

Abstract
Background:
LuxI is a component of the quorum sensing signaling pathway in Vibrio fischeriresponsible for the inducer synthesis that is essential for bioluminescence.
Methods:
Homology modeling of LuxI was carried out using Phyre2 and refined with the GalaxyWEB server. Five models were generated and evaluated by ERRAT, ANOLEA, QMEAN6, and Procheck.
Results:
Five refined models were generated by the GalaxyWEB server, with Model 4 having the greatest quality based on the QMEAN6 score of 0.732. ERRAT analysis revealed an overall quality of 98.9%, while the overall quality of the initial model was 54%. The mean force potential energy, as analyzed by ANOLEA, were better compared to the initial model. Sterochemical quality estimation by Procheck showed that the refined Model 4 had a reliable structure, and was therefore submitted to the protein model database. Drug Discovery Workbench V.2 was used to screen 2700 experimental compounds from the DrugBank database to identify inhibitors that can bind to the active site between amino acids 24 and 110. Ten compounds with high negative scores were selected as the best in binding.
Conclusion:
The model produced, and the predicted acteyltransferase binding site, could be useful in modeling homologous sequences from other microorganisms and the design of new antimicrobials.
Key Words: Docking, Homology modeling, LuxI, Quorum sensing
Introduction
Quorum sensing was first discovered in Vibrio fischeri (V. fischeri), a bacterium that symbiotically produces light with certain marine animals (1-2). It is a signal transduction pathway that provides cell-cell communication and acts as a mechanism for coordinating gene expression in response to cell density (3). This system exists in Gram-positive and Gram-negative bacteria and controls virulence, biofilm formation, and antibiotic production (4, 5, 6).
In V. fischeri, quorum sensing is regulated by two gene products: LuxI and LuxR (7). LuxI is an acylated-homoserine lactone (acyl-HSL) (8). The acyl portion of acyl-HSL is derived from fatty acid precursors conjugated to acyl carrier protein (acyl-ACP), and the HSL moiety is derived from S-adenosylmethionine (SAM). LuxI promotes the formation of an amide bond joining the acyl side chain from acyl-ACP to SAM. Lactonization of the ligated intermediate, with the subsequent release of methylthioadenosine (MTA), results in the formation of acyl-HSL (9).
Homology or comparative modeling of a protein is a method of structure prediction based on amino acid sequence similarity to closely-related known structures (10). Genome-large scale sequencing projects revealed millions of sequences that could not be analyzed by X-ray crystallography and NMR spectroscopy techniques due to time restraints and other technical difficulties (11, 12). Because of these difficulties, researchers utilize bioinformatics to model unknown protein structures (13). These approaches help to identify active sites, design ligands and mutants, predict antigenic epitopes, and determine protein functions (14-17). This study focuses on homology modeling to predict the three-dimensional (3D) structure of LuxI in V. fischeri(strain ATCC 7006 01 / ES114) (18). This protein may be a potential potential target for antimicrobial design.
Materials and Methods
Sequence retrieval, physiochemical properties, and secondary structure
The amino acid sequence of LuxI was obtained from the UniProt database available at http://www.uniprot.org/, (Accession number: P35328) and used in FASTA file format in the analysis. The physiochemical properties of LuxI were characterized using the ProtParam tool of the ExPASy server (Biozentrum, University of Basel, Switzerland) at http://web.expasy.org/protparam/ (19). These parameters include molecular weight, amino acid composition, theoretical isoelectric point (pI), extinction coefficient, and instability index (20-21). The secondary structure was predicted by SSpro8 of SCRATCH, a program specialized to predict secondary and disordered regions (Donald Bren School of Informatics and Computor Sciences, California, USA) at: http://scratch.proteomics.ics.uci.edu/ (22). Structures determined using the method of Kabsch and Sander (23) were alpha-helix, 3-10-helix, extended strand, turn, bend, bridges, and the rest.
