused decreased symmetric arginine dimethylation of WT Lsm4, as monitored with the Sym10 antibody (Fig. 7A). When HeLa cells were depleted of PRMT5 with two different siRNAs, visible PBs were strongly reduced, as monitored with two different PB markers, compared to those in cells treated with a control siRNA (Fig. 7B to D). This demonstrates that PRMT5 is important for PB accumulation and, together with the Lsm4 mutational studies, suggests that symmetric arginine dimethylation of the Lsm4 C-terminal RGG domain contributes to this effect.
FIG 7.

PRMT5 is required for PB formation. (A) WB assay monitoring symmetric arginine dimethylation (with Sym10) in FLAG-tagged WT Lsm4 and KGG immunoisolated from cells treated with siGFP and siPRMT5-1 siRNAs. (B) Indirect IF assays for Dcp1a (red) and Edc4 (IC-6; green) in HeLa cells treated with an siRNA against luciferase (control) or PRMT5 (two different siRNAs). DAPI staining is shown in blue to mark nuclei. Merged images are on the right. (C) Quantification of PBs for Dcp1a and Edc4. Error bars represent standard deviations. (D) WB assay showing PRMT5 depletion with HAT1 serving as a loading control. The first three lanes (from the left) are loaded with 100, 33, and 11% of the total protein in the last two lanes.
DISCUSSION
In this study, we demonstrate that the C-terminal RGG domain of Lsm4 promotes the accumulation of PBs in human cells. This is evidenced by the impaired ability of Lsm4 with the RGG domain deleted or mutated to support PB accumulation (Fig. 1 and 6), despite retaining the ability to associate with Lsm1-7 and decapping factors (Fig. 2 and 4). Two lines of evidence suggest that symmetric arginine dimethylation of the Lsm4 RGG domain stimulates PB accumulation. First, depletion of PRMT5 results in reduced Lsm4 arginine symmetric dimethylation and loss of PBs (Fig. 7). Second, mutant versions of Lsm4 in which all of the arginines in the RGG domain that undergo symmetric dimethylation are mutated to alanines or lysines are impaired in PB accumulation (Fig. 6). The observation that KGG mutant Lsm4 modestly rescues PB formation, as monitored by one marker, suggests a possible contribution from the positive charge of the arginines of the RGG domain to PB accumulation and that methylation of the Lsm4 arginines stimulates PB accumulation but might not be essential. PB components other than Lsm4 might also undergo methylation to stimulate PB accumulation, which could contribute to the dramatic effect of PRMT5 depletion on PB accumulation (Fig. 7); for example, the PB component Lsm14A (also known as RAP55) also contains a C-terminal RGG domain, a likely target of PRMT5-mediated methylation.
What is the molecular mechanism by which the RGG domain of Lsm4 stimulates PB accumulation? PB formation requires the accumulation of translationally repressed mRNAs associated with mRNA decay machinery (17). Several yeast RGG domain proteins were shown to inhibit translation in a manner involving interaction between the RGG domain and eIF4G (39). However, we found no evidence that the human Lsm4 RGG domain plays a role in translation repression, as monitored by tethered-luciferase assays (Fig. 3C and 5C). Additionally, the RGG domain is not limiting for the decay of an ARE reporter mRNA or of histone H2A mRNA (Fig. 3A and B and 5A). This is congruent with the observation that the RGG domain does not affect the interaction of Lsm4 with known decay factors, as evidenced by co-IP and IP–LC-MS/MS experiments (Fig. 2 and 4). Thus, the most likely role of the Lsm4 RGG domain in PB accumulation is that it directly participates in the aggregation of repressed mRNPs. Consistent with this, RGG domains of human RNA-binding proteins (21, 42) and of trypanosome Scd6 (39) (a homolog of human LSM14A) have previously been implicated in granule formation, but it is unknown whether arginine methylation plays a role in those cases. Interestingly, a previous study presented evidence that repeats of (G/S)Y(G/S) can promote the formation of hydrogels that resemble dynamic RNA granules (21). An intriguing possibility is that GRG repeats have the same property by a process that is stimulated by arginine dimethylation. A similar role in PB aggregation was previously identified for the C-terminal domain of budding yeast Lsm4, which contains a Q/N-rich prion-like primary sequence different from the human Lsm4 C-terminal RGG domain (18). Thus, the functionality of the Lsm4 C-terminal domain in PB accumulation may be evolutionarily conserved, despite a great degree of variation in the primary sequence. However, in contrast to observations of the budding yeast Lsm4 C-terminal Q/N domain forming foci when fused to green fluorescent protein (GFP) (19), we observed no granule formation with a GFP-human Lsm4 RGG fusion protein (data not shown), suggesting that in the case of human Lsm4, the RGG domain is not sufficient for protein aggregation.
