

Abstract
Background
Ethanol exposure during brain development causes profound damages to the central nervous system (CNS). The underlying cellular/molecular mechanisms remain unclear. The endoplasmic reticulum (ER) is involved in posttranslational protein processing and transport. The accumulation of unfolded or misfolded proteins in the ER lumen triggers ER stress, which is characterized by translational attenuation, synthesis of ER chaperone proteins, and activation of transcription factors. Sustained ER stress ultimately leads to cell death. ER stress is implicated in various neurodegenerative processes.
Methods
Using a third trimester equivalent mouse model of ethanol exposure, we tested the hypothesis that ethanol induces ER stress in the developing brain. Seven-day-old C57BL/6 mice were acutely exposed to ethanol by subcutaneous injection and the expression of ER stress-inducible proteins (ERSIPs) and signaling pathways associated with ER stress were examined.
Results
Ethanol exposure significantly increased the expression of ERSIPs and activated signaling pathways associated with ER stress; these include ATF6, CHOP/GADD153, GRP78, and mesencephalic astrocyte-derived neurotrophic factor as well as the phosphorylation of IRE1α, eIF2α, PERK, and PKR. The ethanol-induced increase in ERSIPs occurred within 4 hours of ethanol injection, and levels of some ERSIPs remained elevated after 24 hours of ethanol exposure. Ethanol-induced increase in phosphorylated eIF2α, caspase-12, and CHOP was distributed in neurons of specific areas of the cerebral cortex, hippocampus, and thalamus.
Conclusions
Our finding indicates that ethanol induces ER stress in immature neurons, providing novel insight into ethanol’s detrimental effect on the developing CNS.
Keywords: Cell Signaling, Development, Fetal Alcohol Syndrome, Neurodegeneration, Oxidative Stress
Maternal alcohol consumption during pregnancy causes fetal alcohol spectrum disorders (FASDs). The central nervous system (CNS) is particularly susceptible to alcohol exposure and prenatal alcohol exposure induces a spectrum of structural brain anomalies and neuro-cognitive/behavioral disabilities (Riley and McGee, 2005). fetal alcohol syndrome, the most severe form of FASD, is the leading nonhereditary cause of mental retardation (May and Gossage, 2001; Stratton et al., 1996). Ethanol exposure disrupts a variety of developmental events in experimental FASD models; these include neurogenesis, neuron survival, cell migration, cell adhesion, axon outgrowth, synapse formation, and neurotransmitter function (Bearer, 2001; Goodlett et al., 2005; Hoffman et al., 2008; Ikonomidou et al., 2001; Kumada et al., 2007; Luo, 2010; Luo and Miller, 1998; Miller, 1993, 1996; Minana et al., 2000; Olney et al., 2002; Soscia et al., 2006; Swanson et al., 1995; Yanni and Lindsley, 2000). Ethanol-induced neurodegeneration in the developing CNS is the most devastating consequence.
The mechanisms underlying ethanol-induced neurodegeneration are complex. Ethanol exposure produces reactive oxygen species which results in oxidative stress in the brain. The CNS is particularly susceptible to oxidative stress due to its high oxygen consumption rate, elevated levels of polyunsaturated fatty acids, and relatively low content of antioxidative enzymes (Cohen-Kerem and Koren, 2003). Oxidative stress has been proposed as one of the mechanisms for ethanol-induced neurodegeneration (Chen and Luo, 2010; Goodlett et al., 2005). Recent evidence indicates that endoplasmic reticulum (ER) stress may play a role in the pathogenesis of neurological diseases (Scheper and Hoozemans, 2009) and has been implicated in various neurodegenerative processes, such as brain ischemia (Tajiri et al., 2004), Alzheimer’s disease (Katayama et al., 2004), Parkinson’s disease (Chen et al., 2004; Silva et al., 2005; Smith et al., 2005), Huntington’s disease (Hirabayashi et al., 2001), and amyotrophic lateral sclerosis (Turner and Atkin, 2006).
The ER regulates posttranslational protein processing and transport. Approximately one-third of all cellular proteins are translocated into the lumen of the ER where posttranslational modification, folding, and oligomerization occur. The ER is also the site for the biosynthesis of steroids, cholesterol, and other lipids. Under some cellular stress conditions, unfolded or misfolded proteins accumulate in the ER lumen and activate a compensatory response which has been referred to as ER stress response or unfolded protein response (UPR) (Ron, 2002; Xu et al., 2005). The activation of ER stress response or UPR is mediated mainly by 3 transmembrane ER signaling proteins: PERK, IRE1, and ATF6. These proteins straddle ER membranes with their N-terminus in the lumen of the ER and their C-terminus in the cytosol, thus providing a bridge that connects these 2 compartments. Normally, the N-termini of these transmembrane ER proteins are held by the ER chaperone GRP78 (BIP) preventing their aggregation. When misfolded proteins accumulate, GRP78 is released, allowing aggregation of these transmembrane signaling proteins and resulting in their activation. The activation of these ER stress sensors initiates protective responses, resulting in an overall decrease in protein synthesis, enhanced protein degradation, and increased protein-folding capacity of the ER (Ron, 2002). However, sustained ER stress ultimately leads to apoptotic death of the cell (Rasheva and Domingos, 2009; Xu et al., 2005).
In this study, we sought to determine whether or not ethanol causes ER stress in the developing CNS. Administration of ethanol to rodents during the synaptogenesis period (also known as the brain growth spurt period), which occurs in early postnatal days, is equivalent to ethanol exposure during the third trimester of pregnancy in humans. We used a well-established mouse model of developmental ethanol exposure in which ethanol was administered subcutaneously at a total dose of 5 g/kg to 7-day-old mice (Olney et al., 2002). The paradigm of ethanol administration results in widespread neurodegeneration in the developing brain (Liu et al., 2009; Olney et al., 2002). We demonstrate that ethanol activates ER stress sensors and upregulates the expression of ER stress-inducible proteins (ERSIPs), indicating that ethanol causes ER stress in the developing brain.