Functional domain prediction
MOTIF is a program to identify motifs from GenomeNet, Japan using the Pfam and Prosite data bases, at: http://www.genome.jp/tools/motif/ which uses Pfam. Pfam is a data base of protein domain alignment derived from the protein sequence secondary database of the Swiss Institute of Bioinformatics (SWISS-Prot) and translated to nucleic acid. The secondary database is stored in the European Molecular Biology Laboratory database (TrEMBL) (24).
Homology modeling, refinement, and evaluation of the 3D structure
The protein tertiary structure was built by PHYRE2 (Protein Holomogy/analogy Recognition Engine version 2) from Imperial College London available at (http://www.sbg.bio.ic.ac.uk/phyre2/html/page.cgi?id=index) (25). The structures generated were refined by the GalaxyWEB server at the Computational Biology Lab in the Department of Biochemistry, Seoul National University (http://galaxy.seoklab.org/) (26). The refined models were evaluated by several validation tools to select the best model and assess the quality of that model. ERRAT is a protein structure verification algorithm for evaluating the progress of crystallographic model building and refining maintained by the National Health Institute, University of California, USA (http://services.mbi.ucla.edu/ERRAT/) (27). The Z-score was determined by the PROSA web tool from the Center of Applied Molecular Engineering, Division of Bioinformatics, University of Salzburg, Salzburg, Austria. It measures the deviation of the total energy of the structure with respect to an energy distribution derived from random conformations found in native proteins and available at (28). The SWISS-MODEL workspace server was also used from Biozentrum, University of Basel, Switzerland (29). This workspace contains several evaluation tools integrated within ANOLEA of Pontifical Catholic University, Chile, the QMEAN6 server (Qualitative Model Energy ANalysis), which estimates the global and local quality of the models from Biozentrum, University of Basel, Switzerland, and Procheck from the European Bioinformatics Institute, Wellcome Trust Genome Campus, Hinxton, UK (30-32).
Submission of the model
The best refined 3D model was submitted into the protein model database (PMDB) (http:// bioinformatics.cineca.it/PMDB) (33).
Molecular Docking
The compounds used to screen Acyl-HSL synthases inhibitors were obtained from the DrugBank database (http://www.drugbank.ca). This database is supported by the Canadian Institutes of Health Research, Alberta Innovates - Health Solutions, and by The Metabolomics Innovation Centre (TMIC), Canada (34). Docking of the compounds was performed in CLC Drug Discovery workbench 2.0 (CLC Bio, QIAGEN Company, Denmark). Each compound was subjected to 100 iterations.
Results
To predict functional motifs and domains in LuxI, MOTIF software was used. Results show that LuxI has an autoinducer synthetase family signature domain and contains an acetyltransferase domain between residues 24 and 110 (Fig. 1A).
Fig. 1.

(A) The amino acid sequence of LuxI in V. fischeri ES114 showing the acetyltransferase domain in red (B) The secondary structure predicted by SSpro8 where H: alpha-helix, G: 3-10-helix, E: extended strand, T: turn, S: bend, C: the rest.
SSpro8 adopts full DSSP-8 classification of the secondary structure (23). The secondary structure (Fig. 1B) predicted that 30.05% of the protein is comprised of α-helices, 28.5% of extended β-strands, 15% of β-turns, 2.1% of 3-10 helices, 2.1% of bends, and the remaining 22.28% as random coils.
The 3D structure of the protein was built by Phyre2 and refined by GalaxyWEB server. This server can detect unreliable regions and perform ab initio modeling to improve models (26). Five refined models were generated by GalaxyWEB server. Model 4 (Fig. 2) had the best quality according to the QMEAN6 scores (Table 1) and was submitted into the PMDB with the ID: PM0079876.
Fig. 2.
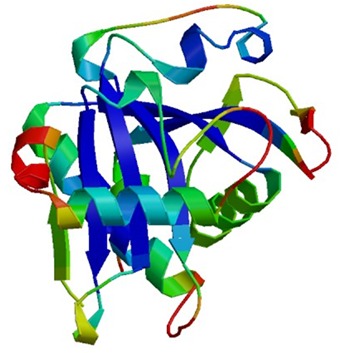
The three-dimensional structure of LuxI, Model 4, produced by Phyre2 and refined by GalaxyWEB servers.