There is increasing evidence that mRNP modification plays an important role in mRNA regulation (43). Posttranscriptional modifications (PTMs) of mRNAs, including nucleoside modification and mRNA tailing, have been found to impact mRNA translation and decay (44, 45). In addition, PTMs are abundant in protein components of mRNPs, but to date, only a few have been found to play regulatory roles (46). Interestingly, our co-IP and IP–LC-MS/MS experiments revealed an RGG-dependent association of Lsm4 with a member of the HAT1-RBBP7 lysine acetylase complex (Fig. 2 and 4), which has previously been implicated in chromatoid body formation in murine germ line cells (35). The observation of a correlation between the association of Lsm4 with the HAT1-RBBP7 complex and the ability to promote PB accumulation suggests a link between these events, although HAT1 and RBBP7 did not appear to be limiting for PB accumulation under the conditions tested here. The identification of an interaction of Lsm4 with PTM machinery via posttranslationally modified residues raises the exciting possibility that mRNP dynamics are regulated by a network of PTMs, similar to the regulation of chromatin dynamics through histone PTMs.
Supplementary Material
ACKNOWLEDGMENTS
We thank Marv Fritzler and Ed Chan for human IC-6 serum. Tilmann Achsel is thanked for anti-Lsm1 and anti-Lsm4 antibodies.
This work was supported by the a New Scholar award from the Ellison Medical Foundation to E.J.B., by National Institutes of Health grants R01 GM077243 to J.L.-A., DP2 GM119132 to E.J.B., F31 GM106655 to M.A.-L., and F31 GM083624 to J.D., and by grants from the Alfred Benzon Foundation, Copenhagen, Denmark, and The Lundbeck Foundation (R140-2013-13425) to C.K.D.
Funding Statement
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.01102-15.
REFERENCES
- 1.Guhaniyogi J, Brewer G. 2001. Regulation of mRNA stability in mammalian cells. Gene 265:11–23 . doi: 10.1016/S0378-1119(01)00350-X. [DOI] [PubMed] [Google Scholar]
- 2.Parker R, Sheth U. 2007. P bodies and the control of mRNA translation and degradation. Mol Cell 25:635–646. doi: 10.1016/j.molcel.2007.02.011. [DOI] [PubMed] [Google Scholar]
- 3.Wells SE, Hillner PE, Vale RD, Sachs AB. 1998. Circularization of mRNA by eukaryotic translation initiation factors. Mol Cell 2:135–140. doi: 10.1016/S1097-2765(00)80122-7. [DOI] [PubMed] [Google Scholar]
- 4.Chen C-YA, Shyu A-B. 2011. Mechanisms of deadenylation-dependent decay. Wiley Interdiscip Rev RNA 2:167–183. doi: 10.1002/wrna.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huch S, Nissan T. 2014. Interrelations between translation and general mRNA degradation in yeast. Wiley Interdiscip Rev RNA 5:747–763. doi: 10.1002/wrna.1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Beelman CA, Parker R. 1995. Degradation of mRNA in eukaryotes. Cell 81:179–183. doi: 10.1016/0092-8674(95)90326-7. [DOI] [PubMed] [Google Scholar]
- 7.Ozgur S, Chekulaeva M, Stoecklin G. 2010. Human Pat1b connects deadenylation with mRNA decapping and controls the assembly of processing bodies. Mol Cell Biol 30:4308–4323. doi: 10.1128/MCB.00429-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tharun S, Parker R. 2001. Targeting an mRNA for decapping: displacement of translation factors and association of the Lsm1p-7p complex on deadenylated yeast mRNAs. Mol Cell 8:1075–1083. doi: 10.1016/S1097-2765(01)00395-1. [DOI] [PubMed] [Google Scholar]
- 9.Song M-G, Kiledjian M. 2007. 3′ terminal oligo U-tract-mediated stimulation of decapping. RNA 13:2356–2365. doi: 10.1261/rna.765807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu D, Muhlrad D, Bowler MW, Jiang S, Liu Z, Parker R, Song H. 2014. Lsm2 and Lsm3 bridge the interaction of the Lsm1-7 complex with Pat1 for decapping activation. Cell Res 24:233–246. doi: 10.1038/cr.2013.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tharun S. 2009. Lsm1-7–Pat1 complex: a link between 3′ and 5′-ends in mRNA decay? RNA Biol 6:228–232. doi: 10.4161/rna.6.3.8282. [DOI] [PubMed] [Google Scholar]