MATERIALS AND METHODS
Materials
Anti-MANF (mesencephalic astrocyte-derived neurotrophic factor) antibody was produced as previously described (Apostolou et al., 2008). Anti-GRP78, anti-CHOP, anti-eIF2α, anti-PERK, anti-ATF6, and anti-PKR antibodies were obtained from Santa Cruz Biotech (Santa Cruz, CA). Anti-caspase-3, anti-phospho-eIF2α (Ser51), anti-IRE1α, anti-phospho-PERK (Thr980), and anti-phospho-PKR (Thr446 and Thr451) antibodies were obtained from Cell Signaling Technology, Inc. (Beverly, MA). Anti-caspase-12 antibody was obtained from Abcam (Cambridge, MA). Anti-phospho-IRE1α (Ser724) was obtained from Novus Biologicals (Littleton, CO). Antineuronal nuclei (NeuN) antibody was obtained from Millipore Corporate (Billerica, MA). Anti-glial fibrillary acidic protein (GFAP) antibody and other chemicals were obtained from Sigma Chemical Co. (St. Louis, MO).
Animals and Treatment
C57BL/6 mice were obtained from Harlan Laboratories (Indianapolis, IN) and maintained at the Animal Facility of the University of Kentucky Medical Center. CHOP knockout (CHOP−/−) mice with C57BL/6 mice background were obtained from the Jackson Laboratory (Bar Harbor, ME). All procedures were performed in accordance with the guidelines set by the NIH and the Animal Care and Use Committee of the University of Kentucky. An acute ethanol exposure paradigm, which had been shown to induce robust neurodegeneration in infant mice (Liu et al., 2009; Olney et al., 2002), was employed. Seven-day-old mice (postnatal day 7 [PD7]) were injected subcutaneously with saline or ethanol (2.5 g/kg, 20% solution in saline) twice at time 0 and 2 hours. At specified times after ethanol injection the brains were removed and processed for immunoblotting or immunohistochemical analysis of ERSIPs.
Analysis of Blood Ethanol Concentrations
Blood was collected 8 hours after the first ethanol injection from the tail veins of mice. The timing for determining blood ethanol concentrations (BECs) was consistent with the biochemical analysis performed in this study. BECs were assayed from 12 pups (6 control and 6 ethanol-exposed) using an Analox AM1 Alcohol Analyzer (Analox Instruments, Lunenburg, MA).
Preparation of Brain Tissue and Immunoblotting
After treatment, mice were anesthetized by an intraperitoneal injection of ketamine (20 mg/ml)/xylazine (3 mg/ml) (Sigma-Aldrich, Inc., St. Louis, MO) at a dose of 1 ml/kg body weight and the brain was immediately dissected. The tissues were frozen in liquid nitrogen and stored at −80°C. Proteins were extracted as previously described (Wang et al., 2007). In brief, tissues were homogenized using an ice-cold lysis buffer containing 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1 mM EGTA, 1 mM PMSF, 0.5% NP-40, 0.25% SDS, 5 μg/ml leupeptin, and 5 μg/ml aprotinin. Homogenates were centrifuged at 20,000×g for 30 min at 4°C and the supernatant fraction was collected.
The immunoblotting procedure has been previously described (Chen et al., 2004). In brief, aliquots of the protein samples (30 μg) were separated on an SDS-polyacrylamide gel by electrophoresis. The separated proteins were transferred to nitrocellulose membranes. The membranes were blocked with either 5% bovine serum albumin (BSA) or 5% nonfat milk in 0.01 M PBS (pH 7.4) and 0.05% Tween-20 (TPBS) at room temperature for 1 hour. Subsequently, the membranes were probed with primary antibodies directed against target proteins overnight at 4°C. After 3 quick washes in TPBS, the membranes were incubated with a secondary antibody conjugated to horseradish peroxidase (Amersham, Arlington Hts, IL). The immune complexes were detected by the enhanced chemiluminescence method (Amersham). In some cases, the blots were stripped and re-probed with either an anti-tubulin or anti-actin antibody. The density of immunoblotting was quantified with the software of Quantity One (Bio-Rad Laboratories, Hercules, CA).
Immunohistochemical and Immunofluorescent staining
After treatments, the mice were deeply anesthetized with chloral hydrate (350 mg/kg), and then perfused with saline followed by 4% paraformaldehyde in 0.1 M potassium phosphate buffer (pH 7.2). The brains were removed and postfixed in 4% paraformaldehyde for an additional 24 hours and then transferred to 30% sucrose. The brain was sectioned at 40 μm with a sliding microtome (Leica Microsystems, Wetzlar, Germany).
The immunohistochemical staining for active caspase-3 and ER-SIPs was performed as previously described (Liu et al., 2009). In brief, free-floating sections were incubated in 0.3% H2O2 in methanol for 30 minutes at room temperature and then treated with 0.1% Tri-tonX-100 for 10 minutes in phosphate buffered saline (PBS). The sections were washed with PBS 3 times and then blocked with 1% BSA and 0.01% TritonX-100 for 1 hour at room temperature. The sections were incubated with primary antibodies: anti-cleaved caspase-3 antibody (1:10,000), anti-p-eIF2α (1:500), CHOP (1:1,000), and cas-pase-12 (1:8,000) overnight at 4°C. Negative controls were performed by omitting the primary antibody. After rinsing in PBS, sections were incubated with a biotinylated goat anti-rabbit IgG (1:200; Vector Laboratories Inc., Burlingame, CA) for 1 hour at room temperature. The sections were washed 3 times with PBS, then incubated in avidin–biotin–peroxidase complex (1:100 in PBS; Vector Laboratories Inc.) for 1 hour and developed in 0.05% 3,3′-diaminobenzidine (DAB) (Sigma-Aldrich, Inc.) containing 0.003% H2O2 in PBS.