Table 1.
The refined models produced by GalaxyWEB with their scores
| Model | ProSA Z-scores | ERRAT Quality (%) | QMEAN6 score | Procheck Ramachandran plot | |||
|---|---|---|---|---|---|---|---|
| Co1 (%) | AA2 (%) | GA3(%) | DA4 (%) | ||||
| Initial | -7.12 | 54.6 | 0.717 | 87.4 | 11.4 | 0.6 | 0.6 |
| Model 1 | -7.45 | 95.7 | 0.710 | 89.7 | 9.1 | 0.6 | 0.6 |
| Model 2 | -7.41 | 94.0 | 0.706 | 90.3 | 8.6 | 0.6 | 0.6 |
| Model 3 | -7.35 | 91.9 | 0.701 | 89.1 | 9.7 | 1.1 | 0.0 |
| Model 4 | -7.25 | 98.9 | 0.732 | 89.1 | 9.7 | 1.1 | 0.0 |
| Model 4 | -7.25 | 98.9 | 0.732 | 89.1 | 9.7 | 1.1 | 0.0 |
| Model 5 | -7.23 | 93.5 | 0.699 | 89.1 | 9.7 | 0.6 | 0.0 |
Residues in the most-favored regions
residues in the additionally-allowed regions
residues in the generously-allowed regions
residues in the disallowed regions
ERRAT is a novel method that can detect incorrect regions of protein structures according to errors leading to random distributions of atoms, which can be distinguished from correct distributions (27). Fig. 3) shows the refined Model 4 with quality of 98.913%, which is greater than the initial model containing many erroneous regions and a quality of 54.595%.
Fig. 3.
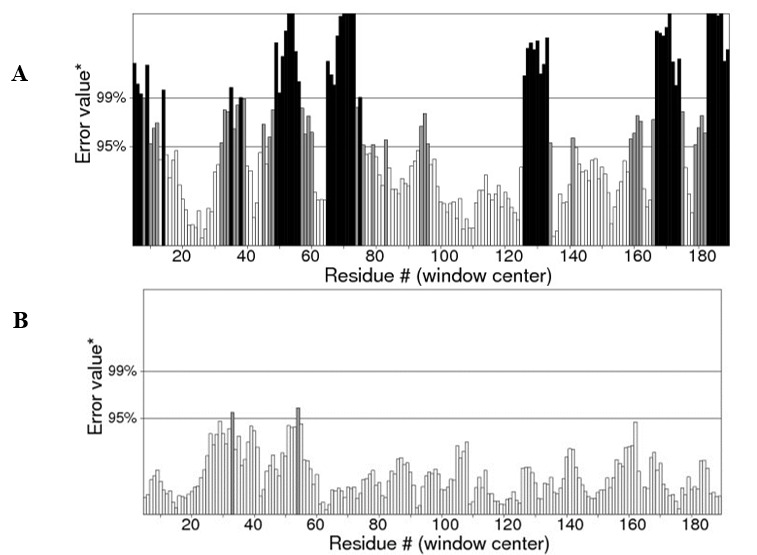
ERRAT plot shows error values for residues. The Y-axis represents the error value and the X-axis represents the amino acid residues of the protein model. An error value exceeding 99% confidence level indicates a poorly-modeled region (A) The initial model with quality of 54.595% (B) the refined Model 4 with quality of 98.913%.
In ANOLEA profile (Fig. 4), the initial model had many areas of high energy, which were greatly improved in the refined model, suggesting greater reliability. The Z-scores of all the models are similar to the normal values commonly found in native structures determined by NMR spectroscopy and X-ray crystallography (Fig. 5).
Fig. 4.

ANOLEA plots showing high energy zones as red (A) The initial model (B) The refined Model 4 with the high energy zones greatly minimized and improved.
Fig. 5.

Z-score plot of LuxI (dot) determined by ProSA. The Z-score is -7.25, within the range of experimental native structures of similar sizes.