- 12.Erickson SL, Lykke-Andersen J. 2011. Cytoplasmic mRNP granules at a glance. J Cell Sci 124:293–297. doi: 10.1242/jcs.072140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Teixeira D, Sheth U, Valencia-Sanchez Ma Brengues M, Parker R. 2005. Processing bodies require RNA for assembly and contain nontranslating mRNAs. RNA 11:371–382. doi: 10.1261/rna.7258505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sheth U, Parker R. 2003. Decapping and decay of messenger RNA occur in cytoplasmic processing bodies. Science 300:805–808. doi: 10.1126/science.1082320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kedersha N, Anderson P. 2007. Mammalian stress granules and processing bodies. Methods Enzymol 431:61–81. doi: 10.1016/S0076-6879(07)31005-7. [DOI] [PubMed] [Google Scholar]
- 16.Zheng D, Ezzeddine N, Chen C-YA, Zhu W, He X, Shyu A-B. 2008. Deadenylation is prerequisite for P-body formation and mRNA decay in mammalian cells. J Cell Biol 182:89–101. doi: 10.1083/jcb.200801196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Franks TM, Lykke-Andersen J. 2008. The control of mRNA decapping and P-body formation. Mol Cell 32:605–615. doi: 10.1016/j.molcel.2008.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Decker CJ, Teixeira D, Parker R. 2007. Edc3p and a glutamine/asparagine-rich domain of Lsm4p function in processing body assembly in Saccharomyces cerevisiae. J Cell Biol 179:437–449. doi: 10.1083/jcb.200704147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reijns MAM, Alexander RD, Spiller MP, Beggs JD. 2008. A role for Q/N-rich aggregation-prone regions in P-body localization. J Cell Sci 121:2463–2472. doi: 10.1242/jcs.024976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kedersha N, Stoecklin G, Ayodele M, Yacono P, Lykke-Andersen J, Fritzler MJ, Scheuner D, Kaufman RJ, Golan DE, Anderson P. 2005. Stress granules and processing bodies are dynamically linked sites of mRNP remodeling. J Cell Biol 169:871–884. doi: 10.1083/jcb.200502088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kato M, Han TW, Xie S, Shi K, Du X, Wu LC, Mirzaei H, Goldsmith EJ, Longgood J, Pei J, Grishin NV, Frantz DE, Schneider JW, Chen S, Li L, Sawaya MR, Eisenberg D, Tycko R, McKnight SL. 2012. Cell-free formation of RNA granules: low complexity sequence domains form dynamic fibers within hydrogels. Cell 149:753–767. doi: 10.1016/j.cell.2012.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brahms H, Meheus L, de Brabandere V, Fischer U, Lührmann R. 2001. Symmetrical dimethylation of arginine residues in spliceosomal Sm protein B/B' and the Sm-like protein LSm4, and their interaction with the SMN protein. RNA 7:1531–1542. doi: 10.1017/S135583820101442X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lykke-Andersen J. 2002. Identification of a human decapping complex associated with hUpf proteins in nonsense-mediated decay. Mol Cell Biol 22:8114–8121. doi: 10.1128/MCB.22.23.8114-8121.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lykke-Andersen J, Shu MD, Steitz JA. 2000. Human Upf proteins target an mRNA for nonsense-mediated decay when bound downstream of a termination codon. Cell 103:1121–1131. doi: 10.1016/S0092-8674(00)00214-2. [DOI] [PubMed] [Google Scholar]
- 25.Franks TM, Lykke-Andersen J. 2007. TTP and BRF proteins nucleate processing body formation to silence mRNAs with AU-rich elements. Genes Dev 21:719–735. doi: 10.1101/gad.1494707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Singh G, Rebbapragada I, Lykke-Andersen J. 2008. A competition between stimulators and antagonists of Upf complex recruitment governs human nonsense-mediated mRNA decay. PLoS Biol 6:e111. doi: 10.1371/journal.pbio.0060111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lykke-Andersen J, Wagner E. 2005. Recruitment and activation of mRNA decay enzymes by two ARE-mediated decay activation domains in the proteins TTP and BRF-1. Genes Dev 19:351–361. doi: 10.1101/gad.1282305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fenger-Grøn M, Fillman C, Norrild B, Lykke-Andersen J. 2005. Multiple processing body factors and the ARE binding protein TTP activate mRNA decapping. Mol Cell 20:905–915. doi: 10.1016/j.molcel.2005.10.031. [DOI] [PubMed] [Google Scholar]