Double immunofluorescent staining on the brain sections was performed as previously reported (Ayoub et al., 2005). In brief, sections were pretreated with 0.3% H2O2 in methanol for 30 minutes at room temperature, then treated with 0.1% TritonX-100 for 10 minutes in PBS, pH 7.2 containing 1% BSA for 30 minutes and finally incubated overnight with rabbit anti-p-eIF2α polyclonal antibody (1:200), rabbit anti-caspase-12 polyclonal antibody (1:2,000), or rabbit anti-CHOP polyclonal antibody (1:500) at 4°C. After rinsing in PBS, sections were incubated with Alexa488-conjugated goat anti-rabbit IgG (1:1,000; Invitrogen, Carlsbad, CA) for 1 hour and rinsed in PBS. Sections were then incubated with mouse anti-NeuN monoclonal antibody (1:1,000) or mouse anti-GFAP monoclonal antibody (1:2,000) at 4°C overnight. After rinsing in PBS, sections were treated with Alexa594-conjugated goat anti-mouse or rat (1:1,000) antibodies for 1 hour. The images were recorded using an Olympus microscope (BX 61) equipped with a DP70 digital camera (Olympus, Center Valley, PA).
RT-PCR
DNA was extracted from tail snips using a REDExtract-N-Ampä Tissue PCR kit (Sigma-Aldrich). The information for CHOP primers used in the polymerase chain reaction (PCR) was provided by Jackson Laboratory. All PCR products were separated by electrophoresis on a 1.5% agarose gel. The expected products include mutant CHOP (approximately 320 bp), heterozygote (approximately 320 and 545 bp), and wild type (545 bp).
Statistical Analysis
Differences among treatment groups were tested using analysis of variance (ANOVA). Differences in which p was <0.05 were considered statistically significant. In cases where significant differences were detected, specific post hoc comparisons between treatment groups were examined with Student-Newman–Keuls tests.
RESULTS
The paradigm of ethanol exposure resulted in sustained high BECs in PD7 mouse pups. Our results indicated the BEC was 338.8 ± 26.5 mg/dl at 8 hours after the first ethanol injection; this was generally consistent with previous reports using this paradigm (Olney et al., 2002). Using this model, previous studies indicate that ethanol exposure caused a wide spread neuroapoptosis in the brain (Liu et al., 2009; Olney et al., 2002). We confirmed this finding by showing an increased expression of active caspase-3 following 8 hours of ethanol exposure (Fig. 1A).
Fig. 1.
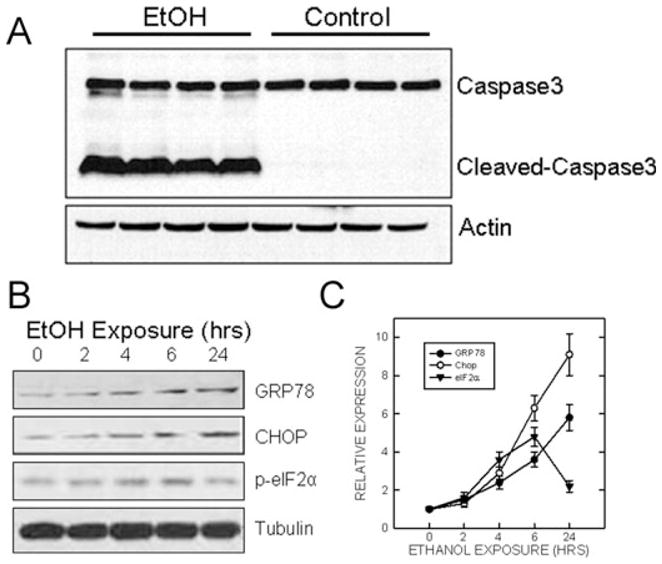
The time sequence of ethanol-induced expression of endoplasmic reticulum (ER) stress-inducible proteins (ERSIPs). Postnatal day 7 mice were injected subcutaneously with ethanol (2.5 g/kg, or saline) at 0 and 2 hours as described under Materials and Methods. (A) At 8 hours after the first injection, the cerebral cortex was removed, protein was extracted and the expression of caspase-3 was evaluated by immunoblotting. The results represent samples obtained from 4 control and 4 ethanol-exposed mouse pups. (B) At the specified time after ethanol injection, the cerebral cortex was dissected and protein was extracted. The expression of GRP78, CHOP, and the phosphorylation of eIF2α was determined with immunoblotting. The experiment was replicated 3 times. (C) The expression of GRP78, CHOP, and the phosphorylation of eIF2α was quantified by densitometry and normalized to the expression of actin as described under Materials and Methods. The results were the mean ± SEM of 3 independent experiments. EtOH, ethanol.
We examined the effect of ethanol on the expression of ERSIPs. As shown in Fig. 1B and 1C, starting after 4 hours of ethanol exposure, a significant increase in the expression of GRP78, CHOP, and the phosphorylation of eIF2α was observed, and longer ethanol exposure caused more up-regulation. Ethanol-stimulated GRP78 and CHOP expression remained at a high level after 24 hours of exposure. However, ethanol-induced phosphorylation of eIF2α started to decline after 6 hours of exposure.
To confirm that ethanol induced ER stress, we investigated the effect of ethanol on additional ERSIPs. As shown in Fig. 2A and 2B, 8 hours of ethanol exposure significantly increased the expression of ERSIPs and activated signaling pathways associated with ER stress. These include the expression GRP78, ATF6, CHOP, and MANF as well as the phosphorylation of eIF2α, IRE1α, PKR, and PERK. The magnitude of the ethanol-induced increase of these ERSIPs was different. The increase of CHOP was the highest (approximately 13-fold), while the up-regulation of phosphorylated PERK was the lowest (2.2-fold). The high relative levels of CHOP expression following ethanol exposure was due to a low basal CHOP expression in the control animals.
Fig. 2.

Effect of ethanol on the expression of endoplasmic reticulum stress-inducible proteins (ERSIPs). (A) Postnatal day 7 mice were injected with ethanol as described. At 8 hours after the first injection, the cerebral cortex was removed, protein was extracted, and the expression of GRP78, ATF6, mesencephalic astrocyte-derived neurotrophic factor (MANF), CHOP, caspase-12, and the phosphorylation of eIF2α, IRE1α, PKR, and PERK was evaluated by immunoblotting. The results represent samples obtained from 4 control and 4 ethanol-exposed mouse pups. (B) The expression of ERSIPs was quantified by densitometry and normalized as described. The results were the mean ± SEM of 3 independent experiments. *Statistically significant difference from controls. EtOH, ethanol.