Table 3 compares stereochemical parameters of the initial model and Model 4 where all Ramachandrans and Chi1-chi2 plots were better in Model 4 and no bad contacts. Ramachandran plot of the Model 4 is shown in Fig. 6.
Table 3.
Stereochemical qualities of the initial and forth models by Procheck
| Parameters | Initial Model | Model 4 |
|---|---|---|
| All Ramachandrans, out of 191 | 5 labeled residues | 3 labeled |
| Chi1-chi2 plots, out of 121 | 3 labeled residues | 0 labeled |
| Main-chain parameters | 6 better | 6 better |
| Side-chain parameters | 5 better | 5 better |
| Residue properties, bad contacts | 5 | 0 |
| Overall G-factor | -0.09 | 0.08 |
| Main chain bond lengths | 99.1% within limits | 97.5% within limits |
| Main chain bond angles | 89.9% within limits | 88.6% within limits |
| Planar groups | 100.0% within limits | 97.3% within limits |
Fig. 6.
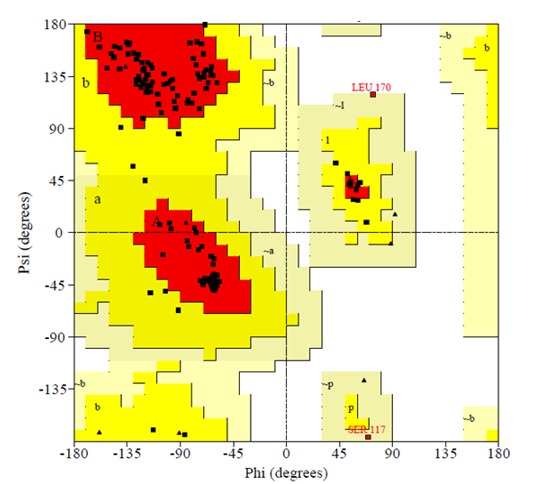
Ramachandran plot of the fourth model is determined by Procheck. The most favoured regions are marked as A, B, and L. The additional allowed regions are marked as a, b, l, and p. All non-glycine and proline residues are shown as filled black squares, whereas glycines (non-end) are shown as filled black triangles. Disallowed residues are coloured red.
Table 4.
Docking results of the two inducers against the experimental compounds in DrugBank
| Drug | DrugBank ID | Weight | Score |
|---|---|---|---|
| K2C | DB08036 | 311.34 | -98.74 |
| Approved 873 | DB03031 | 539.78 | -95.90 |
| 740 | DB07220 | 453.56 | -88.63 |
| Approved 1890 | DB04502 | 600.73 | -86.88 |
| Approved 1155 | DB03471 | 395.54 | -86.74 |
| Approved 292 | DB02089 | 503.38 | -83.62 |
| 4CP | DB07105 | 405.90 | -80.82 |
| 4HD | DB07111 | 344.49 | -79.95 |
| GSK | DB07847 | 465.95 | -75.82 |
During the last decade, quorum sensing system has been proposed as a target for antimicrobial agents and controlling the expression of virulence factors in bacteria (35). About 2700 compounds from the experimental library of DrugBank database were screened for ligands inhibitory to LuxI. The compounds with the ten highest scores are shown in Table 3.
Discussion
Many different LuxI-type proteins have been identified in Proteobacteria; these proteins are 190-230 amino acids in length and share 30-35% similarity. Ten residues are conserved in the terminal 110 amino acids with a conserved threonine at position 10 of the 110c-terminal amino acids. This threonine might be involved in stabilizing interactions with the fatty acyl biosynthetic precursors (36, 37).
The physiochemical properties of LuxI were computed using ProtParam. LuxI is comprised of 193 amino acids with a molecular weight of 22.014 kDa and a calculated pI of 5.7, allowing purification of the protein by isoelectric focusing ( 38). The extinction coefficient of LuxI at 280 nm is 23045 M-1 cm-1, assuming all cysteine pairs come from cystines, and 22920 M-1 cm-1 when all cysteines are reduced. The extinction coefficient indicates how much light a protein can absorb at a given wavelength. Estimation of this parameter is useful in spectrophotometric analysis of the protein ( 20). The instability index provides an estimate of the stability of the protein in a test tube. The instability index of this protein is 44.95, suggesting that it is slightly unstable ( 21).