- 29.Franks TM, Singh G, Lykke-Andersen J. 2010. Upf1 ATPase-dependent mRNP disassembly is required for completion of nonsense-mediated mRNA decay. Cell 143:938–950. doi: 10.1016/j.cell.2010.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gonsalvez GB, Tian L, Ospina JK, Boisvert F-M, Lamond AI, Matera AG. 2007. Two distinct arginine methyltransferases are required for biogenesis of Sm-class ribonucleoproteins. J Cell Biol 178:733–740. doi: 10.1083/jcb.200702147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Scoumanne A, Zhang J, Chen X. 2009. PRMT5 is required for cell-cycle progression and p53 tumor suppressor function. Nucleic Acids Res 37:4965–4976. doi: 10.1093/nar/gkp516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Erickson SL, Corpuz EO, Maloy JP, Fillman C, Webb K, Bennett EJ, Lykke-Andersen J. 2015. Competition between decapping complex formation and ubiquitin-mediated proteasomal degradation controls human Dcp2 decapping activity. Mol Cell Biol 35:2144–2153. doi: 10.1128/MCB.01517-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Carpenter AE, Jones TR, Lamprecht MR, Clarke C, Kang IH, Friman O, Guertin DA, Chang JH, Lindquist RA, Moffat J, Golland P, Sabatini DM. 2006. CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol 7:R100. doi: 10.1186/gb-2006-7-10-r100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Parthun MR, Widom J, Gottschling DE. 1996. The major cytoplasmic histone acetyltransferase in yeast: links to chromatin replication and histone metabolism. Cell 87:85–94. doi: 10.1016/S0092-8674(00)81325-2. [DOI] [PubMed] [Google Scholar]
- 35.Nagamori I, Cruickshank VA, Sassone-Corsi P. 2011. Regulation of an RNA granule during spermatogenesis: acetylation of MVH in the chromatoid body of germ cells. J Cell Sci 124:4346–4355. doi: 10.1242/jcs.096461. [DOI] [PubMed] [Google Scholar]
- 36.Wu H, Moshkina N, Min J, Zeng H, Joshua J, Zhou M-M, Plotnikov AN. 2012. Structural basis for substrate specificity and catalysis of human histone acetyltransferase 1. Proc Natl Acad Sci U S A 109:8925–8930. doi: 10.1073/pnas.1114117109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stoecklin G, Mayo T, Anderson P. 2006. ARE-mRNA degradation requires the 5′-3′ decay pathway. EMBO Rep 7:72–77. doi: 10.1038/sj.embor.7400572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lyons SM, Ricciardi AS, Guo AY, Kambach C, Marzluff WF. 2014. The C-terminal extension of Lsm4 interacts directly with the 3′ end of the histone mRNP and is required for efficient histone mRNA degradation. RNA 20:88–102. doi: 10.1261/rna.042531.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rajyaguru P, She M, Parker R. 2012. Scd6 targets eIF4G to repress translation: RGG motif proteins as a class of eIF4G-binding proteins. Mol Cell 45:244–254. doi: 10.1016/j.molcel.2011.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bedford MT. 2007. Arginine methylation at a glance. J Cell Sci 120:4243–4246. doi: 10.1242/jcs.019885. [DOI] [PubMed] [Google Scholar]
- 41.Nagaraj N, Wisniewski JR, Geiger T, Cox J, Kircher M, Kelso J, Pääbo S, Mann M. 2011. Deep proteome and transcriptome mapping of a human cancer cell line. Mol Syst Biol 7:548. doi: 10.1038/msb.2011.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang W-H, Yu JH, Gulick T, Bloch KD, Bloch DB. 2006. RNA-associated protein 55 (RAP55) localizes to mRNA processing bodies and stress granules. RNA 12:547–554. doi: 10.1261/rna.2302706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee SR, Lykke-Andersen J. 2013. Emerging roles for ribonucleoprotein modification and remodeling in controlling RNA fate. Trends Cell Biol 23:504–510. doi: 10.1016/j.tcb.2013.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang X, Lu Z, Gomez A, Hon GC, Yue Y, Han D, Fu Y, Parisien M, Dai Q, Jia G, Ren B, Pan T, He C. 2014. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature 505:117–120. doi: 10.1038/nature12730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lim J, Ha M, Chang H, Kwon SC, Simanshu DK, Patel DJ, Kim VN. 2014. Uridylation by TUT4 and TUT7 marks mRNA for degradation. Cell 159:1365–1376. doi: 10.1016/j.cell.2014.10.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brook M, McCracken L, Reddington JP, Lu Z-L, Morrice NA, Gray NK. 2012. The multifunctional poly(A)-binding protein (PABP) 1 is subject to extensive dynamic post-translational modification, which molecular modelling suggests plays an important role in co-ordinating its activities. Biochem J 441:803–812. doi: 10.1042/BJ20111474. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.