We then examined the distribution of 3 ERSIPs that are well-known for their involvement in cell apoptosis, namely eIF2α, caspase-12, and CHOP, in the developing brain. As shown in Fig. 3, ethanol-induced phosphorylation of eIF2α (p-eIF2α) was observed in some regions of the cerebral cortex, hippocampus, and thalamus. An increased p-eIF2α expression was observed in the lateral parietal association cortex (LPtA) and retrosplenial granular cortex (RSGc) in the cerebral cortex. Ethanol also induced p-eIF2α in the hippocampus; elevated p-eIF2α was observed in CA1 and CA3 regions as well as the dentate gyrus. High p-eIF2α expression was spotted in some interneurons at the molecular layer of the hippocampus. The levels of p-eIF2α were elevated in several regions of the thalamus, including the dorsal lateral geniculate nucleus (DLG), lateral posterior thalamic nucleus (LPMR), mediodorsal thalamic nucleus (MD), ventral posteromedial thalamic nucleus (VPM), and ventromedial thalamic nucleus (VM). The distribution of ethanol-induced increase in caspase-12 expression was quite similar to that of p-eIF2α (Fig. 4).
Fig. 3.

Ethanol-induced phosphorylation of eIF2α in the developing brain. Postnatal day 7 mice were injected with ethanol as described. At 8 hours after the first ethanol injection, the brain was removed and the expression of phosphorylated eIF2α (p-eIF2α) was analyzed with immunohistochemistry as described under Materials and Methods. Bar = 100 μm. Con, control; EtOH, ethanol; LPtA, lateral parietal association cortex; RSGc, retrosplenial granular cortex; CA1, field CA1 hippocampus; CA3, field CA3 hippocampus; DG, dentate gyrus; DLG, dorsal lateral geniculate nucleus; LPMR, lateral posterior thalamic nucleus; MD, mediodorsal thalamic nucleus; VPM, ventral posteromedial thalamic nucleus; VM, ventromedial thalamic nucleus.
Fig. 4.

Effect of ethanol on caspase-12 expression in the developing brain. (A) Ethanol-induced expression of caspase-12 was examined with immunohistochemistry. The notations are the same as Fig. 3. (B) The expression of caspase-12 in the RSGc, pyramidal layer of parietal cortex, and CA1 area of the hippocampus is shown with higher magnification. Bar = 100 μm.
In control animals, CHOP immunoreactivity was barely detectable in the brain; this was consistent with the results from immunoblotting. In ethanol-treated animals, CHOP-positive cells were distributed in the outer layer of RSGc and LPtA of the cerebral cortex (Fig. 5). The distribution of CHOP-positive cells was relatively sporadic compared to that of p-eIF2α and caspase-12. In the hippocampus, CHOP-positive cells were scattered in the molecular layer of the CA1 region and some cells in the CA3 region were positive for CHOP immunoreactivity. In the thalamus, CHOP-positive cells were distributed at the VPM, VM, LPMR, and MD.
Fig. 5.

Effect of ethanol on CHOP expression in the developing brain. Ethanol-induced expression of CHOP was examined with immunohistochemistry. The notations are the same as Fig. 3.
It appeared that ethanol-induced expression of p-eIF2α, caspase-12, and CHOP was distributed in neurons, but not in glial cells. As shown in Fig. 6, the expression of p-eIF2α, CHOP, and caspase-12 was localized in NeuN-positive cells, but not in GFAP-positive cells. We sought to determine whether or not there was a correlation between the expression of ERSIPs and caspase-3 activation. With currently available antibodies, we were unable to double-label the ERSIPs (p-eIF2α, caspase-12, and CHOP) and active caspase-3. By comparing the distribution of these proteins in response to ethanol exposure, we noted that the distribution pattern of p-eIF2α and caspase-12 was generally consistent with that of active caspase-3, while the CHOP-positive cells were confined to the outer layers of RSGc and LPtA of the cerebral cortex and sporadically scattered in the hippocampus and thalamus (Fig. 7).
Fig. 6.

Localization of endoplasmic reticulum stress-inducible proteins (ERSIPs). Postnatal day 7 mice were injected with ethanol as described. At 8 hours after the first ethanol injection, the brain was removed and the expression of p-eIF2α (A), caspase-12 (B) and CHOP (C) was examined by immunofluorescent staining. The brain sections were double-labeled with either an anti-NeuN (neuron marker) antibody or an anti-GFAP (astrocyte marker) antibody. Merged images show the co-localization of ERSIPs with markers for neurons or astrocytes. Bar = 100 μm.
Fig. 7.

Effect of ethanol on the expression of caspase-3 and endoplasmic reticulum stress-inducible proteins. Postnatal day 7 mice were injected with ethanol as described. At 8 hours after the first ethanol injection, the brain was removed and the expression of active caspase-3, p-eIF2α, caspase-12, and CHOP was analyzed with immunohistochemistry as described. Bar = 1 mm.
Since CHOP is a critical mediator of apoptosis and previous studies indicate that CHOP may contribute to ethanol-induced hepatic damage (Ji et al., 2005), we investigated the role of CHOP in ethanol-induced neuroapoptosis using a CHOP knockout mouse model. As shown in Fig. 8, ethanol-induced caspase-3 and Bax activation was similar between CHOP+/+ and CHOP−/− mice.
Fig. 8.