In homology modeling, sequence identities greater than 40% can produce good overall quality models; however, if the target-template sequence identity is less than 40%, the predicted models will deviate significantly (39-40). Low sequence similarity and high structural divergence indicate the models may contain errors (41-42). Typical error sources are misplaced side chains, backbone distortions, alignment errors, or selecting a template of incorrect fold (43-45). Melo et al. (30) suggested that errors in the model are either in or close to regions that connect secondary structure core components and are of high energy.
In the process of protein structure prediction alternative models are generated, from which the most accurate model is selected using a scoring function (46). The scoring function relies on the principle that the native state of a protein has a minimum free energy (47). Various assessment tools have been developed (48), which aid in the assessment of problems that may arise in evaluation, such as whether the model has the correct fold, the overall geometric accuracy of the protein, and the geometric accuracy of individual protein regions (40).
The QMEAN6 scoring function consists of a linear combination of six descriptors. Two distance-dependent interaction potentials of mean force based on Cβ atoms and all atom types are used to assess long range interactions; a torsion angle potential over three consecutive amino acids is applied to analyze the local backbone geometry of the model, a solvation potential to estimate the burial status of the residues, the agreement of the predicted and the calculated secondary structures, and solvent accessibility prediction. The raw score is between 0-1 (31). The QMEAN6 score of Model 4 was the greatest of the models tested. Because QMEAN6 can provide both global and local (per residue) estimates of model quality (46), this model can be regarded as the best model obtained. Table 2 shows the contribution of each descriptor to the overall score. The pseudo-energies of the contributing terms are given together with their Z-scores with respect to scores obtained for high-resolution experimental structures of similar sizes solved by X-ray crystallography.
Table 2.
The QMEAN6and component scores and their Z-scores of the initial and fourth models with respect to experimental structures of similar sizes
| Scoring function term | Initial Model | Model 4 | ||
|---|---|---|---|---|
| Raw score | Z-score | Z-score | Z-score | |
| Cβ interaction energy | -69.61 | -1.38 | -72.97 | -1.27 |
| All-atom pairwise energy | -4084.92 | -1.55 | -4620.88 | -1.22 |
| Solvation energy | -21.78 | 0.28 | -21.58 | 0.25 |
| Torsion angle energy | -36.60 | -1.22 | -39.50 | -1.02 |
| Secondary structure agreement | 83.9% | 0.13 | 85.0% | 0.29 |
| Solvent accessibility agreement | 80.3% | 0.19 | 80.3% | 0.19 |
| QMEAN6 score | 0.717 | -0.53 | 0.732 | -0.37 |
In the ProSA web tool the Z-score of a protein is defined as the energy separation between the native fold and the average of an ensemble of the misfolds in the units of standard deviation of the ensemble (49). A Z-score outside a range characteristic for native proteins of similar sizes indicates erroneous structure.
This score is displayed in a plot that contains the Z-scores of all experimentally-determined proteins (28).
A Ramachandran plot is an x-y plot of phi/psi dihedral angles between N-Cα and Cα-C planar peptide bonds in a protein’s backbone. According to Ramachandran plots analyzed for over 118 structures at 2.0 Å resolution, a good quality model can obtained when greater than 90% of residues fall into the most favored region (32).
The 3D structure of LuxI could be used to model inducers with homologous sequences in other microorganisms. This study also shows that the acteyltransferase site of action may be exploited in ligand design to inhibit quorum sensing in pathogenic organisms with homologous systems.
Acknowledgements
The author wishes to thank Dr. Raywa Zaghlool said at Technical institute of Mosul for advice and notes.