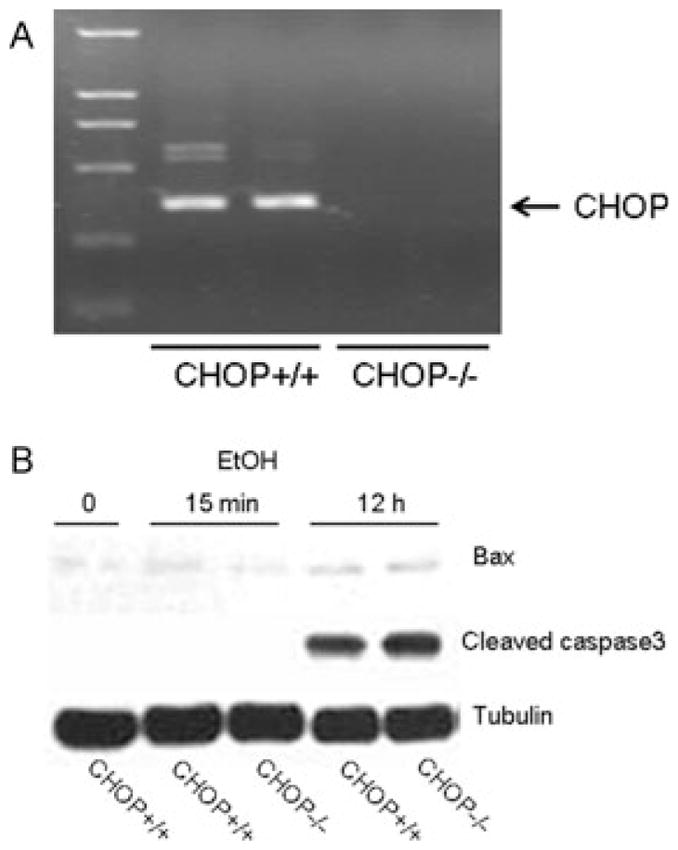
Effect of ethanol on caspase-3 and Bax activation in CHOP knockout mice. (A) The deletion of CHOP was verified by RT-PCR. (B) Postnatal day 7 CHOP knockout mice (CHOP−/−) and wild-type mice (CHOP+/+) were injected with ethanol as described. At specified times after ethanol injection, the brain was removed and the expression of active caspase-3 and Bax was analyzed with immunoblotting as described. The experiment was replicated 3 times. EtOH, ethanol.
DISCUSSION
For the first time, we demonstrate that ethanol induces ER stress in the developing CNS. Acute ethanol administration upregulates ERSIPs and activates 3 transmembrane ER stress sensors (PERK, IRE1, and ATF6) in the brain of 7-day-old mice.
PERK is a serine/threonine protein kinase. Upon removal of GRP78, PERK oligomerizes in ER membranes, inducing autophosphorylation and activation. Activated PERK phosphorylates and inactivates eIF2α, thereby globally inhibiting mRNA translation and reducing the protein load on the ER. However, PERK-mediated phosphorylation of eIF2α leads to the translational up-regulation of specific mRNAs, such as the transcription factor ATF4 that activates the transcription of pro-apoptotic factors CHOP/GADD153 and GADD34. The activation of IRE1 results in XBP-1 mRNA splicing; spliced XBP-1 is an active transcription factor and regulates the transcription of many target genes including ER chaperones and genes encoding the components of ER-associated degradation. Upon UPR, ATF6 translocates to the golgi apparatus, here, it is cleaved and then released into the cytosol, allowing migration into the nucleus to regulate gene transcription. Among targets of ATF6 signaling are XPB-1, GRP78, and CHOP/GADD153. ATF6 collaborates with IRE1, where ATF6 induces transcription to increase XBP-1 mRNA, and IRE1 processes XBP-1 mRNA to produce active XBP-1 (Ron, 2002; Xu et al., 2005).
The precise mechanisms underlying ethanol-induced ER stress are unknown. Many disturbances, such as perturbed calcium homeostasis or redox status, elevated secretory protein synthesis rates, altered glycosylation levels, and cholesterol overloading, can interfere with oxidative protein folding, leading to the accumulation of unfolded or misfolded proteins in the ER lumen and inducing ER stress (Ron, 2002; Xu et al., 2005). It has been shown that ethanol alters cellular homeostasis of calcium, lipid profile, and cholesterol as well as redox status in neurons (Chen and Luo, 2010; Chen et al., 2008; Guizzetti and Costa, 2007; Saito et al., 2007; Webb et al., 2003). These cellular disturbances may be the underlying mechanisms of ethanol-induced ER stress.
It remains unclear how ER stress leads to apoptosis. CHOP/GADD153, a member of the C/EBP family of bZIP transcription factors, appears to play an important role in ER stress-induced apoptosis (Rasheva and Domingos, 2009; Xu et al., 2005; Zinszner et al., 1998). The CHOP gene promoter contains binding sites for all of the major inducers of the UPR, including ATF4, ATF6, and XBP-1. A previous study indicates that intragastric ethanol feeding in mice induces ER stress and hepatocellular apoptosis in the liver (Kaplowitz and Ji, 2006). CHOP null (−/−) mice have remarkable resistance to ethanol-induced death of hepatocytes (Ji et al., 2005). The expression of CHOP is barely detectable in the brain of control animals, but its levels are robustly increased by ethanol exposure. However, the distribution of CHOP is sporadic and not consistent with the pattern of active caspase-3. Furthermore, CHOP−/− and CHOP+/+ mice display similar susceptibility to ethanol-induced neuroapoptosis. These results suggest that CHOP/GADD153 is minimally involved in ethanol-induced neurodegeneration.
Ethanol increases the phosphorylation of PERK [p-PERK (Thr980)] and eIF2α [p-eIF2α (Ser51)]. Activation of the PERK/eIF2α pathway not only causes inhibition of mRNA translation, but also enhances apoptosis (Wek et al., 2006). p-eIF2α (Ser51) is also subjected to the regulation of PKR. PKR, a serine/threonine protein kinase, has been well-known for its antiviral role. Upon activation by viral infection, PKR phosphorylates eIF2α, leading to inhibition of translation, initiation, and apoptosis. PKR can also be activated by direct interactions with its protein activators, PACT, or its mouse homolog, RAX, and regulates protein synthesis and cell survival (Bennett et al., 2004). We have previously shown that ethanol induces p-eIF2α (Ser51) in a PKR-dependent manner in cultured neurons and in the developing cerebellum (Chen et al., 2006). It is therefore likely that both PERK and PKR pathways mediate ethanol-induced p-eIF2α (Ser51). The distribution pattern of p-eIF2α and active caspase-3 is similar (Fig. 7), suggesting that the PERK/PKR/eIF2α pathway may participate in ethanol-induced neuroapoptosis. However, a direct association remains to be determined.