References
- 1.Nealson KH, Hastings JW. Bacterial bioluminescence: its control and ecological significance. Micrbiol Rev. 1979;43:469–518. doi: 10.1128/mr.43.4.496-518.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ruby EG. Lessons from a cooperative bacterial-animal association: the Vibrio fischeri-Euprymna scolopes light organ symbioses. . Annu Rev Microbiol . 1996;50:591–624. doi: 10.1146/annurev.micro.50.1.591. [DOI] [PubMed] [Google Scholar]
- 3.Fuqua WC, Winans SC, Greenberg EP. Quorum sensing in bacteria: the LuxR-LuxI family of cell density-responsive transcriptional regulators. J Bacteriol . 1994;176:269–275. doi: 10.1128/jb.176.2.269-275.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Henke JM, Bassler BL. Bacterial social engagements. Trends cell Biol . 2004;14:648–656. doi: 10.1016/j.tcb.2004.09.012. [DOI] [PubMed] [Google Scholar]
- 5.Waters CM, Bassler BL. Quorum sensing: cell-to-cell communication in bacteria. Annu Rev Cell Dev Biol. 2005;21:319–346. doi: 10.1146/annurev.cellbio.21.012704.131001. [DOI] [PubMed] [Google Scholar]
- 6.Bassler BL, Losick R. Bacterial speaking. Cell. 2006;125:237–246. doi: 10.1016/j.cell.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 7.Engebrecht J, Silverman M. Nucleotide sequence of the regulatory locus controlling expression of bacterial genes for bioluminescence. . Nucleic acids Res . 1987;15:10455–10467. doi: 10.1093/nar/15.24.10455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kaplan HB, Greenberg EP. Diffusion of autoinducer is involved in regulation of the Vibrio fischeri luminescence system. J Bacteriol. 1985;163:1210–1214. doi: 10.1128/jb.163.3.1210-1214.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moré MI, Finger LD, Stryker JL, Fuqua C, Eberhard A, Winans SC. Enzymatic synthesis of a quorum-sensing autoinducer through use of defined substrates. Science . 1996;272:1655–1658. doi: 10.1126/science.272.5268.1655. [DOI] [PubMed] [Google Scholar]
- 10.Mihăṣan M. Basic protein structure prediction for the biologist: a review. Arch Biol Sci Belgrade . 2010;62(4):857–871. [Google Scholar]
- 11.Dutta S, Burkhardt K, Young J, Swamiathan J, Mastsura T, Henrick K, Nakamura H, Berman M. Data deposition and annotation at the worldwide protein data bank. Mol Biotechnol . 2009;42(1):1–13. doi: 10.1007/s12033-008-9127-7. [DOI] [PubMed] [Google Scholar]
- 12.Benson DA, Karsch-Mizrachi I, Lipman J, Ostell J, Sayers W. Genebank. Nucleic Acids Res . 2010;38:D46–51. doi: 10.1093/nar/gkp1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Seniya C, Mishra H, Chaturvedi B. Structure prediction and analysis of hydrogenase enzyme of Rhodobacter capsulatus. Inter J Adv Tech Eng Res . 2012;2(6):24–29. [Google Scholar]
- 14.Sheng Y, Sali A, Herzog H, Lahnstein J, Krilis SA. Site-directed mutagenesis of recombinant human beta 2-glycoprotein I identifies a cluster of lysine residues that are critical for phospholipid binding and anti-cardiolipin antibody activity. . J Immunol . 1996;157(8):3744–3751. [PubMed] [Google Scholar]
- 15.Ring CS, Sun E, McKerrow JH, Lee GK, Rosenthal PJ, Kuntz ID, Cohen FE. Structure-based inhibitor design by using protein models for the development of antiparasitic agents. . Proc Natl Acad USA . 1993;90:3583–3587. doi: 10.1073/pnas.90.8.3583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sali A, Matsumoto R, McNeil HP, Karplus M, Stevens RL. Three-dimensional models of four mouse mast cell chymases. Identification of proteoglycan binding regions and protease-specific antigenic epitopes. J Biol Chem . 1993;268:9023–9034. [PubMed] [Google Scholar]
- 17.Vernal J, Fiser A, Sali A, Muller M, Jose CJ, Nowicki C. Probing the specificity of a trypanosomal aromatic alpha-hydroxy acid dehydrogenase by site-directed mutagenesis. Biochem Biophys Res Commun . 2002;293:633–639. doi: 10.1016/S0006-291X(02)00270-X. [DOI] [PubMed] [Google Scholar]
- 18.Gary KM, Greenberg EP. Sequencing and analysis of luxI and luxR, the luminescence regulatory genes from the squid light organ symbiont Vibrio fischeri ES114. Mol Mar Biol Biotechnol . 1992;1:414–419. [Google Scholar]
- 19.Gasteiger E, Hoogland C, Gattiker A, Duvaud S, Wilkins MR, Appel RD, Bairoch A. Protein Identification and Analysis Tools on the ExPASy Server. In: Walker John M., editor. The Proteomics Protocols Handbook. Humana Press; 2005. pp. pp.571–607. [Google Scholar]
- 20.Gill SC, von Hippel PH. Calculation of protein extinction coefficients from amino acid sequence data. Anal Biochem . 1989;182:319–326. doi: 10.1016/0003-2697(89)90602-7. [DOI] [PubMed] [Google Scholar]
- 21.Guruprasad K, Reddy BVB, Pandit MW. Correlation between stability of a protein and its dipeptide composition: a novel approach for predicting in vivo stability of a protein from its primary sequence. Protein Eng . 1990;4:155–161. doi: 10.1093/protein/4.2.155. [DOI] [PubMed] [Google Scholar]
- 22.Cheng J, Randall A, Sweredoski M, Baldi P. SCRATCH: a protein structure and structural feature prediction server. Nucleic Acids Res . 2005;33:W72–76. doi: 10.1093/nar/gki396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kabsch W, Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers . 1983;22(12):2577–637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- 24.Bateman A, Birney E, Cerrruti L, Durbin R, Etwiller L, Eddy SR, Griffiths-Jones S, Howe KL, Marshal M, Sonnhammer ELL. The Pfam protein families’ database. Nucleic Acids Res . 2000;30(1):276–280. doi: 10.1093/nar/30.1.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kelley LA, Sternberg MJ. Protein structure prediction on the Web: a case study using the Phyre server. . Nat Protocols . 2009;4(3):363–371. doi: 10.1038/nprot.2009.2. [DOI] [PubMed] [Google Scholar]
- 26.Ko J, Park H, Heo L, Seok C. GalaxyWEB server for protein structure prediction and refinement. . Nucleic Acids Res . 2012;40:w294–297. doi: 10.1093/nar/gks493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Colovos C, Yeates TO. Verification of protein structures: Patterns of non-bonded atomic interactions. Protein Sci . 1993;2:1511–1519. doi: 10.1002/pro.5560020916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wiederstein M, Sippl MJ. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. . Nucleic Acids Res . 2007;35:W407–410. doi: 10.1093/nar/gkm290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Arnold K, Bordoli L, Kopp J, Schwede T. The SWISS-MODEL Workspace: A web-based environment for protein structure homology modeling. . Bioinformatics. 2006;22:195–201. doi: 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]
- 30.Melo F, Devos D, Depiereux E, Feytmans E. ANOLEA: a www server to assess protein structures. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1997;5:187–90. [PubMed] [Google Scholar]
- 31.Benkert P, Biasini M, Schwede T. Toward the estimation of the absolute quality of individual protein structure models. . Bioinformatics. 2011;27(3):343–350. doi: 10.1093/bioinformatics/btq662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Laskowski RA, MacArthur MW, Moss D, Thornton J M. PROCHECK: a program to check the stereochemical quality of protein structures. J App Cryst. 1993;26(2):283–291. [Google Scholar]