Caspase-12 is believed to be another major mediator of apoptosis downstream of ER stress (Szegezdi et al., 2003). Prolonged ER stress results in the activation of caspase-12 which acts on downstream effectors including caspase-3, leading to apoptosis (Szegezdi et al., 2003). A recent study shows that a caspase-12 inhibitor significantly protects Chinese hamster ovary cells against ethanol-induced cell death (Balan et al., 2010). A similar distribution pattern of caspase-12 and active caspase-3 suggests that caspase-12 may be involved in ethanol-induced neuroapoptosis. However, further experiments are necessary to establish the association.
We note that ethanol increases the expression of MANF in the developing brain. MANF, also known as arginine-rich mutated in early stage tumors, has been recently identified as an ERSIP; up-regulation of MANF expression can alleviate ER stress (Apostolou et al., 2008). Cerebral ischemia-induced ER stress is accompanied by the up-regulation of MANF in affected brain areas; intracortical delivery of recombinant MANF protein protects the brain from ischemic brain injury (Airavaara et al., 2010; Yu et al., 2010). Furthermore, intrastriatal injection of MANF ameliorates 6-hydroxydopamine-induced degeneration of nigrostriatal dopaminergic neurons in rats in an experimental model of Parkinson’s disease (Voutilainen et al., 2009). 6-Hydroxydopamine has been shown to induce ER stress in neurons (Chen et al., 2004). It is therefore likely that the induction of MANF is an early protective response to ethanol-induced ER stress in the developing CNS. Therefore, administration of exogenous MANF is a potential approach to ameliorate ethanol-induced neuroapoptosis.
In summary, we have demonstrated that ethanol can cause ER stress in the developing brain. Although the linkage between ER stress and neurodegeneration remains to be established, this provides a novel insight into the potential mechanisms of ethanol-induced damage to immature neurons. In addition to neurodegeneration, ER stress may affect other developmental events in the CNS. For example, ER stress has been shown to affect the expression of neuronal cell cycle proteins, suggesting that ER stress may affect the proliferation of neuronal precursors (Hoozemans et al., 2006). A recent study suggests that UPR may regulate neuronal differentiation (Cho et al., 2009). Therefore, ethanol-induced ER stress may have a profound impact on the developing CNS.
Acknowledgments
This work is supported by grants from the National Institutes of Health (AA015407 and AA019693). ZK is also supported by grants from the Ministry of Science and Technology of China (2010CB912000CB912007CB947100), the National Natural Science Foundation of China (30870812), and the Chief Scientist Program of Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences (SIBS2008006).
References
- Airavaara M, Chiocco MJ, Howard DB, Zuchowski KL, Peränen J, Liu C, Fang S, Hoffer BJ, Wang Y, Harvey BK. Widespread cortical expression of MANF by AAV serotype 7: localization and protection against ischemic brain injury. Exp Neurol. 2010;225:104–113. doi: 10.1016/j.expneurol.2010.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apostolou A, Shen Y, Liang Y, Luo J, Fang S. Armet, a UPR-upregulated protein, inhibits cell proliferation and ER stress-induced cell death. Exp Cell Res. 2008;314:2454–2467. doi: 10.1016/j.yexcr.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayoub AE, Cai TQ, Kaplan RA, Luo J. Developmental expression of matrix metalloproteinases 2 and 9 and their potential role in the histogenesis of the cerebellar cortex. J Comp Neurol. 2005;481:403–415. doi: 10.1002/cne.20375. [DOI] [PubMed] [Google Scholar]
- Balan AG, Myers BJ, Maganti JL, Moore DB. ER-targeted Bcl-2 and inhibition of ER-associated caspase-12 rescue cultured immortalized cells from ethanol toxicity. Alcohol. 2010;44:553–563. doi: 10.1016/j.alcohol.2010.07.003. [DOI] [PubMed] [Google Scholar]
- Bearer CF. L1 cell adhesion molecule signal cascades: targets for ethanol developmental neurotoxicity. Neurotoxicology. 2001;22:625–633. doi: 10.1016/s0161-813x(01)00034-1. [DOI] [PubMed] [Google Scholar]
- Bennett RL, Blalock WL, May WS. Serine 18 phosphorylation of RAX, the PKR activator, is required for PKR activation and consequent translation inhibition. J Biol Chem. 2004;279:42687–42693. doi: 10.1074/jbc.M403321200. [DOI] [PubMed] [Google Scholar]
- Chen G, Bower KA, Ma C, Fang S, Thiele CJ, Luo J. Glycogen synthase kinase 3beta (GSK3beta) mediates 6-hydroxydopamine-induced neuronal death. FASEB J. 2004;18:1162–1164. doi: 10.1096/fj.04-1551fje. [DOI] [PubMed] [Google Scholar]
- Chen G, Luo J. Anthocyanins: are they beneficial in treating ethanol neurotoxicity? Neurotox Res. 2010;17:91–101. doi: 10.1007/s12640-009-9083-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Ma C, Bower KA, Ke Z, Luo J. Interaction between RAX and PKR modulates the effect of ethanol on protein synthesis and survival of neurons. J Biol Chem. 2006;281:15909–15915. doi: 10.1074/jbc.M600612200. [DOI] [PubMed] [Google Scholar]
- Chen G, Ma C, Bower KA, Shi X, Ke Z, Luo J. Ethanol promotes endoplasmic reticulum stress-induced neuronal death: involvement of oxidative stress. J Neurosci Res. 2008;86:937–946. doi: 10.1002/jnr.21540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho YM, Jang YS, Jang YM, Chung SM, Kim HS, Lee JH, Jeong SW, Kim IK, Kim JJ, Kim KS, Kwon OJ. Induction of unfolded protein response during neuronal induction of rat bone marrow stromal cells and mouse embryonic stem cells. Exp Mol Med. 2009;41:440–452. doi: 10.3858/emm.2009.41.6.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen-Kerem R, Koren G. Antioxidants and fetal protection against ethanol teratogenicity. I. Review of the experimental data and implications to humans. Neurotoxicol Teratol. 2003;25:1–9. doi: 10.1016/s0892-0362(02)00324-0. [DOI] [PubMed] [Google Scholar]
- Goodlett CR, Horn KH, Zhou FC. Alcohol teratogenesis: mechanisms of damage and strategies for intervention. Exp Biol Med (Maywood) 2005;230:394–406. doi: 10.1177/15353702-0323006-07. [DOI] [PubMed] [Google Scholar]
- Guizzetti M, Costa LG. Cholesterol homeostasis in the developing brain: a possible new target for ethanol. Hum Exp Toxicol. 2007;26:355–360. doi: 10.1177/0960327107078412. [DOI] [PubMed] [Google Scholar]
- Hirabayashi M, Inoue K, Tanaka K, Nakadate K, Ohsawa Y, Kamei Y, Popiel AH, Sinohara A, Iwamatsu A, Kimura Y, Uchiyama Y, Hori S, Kakizuka A. VCP/p97 in abnormal protein aggregates, cytoplasmic vacuoles, and cell death, phenotypes relevant to neurodegeneration. Cell Death Differ. 2001;8:977–984. doi: 10.1038/sj.cdd.4400907. [DOI] [PubMed] [Google Scholar]
- Hoffman EJ, Mintz CD, Wang S, McNickle DG, Salton SR, Benson DL. Effects of ethanol on axon outgrowth and branching in developing rat cortical neurons. Neuroscience. 2008;157:556–565. doi: 10.1016/j.neuroscience.2008.08.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoozemans JJ, Stieler J, van Haastert ES, Veerhuis R, Rozemuller AJ, Baas F, Eikelenboom P, Arendt T, Scheper W. The unfolded protein response affects neuronal cell cycle protein expression: implications for Alzheimer’s disease pathogenesis. Exp Gerontol. 2006;41:380–386. doi: 10.1016/j.exger.2006.01.013. [DOI] [PubMed] [Google Scholar]
- Ikonomidou C, Bittigau P, Koch C, Genz K, Hoerster F, Felderhoff-Mueser U, Tenkova T, Dikranian K, Olney JW. Neurotransmitters and apoptosis in the developing brain. Biochem Pharmacol. 2001;62:401–405. doi: 10.1016/s0006-2952(01)00696-7. [DOI] [PubMed] [Google Scholar]
- Ji C, Mehrian-Shai R, Chan C, Hsu YH, Kaplowitz N. Role of CHOP in hepatic apoptosis in the murine model of intragastric ethanol feeding. Alcohol Clin Exp Res. 2005;29:1496–1503. doi: 10.1097/01.alc.0000174691.03751.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplowitz N, Ji C. Unfolding new mechanisms of alcoholic liver disease in the endoplasmic reticulum. J Gastroenterol Hepatol. 2006;21:S7–S9. doi: 10.1111/j.1440-1746.2006.04581.x. [DOI] [PubMed] [Google Scholar]
- Katayama T, Imaizumi K, Manabe T, Hitomi J, Kudo T, Tohyama M. Induction of neuronal death by ER stress in Alzheimer’s disease. J Chem Neuroanat. 2004;28:67–78. doi: 10.1016/j.jchemneu.2003.12.004. [DOI] [PubMed] [Google Scholar]
- Kumada T, Jiang Y, Cameron DB, Komuro H. How does alcohol impair neuronal migration? J Neurosci Res. 2007;85:465–470. doi: 10.1002/jnr.21149. [DOI] [PubMed] [Google Scholar]
- Liu Y, Chen G, Ma C, Bower KA, Xu M, Fan Z, Shi X, Ke ZJ, Luo J. Overexpression of glycogen synthase kinase 3beta sensitizes neuronal cells to ethanol toxicity. J Neurosci Res. 2009;87:2793–2802. doi: 10.1002/jnr.22098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J. Mechanisms of Ethanol-Induced Death of Cerebellar Granule Cells. Cerebellum. 2010 doi: 10.1007/s12311-010-0219-0. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J, Miller MW. Growth factor-mediated neural proliferation: target of ethanol toxicity. Brain Res Brain Res Rev. 1998;27:157–167. doi: 10.1016/s0165-0173(98)00009-5. [DOI] [PubMed] [Google Scholar]
- May PA, Gossage JP. Estimating the prevalence of fetal alcohol syndrome. Alcohol Res Health. 2001;25:159–167. [PMC free article] [PubMed] [Google Scholar]
- Miller MW. Migration of cortical neurons is altered by gestational exposure to ethanol. Alcohol Clin Exp Res. 1993;17:304–314. doi: 10.1111/j.1530-0277.1993.tb00768.x. [DOI] [PubMed] [Google Scholar]
- Miller MW. Limited ethanol exposure selectively alters the proliferation of precursor cells in the cerebral cortex. Alcohol Clin Exp Res. 1996;20:139–143. doi: 10.1111/j.1530-0277.1996.tb01056.x. [DOI] [PubMed] [Google Scholar]
- Minana R, Climent E, Barettino D, Segui JM, Renau-Piqueras J, Guerri C. Alcohol exposure alters the expression pattern of neural cell adhesion molecules during brain development. J Neurochem. 2000;75:954–964. doi: 10.1046/j.1471-4159.2000.0750954.x. [DOI] [PubMed] [Google Scholar]
- Olney JW, Tenkova T, Dikranian K, Qin YQ, Labruyere J, Ikonomidou C. Ethanol-induced apoptotic neurodegeneration in the developing C57BL/6 mouse brain. Brain Res Dev Brain Res. 2002;133:115–126. doi: 10.1016/s0165-3806(02)00279-1. [DOI] [PubMed] [Google Scholar]