- 33.Castrignano T, De Meo PD, Cozzetto D, Talamo IG, Tramontano A. The PMDB Protein Model Database. Nucleic Acids Res. 2006;34(1):D306–309. doi: 10.1093/nar/gkj105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wishart DS, Knox C, Guo AC, Cheng D, Shrivastava S, Tzur D, Gautam B, Hassanali M. Drug Bank: a knowledgebase for drugs, drug actions and drug targets. Nucleic Acids Res . 2008;36:901–906. doi: 10.1093/nar/gkm958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tan S Y-Y, Chua S-L, Chen Y, Rice SA, Kjelleberg S, Nielsen TE, Yang L, Givskov M. Identification of five structurally unrelated quorum-sensing inhibitors of Pseudomonas aeruginosa from a natural-derivative database. Antimicrob Agents Chemother . 2013;57(11):5629–5641. doi: 10.1128/AAC.00955-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fuqua C, Greenberg EP. Listening in on bacteria: Acylhomoserine lactone signaling. Mol Cell Biol . 2002;3:685–695. doi: 10.1038/nrm907. [DOI] [PubMed] [Google Scholar]
- 37.Watson WT, Minogue TD, Val DL, von Bodman SB, Churchill MEA. Structural basis and specificity of acyl-homoserine lactone signal production in bacterial quorum sensing. Mol Cell . 2002;9:685–694. doi: 10.1016/s1097-2765(02)00480-x. [DOI] [PubMed] [Google Scholar]
- 38.Prabhavathi M, Ashokkumar K, Geetha N, Saradha Devi KM. Homology modeling and structure prediction of thioredoxin (TRX) protein in wheat (Triticum aestivum L.) Inter J Biosci. 2011;1(1):20–32. [Google Scholar]
- 39.Sanchez R, Sali A. Large-scale protein structure modeling of the Saccharomyces cerevisiae genome . Proc Natl Acad Sci USA . 1998;95:13597–13602. doi: 10.1073/pnas.95.23.13597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Melo F, Sali A. Fold assessment for comparative protein structure modeling. Protein Sci . 2007;16:2412–2426. doi: 10.1110/ps.072895107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chothia C, Lesk AM. The relation between the divergence of sequence and structure in proteins. EMBO J. 1986;5:823–826. doi: 10.1002/j.1460-2075.1986.tb04288.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rost B. Twilight zone of protein sequence alignments. Protein Eng . 1999;12:85–94. doi: 10.1093/protein/12.2.85. [DOI] [PubMed] [Google Scholar]
- 43.Baker D, Sali A. Protein structure prediction and structural genomics. Science. 2001;294:93–96. doi: 10.1126/science.1065659. [DOI] [PubMed] [Google Scholar]
- 44.Bordoli L, Kiefer F, Arnold K, Benkert P, Battey J, Schwede T. Protein structure homology modeling using SWISS-MODEL workspace. Nature Protocols . 2009;4(1):13–371. doi: 10.1038/nprot.2008.197. [DOI] [PubMed] [Google Scholar]
- 45.Koh YY, Eyrich A, Marti-Renom A, Przybylski D, Madhusudhan S, Eswar N, Grana O, Pazos F, Valencia A, Sali A, Rost B. EVA: Evaluation of protein structure prediction servers. Nucleic Acids Res . 2003;31(13):3311–3315. doi: 10.1093/nar/gkg619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Benkert P, Kunzli M, Schwede T. QMEAN server for protein model quality estimation. Nucleic Acids Res. 2009;37:W510–514. doi: 10.1093/nar/gkp322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lazaridis T, Karplus M. Effective energy functions for protein structure prediction. Curr Opin Struct Biol. 2000;10:139–145. doi: 10.1016/s0959-440x(00)00063-4. [DOI] [PubMed] [Google Scholar]
- 48.Marti-Renom MA, Stuart A, Fiser A, Sanchez R, Melo F, Sali A. Comparative protein structure modeling of genes and genomes. Annu Rev Biophys Biomol Struct. 2000;29:291–325. doi: 10.1146/annurev.biophys.29.1.291. [DOI] [PubMed] [Google Scholar]
- 49.Zhang L, Skolinck J. What should the Z-scores of native protein structures be? Protein Sci . 1998;7:1201–1207. doi: 10.1002/pro.5560070515. [DOI] [PMC free article] [PubMed] [Google Scholar]