- Rasheva VI, Domingos PM. Cellular responses to endoplasmic reticulum stress and apoptosis. Apoptosis. 2009;14:996–1007. doi: 10.1007/s10495-009-0341-y. [DOI] [PubMed] [Google Scholar]
- Riley EP, McGee CL. Fetal alcohol spectrum disorders: an overview with emphasis on changes in brain and behavior. Exp Biol Med (Maywood) 2005;230:357–365. doi: 10.1177/15353702-0323006-03. [DOI] [PubMed] [Google Scholar]
- Ron D. Translational control in the endoplasmic reticulum stress response. J Clin Invest. 2002;110:1383–1388. doi: 10.1172/JCI16784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito M, Chakraborty G, Mao RF, Wang R, Cooper TB, Vadasz C, Saito M. Ethanol alters lipid profiles and phosphorylation status of AMP-activated protein kinase in the neonatal mouse brain. J Neurochem. 2007;103:1208–1218. doi: 10.1111/j.1471-4159.2007.04836.x. [DOI] [PubMed] [Google Scholar]
- Scheper W, Hoozemans JJ. Endoplasmic reticulum protein quality control in neurodegenerative disease: the good, the bad and the therapy. Curr Med Chem. 2009;16:615–626. doi: 10.2174/092986709787458506. [DOI] [PubMed] [Google Scholar]
- Silva RM, Ries V, Oo TF, Yarygina O, Jackson-Lewis V, Ryu EJ, Lu PD, Marciniak SJ, Ron D, Przedborski S, Kholodilov N, Greene LA, Burke RE. CHOP/GADD153 is a mediator of apoptotic death in substantia nigra dopamine neurons in an in vivo neurotoxin model of parkinsonism. J Neurochem. 2005;95:974–986. doi: 10.1111/j.1471-4159.2005.03428.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith WW, Jiang H, Pei Z, Tanaka Y, Morita H, Sawa A, Dawson VL, Dawson TM, Ross CA. Endoplasmic reticulum stress and mitochondrial cell death pathways mediate A53T mutant alpha-synuclein-induced toxicity. Hum Mol Genet. 2005;14:3801–3811. doi: 10.1093/hmg/ddi396. [DOI] [PubMed] [Google Scholar]
- Soscia SJ, Tong M, Xu XJ, Cohen AC, Chu J, Wands JR, de la Monte SM. Chronic gestational exposure to ethanol causes insulin and IGF resistance and impairs acetylcholine homeostasis in the brain. Cell Mol Life Sci. 2006;63:2039–2056. doi: 10.1007/s00018-006-6208-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stratton K, Howe C, Battagila F, editors. Fetal Alcohol Syndrome: Diagnosis, Epidemiology, Prevention, and Treatment. National Academy Press; Washington, DC: 1996. [Google Scholar]
- Swanson DJ, King MA, Walker DW, Heaton MB. Chronic prenatal ethanol exposure alters the normal ontogeny of choline acetyltransferase activity in the rat septohippocampal system. Alcohol Clin Exp Res. 1995;19:1252–1260. doi: 10.1111/j.1530-0277.1995.tb01608.x. [DOI] [PubMed] [Google Scholar]
- Szegezdi E, Fitzgerald U, Samali A. Caspase-12 and ER-stress-mediated apoptosis: the story so far. Ann N Y Acad Sci. 2003;1010:186–194. doi: 10.1196/annals.1299.032. [DOI] [PubMed] [Google Scholar]
- Tajiri S, Oyadomari S, Yano S, Morioka M, Gotoh T, Hamada JI, Ushio Y, Mori M. Ischemia-induced neuronal cell death is mediated by the endoplasmic reticulum stress pathway involving CHOP. Cell Death Differ. 2004;11:403–415. doi: 10.1038/sj.cdd.4401365. [DOI] [PubMed] [Google Scholar]
- Turner BJ, Atkin JD. ER stress and UPR in familial amyotrophic lateral sclerosis. Curr Mol Med. 2006;6:79–86. doi: 10.2174/156652406775574550. [DOI] [PubMed] [Google Scholar]
- Voutilainen MH, Bäck S, Pörsti E, Toppinen L, Lindgren L, Lindholm P, Peränen J, Saarma M, Tuominen RK. Mesencephalic astrocyte-derived neurotrophic factor is neurorestorative in rat model of Parkinson’s disease. J Neurosci. 2009;29:9651–9659. doi: 10.1523/JNEUROSCI.0833-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Fan Z, Wang B, Luo J, Ke ZJ. Activation of double-stranded RNA-activated protein kinase by mild impairment of oxidative metabolism in neurons. J Neurochem. 2007;103:2380–2390. doi: 10.1111/j.1471-4159.2007.04978.x. [DOI] [PubMed] [Google Scholar]
- Webb B, Walker DW, Heaton MB. Nerve growth factor and chronic ethanol treatment alter calcium homeostasis in developing rat septal neurons. Brain Res Dev Brain Res. 2003;143:57–71. doi: 10.1016/s0165-3806(03)00100-7. [DOI] [PubMed] [Google Scholar]
- Wek RC, Jiang HY, Anthony TG. Coping with stress: eIF2 kinases and translational control. Biochem Soc Trans. 2006;34:7–11. doi: 10.1042/BST20060007. [DOI] [PubMed] [Google Scholar]
- Xu C, Bailly-Maitre B, Reed JC. Endoplasmic reticulum stress: cell life and death decisions. J Clin Invest. 2005;115:2656–2664. doi: 10.1172/JCI26373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanni PA, Lindsley TA. Ethanol inhibits development of dendrites and synapses in rat hippocampal pyramidal neuron cultures. Brain Res Dev Brain Res. 2000;120:233–243. doi: 10.1016/s0165-3806(00)00015-8. [DOI] [PubMed] [Google Scholar]
- Yu YQ, Liu LC, Wang FC, Liang Y, Cha DQ, Zhang JJ, Shen YJ, Wang HP, Fang S, Shen YX. Induction profile of MANF/ARMET by cerebral ischemia and its implication for neuron protection. J Cereb Blood Flow Metab. 2010;30:79–91. doi: 10.1038/jcbfm.2009.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinszner H, Kuroda M, Wang X, Batchvarova N, Lightfoot RT, Remotti H, Stevens JL, Ron D. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev. 1998;12:982–995. doi: 10.1101/gad.12.7.982. [DOI] [PMC free article] [PubMed] [Google Scholar]